 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of the NAMI-A complex with nitric oxide under physiological conditions†
Maria
Oszajca
,
Ewa
Kuliś
,
Grażyna
Stochel
and
Małgorzata
Brindell
*
Faculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Krakow, Poland. E-mail: brindell@chemia.uj.edu.pl; Fax: +48 12 6340515
First published on 19th February 2014
Abstract
The interference of NAMI-A ([ImH][RuCl4(Im)(DMSO)], Im – imidazole, DMSO – dimethyl sulfoxide) with the metabolism of nitric oxide (NO) has been proposed as one of the possible pathways of the antimetastatic activity of this complex. With regard to this observation we present herein detailed spectrophotometric studies on interaction of the NAMI-A complex with NO. The reactivity of NAMI-A toward NO has been studied in aqueous solution under physiological-like conditions (pH = 7.4, [NaCl] = 0.1 M, T = 37 °C). The ability of NAMI-A as well as its hydrolytic products to bind NO has been confirmed spectrophotometrically and separation of reaction products was performed with application of the HPLC technique. The relatively slow NO binding requires opening up a coordination site in the parent NAMI-A complex via simultaneously occurring hydrolysis. The studies in the presence of albumin showed that NO can coordinate to NAMI-A–albumin adducts. The capability of nitrosyl derivatives (Ru2+–NO+) to undergo reduction of the NO+ moiety in the presence of ascorbic acid, glutathione and dithionite has been studied with application of the NO sensor. The obtained results showed that under selected conditions, nitrosyl complexes cannot liberate nitric oxide via one electron reduction using applied reductants. This is due to the relatively low reduction potential of the NO+ group bound to Ru(II) (−0.69 V), determined in electrochemical studies.
Introduction
In recent years extensive interest has been devoted to the study of ruthenium-based complexes as anticancer drugs, due to their lower toxicity when compared with conventional platinum drugs and their ability to overcome drug resistance.2–4 Among the wide group of synthesized ruthenium complexes is imidazolium trans-imidazoledimethylsulfoxidetetrachlorideruthenate ([ImH][RuCl4(Im)(DMSO)], NAMI-A), a very promising antitumor agent due to its selective inhibiting properties in the formation as well as growth of metastases with the negligible cytotoxic effect.5–7 The NAMI-A complex has been found to be active in inhibiting formation and growth of lung, mammary and B16F10 melanoma metastasis.8–12 However, this effect still remains unclear and raises the question of why metastases and not the corresponding primary tumours are sensitive to this drug. Numerous in vivo and in vitro studies have shown multiple paths of NAMI-A action such as: arrest of the cell cycle at the G2M pre-mitotic phase, apoptosis in endothelial cells, interaction with actin-type proteins on the cell surface or extracellular matrix collagen, leading to inhibition of invasiveness.3 However, the main molecular target of this complex is still unknown and literature reports suggest that the overall effect may be resulting from multiple interactions outside and inside the cells. The confirmed antimetastatic properties of ruthenium(III)-dimethyl sulfoxide complexes bring about the need to understand the mechanism behind the therapeutic effect of these complexes. Due to involvement of NO in the several events contributing to tumour progression and the ability of ruthenium complexes to interact with NO,13 a theory that this signalling molecule is one of the targets of the antimetastatic activity of NAMI-A has been proposed.1,14NO plays an important role in progression of several human tumours as evidenced by the increased expression and activity of nitric oxide synthases (NOS).15 The ruthenium complexes capable of acting both as NO scavengers and donors may interfere with the NO-related angiogenic process being a crucial step in the formation of metastases. Thus, the antimetastatic activity of NAMI-A and NAMI-A-type complexes was suggested to be related to their interaction with NO in vivo.1 Indeed, it was shown that NAMI-A, KP1339 (Na[trans-RuCl4(Ind)2], Ind – indazole)16–18 and Ru(EDTA) complexes inhibit NO-dependent angiogenesis by capture of NO in vivo and in vitro without affecting the intracellular mechanism involved in proliferation.1,19,20
NO binding to a ruthenium centre results in a product which is formally described as [RuII–NO+]. Ruthenium nitrosyl complexes are characterized by a linear Ru–N–O arrangement, NO stretching frequency higher than 1870 cm−1 and susceptibility to nucleophilic attack.21 Change in the oxidation state of the ruthenium centre upon NO binding is reflected in electronic spectra which are characterized by a very weak (ε ∼ 50–60 M−1 cm−1) absorption band at around 500 nm and are remarkably different from parent ruthenium(III) complexes characterized by relatively strong ligand-to-metal charge transfer bands.22 Very recent results published by Bučinský et al.20 show that closed-shell structure [RuIII(NO)0]6 fits better with some physical/spectroscopic properties of mer,trans-[RuCl3(HInd)2(NO)], however the authors also underline that notation [RuII(NO)+]6 is formally still suitable for describing the oxidation state of ruthenium in this entity.
The strongly bound NO molecule can be liberated from the nitrosyl complex via thermal or photochemical redox reaction. This leads to the formation of RuII–NO˙ which is more susceptible to NO release.22–25 The ability of Ru(II)-dimethylsulfoxide nitrosyls of general formula [RuIICl5−x(DMSO)x(NO+)](x−2) as well as complexes containing a N-heterocyclic ligand such as imidazole ([RuIICl4(Im)(NO+)]−) or pyridine ([RuIICl2(py)3(NO+)]+) to undergo reduction on the side of NO+ and NO release in organic solvents (DMF, DMSO) has been shown by Serli et al.14,22
Since temporary concentration and duration of NO exposure seem to have a crucial role in the tumorigenesis (stimulating or inhibiting) process, it is reasonable to assume that these complexes may have multiple roles in controlling NO levels. They may act either as NO scavengers or as NO donors by releasing NO in a subsequent reduction process. It is important to note that all studies concerning NO release via one electron reduction by nitrosyl ruthenium complexes similar in structure to NAMI-A have been performed so far in non-aqueous, aprotic media, thus the reaction conditions are significantly different from physiological ones. Detailed studies on chemical behaviour of NAMI-A showed that the complex is relatively stable at pH = 5.0 whereas at physiological pH (7.4) NAMI-A undergoes a series of hydrolysis reactions resulting in chloride dissociation and partial DMSO dissociation.26,27 Since reduction potential strongly depends on the nature of coordinated ligands, the aquation process occurring under physiological conditions leading to chlorides and DMSO release from ruthenium complexes will immensely affect the redox properties. Therefore, we decided to complete this missing part of the puzzle by investigating the reactivity of NAMI-A toward NO under physiological-like conditions. In this context, we report herein detailed spectroscopic, chromatographic and electrochemical characterization of the obtained ruthenium–nitrosyl derivatives. Furthermore, we studied the ability of NAMI-A – nitrosyl derivatives to release NO in consequence of reduction by biologically relevant reductants under physiologically mimicking conditions. Additional studies of NO binding to ruthenium complex were performed in the presence of albumin.
Experimental section
Materials
All chemicals used in this study were of analytical reagent grade. NAMI-A was prepared following the published procedure,28 and its purity was checked by elemental analyses and UV-vis spectroscopy. NO gas was purchased from Linde Gaz Polska Sp. z o.o. and bubbled through concentrated KOH solution to remove higher nitrogen oxides (N2O3, NO2), and then passed through an Ascarite II column (NaOH on silica gel, Sigma Aldrich). Tris buffer (2-amino-2-(hydroxymethyl)-1,3-propanediol, > 99.8%), glutathione, ascorbic acid, NaNO2 and NaCl were purchased from Sigma Aldrich. All solutions were prepared in deionized water.Solution preparation
Samples of NAMI-A were prepared in water. Since experiments with NO require an inert atmosphere, all solutions were deoxygenated by argon and handled using gastight syringes. In a typical experiment water solution of NAMI-A was rapidly mixed with a buffered solution (Tris 0.2 M, pH 7.4 at 37 °C, 0.4 M NaCl) and/or a buffered solution saturated with gaseous nitric oxide ([NO] = 2 mM) in the volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and samples were incubated at 37 °C for a defined period of time.
:1 and samples were incubated at 37 °C for a defined period of time.
Measurements
Time-resolved spectra were recorded with the application of the thermostated (±0.1 °C) Applied Photophysics stopped-flow apparatus SX20 equipped with a sequential mixing mode. In a typical experiment, water solutions of the NAMI-A complex were rapidly mixed with buffered solution (Tris 0.2 M, pH 7.4 at 37 °C, 0.4 M NaCl) with a first mixing drive. After a defined delay period, the aged solution was mixed with buffered solution saturated with gaseous nitric oxide in the second mixing drive, and the reaction was followed spectrophotometrically.IR spectra were recorded using a FT-IR Spectrometer “Spectrum Two” Perkin Elmer equipped with a diamond universal ATR Accessory. Water solution of the NAMI-A complex was mixed with buffered solution (Tris 0.2 M, pH 7.4 at 37 °C, 0.4 M NaCl) and deoxygenated. In the next step the sample was continuously bubbled with NO for 30 min at 37 °C.
The chromatograms were registered using a Perkin Elmer HPLC Chromera system equipped with a diode-array detector. A Brownlee Validated IBD C18 5 μm, 150 × 4.6 mm column was employed for the HPLC separation and 0.1 M CH3COONH4 was used as a mobile phase at a flow-rate of 1 ml min−1.
Differential pulse voltammetry (DPV) and cyclic voltammetry (CV) measurements were performed using an Autolab PGSTAT 302N device (Eco Chemie). All measurements were carried out using a platinum disk working electrode or glassy carbon electrode (geometric area 0.06 cm2, Bio-logic), a platinum wire counter electrode, and an Ag/AgCl (filled with 3 M KCl) reference electrode (Bio-logic). The Tris/HCl buffer (0.1 M, pH 7.4) with 0.2 M NaCl was used as an electrolyte. Before measurement oxygen was removed from the electrolyte solution by bubbling argon through the solvent for several minutes and an argon atmosphere was continuously maintained above the solution while the experiments were in progress. All measurements were performed in the potential range from 0.6 to −0.4 V, scanned in the negative direction. Potentials are cited versus a normal hydrogen electrode (NHE).
To follow NO release an amino-700 nitric oxide sensor connected to an inNO-T nitric oxide measuring system (Innovative Instruments, Inc.) was used.
Results and discussion
Reaction of NAMI-A with NO – spectroscopic and chromatographic analysis
The reactivity of the NAMI-A complex toward NO was studied under physiological mimicking conditions (pH = 7.4, 37 °C, 0.2 M NaCl). In order to monitor nitrosylation reaction, the time-resolved UV-vis spectral changes were recorded by application of a stopped-flow apparatus equipped with a diode-array detector. The NAMI-A complex freshly dissolved in deionized water and deoxygenated using argon was rapidly mixed with the buffered solution saturated with gaseous nitric oxide ([NO] = 2 mM) in the concentration ratio 1:2. The 2-fold excess of NO over the ruthenium complex was maintained to shift the equilibrium toward formation of nitrosyl product/s. The characteristic spectral changes shown in Fig. 1 indicate the disappearance of the ligand-to-metal charge transfer band at 390 nm, characteristic of the parent [RuCl4(Im)(DMSO)]− complex, with concomitant formation of a new band with maximum at 347 nm, which then disappears within ca. 1600 minutes. It is well known from literature that the NAMI-A complex is very unstable under physiological conditions and undergoes gradual hydrolysis. Indeed, the first step registered spectrophotometrically is comparable with the first stage of the hydrolysis process occurring under similar reaction conditions (see ESI,† Fig. S1). Further spectroscopic changes can comprise two parallel processes: advanced hydrolysis of ruthenium complex and/or its nitrosylation. The entirely different kinetic trace at 350 nm observed in the presence of nitric oxide indicates relatively slow formation of ruthenium–nitrosyl species, in addition to hydrolytic products (Fig. 1, inset). This is due to the fact that coordination of NO to the Ru centre requires opening up a coordination site which is provided by the aquation process. The earlier studies on coordination of NO to the NAMI-A complex in unbuffered aqueous solution indicated a replacement of the DMSO ligand by NO and formation of [ImH][mer-RuCl3(NO2)(Im)(NO)] as a main product and the [(Im)2H][mer-RuCl4(Im)(NO)] as a minor one.22 This is in agreement with the proposed hydrolysis pathway in acidic solution (pH of water solution of NAMI-A is around 5 and decreases with time)29 which leads to release of the DMSO ligand without hydrolysis of chloride ions.26 However, the synthesised complexes were crystallized from nitromethane which can lead to the new equilibrium affecting the properties of the final reaction product. The hydrolysis pathway for a slightly basic solution (pH 7.4) is different and the first product was characterised as RuCl3(Im)(H2O)(DMSO) (Scheme 1) whereas DMSO release is only partial and relatively slow.26,27 On the basis of this, we propose that the first putative nitrosylation product can be ascribed to RuIICl3(NO+)(Im)(DMSO) (Scheme 1), which under studied reaction conditions undergoes subsequent hydrolysis. It is expected that release of the second chloride trans to NO from RuIICl3(NO+)(Im)(DMSO) is much faster than from RuIIICl3(Im)(H2O)(DMSO) species (Scheme 1).30,31
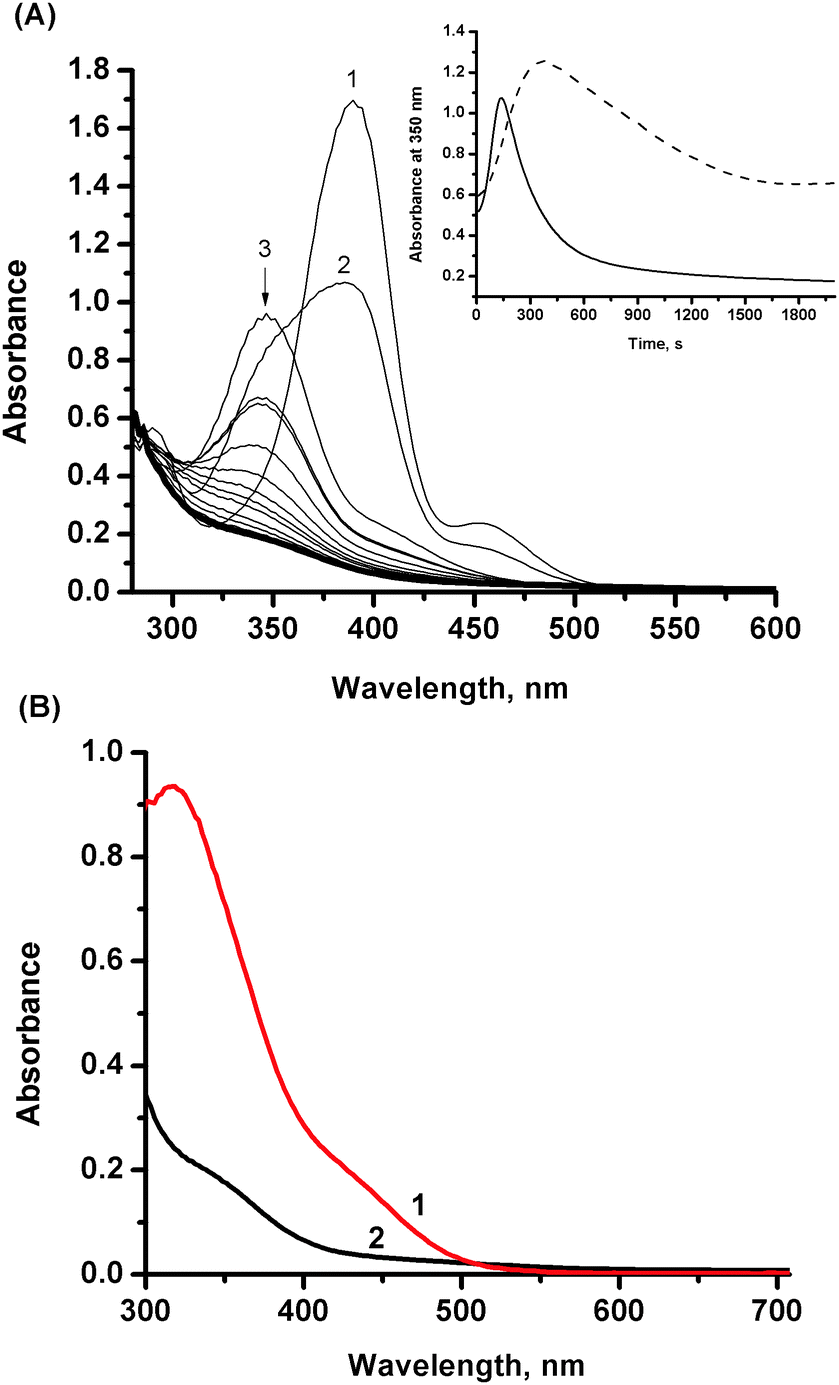 | ||
| Fig. 1 (A) Spectral changes observed after mixing of NO solution in buffer with an aqueous solution of NAMI-A monitored over 30 min. Inset: comparison of absorbance changes at 350 nm as a function of time for reaction of NAMI-A with NO (solid line) and hydrolysis of NAMI-A (dashed line). (B) Comparison of the spectrum of NAMI-A recorded after 2000 s in the absence (red line 1) and in the presence of NO (black line – 2). Experimental conditions: [NAMI-A] = 0.5 mM, [NO] = 1 mM, [NaCl] = 0.2 M, [Tris buffer] = 0.1 M, pH = 7.4, 37 °C. | ||
 | ||
| Scheme 1 Schematic representation of the main nitrosylation pathway of NAMI-A under physiological-like conditions. | ||
This is due to strong π-donation of Cl− which cannot be accepted by the RuII centre. The final reaction mixture is characterized by a broad band centred at ca. 350 nm and relatively intense bands in the UVC (200–280 nm) region. The occurrence of the band at 350 nm was ascribed to the presence of hydrolytic products remaining in the solution.
To obtain more information about the species that are formed during the nitrosylation reaction, the reaction products were analyzed by application of the HPLC technique. A saturated solution of NO (2 mM) in buffer (7.4) was mixed with a deoxygenated solution of NAMI-A (1 mM) in water under an inert atmosphere in the volume ratio 1:1 and incubated for 30 minutes at 37 °C. It was determined by UV-vis measurements that no further changes were observed in the spectra after 30 min. Subsequently, the reaction mixture was bubbled with argon in order to remove excess of unreacted nitric oxide and immediately injected into the column. The NO binding appears to be irreversible as the stream of argon does not shift the equilibrium to the substrates. The resulted elution profile registered at 250 nm is presented in Fig. 2 (red line – 1). In order to differentiate which peaks can be attributed to formation of new products in the reaction with NO, the reference sample was kept under the same conditions but in the absence of NO (Fig. 2, black line – 2). Under these conditions the hydrolysis of NAMI-A is promoted. In the chromatogram obtained after reaction of NAMI-A with NO, at least two major, new peaks are observed at retention times of 3.3 and 5.9 min denoted as products a and b (Fig. 2, red line – 1), respectively. The UV-vis spectra for these products are presented in the inset of Fig. 2, and are characterized by a band at ca. 325 nm and several intensive bands in the UVC (200–280 nm) region. Both spectra are very similar suggesting similar types of complexes. Additionally, several minor peaks are observed in the elution profile of reactants in the presence of NO and some of them overlap with peaks representing hydrolytic products in the reference sample. Analysis of UV-vis spectra of products revealed that except products with retention times at ca. 2.3 (not well-resolved double peak) and 7.3 min, all the other products (tr at 2.9, 3.4, 4.6 and 5.4) have the lowest energy band at ca. 325 nm. These spectra are dominated by the absorbance in the UVC (200–280 nm) region, pointing out to the formation of ruthenium–nitrosyl complexes. This is consistent with the change in the oxidation state of ruthenium upon binding of NO as observed for similar type of complexes.22 The expected d–d bands in the visible spectral range of the ruthenium–nitrosyl complexes were not detected due to too low concentration of the prepared complexes. In contrast a close inspection of the UV-vis spectra for the separated hydrolytic products (see Fig. 2. black line – 2) has revealed that they possess lower energy bands which occur above 340 nm (see ESI,† Fig. S2).
 | ||
| Fig. 2 Comparison of the elution profiles registered at 250 nm for NAMI-A kept for 30 min in buffer solution in the presence (1 – red line) and absence (2 – black line) of nitric oxide. Inset: UV-vis spectra for two major peaks observed on chromatogram 1 (a – tR = 3.3 min, b – tR = 5.9 min). Experimental conditions: [NAMI-A] = 0.5 mM, [NO] = 1 mM, [Tris buffer] = 0.1 M, pH = 7.4, [NaCl] = 0.2 M, 37 °C. | ||
The reaction product/s of NAMI-A with NO were also analyzed by FT-IR spectroscopy. A vibration for coordinated NO was found at 1879 cm−1 which is consistent with a linear diamagnetic (RuII–NO+) moiety (Fig. 3). This value is in good agreement with literature values reported for nitrosylated NAMI-A and other ruthenium–nitrosyl species.1,32
 | ||
| Fig. 3 FT-IR spectra registered for reaction products of NAMI-A with NO. Experimental conditions: [NAMI-A] = 18 mM, [Tris buffer] = 0.1 M, pH = 7.4, 37 °C, 0.2 M NaCl. | ||
It is important to note that nitric oxide undergoes fast oxidation to nitrogen dioxide (NO2) in the presence of oxygen which then undergoes hydrolysis to give nitrous acid (HNO2) under aqueous conditions. All experiments with application of NO were performed under inert conditions, however, to confirm that peaks observed on chromatograms registered after mixing the NAMI-A complex with NO come from nitrosyl derivatives and not from nitrite ones, NAMI-A solution was incubated for 30 min at 37 °C with NaNO2 in the concentration ratio 1:2. Chromatograms registered after a defined period of incubation were identical to the chromatograms obtained after the same time of hydrolysis of NAMI-A (results not shown). This observation indicates that registered spectral changes as well as chromatographically separated reaction products are indeed nitrosylation derivatives of the studied ruthenium complex.
Influence of hydrolysis of NAMI-A on its reactivity toward NO
The results presented in the previous chapter are performed under conditions that inhibit hydrolysis before addition of NO.33 Therefore, the initiated reactions comprise both the hydrolysis (due to introducing of buffer, which immediately changes the pH from ca. 5 to 7.4) and nitrosylation, which can occur simultaneously or consecutively. To assess the role of hydrolysis of the NAMI-A complex in its reactivity toward nitric oxide the nitrosylation reaction was performed at various stages of hydrolysis process development. The ruthenium complex pre-equilibrated with Tris buffer (which corresponds with consecutive hydrolysis steps) for 7 and 30 min was subjected to reaction with NO and the observed UV-vis spectral changes are presented in Fig. 4. In both cases disappearance of bands at 347 and 320 nm for hydrolyzed forms of NAMI-A complex is observed (Fig. 4, insets). Very similar UV-vis spectra obtained for freshly prepared as well as the pre-equilibrated for 7 min ruthenium complex reacting with NO (Fig. S4, ESI†) point out that, the nitrosylation of the NAMI-A complex is preceded by formation of the aqua derivative (small differences can arise from slightly higher amounts of the hydrolytic product in the pre-equilibrated mixture, see Scheme 1). The formation of the same products was also confirmed by chromatographic studies (see further discussion). In contrast, the UV-vis spectrum of the reaction mixture resulted from the addition of NO to the NAMI-A complex, pre-equilibrated with Tris buffer for 30 min, displays bands between 420 and 500 nm characteristic of some products of NAMI-A hydrolysis (see Fig. S1, ESI†). This means that nitrosylation of this mixture takes place only partially, and some products of hydrolysis have a rather weak affinity toward NO. The presence of hydrolytic products was also confirmed by HPLC studies discussed below.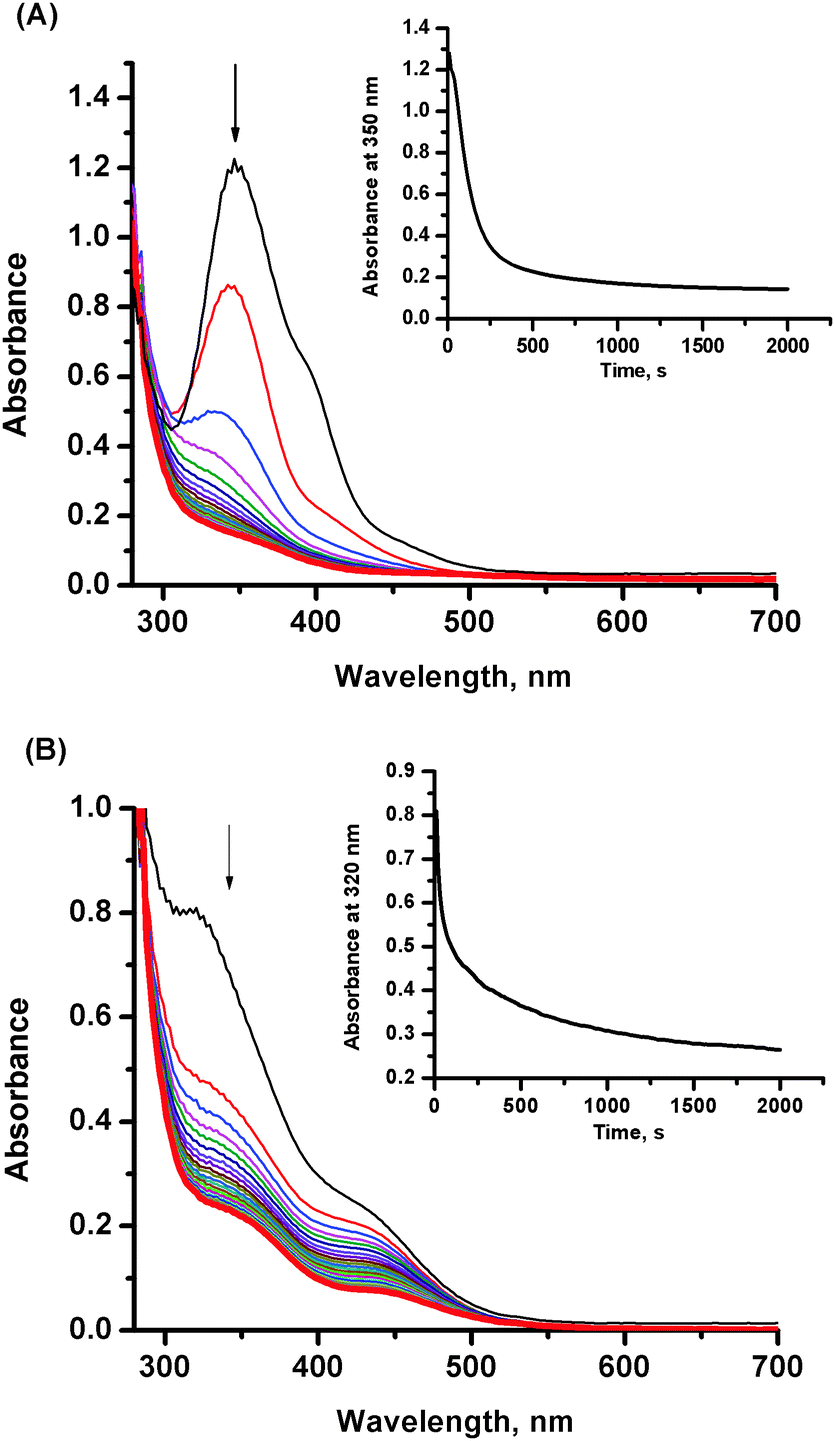 | ||
| Fig. 4 Spectral changes observed upon addition of NO to a solution of the NAMI-A complex after (A) 7 and (B) 30 min of hydrolysis. Experimental conditions: [NAMI-A] = 0.5 mM, [NO] = 1 mM, [NaCl] = 0.2 M, [Tris buffer] = 0.1 M, pH = 7.4, 37 °C. Insets: Absorbance changes as a function of time. | ||
In order to distinguish ruthenium–nitrosyl complexes from products of NAMI-A hydrolysis which occurs simultaneously with nitrosylation, the RP-HPLC analysis was performed for buffered solution of NAMI-A in the absence and the presence of NO (Fig. 5 and 6). An aqueous solution of the NAMI-A complex was mixed in the volume ratio 1:1 with Tris buffered solution (pH 7.4) and incubated at 37 °C. After 7 min of incubation in buffer the peak from the parent complex (at tr 7.3 min) almost completely disappeared, while the peak at a retention time of 3.6 min dominates in the chromatogram. It corresponds to the first product of hydrolysis namely, [RuCl3(DMSO)(H2O)(Im)]27 (Fig. 5B). Additionally, two peaks with much lower intensity appear at retention times of 2.3 and 2.5 min. Fast elution of these products suggests the formation of complexes with the dissociated DMSO ligand, which is in accordance with the suggested hydrolytic pathway.27 A longer incubation time with buffer leads to diminishing of the peak at 3.6 min and increase in the intensity of numerous less intensive peaks. After 30 min of incubation at 37 °C, intensity of the peaks at a retention time of 2.3–2.5 min (not well resolved) meaningfully increased and a new peak at 3.1 min and a broad peak at 4.7 min appeared (Fig. 5C). The obtained chromatographic profile indicates the presence of the complex mixture of ruthenium aqua derivatives upon hydrolysis, which is in good agreement with previous studies.26,27,29,34 The UV-vis spectra of most of the observed hydrolytic species are presented in the insets of Fig. 5. They are all characterized by bands in the region of 300–400 nm, which are not found for ruthenium nitrosyl derivatives (see Fig. S2 and S3, ESI†). This feature can help in identification of ruthenium nitrosyl complexes in the reaction mixture. In order to study the nitrosylation reaction, deoxygenated solutions of Tris buffer (0.1 M, pH 7.4) and unbuffered NAMI-A (2 mM) were mixed in the volume ratio 1:2 and incubated for 30 min at 37 °C. Afterwards, the samples were mixed with NO (resulting in a 1:2 concentration ratio of Ru to NO) and incubated for 30 min at 37 °C. The chromatograms with detection at 250 nm obtained for the sample pre-incubated in buffer together with the sample which was not pre-incubated in buffer are presented in Fig. 6. The reaction of NO with the pre-equilibrated NAMI-A complex for 7 min leads to formation of the same set of products as with freshly prepared one (Fig. S4, ESI†). This observation further supports the assumption, that NO binding to ruthenium in the NAMI-A complex requires formation of an aqua derivative of NAMI-A prior to coordination of NO. This conclusion stays in agreement with the faster nitrosylation reaction in water than in nitromethane observed for trans-[RuCl4(DMSO)2]− by Serli et al.22 The peak at retention time at 5.9 minutes has no equivalent at chromatograms for the NAMI-A complex at various stages of hydrolysis and was ascribed to the main nitrosylation product (see Fig. S3, ESI†). Accumulation of this product, in the reaction mixture, decreases with increasing pre-incubation time since [RuCl3(DMSO)(H2O)(Im)] is no longer available for nitrosylation, which is a consequence of progressive hydrolysis of NAMI-A. Pre-incubation longer than 30 min leads to complete disappearance of the peak at 5.9 min with concomitant increase of the intensity of several other peaks ascribed to nitrosyl derivatives of NAMI-A hydrolytic products (Fig. 6). The nitrosylation of this mixture occurs to a lesser extent as is manifested by the presence of a higher amount of hydrolytic products in comparison to reaction performed with the NAMI-A complex freshly prepared or pre-equilibrated for 7 min (compare Fig. 6 and Fig. S3, ESI†).
 | ||
| Fig. 5 Elution profiles for products of NAMI-A hydrolysis after (A) 0 min, (B) 7 min, (C) 30 min of incubation with buffer. Insets: UV-vis spectra corresponding to separated reaction products. Experimental conditions: [NAMI-A] = 0.5 mM, [Tris buffer] = 0.1 M, pH = 7.4, [NaCl] = 0.2 M, 37 °C. | ||
 | ||
| Fig. 6 Representative chromatograms registered at 250 nm after incubation of the NAMI-A complex with NO for 30 min. Experimental conditions: [NAMI-A] = 0.5 mM, [NO] = 1 mM in [Tris buffer] = 0.1 M, [NaCl] = 0.2 M, pH = 7.4; NAMI-A solution directly mixed with NO (0 min) and incubated with buffer for 7, 30 and 60 min before mixing with NO. | ||
NO-release
Previous studies on NO coordination to various Ru–DMSO-type complexes revealed formation of a diamagnetic Ru(II) centre bound to NO+.14 Additionally, electrochemical measurements performed on a series of Ru–nitrosyl complexes similar in structure to NAMI-A showed that these types of ruthenium complexes are redox active on the NO+ moiety and one electron reduction leads to NO liberation (as an example see Scheme 2).14,22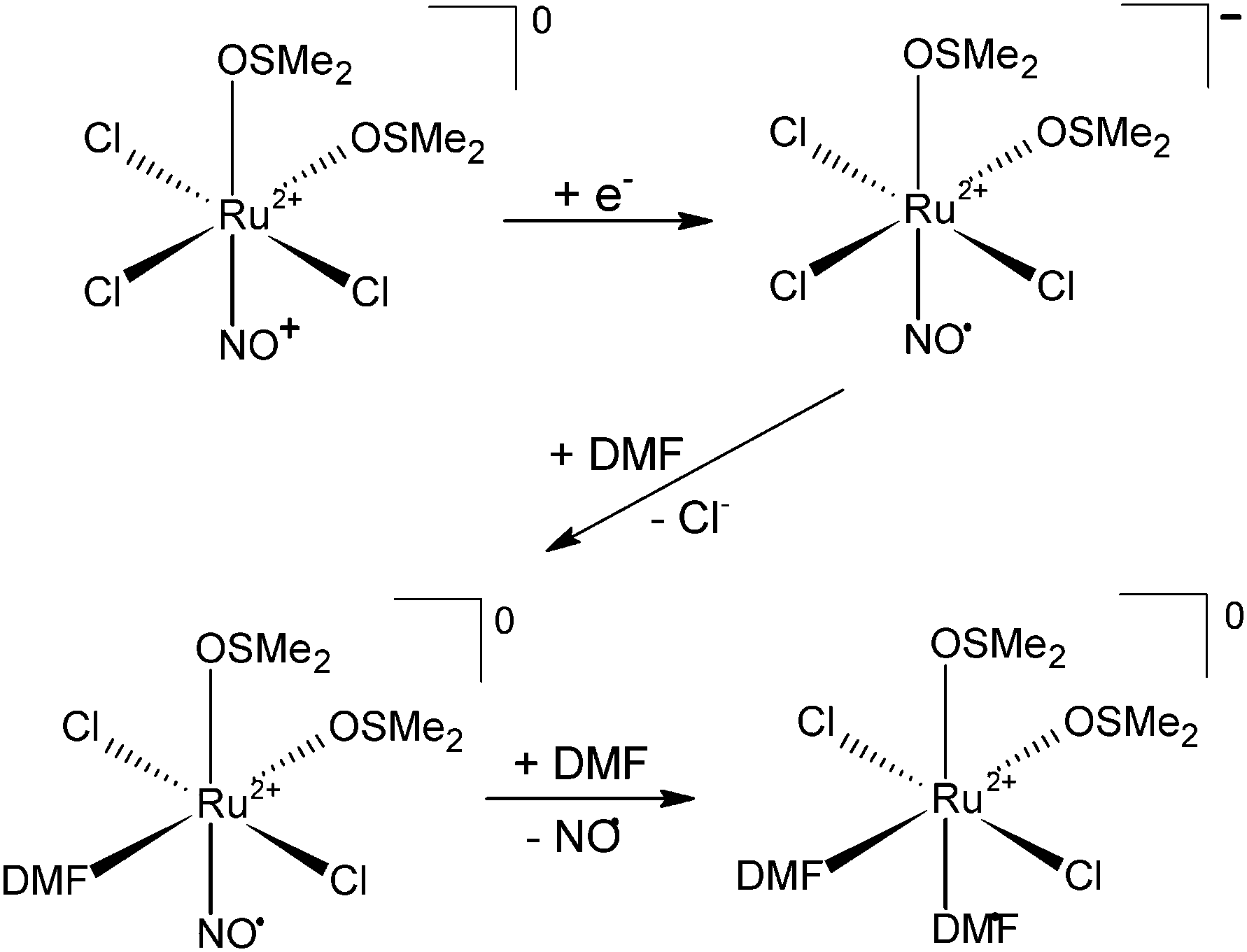 | ||
| Scheme 2 Schematic representation of NO liberation via one electron reduction. | ||
This gave rise to a hypothesis that the antimetastatic mechanism of action of NAMI-A and related Ru–DMSO compounds may be due to interference with NO metabolism through nitric oxide binding and following release upon reduction in vivo. It is important to note that these studies were carried out in non-aqueous aprotic media (DMF) thus these results cannot be directly referred to physiological conditions.14,22 In order to verify this premise we carried out direct reduction studies with application of biologically relevant reductants: ascorbic acid, glutathione and sodium dithionite. It was expected that addition of the reductant into the solution containing (RuII–NO+) species results in the formation of NO, which has much lower affinity to the Ru(II) centre than NO+.13,21 To measure NO production, an amino-700 nitric oxide sensor connected to an inNO-T nitric oxide measuring system (Innovative Instruments, Inc.) was used. The systematic titration of the nitrosyl complex with the selected reductants has been performed. However, addition of relatively high excess of ascorbic acid or glutathione into the studied reaction mixture did not result in increase of electrical current measured by the NO sensor. Similarly the application of a much stronger reductant such as dithionite did not result in the release of coordinated NO. Moreover we performed electrochemical studies of Ru–nitrosyl complexes obtained after reaction of the NAMI-A complex with NO. The DPV (Differential Pulse Voltammetry) and CV (Cyclic Voltammetry) measurements were performed after 30 min. Incubation of NAMI-A with NO. In a typical experiment, an aqueous solution of NAMI-A was mixed with buffered NO solution or pre-incubated in Tris buffer, pH = 7.4, 0.2 M NaCl at 37 °C prior to mixing with NO. The registered CV voltammogram exhibits an irreversible reduction wave (Fig. 7). The peak potential of reduction was measured at −0.69 V vs. NHE and assigned to the reduction of the NO+ group bound to Ru(II). The irreversibility (there is no sign of a return wave even at a scan rate up to 1 V s−1) of the electron transfer reaction indicates that it is followed by rapid chemical reaction.
 | ||
| Fig. 7 Cyclic Voltammetric response after mixing of the NAMI-A complex with Tris buffer (pH = 7.4) containing NO. Experimental conditions: [NAMI-A] = 0.5 mM, [Tris buffer] = 0.1 M, pH = 7.4, [NaCl] = 0.2 M, [NO] = 1 mM, T = 37 °C, scan rate 20 mV s−1. | ||
Previous electrochemical studies showed that products of NAMI-A hydrolysis have redox potentials in the range 0.187–0.597 V.27 None of the waves observed during NAMI-A hydrolysis were registered after complex incubation with NO (see Fig. S5, ESI†) indicating lack of the electrochemically active Ru(III) centre in the detectable concentration range. This indicates that formed product species are not electrochemically active in the potential range accessible for biological reductants such as ascorbic acid or glutathione.
NO binding in the presence of albumin
Since the NAMI-A complex readily binds to the plasma proteins after intravenous administration35 we checked its ability to bind NO when it was present in the form of NAMI-A–albumin adducts. In a typical experiment, a freshly prepared solution of NAMI-A was mixed with a buffered solution of albumin containing 1 mM NO or alternatively NAMI-A was incubated with albumin for 30 min (37 °C, Tris buffer pH 7.4) prior to mixing with NO. After 30 min of incubation of NAMI-A–albumin adducts with NO at 37 °C, the loss of colour was observed. This observation is characteristic of reduction of the ruthenium atom from Ru(III) to Ru(II) and can be ascribed to formation of (RuII–NO+) species. To confirm this assumption the solution after reaction was studied electrochemically. Differential Pulse Voltammetry did not show any significant electrochemical response in the potential range −170–800 mV indicating lack of electrochemically active Ru(III) species in the solution (Fig. S6, ESI†).27 This was in contrast to voltammograms obtained for samples of NAMI-A incubated with albumin without NO which showed typical electrochemical response for products of NAMI-A hydrolysis since the presence of albumin does not considerably modify the trend in the development of voltammograms (Fig. S6, ESI†).27Conclusions
The research presented herein sheds more light on the interaction of nitric oxide with the NAMI-A complex under physiological-like conditions (pH = 7.4, [NaCl] = 0.1 M, T = 37 °C). The performed studies take under consideration the instability of NAMI-A under physiological conditions and include interaction with NO not only of the parent NAMI-A complex but also products of its hydrolysis. The obtained results indicate that (i) the antimetastatic ruthenium(III) complex reacts relatively slowly with NO and requires opening up the coordination site via hydrolysis (Scheme 3); (ii) most of the hydrolytic products of NAMI-A are able to bind NO; (iii) the formation of NAMI-A–albumin adducts does not prevent coordination of NO to the ruthenium centre (Scheme 3); (iv) the Ru–nitrosyl derivatives which are formed are not able to release NO via a reduction process with reductants such as ascorbic acid and glutathione and dithionite (Scheme 3); (v) electrochemically determined reduction potential of the NO+ group bound to Ru(II) is −0.69 mV (vs. NHE).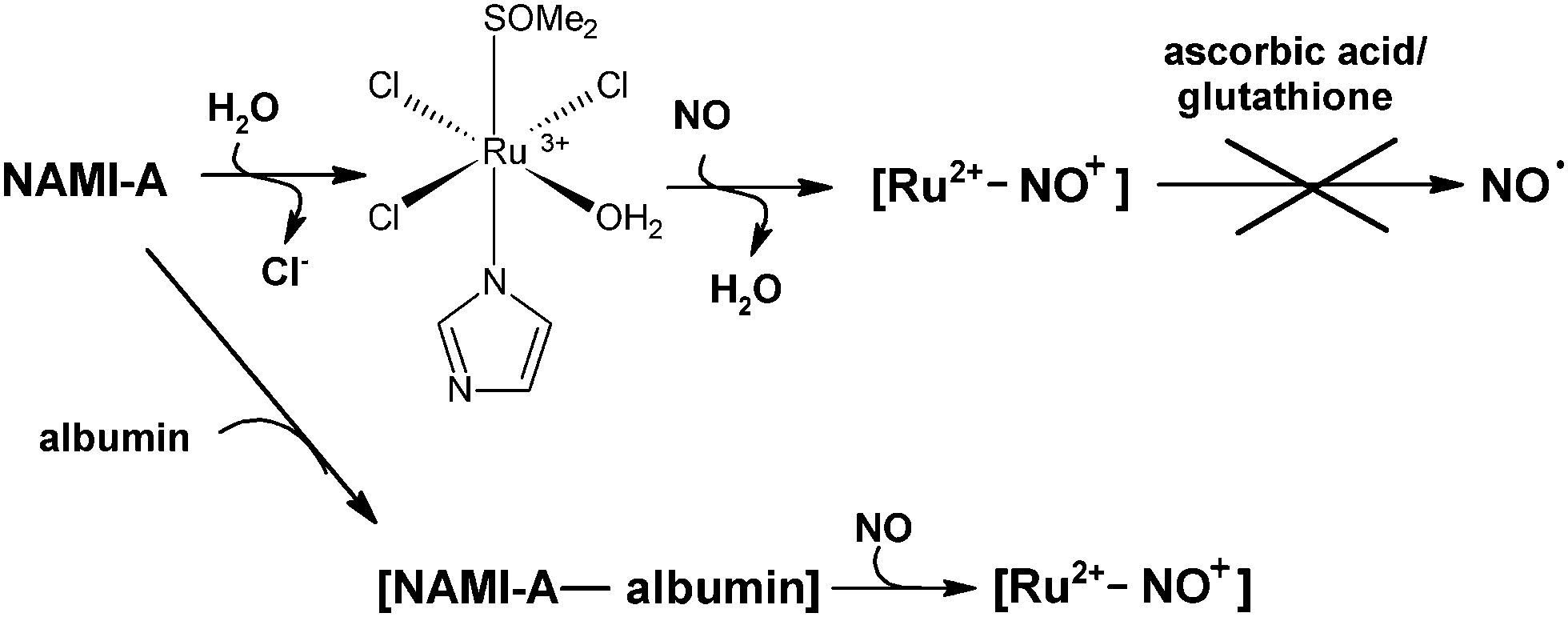 | ||
| Scheme 3 Summary of the reactivity of NAMI-A towards NO. | ||
Presented results pointed out that NAMI-A may interfere under physiological conditions in NO metabolism by coordinating NO. However, a relatively high negative reduction potential for nitrosyl derivatives indicates that reduction of NO+ to NO is rather thermodynamically unfavoured under physiological conditions and generates doubt about the previously suggested ability of NAMI-A nitrosyl derivatives to release NO via one electron reduction. An assumption that NAMI-A may release bonded nitric oxide has been based on electrochemical studies performed in non-aqueous aprotic media and its reconsideration under aqueous conditions was important from the viewpoint of molecular mechanism of action of NAMI-A.
Acknowledgements
This work was supported by a grant from the National Science Centre (2012/05/N/ST5/01125). This work was carried out using the equipment purchased through financial support from the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08). M.O. acknowledges the financial support from the International PhD Study Program at the Faculty of Chemistry, Jagiellonian University, operated within the Foundation for Polish Science MPD Program co-financed by the European Regional Development Fund (MO). We thank Dr hab. Konrad Szaciłowski prof. AGH for support in electrochemical experiments.References
- L. Morbidelli, S. Donnini, S. Filipi, L. Messori, F. Piccioli, G. Sava and M. Ziche, Br. J. Cancer, 2003, 88, 1484–1491 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. L. Lay, Metallomics, 2009, 1, 458–470 RSC , and references therein.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed , and references therein.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 CrossRef CAS.
- G. Sava, E. Alessio, A. Bergamo and G. Mestroni, Top. Biol. Inorg. Chem., 1999, 1, 143 CAS.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, R. Sartor and S. Garbisa, Clin. Cancer Res., 2003, 9, 1898–1905 CAS.
- B. Gava, S. Zorzet, P. Spessotto, M. Cocchietto and G. Sava, J. Pharmacol. Exp. Ther., 2006, 317, 284–291 CrossRef CAS PubMed.
- S. Zorzet, A. Sorc, C. Casarsa, M. Cocchietto and G. Sava, Met.-Based Drugs, 2001, 8, 1–7 CrossRef CAS PubMed.
- G. Sava, I. Capozzi, K. Clerici, G. Gagliardi, E. Alessio and G. Mestroni, Clin. Exp. Metastasis, 1998, 16, 371–379 CrossRef CAS.
- G. Sava, K. Clerici, I. Capozzi, M. Cocchietto, R. Gagliardi, E. Alessio, G. Mestroni and A. Perbellini, Anti-Cancer Drugs, 1999, 10, 129–138 CrossRef CAS PubMed.
- G. Sava, R. Gagliardi, M. Cocchietto, K. Clerici, I. Capozzi, M. Marrella, E. Alessio, G. Mestroni and R. Milanino, Pathol. Oncol. Res., 1998, 4, 30–36 CrossRef CAS.
- E. Tfouni, D. R. Truzzi, A. Tavares, A. J. Gomes, L. E. Figueiredo and D. W. Franco, Nitric Oxide, 2012, 26, 38–53 CrossRef CAS PubMed.
- B. Serli, E. Zangrando, T. Gianferrara, L. Yellowlees and E. Alessio, Coord. Chem. Rev., 2003, 245, 73–83 CrossRef CAS.
- D. Fukumura, S. Kashiwagi and R. K. Jain, Nat. Rev. Cancer, 2006, 6, 521–534 CrossRef CAS PubMed , and references therein.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- A. K. Bytzek, K. Boeck, G. Hermann, S. Hann, B. K. Keppler, C. G. Hartinger and G. Koellensperger, Metallomics, 2011, 3, 1049–1055 RSC.
- T. Storr, B. R. Cameron, R. A. Gossage, H. Yee, R. T. Skerlj, M. C. Darkes, S. P. Fricker, G. J. Bridger, N. A. Davies, M. T. Wilson, K. P. Maresca and J. Zubieta, Eur. J. Inorg. Chem., 2005, 2685–2697 CrossRef CAS.
- L. Bucinský, G. E. Büchel, R. Ponec, P. Rapta, M. Breza, J. Kozísek, M. Gall, S. Biskupic, M. Fronc, K. Schiessl, O. Cuzan, D. Prodius, C. Turta, S. Shova, D. A. Zajc and V. B. Arion, Eur. J. Inorg. Chem., 2013, 2505–2519 CrossRef.
- J. C. Toledo, B. S. L. Neto and D. W. Franco, Coord. Chem. Rev., 2005, 249, 419–431 CrossRef CAS PubMed , and references therein.
- B. Serli, E. Zangrando, E. Iengo, G. Mestroni, L. Yellowlees and E. Alessio, Inorg. Chem., 2002, 41, 4033–4043 CrossRef CAS PubMed.
- D. R. Lang, J. A. Davis, L. G. F. Lopes, A. A. Ferro, L. C. G. Vasconcellos, D. W. Franco, E. Tfouni, A. Wieraszko and M. J. Clarke, Inorg. Chem., 2000, 39, 2294–2300 CrossRef CAS.
- J. M. Slocik, M. S. Ward, K. V. Somayajula and R. E. Shepherd, Transition Met. Chem., 2001, 26, 351–364 CrossRef CAS.
- J. M. Slocik and R. E. Shepherd, Inorg. Chim. Acta, 2000, 311, 80–94 CrossRef CAS.
- M. Bacac, A. C. G. Hotze, K. van der Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402–412 CrossRef CAS PubMed.
- M. Brindell, I. Stawoska, J. Supel, A. Skoczowski, G. Stochel and R. van Eldik, J. Biol. Inorg. Chem., 2008, 13, 909–918 CrossRef CAS PubMed.
- G. Mestroni, E. Alessio and G. Sava, Int. Pat., WO 98/00431 1998 Search PubMed.
- M. Bouma, B. Nuijen, M. T. Jansen, G. Sava, A. Flaibani, A. Bult and J. H. Beijnen, Int. J. Pharm., 2002, 248, 239–246 CrossRef CAS.
- C. B. W. Bezerra, S. C. da Silva, M. T. P. Gambardella, R. H. A. Santos, L. M. A. Plicas, E. Tfouni and D. W. Franco, Inorg. Chem., 1999, 38, 5660–5667 CrossRef CAS.
- J. C. Toledo, H. A. S. Silva, M. Scarpellini, V. Mori, A. J. Camargo, M. Bertotti and D. W. Franco, Eur. J. Inorg. Chem., 2004, 1879–1885 CrossRef CAS.
- D. Griffith, S. Cecco, E. Zangrando, A. Bergamo, G. Sava and C. J. Marmion, J. Biol. Inorg. Chem., 2008, 13, 511–520 CrossRef CAS PubMed.
- O. Mazuryk, K. Kurpiewska, K. Lewiński, G. Stochel and M. Brindell, J. Inorg. Biochem., 2012, 116, 11–18 CrossRef CAS PubMed.
- M. Webb and C. Walsby, Dalton Trans., 2011, 40, 1322–1331 RSC.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral changes observed during the hydrolysis of NAMI-A. Comparison of the chromatograms registered for NAMI-A in buffer solution in the presence and in the absence of nitric oxide and differential pulse voltammograms. See DOI: 10.1039/c3nj01631e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |