 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNH⋯O and OH⋯O interactions of glycine derivatives with squaric acid†
Michalina
Anioła
,
Zofia
Dega-Szafran
*,
Andrzej
Katrusiak
and
Mirosław
Szafran
Faculty of Chemistry, Adam Mickiewicz University, Umultowska 89b, 61-614 Poznań, Poland. E-mail: degasz@amu.edu.pl; Fax: +48-61-8291555; Tel: +48-61-8291216
First published on 28th May 2014
Abstract
Four new hydrogen-bonded complexes of a simple amino acid glycine (GLY) and its methyl derivatives – sarcosine (N-methylglycine, SAR), dimethylglycine (DMG) and betaine (N,N,N-trimethylglycinium, BET) with squaric acid (3,4-dihydroxy-3-cyclobuten-1,2-dione, H2SQ) are synthesized and characterized by X-ray diffraction, FTIR and NMR spectroscopy. The complexes differ in stoichiometry, interaction with H2SQ and the hydrogen-bonding system. Methylation of glycine gradually reverses the dissociation of squaric acid in co-crystals. This process is correlated with the number of N–H⋯O bonds to the squaric acid oxygens and the N–H donor capability of glycine derivatives. The effect of the presence of methyl groups on the proton and carbon-13 chemical shifts is studied. The DFT calculations are performed for one unit of complexes and stable zwitterionic structures of SAR, DMG and BET in complex with H2SQ, except the GLY unit, are suggested, which appears as an uncharged (neutral) form. The experimental and computed vibrational spectra of complexes studied are reported.
1. Introduction
Glycine, GLY, is the simplest protein structure unit and the smallest achiral amino acid that can act as a neuroinhibitor in the mammalian center nervous system.1 Sarcosine, SAR, is the N-methyl derivative of glycine and belongs to a group of biologically important compounds containing a carbon atom participating in transmethylation reactions. In nature, it occurs in starfish, sea urchins and in the antibiotic actinomycin.2N,N-Dimethylglycine, DMG, is a sweet-tasting substance classified as a nutrient. It has been presumed to be a key ingredient of vitamin B-15 or pangamic acid. In the living cell, DMG is present as a product of the metabolism pathway of choline and methionine. DMG has been used in the treatment of mental disturbance conditions, like autism and epileptic seizures. DMG is a popular ingredient of supplementary diet tablets and energetic drinks for sportsmen.2,3 Betaine, BET, trimethylglycinium, is a zwitterion containing an anionic carboxylate group and a positively charged quaternary ammonium group. Betaine is a biological oxidation product of choline and it is widely distributed in plants, algae and animal tissues, where it serves as a methyl transfer agent important in amino acid syntheses.4 GLY,5 SAR,6 DMG7 and BET8 form crystalline adducts mainly with inorganic acids as well as with some organic acids.7b,9 Some of them display interesting physical properties exhibiting phase transitions leading to ferroelectric, antiferroelectric and ferroelastic phases as well as phases with commensurate and incommensurate superstructures.10 The N-alkylation of amino acids is known to increase the population of neutral tautomers in solution.11Since amino acids and their complexes are of considerable chemical and biological interest we have undertaken a systematic investigation of several squaric acid complexes with the simplest amino acid glycine and its N-methyl derivatives, sarcosine, dimethylglycine and betaine (Fig. 1). Squaric acid, H2SQ, is a reagent for chemical synthesis, used for instance to make photosensitive squarate dyes,12 inhibitors of protein tyrosine phosphatases and squaraine-modified DNA.13 The structure of squaric acid is not a perfect square.14 The high acidity of H2SQ with pK1 = 1.5 for the first proton and pK2 = 3.4 for the second one15 is attributable to resonance stabilization of the anion.16 The conjugate base of squaric acid is the hydrogen squarate anion, HSQ−, whereas the conjugate base of the hydrogen squarate anion is the divalent squarate anion, SQ2− (Fig. 1).17
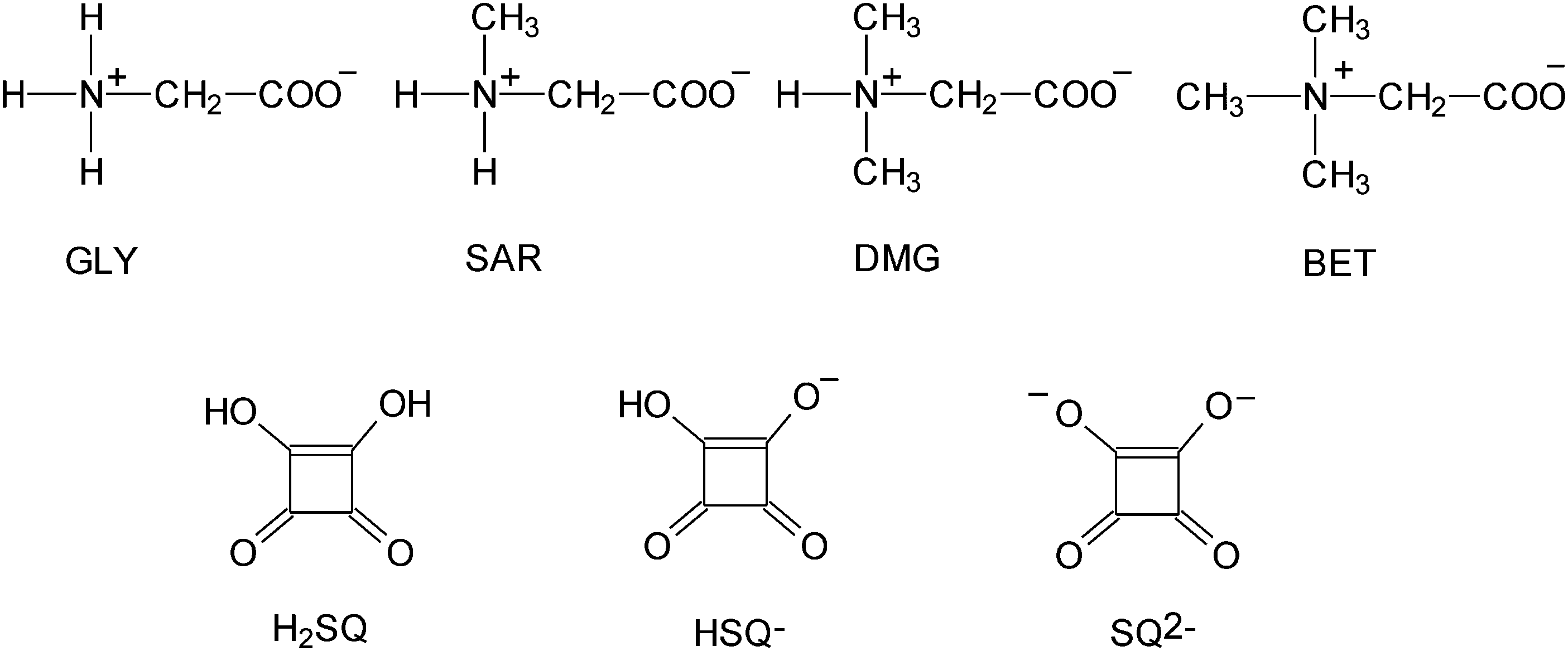 | ||
| Fig. 1 Molecular structures of glycine (GLY), sarcosine (SAR), dimethylglycine (DMG), betaine (BET), squaric acid (H2SQ), hydrogen squarate anion (HSQ−) and squarate dianion (SQ2−). | ||
The proton transfer belongs to the most frequent transformations in Nature, however this process is still not well understood. Squaric acid and amino acids are ideal for investigating the proton transfer. Some earlier papers of the groups of Kolev et al. concern complexes of amino acids and their derivatives with H2SQ.18 In the present work we report on the synthesis of four new complexes of H2SQ with glycine, GLY, and its methyl derivatives, sarcosine (N-methylglycine, SAR), dimethylglycine (DMG), and betaine (trimethylglycinium, BET). The main aim of this work is investigation of the interaction between the zwitterionic compounds and the proton-donor. These complexes differ in their X-ray data, FTIR and NMR spectra. The differences are caused by the presence of different number of acidic protons and methyl groups attached to the nitrogen atoms. From the point of view of materials science, the information on the formation of crystal structures of the complexes of glycine and its methyl derivatives with squaric acid is essential for understanding the role of hydrogen bonds and other intermolecular forces in the association of ions in the crystalline state. Quantum chemical calculations have been performed for one unit of all complexes to study their structures in the isolated molecules and their IR frequencies.
2. Results and discussion
GLY, SAR, DMG and BET form stable, crystalline complexes with H2SQ. Their structures are studied by the single-crystal diffraction method, while the molecular structures of the isolated unit of complexes, without any interactions with the neighboring molecules, have been optimized by the B3LYP/6-311G(d,p) approach. The experimental and calculated bond lengths, bond and torsion angles are given in Tables S1 and S2, respectively (ESI†), while the selected ones are listed in Table 1. The hydrogen bond geometries, calculated energies (HF) and dipole moments are listed in Table 2.| Parameters | 1 | 1a | 2 | 2a | 3 | 3a | 4 | 4a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aa | Bb | Aa | Bb | Aa | Bb | Aa | Bb | Aa | Bb | Aa | Bb | |||
| a Part A - the positions denoted by the unprimed atomic labels. b Part B - the positions denoted by the primed atomic labels. | ||||||||||||||
| Bond lengths | ||||||||||||||
| O(1)–C(1) | 1.242(2) | 1.215 | 1.250(1) | 1.208 | 1.236(2) | 1.235(2) | 1.219 | 1.212 | 1.207(4) | 1.216(4) | 1.210 | 1.217 | ||
| O(2)–C(2) | 1.251(2) | 1.201 | 1.215(1) | 1.219 | 1.218(2) | 1.223(2) | 1.201 | 1.220 | 1.248(4) | 1.240(4) | 1.227 | 1.227 | ||
| O(3)–C(3) | 1.265(2) | 1.305 | 1.247(1) | 1.296 | 1.287(2) | 1.258(2) | 1.309 | 1.302 | 1.314(4) | 1.304(4) | 1.303 | 1.302 | ||
| O(4)–C(4) | 1.249(2) | 1.322 | 1.308(1) | 1.323 | 1.300(2) | 1.318(2) | 1.317 | 1.305 | 1.240(4) | 1.250(4) | 1.285 | 1.285 | ||
| O(5)–C(5) | 1.295(2) | 1.312(2) | 1.215 | 1.217 | 1.314(1) | 1.247 | 1.266(2) | 1.305(2) | 1.229 | 1.242 | 1.298(4) | 1.305(4) | 1.262 | 1.262 |
| O(6)–C(5) | 1.211(2) | 1.197(2) | 1.320 | 1.324 | 1.201(1) | 1.251 | 1.245(2) | 1.213(2) | 1.267 | 1.255 | 1.207(4) | 1.185(4) | 1.233 | 1.233 |
| Bond angles | ||||||||||||||
| C(1)–C(2)–C(3) | 89.9(1) | 86.67 | 87.7(1) | 88.5(1) | 87.7(1) | 88.5(1) | 86.56 | 87.91 | 88.3(3) | 88.9(3) | 88.33 | 88.37 | ||
| C(2)–C(3)–C(4) | 91.2(1) | 92.86 | 89.4(1) | 89.7(1) | 91.4(1) | 89.6(1) | 92.50 | 92.10 | 93.3(3) | 93.2(3) | 92.74 | 92.73 | ||
| C(3)–C(4)–C(1) | 89.7(1) | 93.02 | 93.4(1) | 94.2(1) | 91.9(1) | 94.2(1) | 92.74 | 92.97 | 89.9(3) | 89.1(3) | 91.39 | 91.38 | ||
| C(4)–C(1)–C(2) | 89.3(1) | 87.45 | 89.4(1) | 87.6(1) | 89.0(1) | 87.6(1) | 88.16 | 87.01 | 87.9(3) | 88.7(3) | 87.54 | 87.55 | ||
| Torsion angles | ||||||||||||||
| C(1)–C(2)–C(3)–C(4) | −0.4(1) | 0.16 | −1.3(1) | −0.15 | 1.0(1) | 0.8(1) | 0.42 | −0.31 | −0.1(3) | −1.8(3) | 0.19 | 0.19 | ||
| C(2)–C(3)–C(4)–C(1) | 0.4(1) | −0.17 | 1.3(1) | 0.16 | −1.1(1) | −0.9(1) | −0.43 | 0.32 | 0.1(3) | 1.8(3) | −0.19 | 0.19 | ||
| C(3)–C(4)–C(1)–C(2) | −0.4(1) | 0.16 | −1.3(1) | −0.15 | 1.0(1) | 0.9(1) | 0.42 | −0.30 | −0.1(3) | −1.7(3) | 0.19 | −0.19 | ||
| C(4)–C(1)–C(2)–C(3) | 0.4(1) | −0.15 | 1.3(1) | 0.14 | −1.0(1) | −0.8(1) | −0.39 | 0.28 | 0.1(3) | 1.7(3) | −0.18 | 0.18 | ||
| O(5)–C(5)–C(6)–N(1) | 169.1(1) | 170.4(1) | −176.42 | −160.34 | 178.0(1) | 143.51 | 161.8(1) | −166.4(1) | −176.36 | −174.04 | −168.3(3) | 167.2(3) | 170.91 | −170.90 |
| O(6)–C(5)–C(6)–N(1) | −11.8(2) | 9.2(2) | 4.05 | 20.24 | −0.5(2) | 34.03 | −21.4(2) | 15.4(2) | 4.18 | 4.54 | −10.7(6) | −13.9(6) | −8.03 | 8.04 |
| D–H⋯A | D–H | H⋯D | D⋯A | ∠DHA | ||
|---|---|---|---|---|---|---|
| X-Ray | Symmetry code | |||||
| 1 | O(5)–H(3)⋯O(3) | 1.00(2) | 1.46(2) | 2.444(2) | 167(2) | — |
| O(5′)–H(4)⋯O(4) | 0.80(2) | 1.77(2) | 2.571(2) | 175(3) | — | |
| N(1′)–H(2′N)⋯O(2) | 0.93(2) | 1.90(2) | 2.815(2) | 139(1) | 1.5 − x, 0.5 + y, 1.5 − z | |
| N(1′)–H(3′N)⋯O(5) | 0.85(2) | 2.11(2) | 2.805(2) | 133(1) | 2 − x, y, 1.5 − z | |
| N(1)–H(2N)⋯O(1) | 0.87(2) | 2.08(2) | 2.916(2) | 157(2) | 2 − x, 2 - y, 2 − z | |
| N(1)–H(2N)⋯O(6′) | 0.89(2) | 2.72(2) | 2.828(2) | 88(2) | 2 − x, 2 − y, 2 − z | |
| 2 | O(5)–H(3)⋯O(3) | 0.92(2) | 1.70(2) | 2.612(1) | 171(2) | — |
| O(1W)–H(1W)⋯O(3) | 0.85(2) | 2.00(1) | 2.849(1) | 173(5) | −x, y, 1.5 − z | |
| O(4)–H(4)⋯O(1) | 0.96(2) | 1.60(2) | 2.528(1) | 161(2) | 0.5 + x, 0.5 − y, 2 − z | |
| N(1)–H(2N)⋯O(1) | 0.91(2) | 2.09(2) | 2.924(1) | 152(1) | −0.5 + x, 0.5 − y, −0.5 + z | |
| N(1)–H(1N)⋯O(1W) | 0.97(1) | 1.90(2) | 2.867(1) | 173(2) | −0.5 + x, 0.5 + y, z | |
| 3 | O(3)–H(3)⋯O(5) | 0.82 | 1.65 | 2.444(2) | 163 | — |
| O(4)–H(4)⋯O(6) | 0.88(2) | 1.66(2) | 2.527(2) | 170(2) | — | |
| O(4′)–H(4′)⋯O(1) | 0.94(2) | 1.68(2) | 2.624(2) | 176(2) | — | |
| O(5′)–H(3′)⋯O(3′) | 0.93(2) | 1.59(2) | 2.514(2) | 174(2) | — | |
| N(1)–H(1)⋯O(1′) | 0.90(2) | 2.16(2) | 2.870(2) | 136(1) | 1 − x, 1 − y, 1 − z | |
| N(1′)–H(1′)⋯O(2) | 0.92(2) | 1.94(2) | 2.803(2) | 157(2) | −x, 1 − y, −z | |
| N(1′)–H(1′)⋯O(6′) | 0.92(2) | 2.55(2) | 3.035(2) | 114(1) | −x, −0.5 + y, −0.5 − z | |
| 4 | O(5)–H(4)⋯O(4) | 0.82 | 1.76 | 2.559(4) | 164 | — |
| O(3)–H(3)⋯O(2′) | 0.97(4) | 1.55(4) | 2.503(4) | 167(4) | — | |
| O(3′)–H(3′)⋯O(2) | 0.90(4) | 1.61(4) | 2.501(4) | 166(3) | — | |
| O(5′)–H(4′)⋯O(4′) | 0.82 | 1.73 | 2.546(4) | 178 | — | |
| B3LYP/6-311G(d,p) | Energy/μ | |||||
| 1a | O(3)–H(3)⋯O(5) | 0.997 | 1.648 | 2.644 | 176.57 | −1023.673350/8.574 |
| O(4)–H(4)⋯O(5′) | 0.995 | 1.658 | 2.648 | 172.63 | ||
| O(6)–H(1N)⋯N(1) | 0.990 | 1.862 | 2.588 | 127.64 | ||
| O(6′)–H(1N′)⋯N(1′) | 0.991 | 1.865 | 2.574 | 125.89 | ||
| 2a | O(3)–H(3)⋯O(5) | 1.023 | 1.518 | 2.537 | 173.67 | −854.906921/7.001 |
| O(W)–H(1W)⋯O(6) | 0.973 | 1.868 | 2.815 | 163.74 | ||
| N(1)–H(2N)⋯O(2) | 1.030 | 1.783 | 2.722 | 149.71 | ||
| 3a | O(3)–H(3)⋯O(5) | 0.987 | 1.693 | 2.671 | 170.46 | −1635.534053/25.829 |
| O(4)–H(4)⋯O(6) | 0.993 | 1.673 | 2.658 | 171.35 | ||
| O(4′)–H(4′)⋯O(1) | 0.999 | 1.646 | 2.643 | 175.80 | ||
| O(3′)–H(3′)⋯O(5′) | 1.013 | 1.561 | 2.570 | 172.69 | ||
| N(1)–H(N1)⋯O(6) | 1.064 | 1.669 | 2.495 | 130.49 | ||
| N(1′)–H(N1′)⋯O(6′) | 1.094 | 1.556 | 2.460 | 135.60 | ||
| 4a | O(4)–H(4)⋯O(5) | 1.052 | 1.427 | 2.473 | 171.72 | −1714.166126/0.003 |
| O(3)–H(3)⋯O(2′) | 1.000 | 1.600 | 2.591 | 169.85 | ||
| O(3′)–H(3′)⋯O(2) | 1.000 | 1.601 | 2.591 | 169.87 | ||
| O(4′)–H(4′)⋯O(5′) | 1.052 | 1.427 | 2.473 | 171.71 | ||
2.1. Crystal (1) and optimized (1a) structures of the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 complex of glycine with squaric acid
:1 complex of glycine with squaric acid
Glycine, GLY, is a non-essential, neutral amino acid without a center of chirality.2,19 In an aqueous solution of pH ∼ 7 GLY exists as a zwitterion with the positively charged amino group and the negative carboxylate group. Binding of proton to glycine is shown in Scheme 1.20 The K1 and K2 constants reflect the intrinsic properties of the amino and carboxyl groups and their interactions on protonation (pK(COOH) = 2.35 and pK(NH3+) = 9.78).
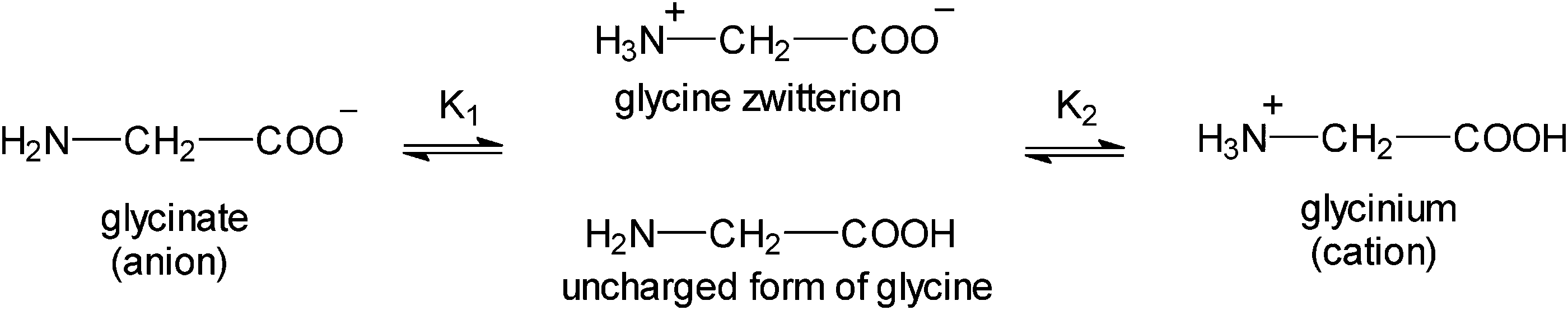 | ||
| Scheme 1 Binding of protons to glycine.20 | ||
GLY with H2SQ forms a crystalline adduct, 1, at a ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Both OH groups of H2SQ are engaged in the short, asymmetric and non-equivalent hydrogen bonds, O(5)–H(3)⋯O(3) and O(5′)–H(4)⋯O(4) of 2.444(2) and 2.571(2) Å, respectively. Both O–H protons from H2SQ are transferred to GLY, generating a squarate dianion, SQ2−, and two glycinium cations (Fig. 2a, Table 2). Unlike in the crystal of GLY,21 the glycine units in the complex exist in the cationic forms. A similar arrangement of the base and SQ2− is observed in the known complexes of H2SQ with bases.17h,22 However, the optimized structure of the 2:1 complex, 1a, is more symmetric than the crystal one, and H2SQ interacts with two uncharged (neutral) forms of GLY units, with the O(3)–H(3)⋯O(5) and O(4)–H(4)⋯O(5′) lengths of 2.644 and 2.648 Å, respectively (Fig. 2b, Table 2). The COOH groups in GLY units in 1a have trans conformations. In 1a there are the O(6)–H(N1)⋯N(1) and O(6′)–H(N1′)⋯N(1′) intramolecular contacts between the carboxylic protons and the nitrogen atoms of GLY moieties, of 2.588 and 2.574 Å, respectively (Table 2), while in the crystal of 1 the N–H atoms form several short N–H⋯O contacts with the squarate dianions of the neighboring molecules. Aggregates of bonded molecules are shown in Fig. 3. The structure of the squarate dianion in 1 is almost square and planar. The C–C–C bond angles vary from 89.3(1)° to 91.2(1)°, whereas the C–C–C–C torsion angles are equal ±0.4(1)° (Table 1).
:1. Both OH groups of H2SQ are engaged in the short, asymmetric and non-equivalent hydrogen bonds, O(5)–H(3)⋯O(3) and O(5′)–H(4)⋯O(4) of 2.444(2) and 2.571(2) Å, respectively. Both O–H protons from H2SQ are transferred to GLY, generating a squarate dianion, SQ2−, and two glycinium cations (Fig. 2a, Table 2). Unlike in the crystal of GLY,21 the glycine units in the complex exist in the cationic forms. A similar arrangement of the base and SQ2− is observed in the known complexes of H2SQ with bases.17h,22 However, the optimized structure of the 2:1 complex, 1a, is more symmetric than the crystal one, and H2SQ interacts with two uncharged (neutral) forms of GLY units, with the O(3)–H(3)⋯O(5) and O(4)–H(4)⋯O(5′) lengths of 2.644 and 2.648 Å, respectively (Fig. 2b, Table 2). The COOH groups in GLY units in 1a have trans conformations. In 1a there are the O(6)–H(N1)⋯N(1) and O(6′)–H(N1′)⋯N(1′) intramolecular contacts between the carboxylic protons and the nitrogen atoms of GLY moieties, of 2.588 and 2.574 Å, respectively (Table 2), while in the crystal of 1 the N–H atoms form several short N–H⋯O contacts with the squarate dianions of the neighboring molecules. Aggregates of bonded molecules are shown in Fig. 3. The structure of the squarate dianion in 1 is almost square and planar. The C–C–C bond angles vary from 89.3(1)° to 91.2(1)°, whereas the C–C–C–C torsion angles are equal ±0.4(1)° (Table 1).
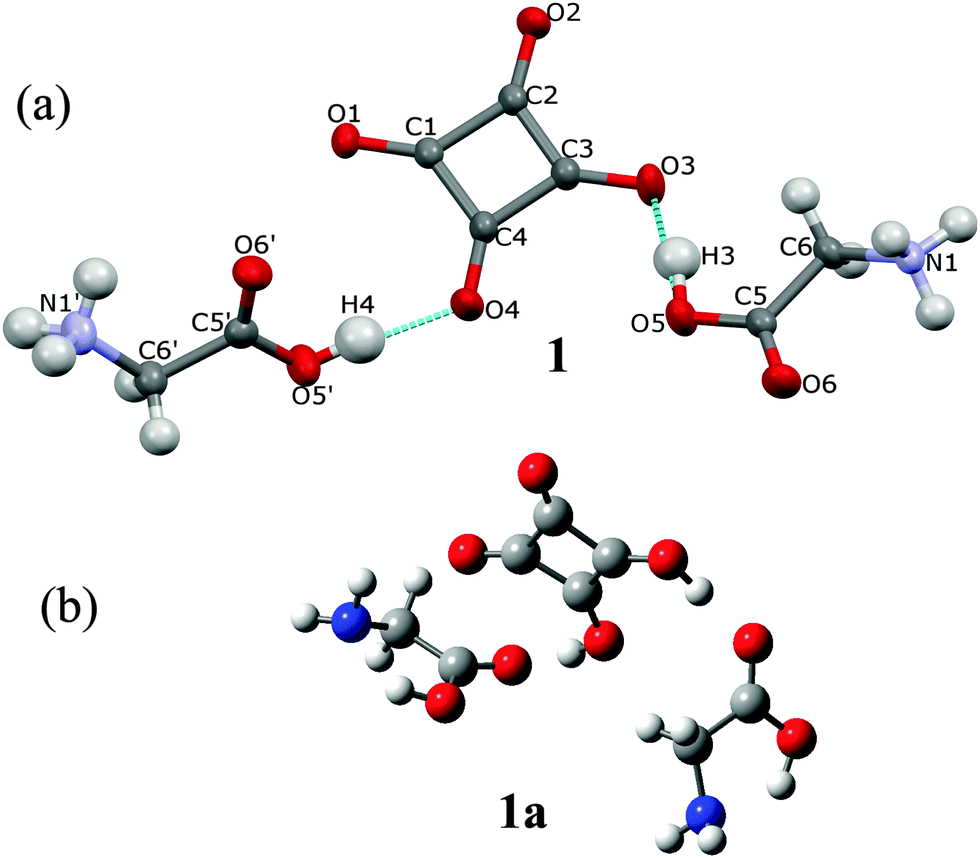 | ||
| Fig. 2 (a) Two glycinium cations and a squarate dianion in the crystal structure (1) and the atomic labeling scheme. Thermal parameters are shown at the 50% probability level and H-bonds are indicated by the dotted lines; (b) the optimized structure of 1a, determined by the B3LYP/6-311G(d,p) approach, showing two uncharged glycine molecules and squaric acid. | ||
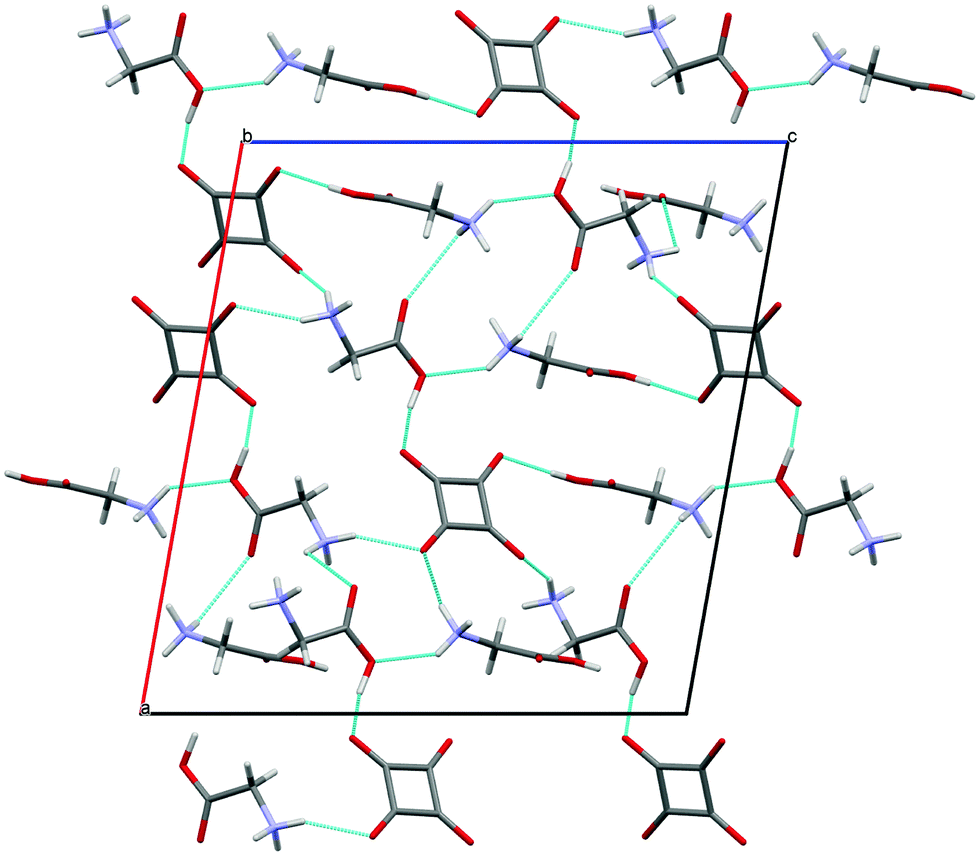 | ||
| Fig. 3 Structure of crystal 1 viewed down the [010] direction. The hydrogen bonds are indicated by the blue dotted lines. | ||
2.2. Crystal (2) and optimized (2a) structures of the complex of sarcosine with squaric acid and water
Sarcosine, also known as N-methylglycine, is an intermediate and by-product in glycine synthesis and degradation.2SAR forms a hydrated complex with H2SQ at a ratio SAR:H2SQ:0.5H2O (2). One proton from squaric acid is transferred to sarcosine and the hydrogen squarate anion, HSQ−, interacts with water (Fig. 4a). The protonation at O(5) is reflected in the C(5)–O(5) bond length of 1.314(1) Å, compared with the C(5)–O(6) bond of 1.201(1) Å. These bond lengths in pure sarcosine are 1.271(2) and 1.239(2) Å.23 The geometry of the HSQ− anion shows that the hydroxyl group is located at the O(4) atom, which is confirmed by (i) the C(4)–O(4) bond length of 1.308(1) Å is much longer than the other ones and (ii) the C(3)–C(4)–C(1) bond angle of 93.4(1)° is more open than the other three ones. The HSQ− anion insignificantly deviates from planarity, as illustrated by the O–C–C–O torsion angles varying from −3.1(1)° to 3.1(1)° (Table 1).
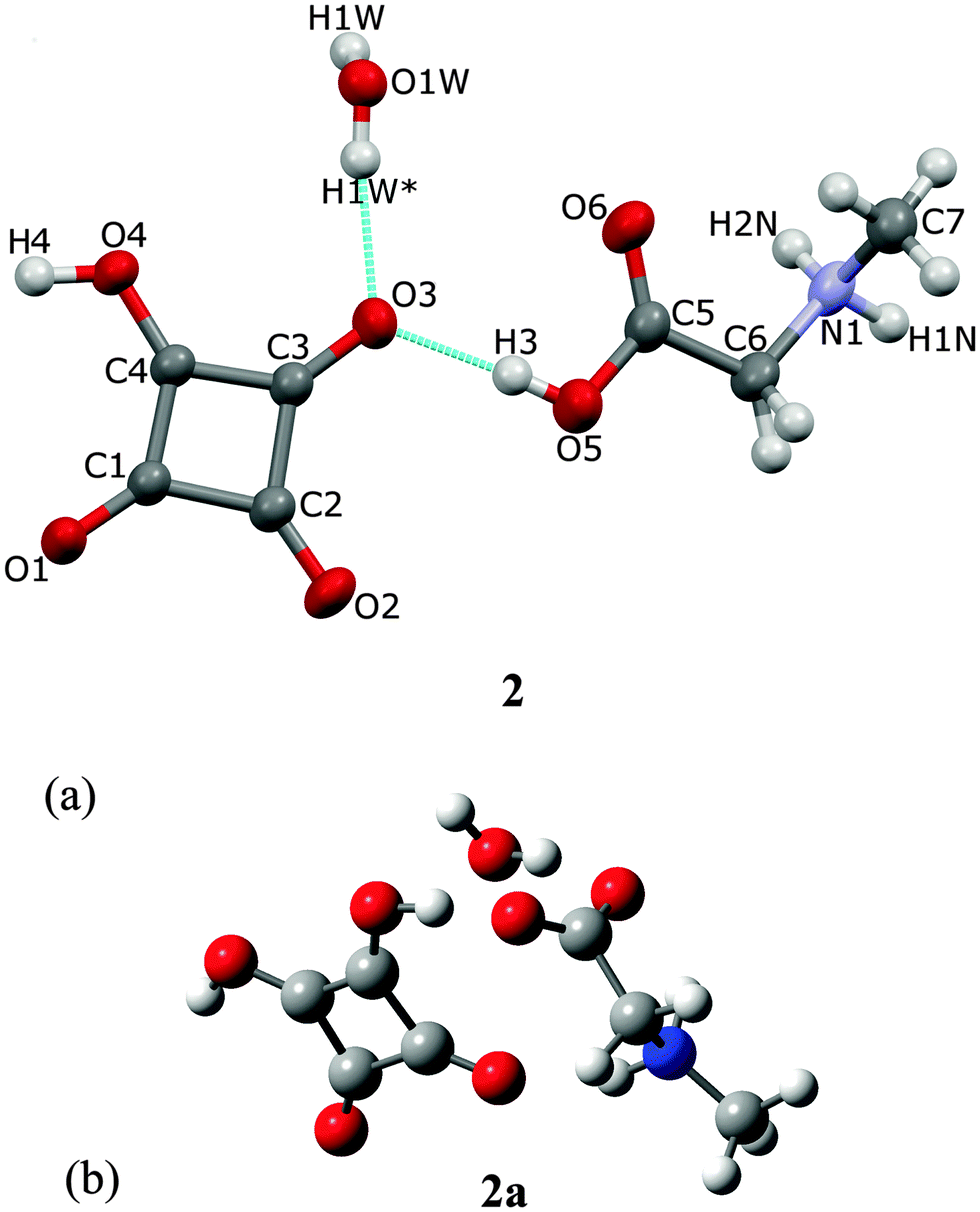 | ||
| Fig. 4 (a) The hydrogen squarate anion, sarcosinium cation and water molecule in the crystal structure 2 and the atomic labeling scheme. All atoms are at general positions, except O(1W) located on the 2-fold axis, hence only H(1W) of the water molecule is symmetry-independent. H1W* is at equivalent position (−x, y, 1.5 − z). Thermal parameters are shown at the 50% probability level and H-bonds are indicated by the dotted lines; (b) the optimized structure of 2a, determined by the B3LYP/6-311G(d,p) approach, showing sarcosine, squaric acid and water molecules. | ||
The O(1W) oxygen atom of the water molecule is located on a two-fold axis, hence only H(1W) of the water molecule is symmetry independent. The water molecule links complexes of SARH+·HSQ− into two cyclamers, R53 (16),24 through four hydrogen bonds: O(5)–H(3)⋯O(3), O(1W)–H(1W)⋯O(3), O(4)–H(4)⋯O(1) and N(1)–H(2N)–O(1) of 2.612(1), 2.849(1), 2.528(1) and 2.924(1) Å, respectively, whereas two squaric units of the neighboring molecules form cyclamer R22 (10)24 through two O(4)–H(4)⋯O(1) hydrogen bonds of 2.528(2) Å (Fig. 5, Table 2).
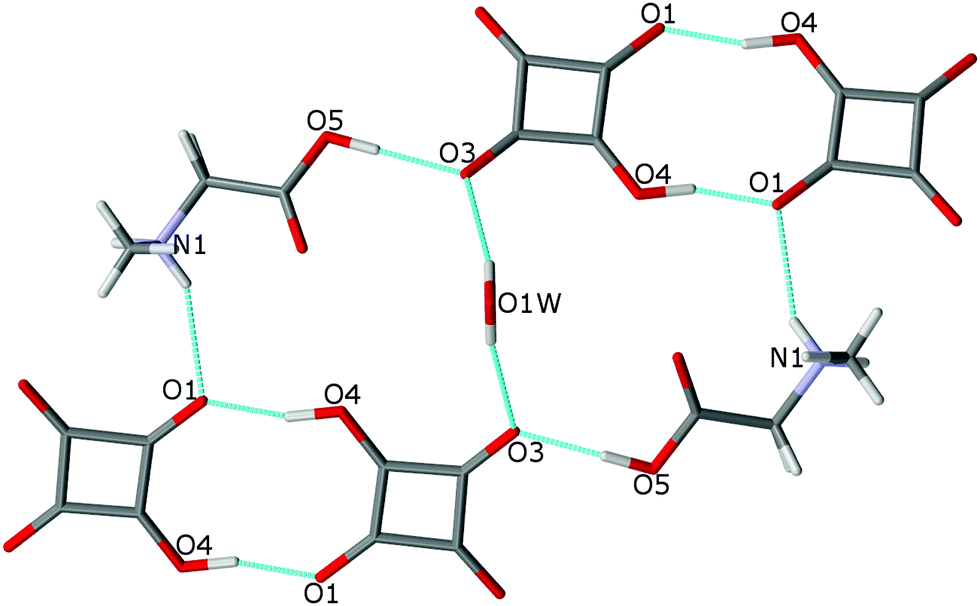 | ||
| Fig. 5 Two hydrogen-bonded cyclamers R53 (16) and R22 (10) in the structure of 2. The O–H⋯O and N–H⋯O bonds are indicated by dotted lines. | ||
The optimized structure at a ratio 1:1:1, SAR:H2SQ:H2O, determined by the B3LYP/6-311G(d,p) approach of the hydrated complex of SAR with H2SQ, 2a, is shown in Fig. 4b. The optimized structure of 2a differs from the crystal of 2, by the lack of interactions between the neighboring molecules. H2SQ interacts with SAR by the O(3)–H(3)⋯O(5) hydrogen bond of 2.537 Å. The zwitterionic character of sarcosine is confirmed by the C(5)–O(5) and C(5)–O(6) bond lengths of 1.247 and 1.251 Å, respectively (Table 1). The water molecule is engaged in the O(W)–H(1W)⋯O(6) hydrogen bonds of 2.815 Å with the carboxylate group of the SAR unit. Additionally, the N(1)–H(2N) group forms an intramolecular hydrogen bond with the O(2) atom of 2.722 Å (Table 2).
2.3. Linear dimer of the 2:2 complex of dimethylglycine with squaric acid (3 and 3a)
The DMG molecule has an amphoteric character and can exist in an anionic or a cationic form, as well as in two uncharged tautomeric forms, one of which is with neutral amino and carboxylic groups, which is more stable in the gas phase and the dipolar zwitterionic form, which is more stable in aqueous solution.11,25 According to a survey of the Cambridge Structural Database there are only two reports of crystal structures of the hydrogen-bonded complexes of DMG with acids,7 where DMG exists in the cationic form.
The investigated complex of DMG with H2SQ, 3, consists of two independent 1:1 complexes linked together into a linear dimer by the O(4′)–H(4′)⋯O(1) hydrogen bond of 2.624(2) Å (Fig. 6a, Table 2). In one of these complexes DMG is bonded to H2SQ through the O(3)–H(3)⋯O(5) and O(4)–H(4)⋯O(6) hydrogen bonds of 2.444(2) and 2.527(2) Å, respectively, with both of its oxygen atoms of the carboxylate group engaged. In the second 1:1 complex, the proton transfer occurs from H2SQ to DMG, and the DMG cation is joined to the hydrogen squarate anion through the O(5′)–H(3′)⋯O(3) hydrogen bond of 2.514(2) Å. The O(6′) atom is not involved in any hydrogen bonds. The optimized structure of dimer 3a (Fig. 6b) differs slightly from the crystal structure of 3. In 3a both DMG units exist in the zwitterionic forms and interact with two H2SQ molecules by the O–H⋯O hydrogen bonds longer than in the crystal of 3 (Table 2). The complex 3 has the extended conformation, whereas the optimized complex 3a is bent. The angles between the plane of the squarate ring in 3 and 3a are 21.37° and 63.18°, respectively.
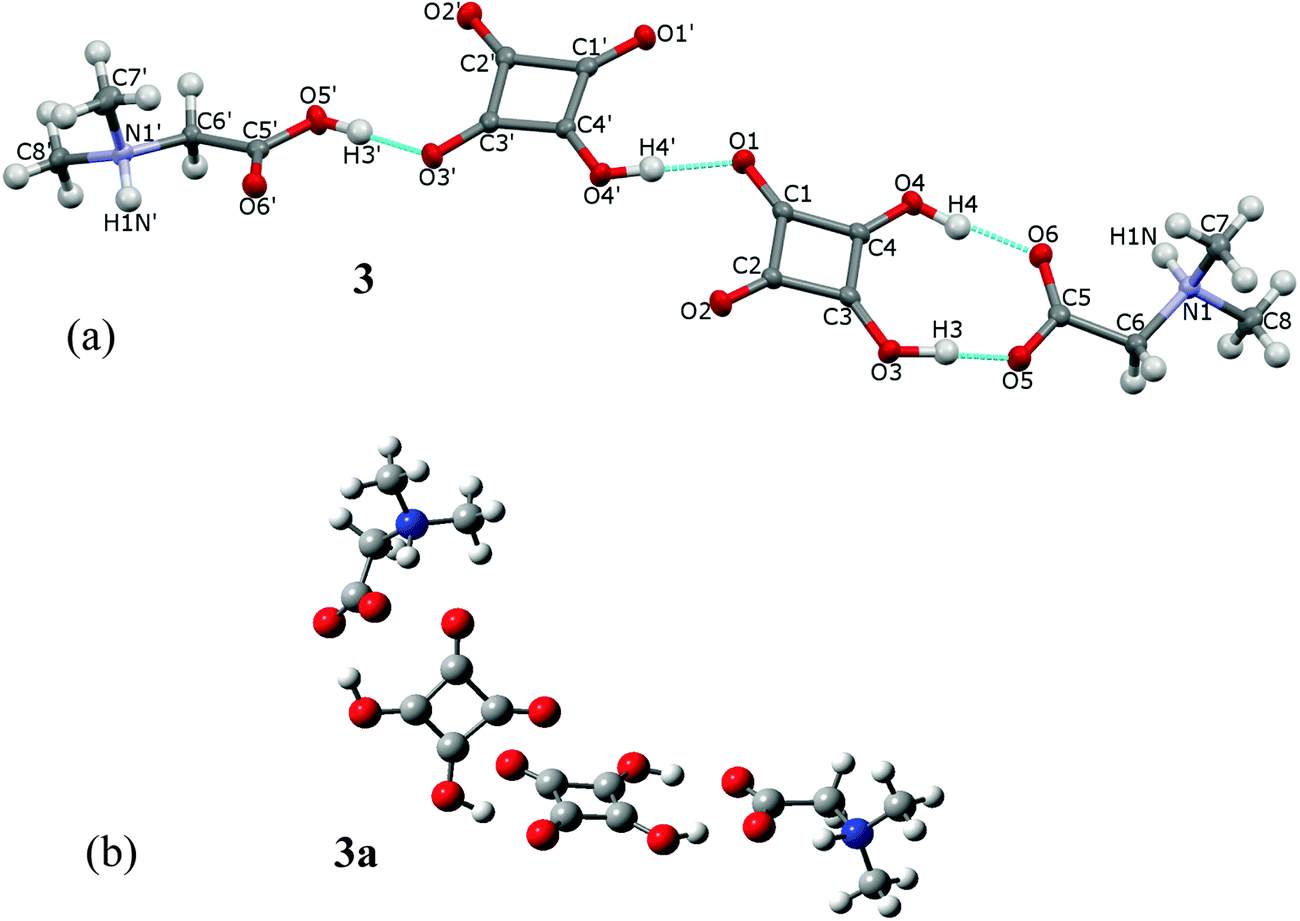 | ||
| Fig. 6 (a) The crystal structure of the dimer of dimethylglycine-squaric acid and dimethylglycinium-hydrogen squarate, 3, and its atomic-labeling scheme. All atoms are at general positions and the asymmetric part of the cell contains hydrogen squarate anion, squaric acid, N-dimethylglycinium cation and N-dimethylglycine as zwitterion. Thermal parameters are shown at the 50% probability level and H-bonds are indicated by the dotted lines; (b) the optimized structure of 3a, determined by the B3LYP/6-311G(d,p) approach, showing two squaric acids and two dimethylglycinium zwitterions linked into a linear dimer. | ||
In the crystal of 3 the dimers are linked to form two cyclamers described by the graph set R66(30),24 where the N–H atoms engaged in the N(1)–H(1)⋯O(1′) and N(1′)–H(1′)⋯O(2) hydrogen bonds of 2.870(2) and 2.803(2) Å, respectively (Fig. 7, Table 2), play the main role.
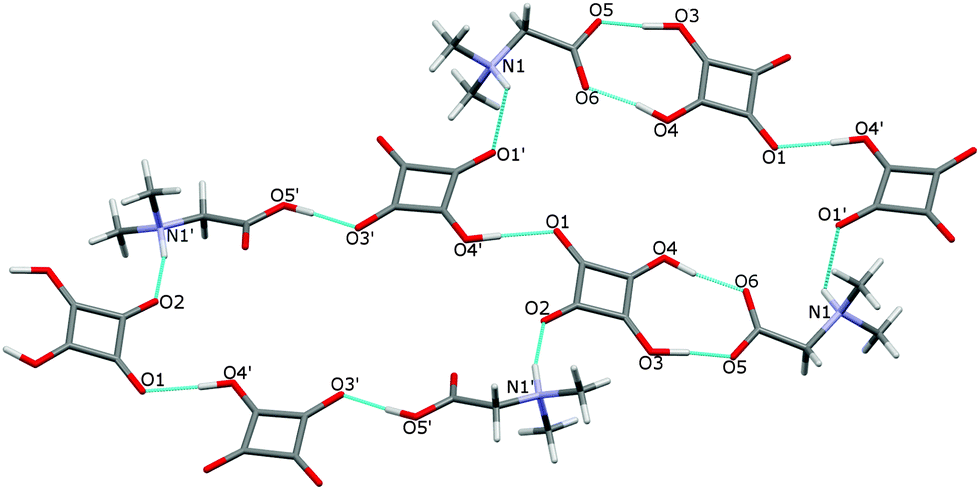 | ||
| Fig. 7 Two hydrogen-bonded cyclamers R66 (30) in the structure of 3. The O–H⋯O and N–H⋯O bonds are indicated by dotted lines. | ||
2.4. Cyclic dimer of betainium hydrogen squarate (4, 4a)
The name betaine (BET) is used, when the bonding proton is not transferred, and betainium (BETH)+ when the proton is transferred from the acid to betaine.8cBET and H2SQ form a crystalline complex 4 at the 2:2 ratio (Fig. 8a). In crystal 4 one proton from each squaric acid molecule is transferred to two BET molecules, generating hydrogen squarate anions, HSQ− and protonated betaine cations, BETH+. The HSQ− anions and the BETH+ cations are linked through the asymmetric O(5)–H(4)⋯O(4) and O(5′)–H(4′)⋯O(4′) hydrogen bonds of 2.559(4) and 2.546(4) Å, respectively. Two 1:1 complexes are joined into a cyclic dimer by two non-equivalent O(3)–H(3)⋯O(2′) and O(3′)–H(3′)⋯O(2) hydrogen bonds of 2.503(4) and 2.501(4), respectively Å (Table 2). The formation of the α-dimer is one of the most common ways of aggregation of HSQ− anions, which are linked by two short O–H⋯O hydrogen bonds of 2.47–2.54 Å.17a,b,d All structures containing α-dimers of HSQ− ions have the interesting property of being completely anhydrous, in spite of the fact that most of them crystallized from water.17c,d,e,g,j,26 The crystal packing of dimers 4 in the unit cell along the [010] axis is shown in Fig. 9.
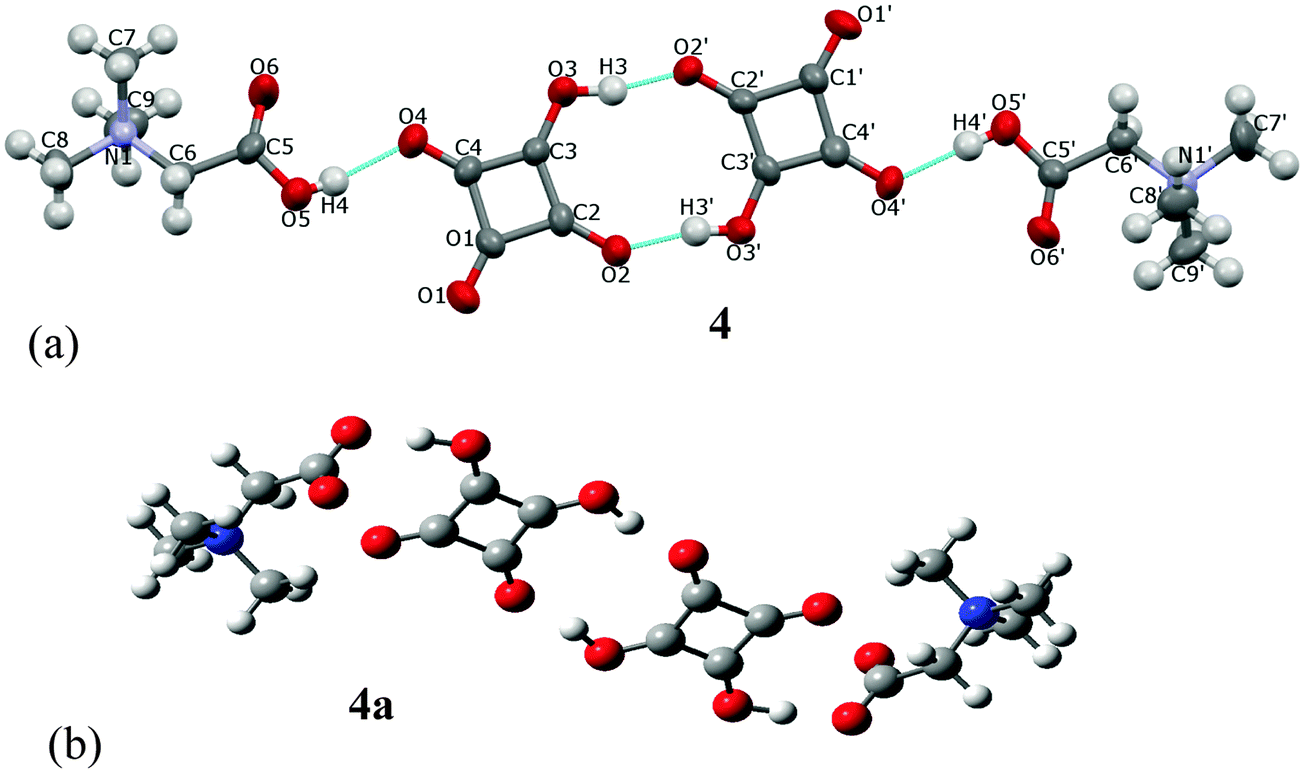 | ||
| Fig. 8 (a) The crystal structure of the H-bonded cyclic dimer of betainium hydrogen squarate, 4, and its labeling scheme. All atoms are at general positions and the asymmetric part of the unit cell contains two squarate anions and two betainium cations. Thermal parameters are shown at the 50% probability level and H-bonds are indicated by the dotted lines; (b) the optimized structure of 4a, determined by the B3LYP/6-311G(d,p) approach, showing two squaric acids and two betaines linked into a centrosymmetrical dimer. | ||
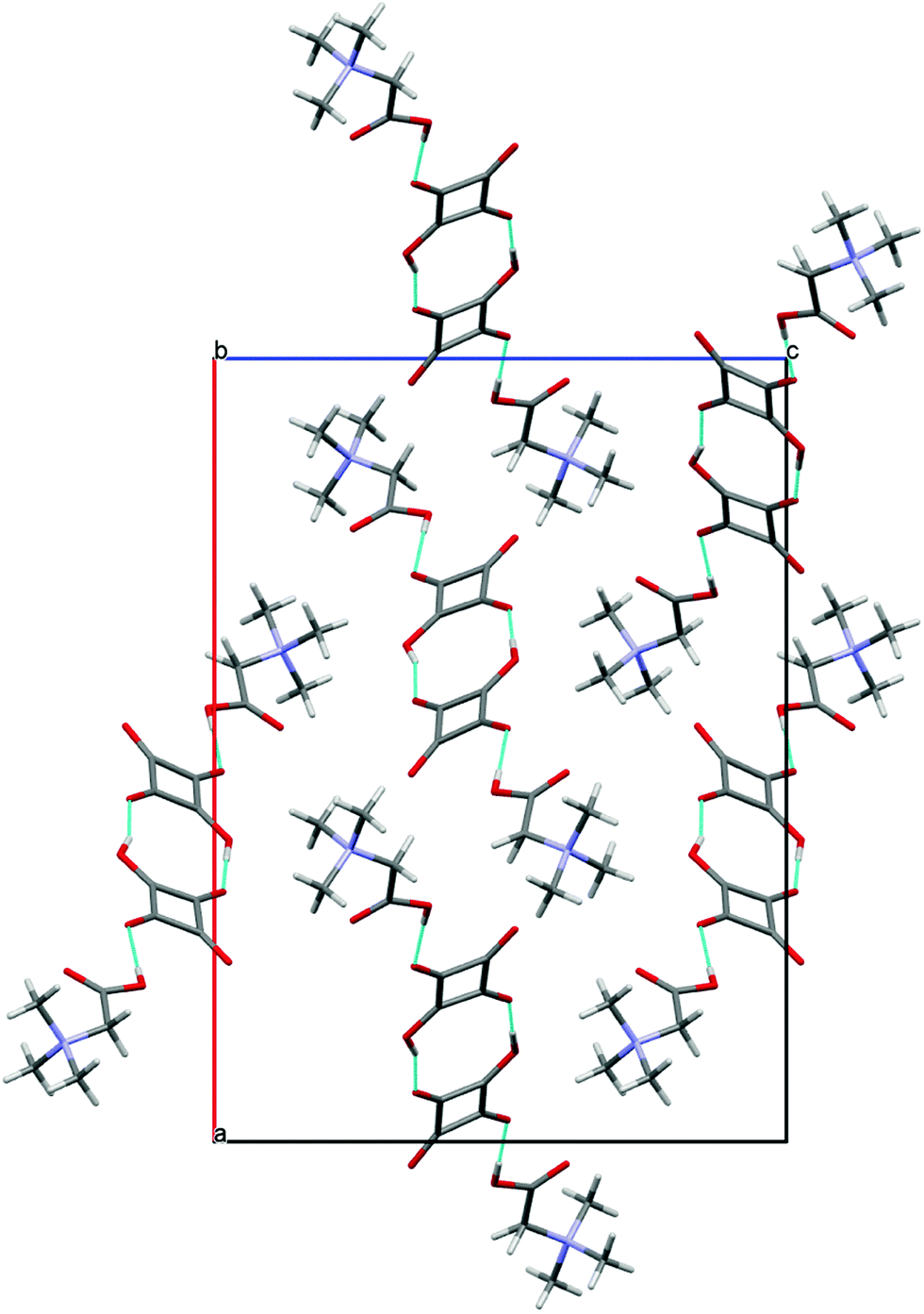 | ||
| Fig. 9 Structure of crystal 4 projected down [010]. The hydrogen bonds are indicated by blue dotted lines. | ||
The optimized structure of the dimer, 4a, determined by the B3LYP/6-311G(d,p) approach is much more symmetric than that of crystal 4 (Fig. 8b). No protons are transferred from squaric acid to betaine and complexes form a centrosymmetric dimer. The calculated dipole moment is 0.003 D. In the optimized complex 4a, the O(4)–H(4)⋯O(5) hydrogen bonds of 2.473 Å, which linked H2SQ and BET molecules, are shorter than those in complex 4, whereas the O(3)–H(3)⋯O(2) bonds of 2.591 Å, which joined 1:1 complexes into a centrosymmetric dimer are longer than in 4 (Table 2). The presence of H2SQ molecules in dimer 4a is confirmed by the C(2)–C(3)–C(4) and C(3)–C(4)–C(1) bond angles of 92.74° and 91.39°, respectively, whereas in 4 the C(2)–C(3)–C(4) bond angles in both HSQ− are ca. 93°, while those of C(3)–C(4)–C(1) are 89.0(3)° and 89.1(3)°. A similar centrosymmetric dimer has been found in the crystal and optimized structures of the complex of quinuclidine betaine with squaric acid.26
In summary the differences between the crystal forms and optimized structures arise from the lack of N–H⋯O interactions in all isolated glycine–squaric acid complexes.
2.5. Correlation between the number of N+H donors and the number of the methyl groups in glycine derivatives with the ionization of squaric acid
The increasing presence of N+H-donors in the glycine derivatives (in the sequence BET → DMG → SAR → GLY) leads to systematic changes in the H-bonding patterns, the molecular arrangement, hydration and, most importantly, in the dissociation of protons transfering from squaric acid to glycine derivatives (Fig. 10). We found that the presence of three N+H donors (in 1) results in the full dissociation of squaric acid. The dissociation reduces through two N+H donors, when even a neutral squaric acid molecule was obtained. When N+H donors are absent (in 4), half-dissociated squaric acid anions (HSQ−) are observed and they aggregate into doubly O–H⋯O bonded dimers. This process cannot be reproduced by calculations involving one isolated structural unit only, as it does not include N+H⋯O bonds to the squaric acid oxygen atoms. Fig. 10 shows the correlation between the frequency of N+H⋯O hydrogen bonds as a function of the number of methyl groups in glycine derivatives (no methyl in GLY, one methyl in SAR, two methyls in DMG and three methyl groups in BET). The squarate dianion, SQ2−, has eight H-acceptor sites in 2GLY+·SQ2−, six of them are used in N+H⋯O bonds and the remaining two in O–H⋯O bonds (8 H-donors to 8-acceptors). The average of the numbers of protons in the symmetry independent H2SQ molecule and the HSQ− anion equal to 1.5 is plotted for DMG structure 3. It can be concluded that each N–H⋯O hydrogen bond somewhat reduces the double-bond character of the squaric acid carbonyls and in this way contributes to the dissociation of H2SQ molecules. This systematically happens for one and two methyl substituting ammonium protons and this relation is valid for GLY, SAR and DMG, however, this relation is reversed for BET, when no ammonium protons are available.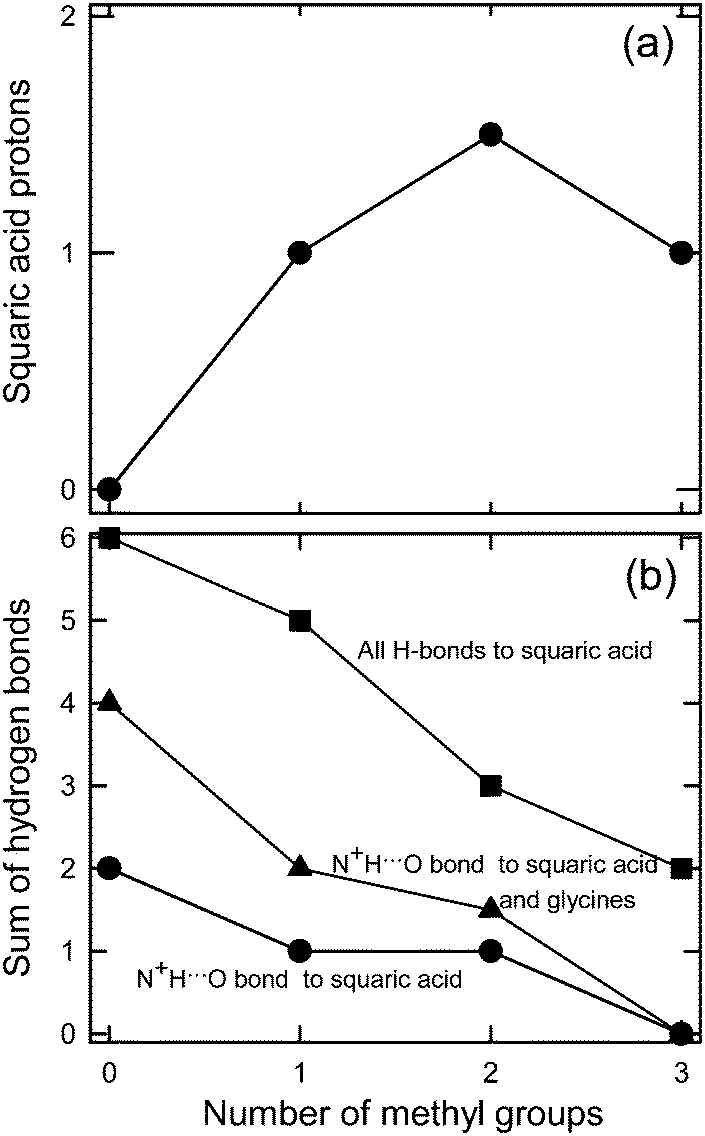 | ||
| Fig. 10 (a) The numbers of protons in a squaric acid moiety as a function of the number of methyl groups in the glycine derivatives, two protons in H2SQ, one in HSQ− and zero in SQ2−; (b) the numbers of H bonds (N–H⋯O, O–H⋯O) to squaric acid and glycine derivatives as a function of the number of methyl groups in the glycine derivatives; ● – N+–H⋯O bonds to squaric acid moiety, ▲ – N+–H⋯O bonds to squaric acid moiety and glycine derivatives; ■ – all H-bonds to squaric acid moiety. | ||
2.6. 1H and 13C NMR spectra
Both proton and carbon-13 NMR spectra of complexes of glycine and its methyl derivatives with squaric acid are very simple, however the effect of the number of methyl groups attached to the quaternary nitrogen atom on the proton and carbon-13 chemical shifts is significant. With increasing number of methyl groups in glycines the resonance signals of the CH2 and CH3 protons and methylene and methyl carbon-13 atoms are linearly shifted to a lower magnetic field, whereas the resonance signal of the COOH carbon atom undergoes an upfield shift (Fig. S1, ESI†). The proton and carbon-13 chemical shifts are given in the Experimental section. The single 13C resonance signal of the hydrogen squarate carbon atoms appear in the lower magnetic field and it is observed between 198.64 and 198.86 ppm. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OH groups can form similarly strong H-bonds to the surrounding water molecules and carboxylate groups, leading to the π-conjugation of their C–C and C–O bonds.17a
O and C–OH groups can form similarly strong H-bonds to the surrounding water molecules and carboxylate groups, leading to the π-conjugation of their C–C and C–O bonds.17a
2.7. Infrared spectra
Amino acids exhibit a great deal of hydrogen bonding as evidenced by the presence of many broad bands in their infrared spectra especially in the region 3000–2000 cm−1.8b,c,27 The solid-state FTIR spectra of complexes 1, 2, 3 and 4 are shown in Fig. 11a, c, e, g, while the frequencies computed by the B3LYP/6-311G(d,p) approach for optimized structures 1a, 2a, 3a and 4a are shown as vertical lines in Fig. 11b, d, f, h, respectively. The proposed assignments of bands were made using the Gauss View molecular visualization program.28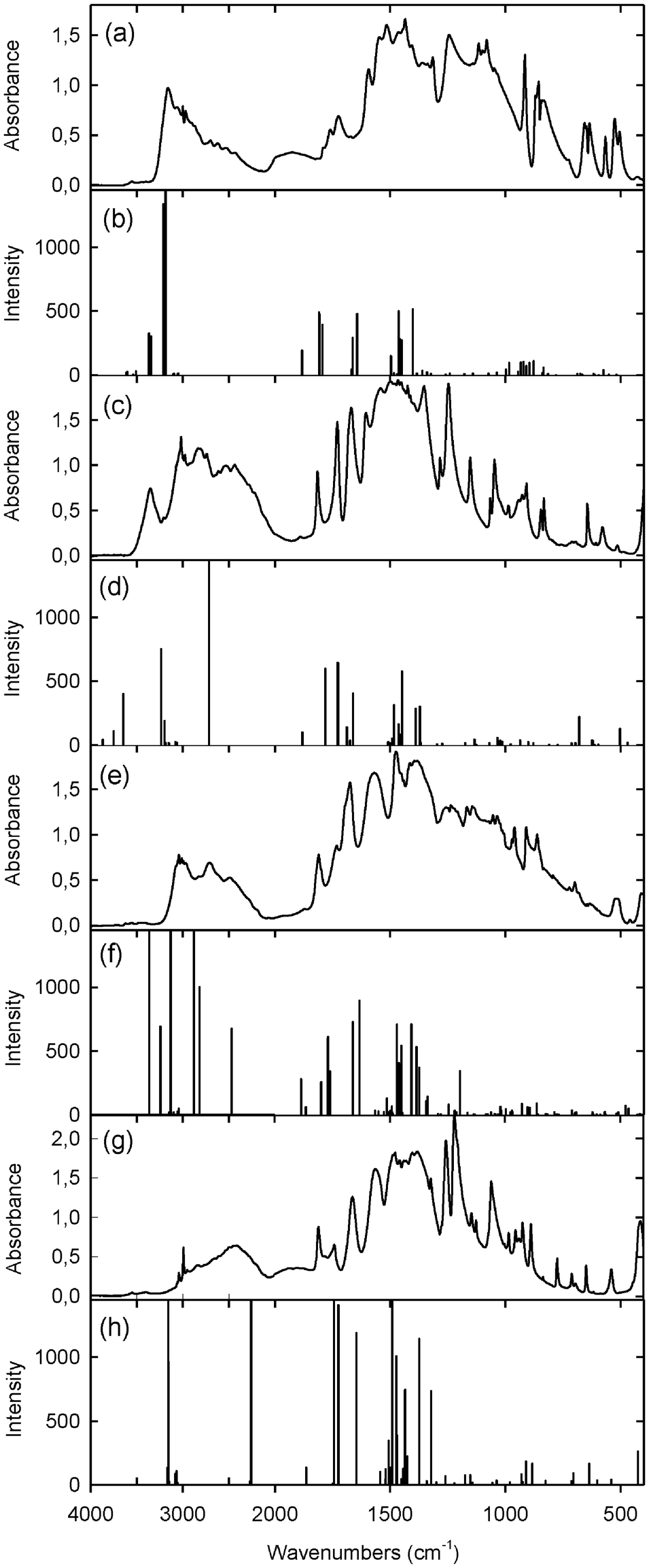 | ||
| Fig. 11 Experimental solid-state spectra and calculated frequencies by the B3LYP/6-311G(d,p) approach (a) and (b) for 1 and 1a; (c) and (d) for 2 and 2a; (e) and (f) for 3 and 3a; (g) and (h) for 4 and 4a, respectively. | ||
In the FTIR spectrum of 1 (Fig. 11a) the νNH band appears at 3160 cm−1, while the νOH vibration is assigned to the broad band in the 3000–2000 cm−1 region. The calculated asym. and sym. stretching, in-plane bending and out-of-plane bending NH vibrations for GLY units in 1a are observed at 3614 and 3603, 3538 and 3507, 1663 and 1643, 984 and 935 cm−1, respectively. The bands at 3367 and 3344 cm−1 are attributed to the νO(6)–H⋯N vibrations, while those at 3212 and 3188 cm−1 to the νO(3,4)–H⋯O(5) modes (Fig. 11b).
In the FTIR spectrum of 2 (Fig. 11c) the bands at 3350, 2830 and 2470 cm−1 are assigned to the νO–H mode in water, νN–H⋯O and νO–H⋯O vibrations, respectively. The calculated νO–H in water, νN–H⋯O and νO–H⋯O vibrations for 2a appear at 3640, 3235 and 2715 cm−1, respectively (Fig. 11d).
The experimental infrared spectrum of 3 (Fig. 11e) shows broad bands at 2720 and 2480 cm−1 attributed to νN–H and νO–H modes, respectively. However, in the calculated spectrum of 3a (Fig. 11f) there are several intensive bands at 3365 and 3243 cm−1 assigned to the νO(3)–H⋯O(5) and νO(4)–H⋯O(6) modes, at 3133 cm−1 to the νO(4′)–H⋯O(1) mode, at 2878 cm−1 to the νO(3′)–H⋯O(5′) vibration and at 2471 cm−1 to the νN–H mode of atoms engaged in the strong intramolecular hydrogen bonds (Table 2).
The FTIR spectrum of 4 (Fig. 11g) is characterized by a broad absorption centered at 2450 cm−1, which is attributed to the νO–H⋯O vibration. A similar absorption is observed in the spectra of betaine complexes with mineral and organic acids,8b,c,9 and its shifts to lower or higher wavenumbers depend on the hydrogen bond strength. The calculated frequencies for 4a at 3160 and 2260 cm−1 are assigned to the stretching vibrations of the O–H groups engaged in the longer O(3)–H⋯O(2) and shorter O(4)–H⋯O(5) hydrogen bonds, respectively (Fig. 11h, Table 2).
The broadening of the IR absorption in the spectra of the complexes investigated is also observed in the region below 1500 cm−1. A similar broad band in this region was observed in the infrared spectrum of H2SQ29 and in the spectra of squaric acid complexes with various bases.17j,30 This broad absorption overlaps the deformation vibrations of the methyl and methylene groups, hydroxyl groups, and ring and skeletal modes. However, the bands in the 1800–1500 cm−1 region are attributed to the νCO, νCC and νCOO vibrations. Two bands at 1759 and 1723 cm−1 are assigned to νCO of glycinium moieties in the spectrum of 1. Similarly, the νCO band at 1740 cm−1 was observed in the spectrum of glycinium monophenylphosphate.5a In the spectrum of 2 the νCO band at 1729 cm−1 of the sarcosinium cation is observed and it appears in a similar frequency as in the spectrum of sarcosinium iodide.6d In the spectrum of the complex of 3, both νasCOO and νCO bands at 1674 and 1731 cm−1 are observed and they are assigned to dimethylglycine and the dimethylglycinium cation, respectively. The band at 1740 cm−1 in the spectrum of 4 confirms the presence of the betainium cation in complex with squaric acid. In the spectrum of the proton-transfer complex of BET with 2,6-dichloro-4-nitrophenol the νCO band appears at 1720 cm−1, while in the spectrum of the molecular complex formed between BET and pentachlorophenol the νasCOO band appears at 1664 cm−1.9b
The frequencies of the squaric acid moiety are much better recognized in the calculated IR spectra than in the experimental ones, where the νCO vibrations appear as a single band in the 1815–1790 cm−1 region. The calculated frequencies from the 1885–1865 cm−1 range are assigned to the νsCO, while those in the 1790–1745 cm−1 region are attributed to the νasCO modes in the H2SQ moiety. The spectral features of the squarate units are similar to those observed earlier.17f,j,18e,27a,31 The experimental and calculated spectra are slightly different, because the experimental spectrum is recorded for the complex in the solid state, while the computations are made for the vibrations of the isolated molecule in the gas phase, and the calculations are based on harmonic frequencies, while the experimental ones are based on the anharmonic frequencies.
The experimental FTIR spectra confirm the crystal structure of the complex investigated, whereas the calculated frequencies correspond well with the structures of the isolated molecules.
The IR data confirm the key information about the proton positions determined in the crystalline complexes by X-ray diffraction.
3. Conclusions
In the crystals of complexes of squaric acid, H2SQ, with glycine, GLY, 1, sarcosine, SAR, 2, dimethylglycine, DMG, 3 and betaine, BET, 4, the proton from H2SQ is transferred to amino acid molecules. The exception is the 2:2 complex of 3 in which one molecule of DMG is protonated, while the other remains in the zwitterionic form. In the optimized structures of one unit of complexes investigated (1a, 2a, 3a, 4a) by the B3LYP/6-311G(d,p) approach, the amino acid moieties are in their zwitterionic forms with the exception of 1a, in which GLY exists in an uncharged (neutral) form. The HSQ− and amino acid molecules are linked by the O–H⋯O hydrogen bonds with the O⋯O distances from 2.444 to 2.624 Å. Depending on methylation of glycine in its co-crystals with squaric acid, its dianion, monoanion, neutral and mixed forms are stabilized. Methylation of glycine gradually reverses the dissociation of squaric acid in crystals. The resonance signals of the proton and carbon-13 of the methylene and methyl groups are shifted to a lower magnetic field with increasing number of methyl groups attached to the amino group, while the resonance signal of the carboxylic group is linearly shifted to a higher magnetic field.
4. Experimental section
4.1. General procedure
Amino acids, GLY, SAR and DMG were purchased from Aldrich Chemical Co, whereas BET from Sigma. FTIR spectra were measured in Nujol and Fluorolube suspensions between KBr plates using a Bruker IFS 66v/S instrument, with a resolution of 2 cm−1. Each spectrum was accumulated for 64 scans. The NMR spectra were recorded using a Varian VNMRS-400 spectrometer operating at 402.6435 and 101.2440 MHz for 1H and 13C, respectively. The spectra were measured in D2O relative to an internal standard of 3-(trimethylsilyl)propionic-d4 acid sodium salt. Elemental analysis was made using an Elemental Model Vario EL III.4.2. Preparations
:1), m.p. 180 °C. Elemental analysis calculated for C8H10N2O8: %C, 36.65; %H, 3.84; %N, 10.68; found: %C, 36.33; %H, 3.75; %N, 10.43. 1H NMR (D2O, ppm) δ 3.89 (CH2); 13C NMR (D2O, ppm) δ 43.07 (CH2), 173.03 (COO), 198.85 (CO, H2SQ).
:1), 0.76 g of H2SQ in 1 mL water was added. The solvents were evaporated and the residue was recrystallized from a mixture of methanol–water (2:1), m.p. 163–164 °C. Elemental analysis calculated for C14H20N2O13: %C, 39.63; %H, 4.75; %N, 6.60; found: %C, 39.47; %H, 4.74; %N, 6.45. 1H NMR (D2O, ppm) δ 2.80 (CH3), 3.93 (CH2); 13C NMR (D2O, ppm) δ 35,67 (CH3), 51.96 (CH2), 172.28 (COO), 198.86 (CO, H2SQ).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 complex of dimethylglycine with squaric acid (3).
0.40 g of DMG and 0.44 g of H2SQ were dissolved in 1 mL of water. The crystals were filtered off and recrystallized from a mixture of methanol–water (3:1), m.p. 182–183 °C. Elemental analysis calculated for C8H11NO6: %C, 44.24; %H, 5.10; %N, 6.45; found: %C, 44.15; %H, 5.03; %N, 6.10. 1H NMR (D2O, ppm) δ 2.99 (CH3), 4.06 (CH2); 13C NMR (D2O, ppm) δ 46.70 (CH3), 60.58 (CH2), 171.41 (COO), 198.64 (CO, H2SQ).
O, H2SQ).
:2 complex of dimethylglycine with squaric acid (3).
0.40 g of DMG and 0.44 g of H2SQ were dissolved in 1 mL of water. The crystals were filtered off and recrystallized from a mixture of methanol–water (3:1), m.p. 182–183 °C. Elemental analysis calculated for C8H11NO6: %C, 44.24; %H, 5.10; %N, 6.45; found: %C, 44.15; %H, 5.03; %N, 6.10. 1H NMR (D2O, ppm) δ 2.99 (CH3), 4.06 (CH2); 13C NMR (D2O, ppm) δ 46.70 (CH3), 60.58 (CH2), 171.41 (COO), 198.64 (CO, H2SQ).
O, H2SQ).
4.3. Single crystal X-ray diffraction
X-ray diffraction data of complexes were collected on a KUMA KM-4 CCD diffractometer.32 The structure was solved by direct methods using SHELXS-97 and refined on F2 by the full-matrix least-squares with SHELXL-97.33 The crystal data, details of data collection and structure refinement are given in Table 3 and the final atomic coordinates in Table S3 of ESI.† CCDC 966748–966751.| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Empirical formula | C8H12N2O8 | C14H18N2O12·H2O | C16H22N2O12 | C9H13NO6 |
| Formula weight | 264.20 | 424.32 | 434.36 | 213.20 |
| Temperature (K) | 296(2) | 296(2) | 296(2) | 296(2) |
| Wavelength (Å) | 1.71073 | 1.54184 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | C2/c | C2/c | P21/c | Pca21 |
| Unit cell dimensions | ||||
| a (Å) | 16.8392(3) | 16.6068(2) | 17.5110(5) | 22.2718(14) |
| b (Å) | 8.3163(2) | 7.21436(8) | 6.5385(2) | 6.0451(3) |
| c (Å) | 15.8267(3) | 15.44410(17) | 17.3475(4) | 16.2765(8) |
| β (°) | 100.063(2) | 97.0249(11) | 108.249(3) | 90.00 |
| Volume (Å3) | 2182.27(8) | 1836.43(4) | 1886.32(9) | 2191.4(2) |
| Z | 8 | 4 | 4 | 8 |
| Calculated density (g cm−3) | 1.608 | 1.535 | 1.529 | 1.402 |
| Absorption coefficient (mm−1) | 0.146 | 1.213 | 0.133 | 0.119 |
| F(000) | 1104 | 888 | 912 | 976 |
| Crystal size (mm) | 0.18 × 0.22 × 0.36 | 0.16 × 0.26 × 0.33 | 0.22 × 0.28 × 0.38 | 0.19 × 0.23 × 0.38 |
| θ range for data collection (°) | 3.13–27.59 | 5.37–75.31 | 2.47–29.13 | 2.22–29.14 |
| Max/min indices h, k, l | −20/21, −10/10, −19/19 | −20/20, −8/7, −18/19 | −23/22, −7/8, −23/22 | −29/20, −5/8, −22/18 |
| Reflections collected/unique | 10909/2371 |
6657/1862 | 11666/4520 |
8662/4068 |
| R int | 0.0156 | 0.0098 | 0.0206 | 0.0353 |
| Completeness (%) | 93.5 | 98.0 | 89.3 | 88.7 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2371/0/199 | 1862/0/147 | 4520/0/341 | 4068/1/297 |
| Goodness-of-fit on F2 | 0.697 | 0.807 | 0.968 | 0.766 |
| Final R1/wR2 indices [I > 2σI] | 0.0382/0.1098 | 0.0331/ 0.1013 | 0.0386/0.0970 | 0.0400/0.0891 |
| R 1/wR2 indices (all data) | 0.0420/0.1159 | 0.0337/0.1020 | 0.0610/0.1042 | 0.0942/0.1069 |
| Largest diff. peak and hole (e Å−3) | 0.296 and −0.278 | 0.234 and −0.141 | 0.297 and −0.328 | 0.156 and −0.185 |
4.4. Computation details
The DFT calculations were performed using the GAUSSIAN 03 program package.34 The calculations employed the B3LYP exchange–correlation functional, which combines the hybrid exchange functional of Becke35 with the gradient-correlation functional of Lee et al.36 and the split-valence polarized 6-311G(d,p) basis set.37 The X-ray geometry was used as a starting point for the calculations. The calculated IR frequencies are positive and confirmed that the optimized structures were in the states of minimum energy.Acknowledgements
The computations were performed at the Poznań Supercomputing and Networking Center.References
- (a) J. H. Caldwell, N. W. Daw and H. J. Wyatt, J. Physiol., 1978, 276, 277–298 CAS; (b) L. Rajendra, J. W. Lynch and P. Schofield, Pharmacol. Ther., 1997, 73, 121–146 CrossRef; (c) P. Schofield, Adv. Neurol., 2002, 89, 263–274 Search PubMed.
- (a) H. D. Jakubke and H. Jeschkeit, Aminosäuren, Peptide, Proteine, Academic-Verlag, Berlin, 1982 Search PubMed; (b) H.-L. Lane, C.-L. Huang, P.-L. Wu, Y.-C. Liu, Y.-C. Chang, P.-y. Lin, P.-W. Chen and G. Tsai, Biol. Psychiatry, 2006, 60, 645–649 CrossRef CAS PubMed.
- (a) R. N. Girandola, R. A. Wiswell and R. Bulbulian, Biochem. Med., 1980, 24, 218–222 CrossRef CAS; (b) W. M. Bolman and J. A. Richmond, J. Autism Dev. Disord., 1999, 29, 191–194 CrossRef CAS; (c) M. E. Tonda and L. L. Hart, Ann. Pharmacother., 1992, 26, 935–937 CAS.
- (a) X. Domingo, in Betaines in Amohoteric Surfactants, ed. E. G. Lomax, Marcel Dekker Inc, New York, 1996, vol. 59, pp. 75–190 Search PubMed; (b) N. Van Thoai, in Nitrogenous Bases in Comprehensive Biochemistry, ed. M. Florkin and E. H. Stotz, Elsevier Publ. Comp. Amsterdam, 1965, vol. 6, pp. 208–252 Search PubMed; (c) G. Blunden and S. M. Gordon, Prog. Phycol. Res., 1986, 4, 39–80 CAS; (d) W. A. Rees, T. D. Yager, J. Korte and P. H. von Hippel, Biochemistry, 1993, 32, 137–144 CrossRef CAS.
- (a) T. Głowiak and A. W. Szemik, J. Crystallogr. Spectrosc. Res., 1986, 16, 79–89 CrossRef; (b) S. Natarajan, K. Ravikumar and S. S. Rajan, Z. Kristallogr., 1984, 168, 75–82 CrossRef CAS PubMed; (c) A. R. Al-Karaghouli, F. E. Cole, M. S. Lehmann, C. F. Miskell, J. J. Verbist and T. F. Koetzle, J. Chem. Phys., 1975, 63, 1360–1366 CrossRef CAS PubMed; (d) J. Baran, K. Łukaszewicz, A. Pietraszko and M. Śledź, J. Mol. Struct., 2002, 611, 155–168 CrossRef CAS; (e) D. Tran Qui, B. Lambert-Andron and J. X. Boucherle, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 907–909 CrossRef; (f) D. Tran Qui, J. Vicat and A. Durif, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 181–184 CrossRef; S. Natarajan, Z. Kristallogr., 1983, 163, 305–306 Search PubMed; (g) T. Głowiak and I. Wnk, J. Crystallogr. Spectrosc. Res., 1985, 15, 157–171 CrossRef.
- (a) S. C. Bhattacharyya and N. N. Saha, J. Cryst. Mol. Struct., 1978, 8, 105–113 CrossRef CAS , 209–215; (b) R. V. Krishnakumar and S. Natarajan, Cryst. Res. Technol., 1995, 30, 825–830 CrossRef CAS; (c) M. T. Averbuch-Pouchot, Z. Kristallogr., 1988, 183, 285–291 CAS; M. T. Averbuch-Pouchot, Z. Kristallogr., 1993, 207, 149–150 Search PubMed; (d) V. V. Ghazaryan, M. Fleck and A. M. Petrosyan, J. Mol. Struct., 2013, 1032, 35–40 CrossRef CAS PubMed; (e) R. V. Krishnakumar, M. P. Rameela and S. Natarajan, Cryst. Res. Technol., 1996, 31, 203–207 CrossRef CAS; (f) A. Mostad and S. Natarajan, Cryst. Res. Technol., 1996, 31, 295–300 CrossRef CAS.
- (a) B. D. Santarsiero and R. E. Marsh, J. Crystallogr. Spectrosc. Res., 1983, 11, 245–251 CrossRef; (b) V. H. Rodrigues, J. A. Paixão, M. M. R. R. Costa and A. M. Matos Beja, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 417–420 CAS.
- (a) J. Baran, M. Drozd, T. Głowiak, M. Śledź and H. Ratajczak, J. Mol. Struct., 1995, 372, 131–144 CAS; (b) M. Iczyszyn, D. Godzisz and M. M. Ilczyszyn, J. Mol. Struct., 2003, 611, 103–118 CrossRef; (c) D. Godzisz, M. M. Ilczyszyn and M. Ilczyszyn, J. Mol. Struct., 2002, 606, 123–137 CrossRef CAS; (d) W. H. Yip, R.-J. Wang and T. C. W. Mak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 717–719 CrossRef; (e) J. Baran, M. Drozd, T. Lis, M. Śledź, A. J. Barnes and H. Ratajczak, J. Mol. Struct., 1995, 354, 109–118 CrossRef; (f) D.-D. Wu and T. W. C. Mak, J. Chem. Crystallogr., 1994, 24, 689–694 CrossRef CAS.
- (a) Z. Dega-Szafran, I. Kowalczyk and M. Szafran, Bull. Pol. Acad. Sci., Chem., 1995, 43, 303–312 CAS; (b) Z. Dega-Szafran, A. Komasa, M. Grundwald-Wyspiańska, M. Szafran, G. Buczak and A. Katrusiak, J. Mol. Struct., 1997, 404, 13–23 CrossRef CAS; (c) M. M. Ilczyszyn, T. Lis and H. Ratajczak, J. Mol. Struct., 1995, 372, 9–27 CrossRef CAS; (d) J. Baran, A. J. Barnes, B. Engelen, M. Panthofer, A. Pietraszko and H. Ratajczak, J. Mol. Struct., 1995, 372, 29–40 CAS.
- (a) S. Haussühl, Z. Kristallogr., 1989, 188, 311–320 CrossRef PubMed; (b) S. Sato, J. Phys. Soc. Jpn., 1968, 25, 185–201 CrossRef CAS; (c) G. Schaack, Ferroelectrics, 1990, 104, 147–158 CrossRef CAS.
- A. D. Headley, R. E. Corona and E. T. Cheung, J. Phys. Org. Chem., 1997, 10, 898–900 CrossRef CAS.
- (a) A. H. Schmidt, Synthesis, 1980, 961–994 CrossRef CAS; (b) M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781–805 CrossRef CAS PubMed.
- L. I. Markova, V. L. Malinovskii, L. D. Patsenker and R. Häner, Org. Biomol. Chem., 2012, 10, 8944–8947 CAS.
- (a) Y. Wang, G. D. Stucky and J. M. Williams, J. Chem. Soc., Perkin Trans. 2, 1974, 35–38 RSC; (b) D. Semmingsen, F. J. Hollander and T. F. Koetzle, J. Chem. Phys., 1977, 66, 4405–4412 CrossRef CAS PubMed; (c) F. J. Hollander, D. Semmingsen and T. F. Koetzle, J. Chem. Phys., 1977, 67, 4825–4831 CrossRef CAS PubMed; (d) D. Semmingsen, Acta Chem. Scand., Ser. A, 1973, 27, 3961–3972 CrossRef CAS PubMed; (e) A. Katrusiak and R. J. Nelmes, J. Phys. C: Solid State Phys., 1986, 19, L765–L772 CrossRef CAS.
- (a) S. Cohen, J. R. Lacher and J. D. Park, J. Am. Chem. Soc., 1959, 81, 3480 CrossRef CAS; (b) D. J. MacDonald, J. Org. Chem., 1968, 33, 4559–4560 CrossRef CAS; (c) D. T. Ireland and H. F. Walton, J. Phys. Chem., 1967, 71, 751–754 CrossRef CAS.
- R. West and D. L. Powell, J. Am. Chem. Soc., 1963, 85, 2577–2579 CrossRef CAS.
- (a) G. Gilli, V. Bertolasi, P. Gilli and V. Ferretti, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 859–865 CrossRef CAS PubMed; (b) G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Outline of a Comprehensive Hydrogen Bond Theory, Oxford University Press, 2009, pp. 251–253 Search PubMed; (c) V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 591–598 CAS; (d) B. B. Koleva, T. Kolev, R. W. Seidel, M. Spiteller, H. Mayer-Figge and W. S. Sheldrick, J. Phys. Chem. A, 2009, 113, 3088–3095 CrossRef CAS PubMed; (e) S. Mathew, G. Paul, K. Shivasankar, A. Choudhury and C. N. R. Rao, J. Mol. Struct., 2002, 641, 263–279 CrossRef CAS; (f) B. B. Koleva, T. Kolev, R. W. Seidel, H. Mayer-Figge, M. Spiteller and W. S. Sheldrick, J. Phys. Chem. A, 2008, 112, 2899–2905 CrossRef CAS PubMed; (g) T. Kolev, R. Wortmann, M. Spiteller, W. S. Sheldrick and M. Heller, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o956–o957 CAS; (h) A. Bulut, O. Z. Yeşilel, N. Dege, H. Icbudak, H. Olmez and O. Bsüyükgüngör, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o727–o729 Search PubMed; (i) I. Uçar, A. Bulut, O. Z. Yeşilel and O. Büyükgüngör, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o585–o588 Search PubMed; (j) B. B. Koleva, T. Kolev, R. W. Seidel, T. Tsanev, H. Mayer-Figge, M. Spiteller and W. S. Sheldrick, Spectrochim. Acta, Part A, 2008, 71, 695–702 CrossRef PubMed; (k) I. L. Karle, D. Ranganathan and V. Haridas, J. Am. Chem. Soc., 1996, 118, 7128–7133 CrossRef CAS.
- (a) T. Kolev, D. Yancheva, M. Spiteller, W. S. Sheldrick and H. Mayer-Figge, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o463–o465 CAS; (b) O. Angelova, R. Petrova, V. Radomirska and T. Kolev, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 2218–2220 CrossRef; (c) O. Angelova, V. Velikova, T. Kolev and V. Radomirska, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 3252–3256 CrossRef; (d) T. Kolev, H. Mayer-Figge, R. W. Seidel, W. S. Sheldrick, M. Spiteller and B. B. Koleva, J. Mol. Struct., 2009, 919, 246–254 CrossRef CAS PubMed; (e) T. Kolev, R. Stahl, H. Preut, V. Koniczek, P. Bleckmann and V. Radomirska, Z. Kristallogr. - New Cryst. Struct., 1998, 213, 167–169 CAS; (f) T. Kolev, R. Stahl, H. Preut, P. Bleckmann and V. Radomirska, Z. Kristallogr. - New Cryst. Struct., 1998, 213, 169–170 CAS; (g) T. Kolev, Z. Glavcheva, R. Stahl, H. Preut, P. Bleckmann and V. Radomirska, Z. Kristallogr. - New Cryst. Struct., 1999, 214, 193–194 CAS; (h) T. Kolev, M. Spiteller, W. S. Sheldrick and H. Mayer-Figge, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o299–o300 Search PubMed; (i) T. Kolev, M. Spiteller, W. S. Sheldrick and H. Mayer-Figge, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o4292–o4294 CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W.H. Freeman and Company, New York, Basingstone, 2007 Search PubMed.
- (a) H. A. Saroff, J. Chem. Educ., 1994, 71, 637–643 CrossRef CAS; (b) H. B. F. Dixon, A. Cornish-Bowden, C. Liébeq, K. L. Loening, G. P. Moss, J. Reeduk, S. F. Velick, J. F. G. Vliegenthart, H. Bielka, N. Sharon, E. C. Webb, P. Karlson, B. Keil, W. E. Cohn, J. T. Edsall, J. S. Morley and G. T. Young, Pure Appl. Chem., 1984, 56, 595–624 Search PubMed.
- (a) Y. Iitaka, Acta Crystallogr., 1960, 13, 35–45 CrossRef CAS; Y. Iitaka, Acta Crystallogr., 1960, 14, 1–10 Search PubMed; (b) K. Ishikawa, M. Tanaka, T. Suzuki, A. Sekine, T. Kawasaki, K. Soai, M. Shiro, M. Lahav and T. Asahi, Chem. Commun., 2012, 48, 6031–6033 RSC , and references cited therein.
- (a) J. A. Kanters, A. Schouten, J. Kroon and E. Grech, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 807–810 CrossRef; (b) P. Barczyński, Z. Dega-Szafran, A. Katrusiak and M. Szafran, J. Mol. Struct., 2012, 1013, 95–101 CrossRef PubMed; (c) U. Korkmaz, I. Uçar, A. Bulut and O. Büyükgüngör, Struct. Chem., 2011, 22, 1249–1259 CrossRef CAS.
- (a) A. Mostad and S. Natarajan, Acta Chem. Scand., 1989, 43, 1004–1006 CrossRef CAS PubMed; (b) B. Dittrich and M. A. Spackman, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 63, 426–436 CAS.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- (a) G. M. Barrow, J. Am. Chem. Soc., 1958, 80, 86–88 CrossRef CAS; (b) A. D. Headley, B. Patel and E. T. Cheung, Tetrahedron Lett., 1996, 37, 6673–6676 CrossRef CAS.
- (a) Z. Dega-Szafran, A. Katrusiak and M. Szafran, J. Mol. Struct., 2012, 1030, 184–190 CrossRef CAS PubMed; (b) E. J. MacLean, P. S. Wheatley, G. Ferguson and C. Glidewell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1892–1896 Search PubMed.
- (a) A. Leiper and E. R. Lippincott, J. Am. Chem. Soc., 1957, 79, 5098–5101 CrossRef; (b) A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS; (c) M. T. Rosado, M. Leonor, T. S. Duarte and R. Fausto, Vib. Spectrosc., 1998, 16, 35–54 CrossRef CAS.
- A. E. Frisch, R. D. Dennington, T. A. Keith, A. B. Neilsen and A. J. Holder, Gauss View, revision 3.0.9, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- (a) F. G. Baglin and C. B. Rose, Spectrochim. Acta, Part A, 1970, 26, 2293–2304 CrossRef CAS; (b) S. L. Georgopoulos, R. Diniz, M. I. Yoshida, N. L. Speziali, H. F. Dos Santos, G. M. A. Junqueira and L. F. C. de Oliveira, J. Mol. Struct., 2006, 794, 63–70 CrossRef CAS PubMed.
- U. Korkmaz and A. Bulut, J. Mol. Struct., 2013, 1050, 61–68 CrossRef CAS PubMed.
- (a) P. Barczyński, Z. Dega-Szafran, A. Katrusiak and M. Szafran, J. Mol. Struct., 2011, 998, 240–245 CrossRef PubMed; (b) T. Kolev, R. W. Seidel, B. B. Koleva, H. Mayer-Figge, M. Spiteller and W. S. Sheldrick, J. Mol. Struct., 2009, 931, 45–49 CrossRef CAS PubMed; (c) B. Koleva, R. W. Seidel, T. Kolev, S. Zareva, H. Mayer-Figge, I. M. Oppel and W. S. Sheldrick, J. Mol. Struct., 2009, 921, 163–171 CrossRef CAS PubMed; (d) T. Głowiak and A. W. Szemik, J. Crystallogr. Spectrosc. Res., 1986, 16, 79–89 CrossRef; (e) J. Nowicka-Scheibe, E. Grech, W. Sawka-Dobrowolska, G. Bator and L. Sobczyk, Pol. J. Chem., 2007, 81, 643–652 CAS; (f) T. M. Kolev, B. A. Stamboliyska, D. Y. Yancheva and V. Enchev, J. Mol. Struct., 2004, 691, 241–248 CrossRef CAS PubMed; (g) P. Barczyński, Z. Dega-Szafran, A. Katrusiak and M. Szafran, J. Mol. Struct., 2012, 1018, 28–34 CrossRef PubMed; (h) P. Barczyński, Z. Dega-Szafran, A. Katrusiak and M. Szafran, J. Mol. Struct., 2012, 1013, 95–101 CrossRef PubMed; (i) Z. Dega-Szafran, A. Katrusiak, A. Komasa and M. Szafran, Supramol. Chem., 2013, 25, 432–440 CrossRef CAS.
- (a) KUMA KM4 CCD Software, version 161, Kuma Diffraction, Wroclaw, Poland, 1999 Search PubMed; (b) CrysAlis 162, Kuma Diffraction, Wroclaw, Poland, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8580 CrossRef PubMed.
- C. Lee, W. Yang and G. R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- W. J. Hehre, L. Random, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3; Fig. S1. CCDC 966748–966751. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj00212a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |