 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and properties of metal complexes of a pyridine based oxazolidinone synthesized by atmospheric CO2 fixation†
Amrita
Sarkar
,
Sudipta
Bhattacharyya
,
Suman Kr
Dey
,
Subhendu
Karmakar
and
Arindam
Mukherjee
*
Department of Chemical Sciences Indian Institute of Science Education & Research Kolkata, Mohanpur Campus, P.O.-BCKV Main Campus, District-Nadia, 741252, India. E-mail: a.mukherjee@iiserkol.ac.in; Fax: +91-033-25873118; Tel: +91-033-25873121
First published on 6th January 2014
Abstract
We present here the synthesis of a pyridine containing oxazolidinone, 3-(pyridin-2-ylmethyl)oxazolidin-2-one (L) through the fixation of atmospheric CO2 and without any added base. L has been used to generate three metal complexes [Cu(L)2(ClO4)2] (1), trans-[Pt(L)(DMSO)Cl2] (2), and trans-[Pd(L)2Cl2] (3). Complexes 1–3 have been structurally characterized using single crystal X-ray diffraction and other analytical techniques. The activity studies of the three metal complexes show that the CuII complex, 1, is more cytotoxic against the MCF-7 cancer cell line (IC50 = 114 ± 2 μM) as compared to a non-carcinogenic mouse fibroblast (NIH 3T3) (IC50 = 198 ± 1 μM). The PtII complex, 2, is the most toxic among the three complexes against both human breast adenocarcinoma (MCF-7) (IC50 = 102 ± 2 μM) and the human lung adenocarcinoma epithelial cell line (A549) (IC50 = 198 ± 2 μM), whereas the PdII complex, 3, is found to be an effective first generation oxazolidinone based PdII catalyst, for the Suzuki–Miyaura cross coupling of aryl halides and phenylboronic acids in aerobic conditions, both in a conventional heating method, as well as a microwave method. The results show that although PtII and CuII were also complexed with oxazolidinone, PdII seems to be the most likely choice of metal when it comes to C–C bond coupling via C–X bond activation.
Introduction
Oxazolidinone compounds are of high interest for their potential as antimicrobial agents. Linezolid, the first oxazolidinone compound to be approved for clinical use, shows excellent antimicrobial activity against many important resistant pathogens. Eperezolid, radezolid, posizolid, and torezolid are some of the oxazolidinone class of compounds (Scheme 1), under clinical investigation, that are potent against bacterial infections. Cycloserine, an oxazolidinone derivative, is used for the treatment of tuberculosis when one or more drugs fails to treat the disease. | ||
| Scheme 1 Structure of oxazolidinone based drugs. | ||
Apart from being antimicrobial compounds, oxazolidinone derivatives also are potential ligands from which to generate metal complexes. Metal complexes of oxazolidinones are known and have been used in catalysis viz. the Diels–Alder reaction,1–3 aldol reaction,4–6 Henry reaction,7 Mukaiyama–Michael reaction,8 diamination of alkenes9,10 or to generate MOFs11 mostly depending on the choice of metal and the coordination environment rendered. Although oxazolidinone based metal complexes have been used in catalysis, C–C bond coupling via C–X bond activation has never been attempted with oxazolidinone metal complexes. In addition, despite being of high therapeutic interest, oxazolidinones metal complexes have not been effectively probed as therapeutic agents (viz. antimicrobial or anticancer agents).12–14
From a synthetic point of view, oxazolidinone compounds can be obtained in various ways viz. using urea or epoxides, along with amines,15–19 or from ethanolamine by purging CO2 gas, using the Mitsunobu reaction.20 There are also a few reports on the syntheses of oxazolidinones from amino alcohols with purging CO2 gas and using electrophiles and bases.21–25 However, the synthesis of oxazolidinone using air (which contains CO2) is not reported, although it should be possible, since air has a significant CO2 content. In addition pyridine containing oxazolidinones are a rare find26,27 and no synthesis of pyridine containing oxazolidinone has been reported through the fixation of atmospheric CO2 (Scheme 2). If by using air, the atmospheric CO2 content could undergo reaction efficiently, then the lack of CO2 pressurized cylinders would represent a more convenient way of fixing CO2, through organic synthesis. Our interest in pyridine based oxazolidinones is due to their potential for metal chelation and possible use in therapeutics.28–34
 | ||
| Scheme 2 Synthetic method of pyridine based oxazolidinone compounds reported earlier and ligand L in this work. | ||
In our attempt to generate a pyridine based oxazolidinone, we synthesized 3-(pyridin-2-ylmethyl)oxazolidin-2-one (L) in the presence of SOCl2 (Scheme 2) using air (atmospheric CO2) and without any added base. L is also a potential metal binder and hence, three metal complexes of the formulae [Cu(L)2(ClO4)2] (1), trans-[Pt(L)(DMSO)Cl2] (2), and trans-[Pd(L)2Cl2] (3) were synthesized, to probe the potential of the newly designed pyridyl oxazolidinone ligand. Herein, we present the synthesis, structure and activity of the above three metal complexes, along with in vitro cytotoxicity studies against cancerous (MCF-7 and A549) and non-cancerous mouse fibroblast (NIH 3T3) cell lines.
Our work shows how the oxazolidinone and metal ions (Cu, Pt, Pd) influence each other’s chemistry, rendering the desired properties to the resultant complexes. 1 and 2 show toxicity against cancer cell lines, whilst the CuII complex (1) is less toxic in the non-carcinogenic NIH 3T3 cell line. The palladium complex (3) is found to be a catalyst for the Suzuki–Miyaura coupling of aryl halides and phenylboronic acids in aerobic conditions, both in a conventional heating method as well as a microwave method. The results show that a metal complex of an oxazolidinone has the potential as a C–C bond cross coupling catalyst, or in therapeutics.
Results
Preparation of ligands and complexes
The three metal complexes synthesized with 3-(pyridin-2-ylmethyl)oxazolidin-2-one (L) bear different coordination environments (Scheme 3). Complexes 1 and 3, having CuII and PdII, react with two mole equivalent of L, rendering complexes of the type ML2. Whereas, the PtII containing complex 2 was formed by reaction with only one mole equivalent of the oxazolidinone ligand, in spite of having two mole equivalents of ligand per metal ion in the reaction solution. Once the composition became known we tuned the stoichiometry accordingly, providing a cleaner reaction and a better yield for 2. | ||
| Scheme 3 Representative scheme for syntheses of metal complexes 1–3. | ||
X-ray crystallographic study of complexes 1–3
The complexes 1–3 were characterized by single crystal X-ray crystallography. Complex 1 (Fig. 1) crystallizes in the monoclinic space group P2(1)/c, and 2 in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . 3 crystallized in the monoclinic space group P2(1). However, although the crystal structure provides information about the structural nature of complex 3, is not publishable, due to relatively poor quality of the data. Single crystals suitable for X-ray crystallography were grown by the slow evaporation of an acetonitrile–methanol mixture (1
. 3 crystallized in the monoclinic space group P2(1). However, although the crystal structure provides information about the structural nature of complex 3, is not publishable, due to relatively poor quality of the data. Single crystals suitable for X-ray crystallography were grown by the slow evaporation of an acetonitrile–methanol mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for the complex 1. For 2 (Fig. 2), layering a dichloromethane solution with hexane gave yellow needle shaped crystals, suitable for X-ray diffraction, whereas complex 3 gave orange coloured, block shaped crystals on the slow evaporation of an acetonitrile solution. The important crystallographic parameters for 1–3 and selected bond distances and angles of 1 and 2 have been summarised in Table S1 (ESI†) and Table 1.
:1) for the complex 1. For 2 (Fig. 2), layering a dichloromethane solution with hexane gave yellow needle shaped crystals, suitable for X-ray diffraction, whereas complex 3 gave orange coloured, block shaped crystals on the slow evaporation of an acetonitrile solution. The important crystallographic parameters for 1–3 and selected bond distances and angles of 1 and 2 have been summarised in Table S1 (ESI†) and Table 1.
 | ||
| Fig. 1 Diagram of complex 1 with 50% probability thermal ellipsoids. Hydrogen atoms have been omitted for clarity. Symmetry transformations used to generate equivalent atoms: A = −x + 1, −y, −z + 2. | ||
 | ||
| Fig. 2 Diagram of 2 showing one of two independent molecules in the asymmetric unit with 50% probability thermal ellipsoids. Hydrogen atoms and solvents have been omitted for clarity. | ||
| 1 | 2 | ||
|---|---|---|---|
| a A = −x + 1, −y, −z + 2 for 1. | |||
| Cu(1)–N(1) | 1.987(2) | Pt(1)–N(1) | 2.057(3) |
| Cu(1)–O(1) | 1.973(2) | Pt(1)–S(1) | 2.2183(9) |
| Cu(1)–O(6) | 2.75(2) | Pt(1)–Cl(1) | 2.3025(8) |
| N(1)–Cu(1)–N(1A)a | 180 | Pt(1)–Cl(2) | 2.2965(9) |
| O(1)–Cu(1)–O(1A)a | 180 | Pt(2)–N(3) | 2.049(3) |
| O(1)–Cu(1)–N(1) | 93.75(9) | Pt(2)–S(2) | 2.2182(8) |
| O(1)–Cu(1)–N(1A)a | 86.25(9) | Pt(2)–Cl(3) | 2.3019(9) |
| N(1)–Cu(1)–O(6) | 95.52(9) | Pt(2)–Cl(4) | 2.3145(9) |
| O(1)–Cu(1)–O(6) | 107.66(8) | N(1)–Pt(1)–S(1) | 178.16(8) |
| N(1A)a–Cu(1)–O(6) | 84.48(9) | N(1)–Pt(1)–Cl(1) | 88.22(8) |
| O(1A)a–Cu(1)–O(6) | 72.34(8) | S(1)–Pt(1)–Cl(1) | 90.66(3) |
| N(1)–Pt(1)–Cl(2) | 88.27(8) | ||
| S(1)–Pt(1)–Cl(2) | 92.97(3) | ||
| Cl(1)–Pt(1)–Cl(2) | 174.09(3) | ||
| N(3)–Pt(2)–S(2) | 179.13(8) | ||
| N(3)–Pt(2)–Cl(3) | 87.30(8) | ||
| S(2)–Pt(2)–Cl(3) | 92.14(3) | ||
| N(3)–Pt(2)–Cl(4) | 88.33(8) | ||
| S(2)–Pt(2)–Cl(4) | 92.26(3) | ||
| Cl(3)–Pt(2)–Cl(4) | 175.23(3) | ||
The copper complex 1 is ML2 type, with the N2O2 coordination bearing two weak axial linkages (ca. 2.75 Å, Fig. 1), with two oxygen atoms of two perchlorate anions making the co-ordination environment an elongated octahedral type (Fig. 1). Whereas, complex 2 showed the PtII to be in a trans geometry, with one L per molecule (Fig. 2). The trans-PtII in complex 2 is coordinated via the pyridine N. Unlike the CuII complex, the carbonyl oxygen is not bound to PtII. There are two trans Cl− ligands along with a DMSO, bound to the PtII centre via the sulphur atom. On the other hand complex 3 is also a trans complex of PdII, with two ligands trans to each other and two chlorides as found from the crystallographic studies, but the structural details are not presented, here since the data obtained do not render a fully publishable structure.
Lipophilicity measurement
The partition coefficients between octanol and an aqueous buffer layer, to determine the lipophilicity of the three complexes (1–3), were calculated using the well known formula of logP = Coct/Caq, where Coct was the concentration of the complex in the organic layer and Caq was the concentration of the complex in the phosphate buffer (20 mM) found through UV-visible spectroscopy. The results (Fig. 3 and Table 2) show the lipophilicity, or the logPo/w for the complexes 1–3. We found that all three complexes are hydrophilic in nature and complex 3 is the most hydrophilic among the three complexes. The lipophilicity order is 1 > 2 > 3.
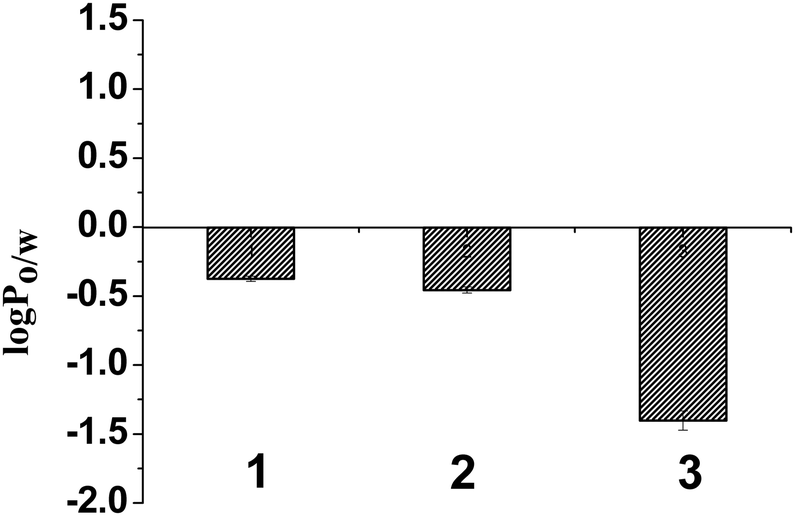 | ||
| Fig. 3 Lipophilicity of complexes 1–3 represented by a bar diagram showing comparative logP values of 1–3 in octanol–water system, where error bars in the graph represent the standard deviation in measurement. | ||
P values of complexes 1–3 in octanol–water system
Cell cytotoxicity
The complexes 1–3 were tested in vitro against a human breast adenocarcinoma cell line (MCF-7) and a human lung adenocarcinoma epithelial cell line (A549) as well as a mouse embryonic fibroblast cell line (NIH 3T3) by the MTT assay, where cisplatin was used as a standard in each 96 well plate. The data show that the ligand L has an IC50 value greater than 1000 μM in the MCF-7 cell line, so it was not probed in the A549 and NIH 3T3 cell lines. Among the three metal complexes, the trans-platinum complex 2 shows the highest cytotoxicity in the MCF-7 cell line (IC50 = 102 ± 2 μM), whereas in the A549 cell line, the IC50 is 198 ± 2 μM. The IC50 of the CuII complex 1 in MCF-7 is 114 ± 2 μM and in A549 it is 455 ± 3 μM, respectively. In the MCF 7 cell line, the palladium complex 3 shows an IC50 260 ± 3 μM, but in the A549 cell line, it is 236 ± 1 μM. In the non-cancerous NIH 3T3 cell line, the copper complex 1 is less toxic, with an IC50 of 198 ± 1 μM, as compared to the MCF-7 cancer cell line. The same is not true for the well known drug cisplatin or the new synthons, complexes 2 and 3 reported here (Table 3). The obtained data were plotted and fitted using GraphPad Prism 5® Ver 5.03 (Fig. S1–S3, ESI†). The IC50 values are summarised in Table 3.| Compounds | IC50 (μM) ± SDa | ||
|---|---|---|---|
| MCF-7 | A549 | NIH 3T3 | |
| a SD = standard deviation; IC50 values were calculated by variable slope model using GraphPad Prism 5®. 6 × 103 cells per well were treated for 48 h with increasing concentrations of tested compounds. b On drug exposure of 72 h.35 | |||
| L | >1000 | n.d. | n.d. |
| 1 | 114 ± 2 | 455 ± 3 | 198 ± 1 |
| 2 | 102 ± 2 | 198 ± 2 | 41 ± 1 |
| 3 | 260 ± 3 | 236 ± 3 | 151 ± 1 |
| Cisplatin | 14 ± 1 | 21 ± 2 | 11 ± 2 |
| Carboplatinb | 62.19 | — | — |
| Cyclophosphamide | >3000 | n.d. | >3000 |
Suzuki–Miyaura cross coupling catalyzed by palladium complex (3)
The Suzuki–Miyaura coupling reactions between various aryl halides and phenylboronic acid catalyzed by the complex 3 were carried out both with a conventional heating method, as well as a microwave method. All the reactions were carried out in atmospheric conditions, as the palladium catalyst is stable in air. For both cases, the temperature was 80 °C. We optimized the solvent system by varying the solvents among water, ethanol and acetonitrile. Ethanol was preferred as the solvent, since the precipitation of palladium black, which may indicate the degradation of the catalyst, was less. The catalytic results were also good in water, but the precipitation of palladium black during catalysis was more in an aqueous medium (Table S2, ESI†). The reaction in the presence of various bases provided the information that the best results were obtained in the presence of Cs2CO3 (Table 4). The test reaction used in these cases was the cross coupling between 4-bromoanisole and phenylboronic acid (Table 4).
Hence forth, Cs2CO3 was used as the base and the coupling reactions were performed using various aryl halides with phenylboronic acid in an EtOH medium, by both the conventional heating method and microwave irradiation methods. The catalyst loading was varied from 0.2 mol% to 1 mol%, with respect to the aryl halides. The results are tabulated in Tables 5 and 6. The results show that the catalytic activity depends on the nature of the aryl halides, which is a well known fact.36 With variation of the substitution in the aryl ring, the rate of reaction also varied significantly i.e. 4-bromoacetophenone gave complete conversion to its corresponding biaryl in an hour using 0.5% catalyst at 80 °C (Table 5, entry 5), whereas 4-bromoanisole needed 2 hours for complete conversion, using 1 mol% catalyst and 80 °C (Table 5, entry 4). A similar trend was observed using the microwave method. We found that the microwave method improved the TOF, since the reaction completion time decreased (Table 6). The TOF (turn over frequency) was calculated using the formula, TOF (h−1) = (no of moles of the product formed)/{(no of moles of the catalyst involved) × (reaction time for completion in hour)}.37 The yields reported in the table are the isolated yields, after individual column chromatography.
| Entry | Aryl halide | Catalyst loading (mol%) | Time | Isolated yield (%) | TOFb |
|---|---|---|---|---|---|
| a Reaction conditions: aryl halides (0.5 mmol), phenylboronic acid (0.0915 g, 0.75 mmol), Cs2CO3 (0.48 g, 1.5 mmol) EtOH (5 mL). b TOF (h−1). | |||||
| 1 |

|
0.2 | 20 min | >99 | 1515 |
| 2 |

|
0.2 | 30 min | >99 | 1000 |
| 3 |

|
0.2 | 30 min | >99 | 1000 |
| 4 |

|
1.0 | 2 h | 92 | 46 |
| 5 |

|
0.5 | 1 h | 97 | 194 |
| 6 |

|
0.5 | 1 h | 99 | 198 |
| 7 |

|
0.5 | 30 min | 95 | 380 |
| 8 |

|
1.0 | 4 h | >99 | 24 |
| 9 |

|
2.0 | 12 h | — | — |
| Entry | Aryl halide | Catalyst loading (mol%) | Time | Isolated yield (%) | TOFb |
|---|---|---|---|---|---|
| a Reaction conditions: aryl halides (0.5 mmol), phenylboronic acid (0.0915 g, 0.75 mmol), Cs2CO3 (0.48 g, 1.5 mmol), EtOH (5 mL). Microwave temperature 80 °C, max pressure = 8 bar, max power = 46 W. b TOF (h−1). | |||||
| 1 |

|
0.2 | 10 min | >99 | 3000 |
| 2 |

|
0.2 | 10 min | >99 | 3000 |
| 3 |

|
0.2 | 10 min | >99 | 3000 |
| 4 |

|
1.0 | 30 min | 84 | 168 |
| 5 |

|
0.5 | 20 min | 98 | 588 |
| 6 |

|
0.5 | 15 min | 92 | 736 |
| 7 |

|
0.5 | 15 min | 98 | 784 |
| 8 |

|
1.0 | 30 min | >99 | 198 |
| 9 |

|
2.0 | 2 h | — | — |
Discussion
There have been several reports where 2-oxazolidinones were formed from amino ethanol derivatives by the purging of CO2 and the addition of electrophiles and bases.20–24 Earlier work to form pyridine based oxazolidinone was mostly done with preformed oxazolidinone derivatives.38,39 A recent report, using non-pyridine substrates, showed that in the presence of SOCl2 and a base, 1,2 amino alcohols can form oxazolidinones with the inversion or retention of the configuration,24 depending on the substituents. Based on that proposed mechanism, we should obtain an inversion of the configuration during product formation and indeed, we obtained the inverted product in a major yield, although unlike the earlier report, we had no added base to our reaction. However, the yield is 60% and hence the pyridine substrate may also be acting as a base.24 We carried out the reaction in SOCl2, which serves as an electrophile and the presence of atmospheric CO2 lead to carbonylation, without any additional base. Based on the earlier proposed mechanism, we propose the formation of a transient carbamate species, by the incorporation of CO2, without any added base and the pyridine moiety in the substrate may stabilise the species. In the presence of SOCl2, the hydroxyl oxygen of the amino alcohol is activated and becomes a good leaving group, which then suffers a nucleophilic attack from a carbamate oxygen, forming the oxazolidinone ring. The mechanistic proposal is depicted in Scheme 4. | ||
| Scheme 4 Mechanistic proposal for preparation of ligand L. | ||
The single crystal structures of 1, 2 and 3 showed that metal complexes of a varying ligand to metal stoichiometry are formed. In complexes 1 and 3, the metal to ligand (L) ratio was 1:2, whereas in 2 it was 1:1. The unique electronic property of the metal leads to a difference in the dissociation of the labile ligands, leading to a change in the reactivity and redox properties.
We probed the reactivity with 3 for C–C bond cross coupling as a proof of concept, since Pd(OAc)2 (5–10 mol%) itself can act as a catalyst for the same purpose. Palladium catalyzed Suzuki–Miyaura cross coupling reactions between organic halides with aryl boronic acid are the most effective and widely used method for the synthesis of various biaryls.40–45 PdII complexes of phosphane based ligands have been among the most effective ones to perform the Suzuki–Miyaura cross coupling reactions.46–53 The search for alternative ligands has seen a surge during the past and present decade,54–58 due to the sensitivity and non-environmentally friendly nature of phosphane based catalysts. Efficient catalysts for the Suzuki–Miyaura reaction have been made using N-heterocyclic carbene ligands,59–67 palladacycles68–70 and other ligand systems.71–73 However, the use of oxazolidinone as a ligand for such catalysis is unknown. Our work shows that oxazolidinone has the potential as a ligand to palladium, to generate catalysts for C–C bond cross coupling, using aryl halides and phenylboronic acids.
Complex 3 act as a good catalyst for the Suzuki–Miyaura cross-coupling reaction of different aryl halides viz. bromides and iodides with different electron donating and withdrawing groups in the para and ortho positions, in considerably good yields. For aryl iodides, with only 0.2 mol% catalyst loading, we obtained almost full conversion, in 20–30 min, whereas for aryl bromides, we had to increase the catalyst loading to 0.5–1.0%, to afford a full conversion, in 0.5–1.0 h. This observation can be well argued, since iodide is a better leaving group than bromide. Electronic factors play an important role for the reaction of aryl halides containing electron donating and electron withdrawing groups. An electron withdrawing substituent present in the ortho or para position of the aryl halide favours the reaction, thus the rate of reaction increases, while in the presence of an electron donating substituent, the rate of reaction decreases as it strengthens the C–X bond. Thus, for 4-acetyl, 4-nitro and 4-cyanophenylbromides, with 0.5 mol% catalyst loading, we obtained excellent yields within 30 minutes to 1.0 hours (Table 5, entries 5–7), but for 4-bromoanisole it needed 2 hours for a complete conversion, using 1 mol% catalyst and 80 °C (Table 5, entry 4). Using a microwave reactor, we observed the same trend, although here the completion of reaction is achieved within 10–30 min. Hence, the microwave method is better than the conventional heating method. However, for aryl chlorides we did not get the desired product, even after 12 hours, hence the Pd complex 3 could not serve as a catalyst for the Suzuki–Miyaura coupling of aryl chlorides (Tables 5 and 6, entry 9). The results are still important and encouraging, since the catalytic C–C bond cross coupling reaction using L is at its infancy. Complex 3 is proposed to follow the general mechanistic pathway via oxidative addition, transmetalation and reductive elimination reported in the literature for C–C bond cross coupling via C–X bond activation (Scheme S1, ESI†).40,41,74
L was also probed for its potential as a ligand in therapeutics. In vitro studies of L complexed with CuII (1) and PtII (2) show anticancer activity, with an IC50 of 114 ± 2 μM for 1 and 102 ± 2 μM for 2 in the MCF-7 cancer cell line. 1 is less toxic in the non-cancerous mouse cell line (NIH 3T3) (IC50 = 198 ± 1 μM) in vitro, as compared to its toxicity over MCF-7, which is encouraging, since the well known drug cisplatin, which has a much lower IC50 value in MCF-7 or A549, does not exhibit this property. However, the PtII complex 2 is relatively more active against the lung cancer cell line (A549), showing an IC50 of 198 ± 1 μM when compared to 1 (IC50 = 455 ± 3 μM). The in vitro toxicity data show that the trans-Pt complex is most potent (IC50 = 102 ± 2 μM in MCF-7) among the three metal complexes. The palladium complex is the least active with IC50 values > 200 μM and it is the most hydrophilic among the three candidates. 3 is more active over the A549 cell line (IC50 = 236 ± 3 μM), compared to MCF-7 (IC50 = 260 ± 3 μM), but at the same time it is also more toxic to the non-cancerous mouse NIH 3T3 cell line (IC50 = 151 ± 1 μM). trans-PtII compounds are, in general, less reactive than cis-PtII compounds and the in vitro data are in accordance with the known literature. The dose required to achieve a 50% killing is high for the series of complexes, as compared to many drugs already in the clinic. However, it is encouraging to see that there are drugs which are widely used in the clinic, or are in clinical trials and have in vitro toxicity data from which they seem apparently much less potent (viz. in vitro IC50 values: cyclophosphamide >1000 μM on MCF-7,75 gallium nitrate in the range 60–250 μM in HCC lines,76 ruthenium based compounds NAMI-A > 500 μM in the MCF-7 cell line77,78 and RAPTA-C > 350 μM in the A2780 cell line79). In fact, when we tested cyclophosphamide against MCF-7, we could not reach an IC50 value, even at a 3000 μM concentration of the drug. Hence, it should be noted that having a lower IC50 does not necessarily mean that the complex would be a better drug candidate, rather the unwanted side effects might also be more. The obtained IC50 values of 1–3, the success of oxazolidinones as antimicrobial and the relatively less toxic nature of L on human cells, on its own (IC50 > 1000 μM in MCF-7), warrants that the complexes are worth tuning with respect to the oxazolidinone and the labile ligands.
Conclusions
We found that the CO2 present in air is able to convert a pyridine containing amino alcohol into an oxazolidinone, in the presence of SOCl2 as an electrophile. The compound obtained, 3-(pyridin-2-ylmethyl)oxazolidin-2-one, may act as a ligand to synthesize late 3d, 4d and 5d transition metal complexes, which would generate potential new candidates in catalysis and therapeutics. The ligand synthetic method is a simple way to fix atmospheric CO2 in organic synthesis. The three metal complexes generated as a proof of concept with the synthesized ligand without tuning it any further, showed that the CuII complex (1) has some degree of selectivity for the MCF-7 cancer cell line. The platinum complex (2) is the most toxic among the three against both the MCF-7 and A549 cell lines, yet it is not as toxic as cisplatin, which might be good to check upon, since it has scope for rendering reduced side effects, due to the ligand being an oxazolidinone, with much less toxicity. The PdII complex (3) is successful in the C–C bond cross coupling reaction of activated and deactivated aryl bromides, and has scope for tuning, with very simple synthetic modifications. The results warrant further investigation, to tune the ligand and generate metal complexes which may have potential in catalysis and therapeutics.Experimental section
Material and methods
All the chemicals and solvents used for this work were purchased from commercial sources. The solvents were distilled and dried, prior to use. Silica gel 60–120 mesh size (Merck-India) was used for the column chromatography. The UV-visible measurements were done using a Perkin Elmer Lambda 35 spectrophotometer. The FT-IR spectra were recorded using a Perkin-Elmer 120-000A, in KBr pellets. The 1H and 13C NMR spectra were measured using either a JEOL ECS 400 MHz or Bruker Avance III spectrometer 500 MHz, at room temperature. The chemical shifts are reported in parts per million (ppm). The 13C NMR spectra recorded are proton decoupled. The elemental analyses were performed on a Perkin Elmer Inc. EA 2400 CHNS series. The electro-spray ionization mass spectra were recorded using a Micromass Q-Tof microTM, Waters, by +ve mode electrospray ionization. The synthetic yields reported are of the isolated, analytically pure compounds. The Suzuki–Miyaura cross-coupling reactions were performed with a conventional heating method, as well as in a microwave (Anton Paar Monowave 300). The microwave reactions were performed in 10 mL Teflon capped glass vials. All the reactions in the microwave were carried out at 80 °C for a certain period of time (based on the completion of the reaction) while the pressure and microwave power were varied. The maximum pressure reached was 8 bar and the microwave power reached up to 46 W.Caution! Perchlorate salts are explosive and corrosive, so special care should be given for handling these compounds.
Synthesis of ligands and their precursors
X-ray crystallographic study
Single crystals of 1–3 were mounted using loops on the goniometer head of a Bruker Kappa Apex II CCD Duo diffractometer with graphite monochromated Mo-Kα radiation (0.71073 Å) and the data were collected at a temperature of 100 K. An empirical multi-scan absorption correction was performed using SADABS.81 The structures were solved by direct methods and all the non-hydrogen atoms were refined anisotropically by full matrix least-squares on F2. The hydrogen atoms were calculated and fixed using SHELXL-97, after hybridization of all the non-hydrogen atoms.82 The ball and stick diagrams were made using Diamond version 2.0f. CCDC 956432 (1) and 956433 (2).Lipophilicity measurement
The partition coefficients of the three complexes in an octanol–water system were determined using a standard shake-flask method.83 Octanol and phosphate buffer (20 μM) (each 3 mL) were pre-equilibrated for 8 hours before the experiment.84 After equilibration, the solid samples were added to the mixture of solvents and shaken on a dancing shaker overnight, at room temperature. After that, the tubes were centrifuged and left undisturbed for an hour. Aliquots of the aqueous and octanol layers were pipetted out separately and the absorbance measured using UV-vis spectroscopy. The concentration of the substances in each layer was calculated using the respective molar extinction coefficients of 1–3.Cell lines and culture condition
The human breast adenocarcinoma cell line (MCF-7), human lung carcinoma cell line (A549) and mouse embryonic fibroblast cell line (NIH 3T3) were kind donations of Dr Jayasri Das Sarma and Dr Tapas Sengupta, Department of Biological Sciences, IISER-Kolkata, India. The cell lines were maintained in the logarithmic phase at 37 °C in a 5% carbon dioxide atmosphere, using a culture media containing DMEM, 10% foetal bovine serum (GIBCO) and antibiotics (100 units mL−1 penicillin and 100 mg mL−1 streptomycin).Cell viability assay
The cytotoxicity of complexes 1–3 on the MCF-7 and A549 cell lines were evaluated by the MTT (tetrazolium salt reduction) assay.85 Briefly, 6 × 103 cells per well were seeded in 96-well plates in growth medium (200 mL) and then incubated at 37 °C in a 5% carbon dioxide atmosphere. After 48 h, the medium was removed and replaced with a fresh one and the compounds to be studied were added at appropriate concentrations. Triplicates for each concentration were used in the wells. The compounds to be added were solubilized in media, or PBS containing DMSO (when needed), such that the concentration of DMSO in each well should not exceed 0.2%. Cisplatin was dissolved in DMSO just before the experiment and a calculated amount of the drug solution was added to the growth medium containing cells, such that the final DMSO concentration in the wells was no more than 0.2%. After 48 h, the media containing compound was removed and fresh media was added to each well and successively treated with 20 μL of a 2 mg mL−1 MTT in saline solution, followed by 3 h of incubation at 37 °C in a 5% carbon dioxide atmosphere. After 3 h, the media were removed and 100 μL of DMSO (molecular biology grade) added to each well. The inhibition of cell growth induced by the tested complexes was detected by measuring the absorbance of each well at 515 nm86,87 using a BIOTEK ELx800 plate reader. All the experiments had the respective controls and standards, as needed. The IC50 values represent the drug concentration that reduces the mean absorbance at 515 nm to 50%, as compared to the untreated control wells.Catalytic study
By conventional heating method. A typical 50 mL round bottom flask containing a magnetic stir bar was charged with phenylboronic acid (0.091 g, 0.75 mmol) and Cs2CO3 (0.488 g, 1.5 mmol). The aryl halide (0.5 mmol) in EtOH (5 mL) was added to it. The catalyst (palladium complex) solution in EtOH was added, varying the loading percentage from 0.2 mol% to 1 mol%, with respect to the aryl halide. The resulting mixture was refluxed and after completion of the reaction, the solvent was evaporated under a reduced pressure. The residue was diluted with water (30 mL) and extracted with dichloromethane (3 × 20 mL). The organic layer was dried with Na2SO4 and the solvent was removed in vacuo. The resulting product was purified by silica gel column chromatography (mesh size 60–100) using either hexane or a dichloromethane–hexane mixture (3
:1) depending on the product polarity. After column chromatography, the isolated yield was calculated.
By microwave method. A typical Teflon capped 10 mL glass vial, containing a magnetic stir bar was charged with phenylboronic acid (0.091 g, 0.75 mmol) and Cs2CO3 (0.488 g, 1.5 mmol). The aryl halide (0.5 mmol) in EtOH (5 mL) was added to it. The catalyst (palladium complex) solution in EtOH was added, varying the loading percentage from 0.2 mol% to 1 mol% with respect to the aryl halide. The resulting mixture was warmed at 80 °C for 10–45 min, under a standard irradiation mode. After the completion of the reaction, the solvent was evaporated under a reduced pressure. The residue was diluted with water (30 mL) and extracted with dichloromethane (3 × 20 mL). The organic layer was dried with Na2SO4 and the solvent was removed in vacuo. The resulting product was purified by silica gel column chromatography (mesh size 60–100) using either hexane or a dichloromethane–hexane mixture (3
:1) depending on the product polarity. After column chromatography, the isolated yield was calculated.
Biphenyl. White solid. 1H NMR (CDCl3, 400 MHz) δ 7.63 (d, J = 7.64 Hz, 4H, ArH), 7.48 (t, J = 7.64 Hz, 4H, ArH), 7.38 (t, J = 6.88 Hz, 2H, ArH) ppm (Fig. S6, ESI†). 13C NMR (CDCl3, 100 MHz) δ 141, 128.9, 127.4, 127.3 ppm (Fig. S7, ESI†).
4-Methylbiphenyl. White solid. 1H NMR (CDCl3, 400 MHz,) δ 7.51 (d, J = 7.64 Hz, 2H, ArH), 7.42 (d, J = 7.64 Hz, 2H, ArH), 7.36 (t, J = 7.64 Hz, 2H, ArH), 7.25 (t, J = 7.64 Hz, 1H, ArH), 7.16 (d, J = 7.6 Hz, 2H, ArH), 2.31 (s, 3H, CH3) ppm (Fig. S8, ESI†). 13C NMR (CDCl3, 100 MHz) δ 141, 138, 136, 129, 128, 126, 21 ppm (Fig. S9, ESI†).
4-Methoxybiphenyl. White solid. 1H NMR (CDCl3, 400 MHz) δ 7.59 (m, 2H, ArH), 7.45 (t, J = 7.64 Hz, 2H, ArH), 7.34 (t, J = 7.64 Hz, 1H, ArH), 7.01 (d, J = 8.4 Hz, 2H, ArH), 3.87 (s, 3H, CH3) ppm (Fig. S10, ESI†). 13C NMR (CDCl3, 100 MHz) δ 159, 141, 133, 129, 128, 126.8, 126.7, 114, 56 ppm (Fig. S11, ESI†).
4-Acetylbiphenyl. White solid. 1H NMR (CDCl3, 400 MHz) δ 8.05 (d, J = 8.4 Hz, 2H, ArH), 7.70 (d, J = 8.4 Hz, 2H, ArH), 7.64 (t, J = 2.28 Hz, 2H, ArH), 7.49 (t, J = 6.88 Hz, 2H, ArH), 7.42 (t, J = 6.88 Hz, 1H, ArH), 2.64 (s, 3H, CH3) ppm (Fig. S12, ESI†). 13C NMR (CDCl3, 100 MHz) δ 197, 146, 140, 135, 129.07, 129.04, 128.9, 127.40, 127.30, 27 ppm (Fig. S13, ESI†).
4-Nitrobiphenyl. Light yellow solid. 1H NMR (CDCl3, 400 MHz) δ 8.31 (d, J = 9.16 Hz, 2H, ArH), 7.75 (d, J = 8.4 Hz, 2H, ArH), 7.64 (d, J = 6.88 Hz, 2H, ArH), 7.52 (m, 3H, ArH) ppm (Fig. S14, ESI†). 13C NMR (CDCl3, 100 MHz) δ 146, 139, 133, 129.2, 128.8, 127.9, 127.4, 119, 111 ppm (Fig. S15, ESI†).
4-Cyanobiphenyl. White solid. 1H NMR (CDCl3, 400 MHz) δ 7.74 (m, 4H, ArH), 7.60(d, J = 6.88 Hz, 2H, ArH), 7.51 (t, J = 6.88 Hz, 2H, ArH), 7.45 (t, J = 7.64 Hz, 1H, ArH) ppm (Fig. S16, ESI†). 13C NMR (CDCl3, 100 MHz) δ 148, 147, 139, 129.3, 129.1, 127.9, 127.9, 127.5, 124 ppm (Fig. S17, ESI†).
2-Phenylpyridine. Yellow oil. 1H NMR (CDCl3, 400 MHz) δ 8.71 (m, 1H, ArH), 7.99 (m, 2H, ArH), 7.77 (m, 2H, ArH), 7.50 (m, 3H, ArH), 7.27 (m, 1H, ArH) ppm (Fig. S18, ESI†). 13C NMR (CDCl3, 100 MHz) δ 157, 150, 139, 137, 128.9, 128.7, 127, 122, 120 ppm (Fig. S19, ESI†).
Acknowledgements
We sincerely acknowledge the DST, India for funding (Vide Project no-SR/S1/IC-36/2010). We are also thankful to the IISER Kolkata, for the financial and infra-structural support, including the NMR and single crystal X-ray facilities. We are thankful to Dr Tapas K. Sengupta and Dr Jayasri Das Sarma for the donation of the MCF-7, NIH 3T3 and A549 cell lines. We also thank Dr Venkataramanan Mahalingam for the microwave reactor used in this work. A. S. sincerely thanks the IISER Kolkata for a research fellowship. S. B. and S. K. D. thank the CSIR-India for research fellowships. S. K. acknowledges the UGC for a research fellowship.References
- D. A. Evans, S. J. Miller and T. Lectka, J. Am. Chem. Soc., 1993, 115, 6460–6461 CrossRef CAS.
- S. Kanemasa, Y. Oderaotoshi, S.-i. Sakaguchi, H. Yamamoto, J. Tanaka, E. Wada and D. P. Curran, J. Am. Chem. Soc., 1998, 120, 3074–3088 CrossRef CAS.
- D. A. Evans, J. S. Johnson and E. J. Olhava, J. Am. Chem. Soc., 2000, 122, 1635–1649 CrossRef CAS.
- C. B. Shinisha and R. B. Sunoj, J. Am. Chem. Soc., 2010, 132, 12319–12330 CrossRef CAS PubMed.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325–335 CrossRef CAS PubMed.
- D. A. Evans, C. W. Downey and J. L. Hubbs, J. Am. Chem. Soc., 2003, 125, 8706–8707 CrossRef CAS PubMed.
- D. A. Evans, D. Seidel, M. Rueping, H. W. Lam, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2003, 125, 12692–12693 CrossRef CAS PubMed.
- D. A. Evans, K. A. Scheidt, J. N. Johnston and M. C. Willis, J. Am. Chem. Soc., 2001, 123, 4480–4491 CrossRef CAS PubMed.
- K. Muniz and M. Nieger, Chem. Commun., 2005, 2729–2731 RSC.
- I. Almodovar, C. H. Hoevelmann, J. Streuff, M. Nieger and K. Muniz, Eur. J. Org. Chem., 2006, 704–712 CrossRef CAS.
- K. Gedrich, M. Heitbaum, A. Notzon, I. Senkovska, R. Froehlich, J. Getzschmann, U. Mueller, F. Glorius and S. Kaskel, Chem.–Eur. J., 2011, 17, 2099–2106 CrossRef CAS PubMed.
- B. A. Howell and E. W. Walles, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1986, 27, 460–461 CAS.
- S. Lundberg, S. Ayesa, O. Belda, I. Dorange, K. Ersmark, K. Hammer, P.-O. Johansson, S. Lindstroem, A. Rosenquist, B. Samuelsson, M. Baeck, I. Kvarnstroem, F. Waangsell and K. Bjoerklund, Medivir AB, Swed., WO 2010042030, 2010, p. 153 Search PubMed.
- B. H. Candon, M. Chessin and W. E. Lange, Purdue Frederick Co., US 3108045, 1963, p. 16 Search PubMed.
- M. E. Dyen and D. Swern, Chem. Rev., 1967, 67, 197–246 CrossRef CAS.
- G. P. Speranza and W. J. Peppel, J. Org. Chem., 1958, 23, 1922–1924 CrossRef CAS.
- G. Y. Lesher and A. R. Surrey, J. Am. Chem. Soc., 1955, 77, 636–641 CrossRef CAS.
- W. J. Close, J. Am. Chem. Soc., 1951, 73, 95–98 CrossRef CAS.
- J. E. Herweh and W. J. Kauffman, Tetrahedron Lett., 1971, 12, 809–812 CrossRef.
- M. Kodaka, T. Tomohiro and H. Okuno, J. Chem. Soc., Chem. Commun., 1993, 81–82 RSC.
- M. A. Casadei, M. Feroci, A. Inesi, L. Rossi and G. Sotgiu, J. Org. Chem., 2000, 65, 4759–4761 CrossRef CAS.
- Y. Du, Y. Wu, A.-H. Liu and L.-N. He, J. Org. Chem., 2008, 73, 4709–4712 CrossRef CAS PubMed.
- B. Gabriele, G. Salerno, D. Brindisi, M. Costa and G. P. Chiusoli, Org. Lett., 2000, 2, 625–627 CrossRef CAS PubMed.
- J. Paz, C. Perez-Balado, B. Iglesias and L. Munoz, J. Org. Chem., 2010, 75, 3037–3046 CrossRef CAS PubMed.
- W. B. Wright, Jr., J. Heterocycl. Chem., 1965, 2, 41–43 CrossRef.
- S. Cutugno, G. Martelli, L. Negro and D. Savoia, Eur. J. Org. Chem., 2001, 517–522 CrossRef CAS.
- A. J. Duplantier, I. Efremov, J. Candler, A. C. Doran, A. H. Ganong, J. A. Haas, A. N. Hanks, K. G. Kraus, J. T. Lazzaro, J. Lu, N. Maklad, S. A. McCarthy, T. J. O'Sullivan, B. N. Rogers, J. A. Siuciak, D. K. Spracklin and L. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 2524–2529 CrossRef CAS PubMed.
- W. A. Gregory, D. R. Brittelli, C. L. J. Wang, M. A. Wuonola, R. J. McRipley, D. C. Eustice, V. S. Eberly, A. M. Slee, M. Forbes and P. T. Bartholomew, J. Med. Chem., 1989, 32, 1673–1681 CrossRef CAS.
- W. A. Gregory, D. R. Brittelli, C. L. J. Wang, H. S. Kezar, III, R. K. Carlson, C. H. Park, P. F. Corless, S. J. Miller, P. Rajagopalan, M. A. Wuonola, R. J. McRipley, V. S. Eberly, A. M. Slee and M. Forbes, J. Med. Chem., 1990, 33, 2569–2578 CrossRef CAS.
- C. H. Park, D. R. Brittelli, C. L. J. Wang, F. D. Marsh, W. A. Gregory, M. A. Wuonola, R. J. McRipley, V. S. Eberly, A. M. Slee and M. Forbes, J. Med. Chem., 1992, 35, 1156–1165 CrossRef CAS.
- J. A. Tucker, D. A. Allwine, K. C. Grega, M. R. Barbachyn, J. L. Klock, J. L. Adamski, S. J. Brickner, D. K. Hutchinson, C. W. Ford, G. E. Zurenko, R. A. Conradi, P. S. Burton and R. M. Jensen, J. Med. Chem., 1998, 41, 3727–3735 CrossRef CAS PubMed.
- S. D. Paget, B. D. Foleno, C. M. Boggs, R. M. Goldschmidt, D. J. Hlasta, M. A. Weidner-Wells, H. M. Werblood, E. Wira, K. Bush and M. J. Macielag, Bioorg. Med. Chem. Lett., 2003, 13, 4173–4177 CrossRef CAS PubMed.
- D. A. Evans, K. A. Scheidt and C. W. Downey, Org. Lett., 2001, 3, 3009–3012 CrossRef CAS PubMed.
- C. Ji, W. Lin, G. C. Moraski, J. A. Thanassi, M. J. Pucci, S. G. Franzblau, U. Mollmann and M. J. Miller, Bioorg. Med. Chem., 2012, 20, 3422–3428 CrossRef CAS PubMed.
- Y.-E. Kwon, J.-Y. Park and W.-K. Kim, Anticancer Res., 2007, 27, 321–326 CAS.
- M. Micksch and T. Strassner, Eur. J. Inorg. Chem., 2012, 5872–5880 CrossRef CAS.
- M. Boudart, Chem. Rev., 1995, 95, 661–666 CrossRef CAS.
- T. P. Shiau, E. D. Turtle, C. Francavilla, N. J. Alvarez, M. Zuck, L. Friedman, D. J. R. O'Mahony, E. Low, M. B. Anderson, R. Najafi and R. K. Jain, Bioorg. Med. Chem. Lett., 2011, 21, 3025–3028 CrossRef CAS PubMed.
- M. L. H. Mantel, A. T. Lindhardt, D. Lupp and T. Skrydstrup, Chem.–Eur. J., 2010, 16, 5437–5442 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, J. Organomet. Chem., 2002, 653, 83–90 CrossRef CAS.
- S. P. Stanforth, Tetrahedron, 1998, 54, 263–303 CrossRef CAS.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS.
- R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS PubMed.
- A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed.
- L. Braun, P. Liptau, G. Kehr, J. Ugolotti, R. Froehlich and G. Erker, Dalton Trans., 2007, 1409–1415 RSC.
- E. J. Garcia Suarez, A. Ruiz, S. Castillon, W. Oberhauser, C. Bianchini and C. Claver, Dalton Trans., 2007, 2859–2861 RSC.
- S. K. Yen, L. L. Koh, H. V. Huynh and T. S. A. Hor, Dalton Trans., 2008, 699–706 RSC.
- B. Punji, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2006, 45, 9454–9464 CrossRef CAS PubMed.
- B. Punji, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2007, 46, 10268–10275 CrossRef CAS PubMed.
- A. Ros, B. Estepa, A. Bermejo, E. Alvarez, R. Fernandez and J. M. Lassaletta, J. Org. Chem., 2012, 77, 4740–4750 CrossRef CAS PubMed.
- P. Stepnicka, J. Schulz, T. Klemann, U. Siemeling and I. Cisarova, Organometallics, 2010, 29, 3187–3200 CrossRef CAS.
- O. Piechaczyk, M. Doux, L. Ricard and P. Le Floch, Organometallics, 2005, 24, 1204–1213 CrossRef CAS.
- T. Mino, Y. Shirae, M. Sakamoto and T. Fujita, J. Org. Chem., 2005, 70, 2191–2194 CrossRef CAS PubMed.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed.
- L. Zhang, L. Wang, H. Li and P. Li, Synth. Commun., 2008, 38, 1498–1511 CrossRef CAS.
- H. Yan, P. Chellan, T. Li, J. Mao, K. Chibale and G. S. Smith, Tetrahedron Lett., 2013, 54, 154–157 CrossRef CAS PubMed.
- M. Basauri-Molina, S. Hernandez-Ortega, R. A. Toscano, J. Valdes-Martinez and D. Morales-Morales, Inorg. Chim. Acta, 2010, 363, 1222–1229 CrossRef CAS PubMed.
- H. Lebel, M. K. Janes, A. B. Charette and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5046–5047 CrossRef CAS PubMed.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- Z.-Y. Wang, G.-Q. Chen and L.-X. Shao, J. Org. Chem., 2012, 77, 6608–6614 CrossRef CAS PubMed.
- L. Ray, M. M. Shaikh and P. Ghosh, Organometallics, 2007, 26, 958–964 CrossRef CAS.
- T. Zhang, W. Wang, X. Gu and M. Shi, Organometallics, 2008, 27, 753–757 CrossRef CAS.
- W. Wei, Y. Qin, M. Luo, P. Xia and M. S. Wong, Organometallics, 2008, 27, 2268–2272 CrossRef CAS.
- H. Turkmen, R. Can and B. Cetinkaya, Dalton Trans., 2009, 7039–7044 RSC.
- H. Ohta, T. Fujihara and Y. Tsuji, Dalton Trans., 2008, 379–385 RSC.
- O. Diebolt, P. Braunstein, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2008, 3190–3192 RSC.
- K.-E. Lee, H.-T. Jeon, S.-Y. Han, J. Ham, Y.-J. Kim and S. W. Lee, Dalton Trans., 2009, 6578–6592 RSC.
- I. J. S. Fairlamb, A. R. Kapdi, A. F. Lee, G. Sanchez, G. Lopez, J. L. Serrano, L. Garcia, J. Perez and E. Perez, Dalton Trans., 2004, 3970–3981 RSC.
- Y.-J. Kim, J.-H. Lee, T. Kim, J. Ham, Z. N. Zheng and S. W. Lee, Eur. J. Inorg. Chem., 2012, 6011–6017 CrossRef CAS.
- A. John, M. M. Shaikh, R. J. Butcher and P. Ghosh, Dalton Trans., 2010, 39, 7353–7363 RSC.
- D. V. Aleksanyan, V. A. Kozlov, Y. V. Nelyubina, K. A. Lyssenko, L. N. Puntus, E. I. Gutsul, N. E. Shepel, A. A. Vasil'ev, P. V. Petrovskii and I. L. Odinets, Dalton Trans., 2011, 40, 1535–1546 RSC.
- M. D. Santana, R. Garcia-Bueno, G. Garcia, G. Sanchez, J. Garcia, A. R. Kapdi, M. Naik, S. Pednekar, J. Perez, L. Garcia, E. Perez and J. L. Serrano, Dalton Trans., 2012, 41, 3832–3842 RSC.
- N. T. S. Phan, D. S. M. Van and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- W. M. Liu, D. W. Fowler, P. Smith and A. G. Dalgleish, Br. J. Cancer, 2010, 102, 115–123 CrossRef CAS PubMed.
- M.-S. Chua, L. R. Bernstein and S. K. S. So, Anticancer Res., 2006, 26, 1739–1744 CAS.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- D. Pluim, R. C. A. M. van Waardenburg, J. H. Beijnen and J. H. M. Schellens, Cancer Chemother. Pharmacol., 2004, 54, 71–78 CrossRef CAS PubMed.
- A. Casini, F. Edafe, M. Erlandsson, L. Gonsalvi, A. Ciancetta, N. Re, A. Ienco, L. Messori, M. Peruzzini and P. J. Dyson, Dalton Trans., 2010, 39, 5556–5563 RSC.
- S. Striegler and M. Dittel, Inorg. Chem., 2005, 44, 2728–2733 CrossRef CAS PubMed.
- G. M. Sheldrick, Z. Kristallogr., 2002, 217, 644–650 CrossRef CAS.
- G. M. Sheldrick, Int. Union Crystallogr., Crystallogr. Symp., 1991, 5, 145–157 CAS.
- C. Zhang, Y. Wang and F. Wang, Bull. Korean Chem. Soc., 2007, 28, 1183–1186 CrossRef CAS.
- J. Sangster and A. D. Pelton, J. Phys. Chem. Ref. Data, 1987, 16, 509–561 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- B. L. Lokeshwar, E. Escatel and B. Zhu, Curr. Med. Chem., 2001, 8, 271–279 CrossRef CAS.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF, schematic representation of catalytic cycle, 1H-NMR, 13C-NMR data of products in catalysis. CCDC 956432 (1) and 956433 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00990d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |