 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescence behaviour in acetonitrile and in the solid state of a series of lanthanide complexes with a single helical ligand†‡
Miki
Hasegawa
*a,
Hideki
Ohtsu
ab,
Daisuke
Kodama
a,
Takeshi
Kasai
a,
Shoya
Sakurai
a,
Ayumi
Ishii
a and
Kengo
Suzuki
c
aCollege of Science and Engineering, Aoyama Gakuin University, 5-10-1 Fuchinobe, Chuo-ku, Sagamihara, Kanagawa 252-5258, Japan. E-mail: hasemiki@chem.aoyama.ac.jp; Fax: +81-42-759-6221
bGraduate School of Science and Engineering, University of Toyama, 3190 Gofuku, Toyama, Toyama 930-8555, Japan
cHamamatsu Photonics K. K., 812 Joko-cho, Higashi-ku, Hamamatsu, Shizuoka 341-3196, Japan
First published on 9th January 2014
Abstract
Luminescence mechanisms of EuIII, TbIII, GdIII and NdIII complexes with a hexadentate ligand (abbreviated to EuL, TbL, GdL, and NdL, respectively), which have two bipyridine moieties bridged by an ethylenediamine unit, have been examined. Our molecular design is that each complex forms a single helical polar structure based on the chelate ring to retain solubility in solutions. EuL and NdL show comparably bright emission from ff transitions both in acetonitrile solution and in the solid state. To understand the mechanism of the emission in detail, the energy level of the triplet (T) state of the ligand L has been estimated based on the phosphorescence measurements of GdL, because GdIII shows no ff emission. The donor level of the T state of L and the acceptor level of EuIII or NdIII can overlap, indicating that the excited photon localized on L has been used for the efficient ff emission, while not for ππ* emission. For TbL, the luminescence quantum yield is significantly dependent on temperature and the state: in the solid state of TbL, the quantum yield of ff emission is over 90% at 77 K, while no luminescence is observed at room temperature, and in solution TbL shows no emission. This observation suggests that the emissive f-level of TbIII and the energy donor level of the excited T state of L are in thermal equilibrium. The described lanthanide complexes are stable and retain their molecular structure even in solutions and show characteristic luminescence behaviour based on the energy relaxation process of each lanthanide ion. Furthermore the HoIII complex with L (HoL) has been prepared and its structure has been analyzed. HoL has a twisted arrangement of the bipyridine moiety surrounding HoIII due to the small ionic radius of HoIII.
Introduction
Highly luminescent lanthanide (Ln) complexes have attracted much attention because they are applied to functional materials such as OLEDs,1–6 solar cell systems,7,8 and sensors in solutions.9–13 Recently, intensive work has focused on the Ln complexes with organic ligands which exhibit significant luminescent properties.14,15The spectral feature of their luminescence originating from ff-electronic transitions of Ln complexes, generally, is specifically compared with that of fluorescence or phosphorescence of neat organic compounds;16,17 the ff emission of the Ln ion appears as narrow bands because of the inner-core transitions being shielded from the outer-sphere, and has been induced by the photo-excitation of π-electronic systems of the organic ligand moieties via an intramolecular energy transfer.18–21 In other words, for these Ln complexes, the π-electronic moieties of the ligands act as photo-antennae and as energy donors, and a splitter of the f-level of LnIII as an acceptor in the complexes. The energy differences between the energy donor and the acceptor based on molecular design affect directly the efficiency of ff-emissions. For instance, there are a number of papers that describe Ln complexes with an organic ligand showing over 50% or 100% efficiency in the luminescence.16
These ligands also exert structural effects on the properties of the Ln complexes. For instance, such structural modifications lead to the tuning of luminescence efficiency and the band appearances of the ff emission.22–26 Moreover, the enhanced ff emission may be induced, if the ligands act as a shield from the outer-sphere with a stable structure in solution23,27–30 or in the solid state.24,31–35
A chelate effect provides a fundamental but most useful technique to design stable metal complexes, and the coordination number of the chelate ring contributes to the stability constant of the coordination in solution. There are some manuscripts for metal complexes, e.g. Co, Cu, Fe, Zn, Ag, Eu ions, with a hexadentate ligand forming the helicate molecular structure.36–38 It is well known that Constable and Tocher et al. reported various metal complexes having d- or f-block ions with a helicate of a polypyridyl ligand. In their evolution of the molecular preparation,38 for instance, six pyridine moieties in the ligand were demonstrated to have the ability to form a single-helical structure by coordinating with a Eu ion.
Our approach is to understand the emission mechanism of a series of lanthanide (Eu, Gd, Tb, and Nd) complexes, in view of photochemistry to realize the efficient luminescent principle in the solid state and in solution. In this strategy, we designed a new hexadentate ligand to wind the Ln ion, and the formed complex would retain the strong dipole moment to dissolve into solution with emitting ability (Fig. 1). The target complexes would have five pentagonal-chelate-rings in a molecule to retain their stability in solution, the so called chelate effect. It is expected that the molecular structure with the organic ligand would form a co-planar structure, and both sides of the structure would give two exchangeable/flexible sites.
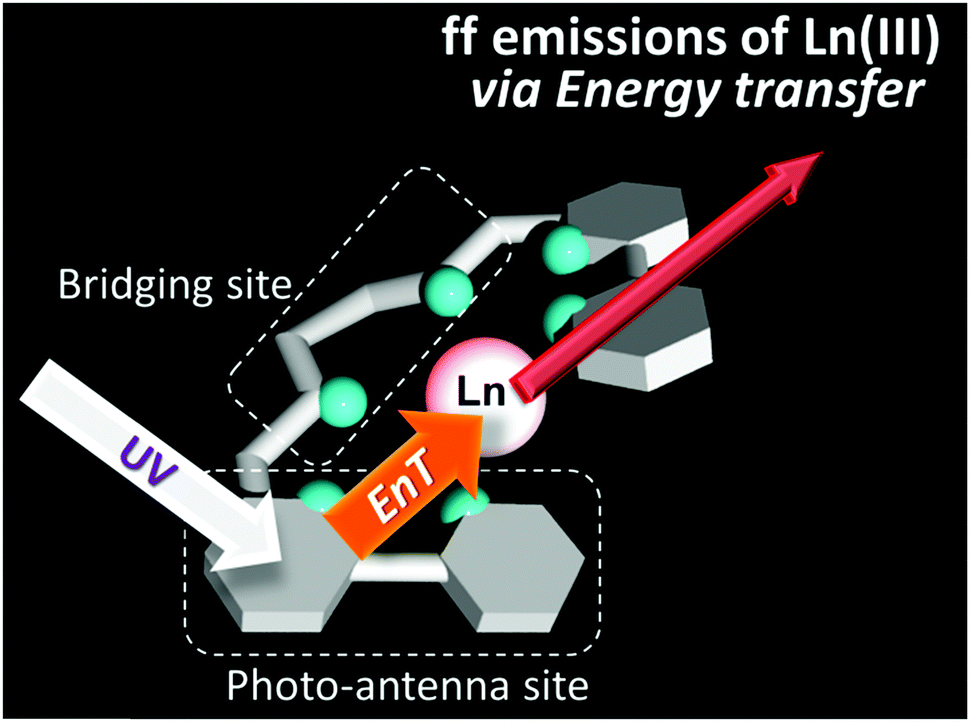 | ||
| Fig. 1 Strategy to design a helical molecular structure that retains a strong dipole moment to induce the ff-emission of Ln complexes. In this figure, EnT means the intramolecular energy transfer. | ||
In the present study, electronic transitions of a series of new Ln complexes (abbreviated to LnL, Ln = Nd, Eu, Gd, Tb and Ho shown in Fig. 2) have been discussed. In particular, we have focused on the emission properties of the Eu and Tb complexes both in solution and the solid state. Here, we use a hexadentate ligand L having two bipyridine (abbreviated to bpy) moieties bridged by an ethylenediamine unit (abbreviated to en) designed based on the above strategy. Two bpy moieties may also act as photo-antennae to transport the excitation energy for the Ln excitation. The en group prevents conjugation of two bpy in LnL, and the excited state of the π electronic system of the bpy can be treated as the same as those of simple bpy derivatives. The X-ray structural analysis of a series of LnL (Ln = Nd, Eu, Gd, Tb and Ho) complexes was also performed to support spectral discussion concerning the molecular structure.
 | ||
| Fig. 2 Top: molecular structure of LnL (a) and the ORTEP drawing of EuL with 70% probability thermal ellipsoids (b) obtained from the single crystal X-ray analysis. Hydrogen atoms, PF6− and acetonitrile are omitted for clarity. Bottom: (c) side views to compare the dihedral angles among two bipyridine moieties and averaged binding distances between a Ln ion and nitrogen atoms localized on bpy moieties for a series of LnL (Ln = Nd, Eu, Gd, Tb and Ho). Gray: carbon, blue: nitrogen, red: oxigen, lilac: neodimium, pink: europium, violet: gadrinium, green: terbium and yellow: hormium. For each of the illustrations, LnL has no NO3− for clarity. | ||
Results and discussion
1. Structural analyses
We have thus synthesized a new ligand L containing two bpy moieties and a series of LnL (Nd, Eu, Gd, Tb and Ho complexes with L are abbreviated to NdL, EuL, GdL, TbL and HoL, respectively).The single crystal structures of LnL have been determined by the single crystal X-ray diffraction analyses. Fig. 2 and Fig. S3 (ESI‡) show the molecular structure of EuL and the comparison of those of NdL, GdL, TbL and HoL. In EuL, six nitrogen atoms of L coordinate to the centre metal Eu, and form five pentagonal-chelate rings equatorially. Each bpy moiety keeps coplanar, and two terminated-pyridine rings take the face-to-face conformation each other in a van der Waals distance, r(C(1)–C(24)) = 3.178 Å in EuL. Furthermore, two nitrate anions bind to the Eu from both apical sites, and the Eu ion has a total coordination number of ten. From the comparison of the molecular shape of a series of LnL (Ln = Nd, Eu, Gd, Tb and Ho), it is found that L can form the same molecular structure with other Ln ions (Fig. S3, ESI‡ and Table 1). These metal ions are in the order corresponding to the ionic radii and atomic number, i.e., the ionic radius of NdIII is larger than that of HoIII known as the lanthanide contraction. The interatomic distance shows the average value of four Ln–Nbpy distances, r(Ln–Nbpy), of each complex (Tables S1 and S2, ESI‡). It is worth noting that the lanthanide contraction affects the length of Ln–Nbpy bonding and the distance of the terminal pyridyl group in the bpy skeleton based on the periodicity of Ln ionic radii. The dihedral angle among two bpy moieties of the complex increases periodically with increasing atomic numbers. It is reflected in their electronic repulsion among the terminal pyridyl group in the bpy skeleton, due to the changes in distances of the Ln–Nbpy bonding. In the case of HoL, the dihedral angle does not show periodicity, because of the distortion of bpy skeletons.
| Ln | Nd | Eu | Gd | Tb | Ho |
|---|---|---|---|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|. b wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2. | |||||
| Formula | C26H23F6N9NdO6P | C26H23EuF6N9O6P | C26H23F6GdN9O6P | C26H23F6N9O6PTb | C28H26F6HoN10O6P |
| Formula weight | 846.74 | 854.46 | 859.75 | 861.42 | 908.49 |
| Crystal size (mm) | 0.13 × 0.09 × 0.02 | 0.15 × 0.11 × 0.08 | 0.18 × 0.16 × 0.10 | 0.22 × 0.10 × 0.08 | 0.13 × 0.09 × 0.02 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P |
| a (Å) | 8.949(2) | 8.9999(9) | 9.0151(7) | 9.0172(11) | 9.961(3) |
| b (Å) | 12.231(3) | 12.2528(12) | 12.2140(9) | 12.2389(15) | 13.593(4) |
| c (Å) | 16.394(4) | 16.2526(16) | 16.1835(12) | 16.133(2) | 13.751(4) |
| α (°) | 112.158(2) | 112.3060(10) | 112.0360(10) | 112.0540(10) | 68.307(3) |
| β (°) | 104.132(2) | 104.4460(10) | 104.2970(10) | 104.2690(10) | 86.035(3) |
| γ (°) | 92.428(3) | 91.8160(10) | 91.8260(10) | 91.6220(10) | 87.405(3) |
| V (Å3) | 1593.4(7) | 1589.6(3) | 1585.2(2) | 1584.7(3) | 1725.5(9) |
| Z value | 2 | 2 | 2 | 2 | 2 |
| D calcd (Mg m−3) | 1.765 | 1.785 | 1.801 | 1.805 | 1.749 |
| μ (Mo Kα) (mm−1) | 1.770 | 2.114 | 2.234 | 2.373 | 2.428 |
| F(000) | 838 | 844 | 846 | 848 | 896 |
| λ (Mo Kα) (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| R 1 (I > 2.00σ(I)) | 0.0308 | 0.0349 | 0.0325 | 0.0294 | 0.0378 |
| wR2b (I > 2.00σ(I)) | 0.0823 | 0.0947 | 0.0821 | 0.0807 | 0.0845 |
| Goodness of fit | 1.055 | 1.082 | 1.061 | 1.073 | 1.072 |
| Largest peak and hole (e Å−3) | 2.110, −1.090 | 2.639, −1.439 | 2.311, −1.227 | 2.870, −1.409 | 3.002, −1.530 |
In a unit cell of EuL, there are two molecules having chiralities; a right- and a left-handed isomer (Fig. 3(a)). A couple of hexafluorophosphate and acetonitrile is also included in the unit cell of EuL (Fig. S4(a), ESI‡). As shown in Fig. 3(b), the molecular packing of EuL suggests that two isomers between the neighboring unit cells interact through the intermolecular ππ interaction of their bpy skeletons at the distance of 3.59–3.83 Å. This interaction results in the formation of their independent column-like structure as shown in Fig. S4(b) and (c) (ESI‡), which is supported by the existence of PF6− ions and acetonitrile molecules.
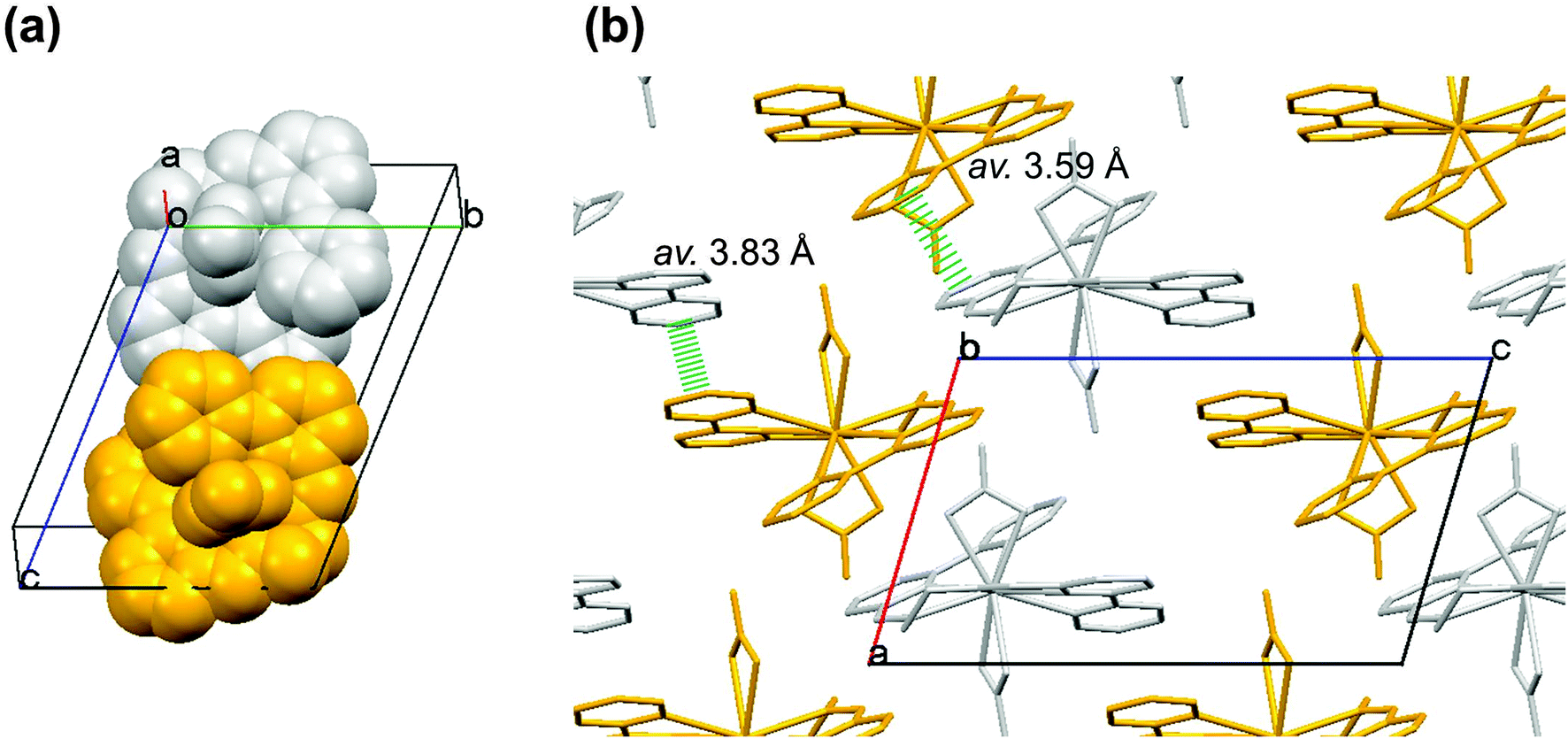 | ||
| Fig. 3 (a) Two isomers in a unit cell of EuL, having left-handed (gold) and right-handed (silver) isomers obtained from X-ray single crystal structural analysis. (b) Molecular packing of EuL projected from along the b-axis. (Hydrogen atoms, PF6− and acetonitrile are omitted for clarity). | ||
X-ray structure analysis reveals that all complexes are isostructural with each lanthanide counterpart, and the Nd, Eu, Gd, and Tb complexes are isomorphous with each other. Remarkably, the detailed crystallographic analysis clarified the void spaces in these complexes, which are placed around the inversion centre of the unit cell as shown in Fig. S5 and Table S3 (ESI‡), originating from the desolvation of one acetonitrile. This means that 1.5 acetonitrile molecules were in an asymmetric unit and totally three acetonitrile molecules were in the unit cell before desolvation occurred. In this case, the space group of the initial crystal should have lower symmetry, P1, compared to that of the desolvated crystal, P, because acetonitrile is not a symmetric molecule. The channel structure of the solvents makes it possible to desolvate easily from the crystal.
Among those complexes, HoL is the only one complex that has two molecules of acetonitrile in an asymmetric unit, meaning that there are totally four acetonitrile molecules in a unit cell.
2. Luminescence properties of LnL
To discuss the luminescence spectra of LnL, the electronic absorption spectra should be observed, because they generally give much information on the electronic and molecular structure. Fig. 4 shows the electronic spectra localized on the ligand moieties of EuL, GdL and TbL in the solid state and in acetonitrile. All complexes LnL show the ππ* absorption bands of the ligand mainly at 280, 305 and 330 nm in the solid state. Their corresponding bands appear at 275, 315 and 330 nm, respectively, in solution. This observation suggests that a series of LnL form molecular structures similar to those in solution and similar molecular arrangements in the solid state. ESI-TOF-MS spectra of these complexes in acetonitrile show a molecular ion for each LnL unit (Fig. S2, ESI‡). The shape of the broadened absorption band in the solid state compared with that in the solution is attributed to the intermolecular interaction as above mentioned. The electronic absorption bands of EuL are observed in acetonitrile, and the band at 330 nm can be ascribed as the lowest ππ* transition experimentally. The ππ* electronic absorption bands of NdL and HoL also appear at exactly corresponding positions of those of EuL (Fig. S6, ESI‡). Additionally, NdL or HoL shows sharp absorption bands originating from the ff-transitions of NdIII or HoIII as shown below.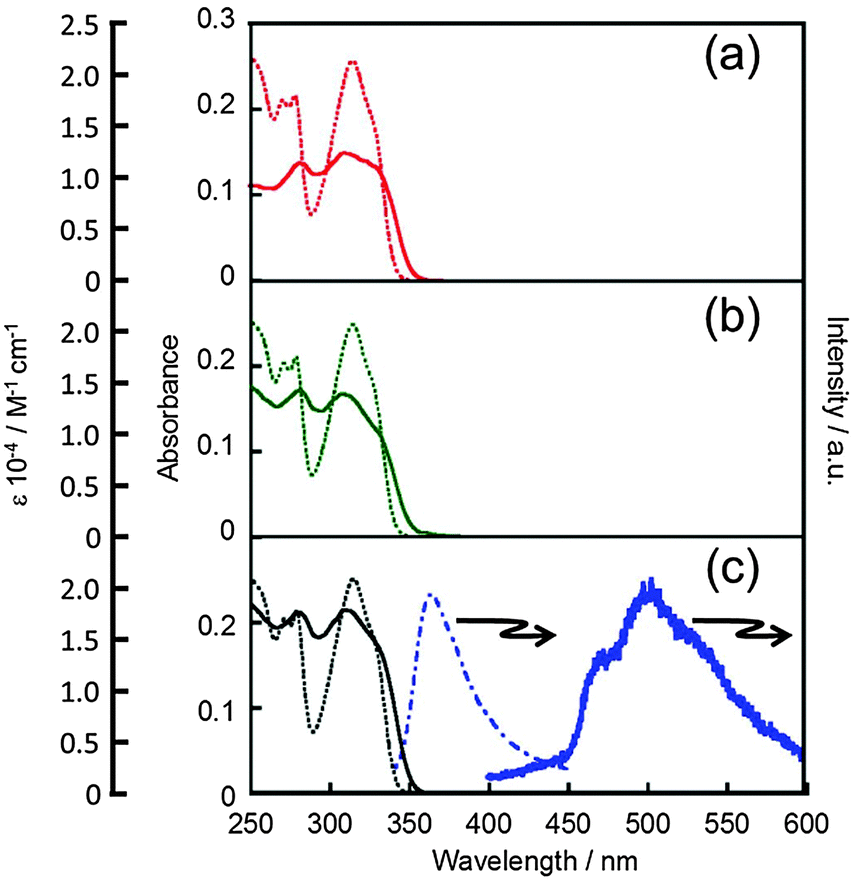 | ||
| Fig. 4 Electronic spectra of EuL (a), TbL (b), and GdL (c) localized on the π electronic systems. Absorption spectra in acetonitrile (dotted line) and in the solid state (solid line) use the left vertical scales; emission spectra in the solid state at rt (dotted-dash) and 77 K (thick) the right one (λex = 330 nm). | ||
To estimate the energy donor level of L of LnL, luminescence spectra of GdL have been examined, since a split upper f-level of GdIII, 6PJ, locates in the vacuum ultra violet region meaning that GdIII shows no ff emission. Emission bands of GdL are observed at 361 (27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500) and ca. 500 nm (as a broadened band 21700–16600 cm−1) mainly, at rt and 77 K, respectively (Fig. 4c).
500) and ca. 500 nm (as a broadened band 21700–16600 cm−1) mainly, at rt and 77 K, respectively (Fig. 4c).
The former transition and latter are assigned to the fluorescence 1S and phosphorescence T band, respectively, localized on the bpy moiety of GdL. It means that the excited T level of L is suitable to act as an energy donor to EuIII or TbIII. The complex, EuL or TbL (also, NdL), shows no ππ* emissions, because the excited ligand-centred photon prevails for energy transfer to EuIII or TbIII relative to the ππ* relaxations. It is notable that these ff emission properties of our present systems are unique, because the complexes have no ππ* emission due to the efficient intramolecular energy transfer from L to LnIII.
The luminescence spectra of EuL and TbL in acetonitrile and in the solid state are shown in Fig. 5. EuL in the solid state at rt shows the ff emission of the trivalent Eu ion at 580, 595, 615, 650 and 685 nm, which is assigned to the 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2, 5D0 → 7F3 and 5D0 → 7F4 transitions, respectively. These bands of EuL appear at the same position at 77 K. These corresponding bands are also observed at almost the same position in acetonitrile. Each excitation spectrum monitored at the ff emission band position reproduces well each electronic absorption spectrum assigned to the lowest excited state of L (Fig. S7, ESI‡).
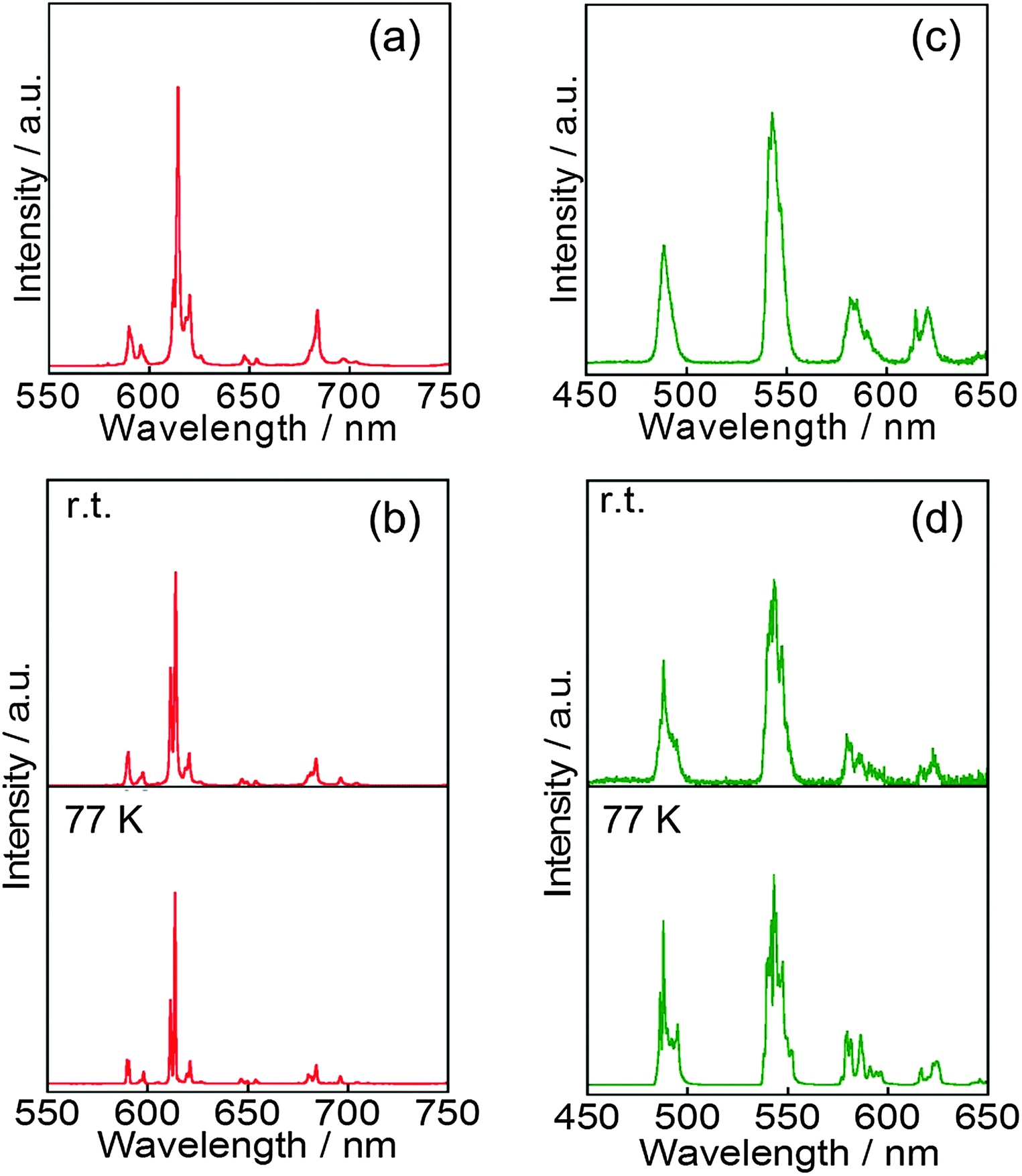 | ||
| Fig. 5 Luminescence spectra of EuL ((a) and (b)) and TbL ((c) and (d)). (a) and (c) are in acetonitrile at rt, and (b) and (d) in the solid state at rt and 77 K (λex = 330 nm). | ||
Absolute luminescence quantum yields QLLn and luminescence lifetimes τobs of EuL and TbL are estimated and the values are given in Table 2. The τobs values for ff-emissions of EuL in the solid state and in acetonitrile as well as TbL in the solid state at 77 K are closer to each other in the 1.27–1.55 ms range (Fig. S8, ESI‡). The QLLn values of EuL in the solid state preserve over 50% at ambient and low temperatures. Furthermore, the QLLn value of EuL in acetonitrile is 12%, which is not all that high while enough to discuss photoproperties even in solutions as same as those of other systems.39
| Temp. | τ obs [ms] (amp.) | Q LLn [%] | ||
|---|---|---|---|---|
| a The values of Ln emission were based on the ligand excitation. | ||||
| EuL | In the solid state | rt | 1.27 (1.0) | 52.6 (±1.4) |
| 77 K | 1.35 (1.0) | 63.5 (±2.7) | ||
| In acetonitrile | rt | 1.55 (1.0) | 12.0 (±0.5) | |
| TbL | In the solid state | rt | 0.0153 (0.96) | 1.0 (±0.2) |
| 0.00234 (0.04) | ||||
| 77 K | 1.49 (1.0) | 91.5 (±1.4) | ||
| In acetonitrile | rt | n.d. | ≈0 | |
It is generally known that the ff emissions of TbIII show thermal sensitivity, which is caused by the thermal equilibrium on the energy transfer pathway between the T level of L and the energy acceptor level of TbIII.40 It is noteworthy that TbL also leads to the efficient luminescence of TbL at 77 K. Luminescence bands of TbL in acetonitrile appear at 490, 545, 587 and 623 nm, which are assigned to the 5D4 → 7F6, 5D4 → 7F5, 5D4 → 7F4 and 5D4 → 7F3 transitions, respectively (Fig. 5c). These band positions are almost the same as those in the solid state (Fig. 5d). Based on the estimation of QLLn values of TbL (Table 2), the values at ambient temperature both in the solid state and in acetonitrile show negligible low values. While it is significant that at 77 K in the solid state, the QLLn value of TbL drastically increases over 90%. As far as we know, this value is the remarkably high value of quantum yields in Tb complexes,16a,41 and thus drastic thermal effect on the value is unusual. The energy relaxation mechanism of TbL is shown in Fig. 6; the T level of L acts as an energy donor via intersystem crossing after the internal conversion. That is, this thermal sensing effect of TbL is caused by the equilibrium between the emissive level of TbIII and the energy donating level of L. Also, it is known that molecular oxygen acts as a quencher for the aromatic triplet excited state and similarly affects the lanthanide luminescence.42 Thus, the significantly low QLLn value of TbL in acetonitrile might be affected by such oxygen molecule effects with thermal relaxation.
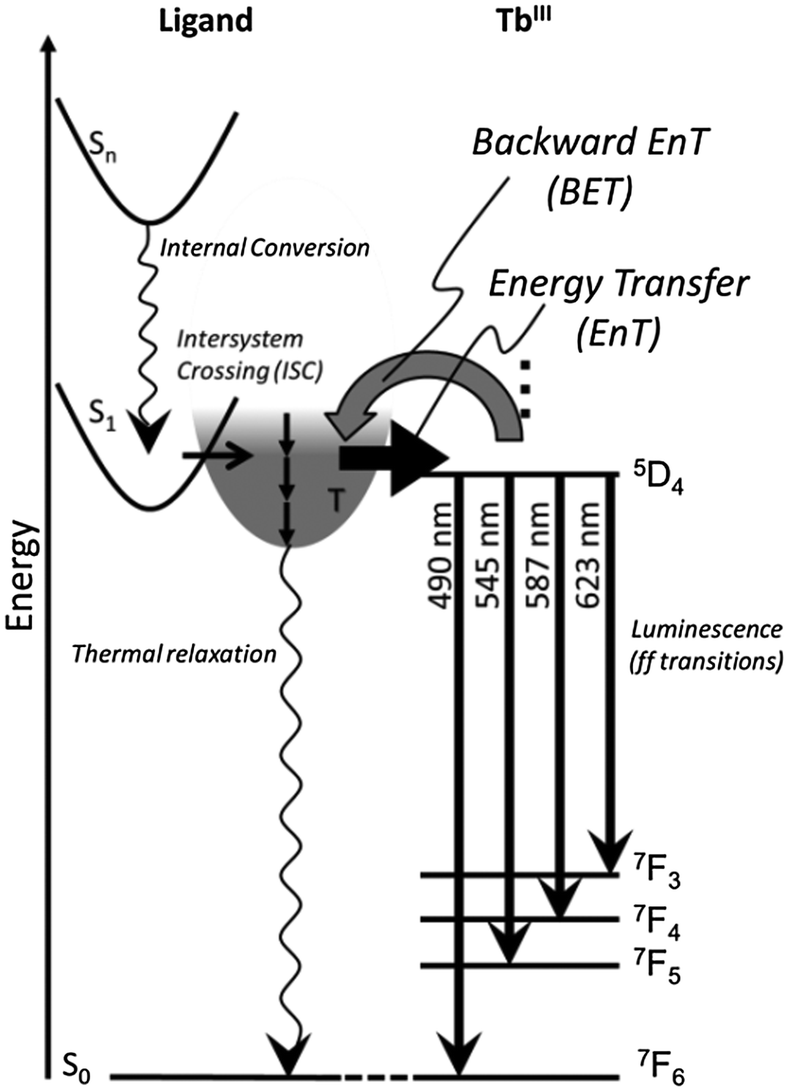 | ||
| Fig. 6 Schematic representation of a plausible energy diagram of the electronic transitions of TbL. | ||
Emission and excitation spectra of NdL under various conditions are shown in Fig. 7 and Fig. S9 (ESI‡), respectively. The ff emission bands of NdIII appearing at 906, 1055 and 1345 nm are assigned to the 4F3/2 → 4I9/2, 4F3/2 → 4I11/2 and 4F3/2 → 4I13/2 transition, respectively. Former two transitions are divided into two or more by the Stark effect. The emission efficiency is quite low and it is difficult to estimate a quantitative value. However, it is found that the ligand L also acts as an energy donor and sensitizes the ff emissions of NdIII in the NIR region, since the excitation spectrum shows good correspondence with the absorption bands of L, not ff transitions of NdIII (Fig. 8a and b). In the solid state, the ff-absorption bands of NdL in the visible-NIR region appear at 468 (the 4I9/2 → 4G9/2 transition), 526 (4I9/2 → 4G7/2), 584 (4I9/2 → 4G5/2), 626 (4I9/2 → 2H11/2), 679 (4I9/2 → 4F9/2), 739 (4I9/2 → 2H9/2), 800 (4I9/2 → 4S3/2) and 874 (4I9/2 → 4F3/2) nm. Corresponding ff-absorption bands of NdL in the solid state were clearly observed in acetonitrile.
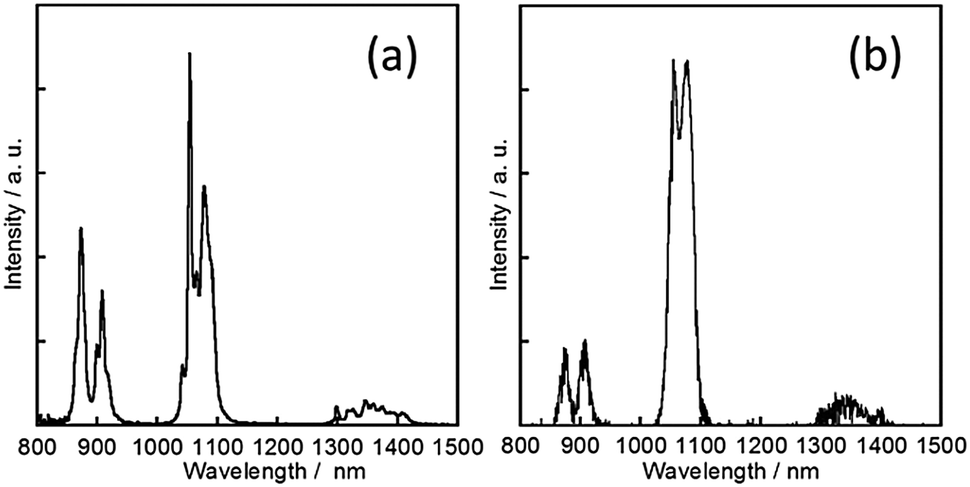 | ||
| Fig. 7 Luminescence spectra of NdL (a) in the solid state and (b) in acetonitrile (λex = 330 nm). | ||
 | ||
| Fig. 8 Electronic absorption spectra of NdL ((a) and (b)) and HoL ((c) and (d)) in acetonitrile (upper) and in the solid state (bottom) (* due to apparatus). | ||
HoL also shows the bands at 420 (5I8 → 5G5), 450 (5I8 → 5G6), 472 (5I8 → 3K8), 486 (5I8 → 5F2), 537 (overlap of 5I8 → 5F4 and 5I8 → 4S2) and 644 (5I8 → 5F5) nm in acetonitrile and in the solid state (Fig. 8c and d). Due to small differences among the split f-levels of HoIII, thermo-relaxation prevails over photo-relaxation (luminescence).
Energy diagrams of the relaxation process of EuL, GdL and NdL are illustrated in Fig. S10 (ESI‡). The phosphorescence band position and width of GdL refer to the energy donor level against Ln ions. Since the structural analyses and the electronic absorption spectra localized on L for these complexes are in accordance with each other, it is considered that the donor level of L (triplet) of all complexes exists at the same energy level. The acceptor level of EuIII is more overlapped with the donor (Fig. S10(a), ESI‡) than that of NdIII (Fig. S10(c), ESI‡). These differences of EuL and NdL would affect the luminescence efficiencies.
Conclusions
The above presented new organic ligand forms stable Ln complexes with Nd, Eu, Gd, Tb and Ho. All of them formed a similar helical molecular structure in the crystalline state. Based on the above spectral results, TbL is preferred to thermal energy relaxation, since the thermal equilibrium between the energy donor and the acceptor level has been more preferential than the vibration effect of intermolecular interactions among neighbouring complexes. Furthermore, in solution, the ff-emission property of EuL is still present although it is less pronounced than in the solid state. This seems to be due to its stable molecular structure. Also NdL shows the respective ff-emission in the NIR region even in solution, despite its low efficiency probably due to its electronic structure. All complexes LnL with ff-emission properties do not show ππ* luminescence, meaning that the photo-antenna acts efficiently.Experiments
Materials and synthesis
Synthesis of ligand (L). Most reagents and spectra-grade solvents were used without further purification. Bipyridine-6-aldehyde was obtained by mixing 6-bromo-bipyridine 2.10 g (8.93 mmol; Isotech Laboratories, Inc.) and 1.6 M n-hexane solution of n-butyl-lithium 6.80 ml (10.9 mmol) in THF at −80 °C for 30 min, then stirred for 1 h after addition of dimethylformamide43 (0.670 mg, 3.64 mmol, 40.8% yield). 1H-NMR (500.00 MHz, CDCl3); δ 10.2 (s, 1H), 8.72 (d, 3J = 4.1 Hz, 1H), 8.64 (m, 1H), 8.57 (d, 3J = 8.2 Hz, 1H), 8.00 (m, 2H), 7.86 (m, 1H), 7.23 (m, 2H), 7.38 (m, 1H).The hexadentate ligand L was prepared by the reaction of bipyridine-6-aldehyde 200 mg and ethylene-diamine (Kanto Chemicals Co., Inc.) in methanol (160 mg, 0.408 mmol, 74.9% yield). 1H-NMR (500.00 MHz, CDCl3); δ 8.64 (d, 3J = 4.1 Hz, 2H), 8.53 (s, 2H), 8.41 (dd, 3J = 7.8 and 7.3 Hz, 4H), 8.04 (d, 3J = 6.9 Hz, 2H), 7.80 (t, 3J = 7.8 Hz, 2H), 7.73 (m, 2H), 7.23 (m, 2H), 4.10 (s, 4H). 1H-NMR chart of above compounds are shown in Fig. S1 (ESI‡).
NdL. 50.4 mg (0.115 mmol) Nd(NO3)3·6H2O, 45.0 mg (0.115 mmol) L and NH3PF6 18.7 mg (0.115 mmol). Yield 77.2 mg (80.1%). Elemental analysis, calcd for [Nd(L)(NO3)2](PF6) (C24H20N8O6NdPF6); C 35.78, H 2.50, N 13.91; found; C 35.48, H 2.56, N 14.07; MS (ESI-TOF, acetonitrile); m/z, 657.94 [M − PF6−]+ (calcd 658.06).
EuL. 51.3 mg (0.115 mmol) Eu(NO3)3·6H2O, 45.0 mg (0.115 mmol) L and NH3PF6 18.7 mg (0.115 mmol). Yield 51.3 mg (55%). Elemental analysis, calcd for [Eu(L)(NO3)2](PF6) (C24H20N8O6EuPF6); C 35.44, H 2.48, N 13.78; found; C 35.27, H 2.62, N 13.70; MS (ESI-TOF, acetonitrile); m/z, 668.97 [M − PF6−]+ (calcd 669.07).
GdL. 121 mg (0.270 mmol) Gd(NO3)3·6H2O, 105.6 mg (0.270 mmol) L and NH3PF6 44.0 mg (0.270 mmol). Yield 58.4 mg (31%). Elemental analysis, calcd for [Gd(L)(NO3)2](PF6) (C24H20N8O6GdPF6); C 35.21, H 2.46, N 13.69; found; C 35.12, H 2.68, N 13.62; MS (ESI-TOF, acetonitrile); m/z, 674.06 [M − PF6−]+ (calcd 674.08).
TbL. 52.1 mg (0.115 mmol) Tb(NO3)3·6H2O, 45.0 mg (0.115 mmol) L and NH3PF6 18.7 mg (0.115 mmol). Yield 68.3 mg (62%). Elemental analysis, calcd for [Tb(L)(NO3)2](PF6) (C24H20N8O6TbPF6); C 35.14, H 2.46, N 13.66; found; C 35.26, H 2.70, N 13.50; MS (ESI-TOF, acetonitrile); m/z, 675.02 [M − PF6−]+ (calcd 675.08).
HoL. 50.7 mg (0.115 mmol) Ho(NO3)3·5H2O, 45.0 mg (0.115 mmol) L and NH3PF6 18.7 mg (0.115 mmol). Yield 79.4 mg (80.4%). Elemental analysis, calcd for [Ho(L)(NO3)2](PF6)·H2O (C24H22N8O7HoPF6); C 34.14, H 2.63, N 13.27; found; C 33.95, H 2.93, N 13.19; MS (ESI-TOF, acetonitrile); m/z, 680.97 [M − PF6−]+ (calcd 681.08).
Instrumentation
Electronic absorption and luminescence spectra were recorded on a Shimadzu UV3101PC and a Horiba Jobin-Ybon Fluorolog 3-22. The NIR emission was detected by the attachment C1452-AU on the above apparatus. The emission decay curves were measured using a Quantaurus-Tau C11367-12 (Hamamatsu Photonics K. K.) excited by a Xenon flash lamp with a band-path filter (λex = 340 nm). The fluorescence quantum yields were measured using The C9920-02 Absolute PL Quantum Yield Measurement System (Hamamatsu Photonics K. K.).44Electrospray ionization time of flight (ESI-TOF) mass spectra and elemental analyses for CHN were recorded on a LCT ESI-TOF spectrometer (Micromass), and MICRO CORDER (J-SCIENCE LAB), respectively.
X-ray crystallography
X-ray structural data for LnL (Ln = Nd, Eu, Gd, Tb and Ho) were collected on a Bruker Smart APEX-II CCD diffractometer equipped with graphite monochromated Mo Kα radiation at 100 K. The data were collected to a maximum 2 h value of 55° and processed using the Bruker Apex2 software package.45 The structures were solved by direct methods and refined by full-matrix least-squares calculations using SHELX-97.46 All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were located at idealized positions. Summaries of the fundamental crystal data and experimental parameters used to determine the structures of complexes LnL are given in ESI.‡ CCDC 919427 for EuL, 919428 for GdL, 919429 for HoL, 919430 for NdL and 919431 for TbL.Acknowledgements
We thank Prof. Takashi Kato (the University of Tokyo) and Prof. Wolfgang Linert (Technical University of Vienna) for their kind discussion. Prof. Yoshiki Ohgo (Teikyo University) could help to refine structural analyses of a series of single crystals. Prof. Yasuteru Shigeta (Osaka University) could suggest important points of view for the energy diagram of electronic transitions. Dr Isao Takahashi, Yu Yoshimura and Sae Enomoto (AGU) also helped to synthesize the related complexes. Prof. Hideki Masuda, Prof. Tomohiro Ozawa and Dr Tomohiko Inomata (Nagoya Institute of Technology) could kindly help to measure ESI-TOF-MS in solutions. This work was partly supported by the Grants-in-Aid for Young Scientists A (No. 20685011), Scientific Research on Innovative Areas of “Fusion Materials: Creative Development of Materials and Exploration of Their Function through Molecular Control (Area Number: 2206)” (No. 23107528 and 25107730), Challenging Exploratory Research (No. 23656544) and The High-Tech Research Centre Project for Private Universities with a matching fund subsidy from MEXT.Notes and references
- (a) M. A. Katkova and M. N. Bochkarev, Dalton Trans., 2010, 39, 6599 RSC; (b) G. Zucchi, Int. J. Inorg. Chem., 2011, 2011, 92435 Search PubMed; (c) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357 CrossRef CAS PubMed.
- J. F. Wang, R. Y. Wang, J. Yang, Z. P. Zheng, M. D. Carducci, T. Cayou, N. Pwyghambarian and G. E. Jabbour, J. Am. Chem. Soc., 2001, 123, 6179 CrossRef CAS.
- Z. Chen, F. Ding, F. Hao, M. Guan, Z. Bian, B. Ding and C. Huang, New J. Chem., 2010, 34, 487 RSC.
- S.-J. Yeh, M.-F. Wu, C.-T. Chen, Y.-H. Song, Y. Chi, M.-H. Ho, S.-F. Hsu and C.-H. Chen, Adv. Mater., 2005, 17, 285 CrossRef CAS.
- H. Xin, F. Y. Li, M. Shi, Z. Q. A. Bian and C. H. Huang, J. Am. Chem. Soc., 2003, 125, 7166 CrossRef CAS PubMed; M. A. Katkova, A. P. Pushkarev, T. V. Balashova, A. N. Konev, G. K. Fukin, S. Y. Ketkov and M. N. Bochkarev, J. Mater. Chem., 2011, 21, 16611 RSC.
- S. F. Wuister, C. Donegá and A. Meijerink, Phys. Chem. Chem. Phys., 2004, 6, 1633 RSC.
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081 RSC.
- (a) R. Pal, D. Parker and L. C. Costello, Org. Biomol. Chem., 2009, 7, 1525 RSC; (b) G.-L. Law, R. Pal, L. O. Palsson, D. Parker and K.-L. Wong, Chem. Commun., 2009, 7321 RSC; (c) D. G. Smith, R. Pal and D. Parker, Chem.–Eur. J., 2012, 18, 11604 CrossRef CAS PubMed; (d) P. K. Senanayake, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2129 Search PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS PubMed.
- (a) S. Shinoda, K. Terada, M. Eiraku Masaki, Y. Kataoka and H. Tsukube, New J. Chem., 2012, 36, 1545 RSC; (b) H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977 RSC; (c) K. Ariga, H. Ito, J. P. Hill and H. Tsukube, Chem. Soc. Rev., 2012, 41, 5800 RSC.
- (a) J. W. Walton, A. Bourdolle, S. J. Butler, M. Soulie, M. Deldianco, B. K. McMahon, R. Pal, H. Puschmann, J. M. Zwier, L. Lamarque, O. Maury, C. Andraud and D. Parker, Chem. Commun., 2013, 49, 1600 RSC; (b) R. A. Poole, G. Bobba, M. J. Cann, J. C. Frias, D. Parker and R. D. Peacock, Org. Biomol. Chem., 2005, 3, 1013 RSC.
- (a) T. Gunnlaugsson, J. P. Leonard, K. Sénéchal and A. J. Harte, J. Am. Chem. Soc., 2003, 125, 12062 CrossRef CAS PubMed; (b) O. Kotova, S. Comby and T. Gunnlaugsson, Chem. Commun., 2011, 47, 6810 RSC.
- (a) F. S. Richardson, Chem. Rev., 1982, 82, 541 CrossRef CAS; (b) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487 CrossRef CAS PubMed.
- (a) S. V. Eliseeva and J.-C. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC; (b) J.-C. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- (a) M. Brebol, U. Kynast and C. Ronda, Adv. Mater., 1991, 3, 361 CrossRef; (b) N. Filipescu, G. W. Mushrush, C. R. Hurt and N. McAvoy, Nature, 1966, 211, 960 CrossRef CAS; (c) S. Sato and M. Wada, Bull. Chem. Soc. Jpn., 1970, 43, 1955 CrossRef CAS.
- (a) M. Hasegawa, A. Nakao, M. Masui, T. Tamura, D. Suzuki, W. Linert, Y. Fukuda and T. Hoshi, Chem. Phys., 2001, 269, 323 CrossRef CAS; (b) M. Hasegawa, A. Ishii, T. Yamazaki, S. Kishi and I. Yamazaki, Chem. Lett., 2005, 1418 CrossRef CAS; (c) M. Hasegawa, A. Ishii and S. Kishi, J. Photochem. Photobiol., A, 2006, 178, 220 CrossRef CAS PubMed.
- (a) J.-C. G. Bünzli, A.-S. Chauvin, H. K. Kim, E. Deiters and S. V. Eliseeva, Coord. Chem. Rev., 2010, 254, 2623 CrossRef PubMed; (b) A. Beeby, L. M. Bushby, D. Maffeo and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2002, 48 RSC; (c) G. Adachi, N. Imanaka and S. Tamura, J. Rare Earths, 2010, 28, 843 CrossRef CAS.
- (a) S. Quici, G. Marzanni, A. Forni, G. Accorsi and F. Barigelletti, Inorg. Chem., 2004, 43, 1294 CrossRef CAS PubMed; (b) O. L. Malta, H. F. Brito, J. F. S. Menezes, F. R. Gonçalves e Silva, C. de Mello Donegá and S. Alves Jr., Chem. Phys. Lett., 1998, 282, 233 CrossRef CAS; (c) E. G. Moore, J. Jocher, J. Xu, E. J. Werner and K. N. Raymond, Inorg. Chem., 2007, 46, 5468 CrossRef CAS PubMed; (d) R. Zong, G. Zhang, S. V. Eliseeva, J.-C. G. Bünzli and R. P. Thummel, Inorg. Chem., 2010, 49, 4657 CrossRef CAS PubMed; (e) J.-F. Lemonnier, L. Guénée, C. Beuchat, T. A. Wesolowski, P. Mukherjee, D. H. Waldeck, K. A. Gogick, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2011, 113, 16219 CrossRef PubMed.
- (a) S. Quici, M. Cavazzini, G. Marzanni, G. Accorsi, N. Armaroli, B. Ventura and F. Barigelletti, Inorg. Chem., 2005, 44, 529 CrossRef CAS PubMed; (b) S. I. Klink, G. A. Hebbink, L. Grave, P. G. B. O. Alink, F. C. J. M. van Veggel and M. H. V. Werts, J. Phys. Chem. A, 2002, 106, 3681 CrossRef CAS; (c) S. Lis, Z. Hnatejko, P. Barczynski and M. Elbanowski, J. Alloys Compd., 2002, 344, 70 CrossRef CAS.
- V.-M. Mukkala and J. J. Kankare, Helv. Chim. Acta, 1992, 75, 1578 CrossRef CAS.
- A. Bourdolle, M. Allali, J.-C. Mulatier, B. Le Guennic, J. M. Zwier, P. L. Baldeck, J.-C. G. Bünzli, C. Andraud, L. Lamarque and O. Maury, Inorg. Chem., 2011, 50, 4987 CrossRef CAS PubMed.
- Z. Pan, G. Jia, C.-K. Duan, W.-Y. Wong, W.-T. Wong and P.-A. Tanner, Eur. J. Inorg. Chem., 2011, 637 CrossRef CAS.
- N. M. Shavaleev, S. V. Eliseeva, R. Scopelliti and J.-C. Bünzli, Chem.–Eur. J., 2009, 15, 10790 CrossRef CAS PubMed.
- K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem.–Eur. J., 2011, 17, 521 CrossRef CAS PubMed.
- L. J. Charbonnière, N. Wiebel, P. Retailleau and R. Ziessel, Chem.–Eur. J., 2007, 13, 346 CrossRef PubMed.
- C. Butler, S. Goetz, C. M. Fitchett, P. E. Kruger and T. Gunnlaugsson, Inorg. Chem., 2011, 50, 2723 CrossRef CAS PubMed.
- S. Quici, M. Cavazzini, G. Marzanni, G. Accorsi, N. Armaroli, B. Ventura and F. Barigelletti, Inorg. Chem., 2005, 44, 529 CrossRef CAS PubMed.
- C. Kachi-Terajima, K. Yanagi, T. Kazuki, T. Kitazawa and M. Hasegawa, Dalton Trans., 2011, 40, 2249 RSC.
- Y. Hasegawa, R. Hieda, K. Miyata, T. Nakagawa and T. Kawai, Eur. J. Inorg. Chem., 2011, 4978 CrossRef CAS.
- J. L. Bender and C. L. Fraser, ACS Symposium Series “Chromogenic Phenomena in Polymers”, 2005, ch. 18, vol. 888, p. 233 Search PubMed.
- (a) Y.-L. Huang, M.-Y. Huang, T.-H. Chan, B.-C. Chang and K.-H. Lii, Chem. Mater., 2007, 19, 3232 CrossRef CAS; (b) B. D. Chandler, J. O. Yu, D. T. Cramb and G. K. H. Shimizu, Chem. Mater., 2007, 19, 4467 CrossRef CAS.
- (a) A. Kobayashi, H.-C. Chang, Y. Fukuzawa and M. Kato, Inorg. Chem., 2012, 51, 7508 CrossRef CAS PubMed; (b) T. K. Maji, G. Mostafa, H.-C. Chang and S. Kitagawa, Chem. Commun., 2005, 2436 RSC; (c) L. J. Charbonnière, R. Ziessel, M. Montalti, L. Prodi, N. Zaccheroni, C. Boehme and G. Wipff, J. Am. Chem. Soc., 2002, 124, 7779 CrossRef PubMed.
- T. Kajiwara, M. Hasegawa, A. Ishii, K. Katagiri, M. Baater, S. Takaishi, N. Iki and M. Yamashita, Eur. J. Inorg. Chem., 2008, 5565 CrossRef CAS.
- L. Charbonnière, S. Mameri, P. Kadjane, C. Platas-Iglesias and R. Ziessel, Inorg. Chem., 2008, 47, 3748 CrossRef PubMed.
- (a) E. C. Constable, R. Chotalia and D. A. Tocher, J. Chem. Soc., Chem. Commun., 1992, 771 RSC; (b) M. H. W. Lam, D. Y. K. Lee, S. S. M. Chiu, K. W. Man and W. T. Wong, Eur. J. Inorg. Chem., 2000, 1483 CrossRef CAS.
- (a) R. Zong and R. P. Thummel, Inorg. Chem., 2005, 44, 5984 CrossRef CAS PubMed; (b) L. Aboshyan-Sorgho, H. Nozary, A. Aebischer, J.-C. G. Bünzli, P.-Y. Morgantini, K. R. Kittilstved, A. Hauser, S. V. Eliseeva, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2012, 134, 12675 CrossRef CAS PubMed; (c) S. Petoud, J.-C. G. Bünzli, F. Renaud, C. Piguet, K. J. Schenk and G. Hopfgartner, Inorg. Chem., 1997, 36, 5750 CrossRef CAS PubMed.
- (a) E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and J. A. Zampese, CrystEngComm, 2010, 12, 3724 RSC; (b) E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and S. Schaffner, Dalton Trans., 2009, 8165 RSC; (c) E. C. Constable, G. Zhang, C. E. Housecroft and J. A. Zampese, Dalton Trans., 2010, 39, 5332 RSC; (d) E. C. Constable, G. Zhang, C. E. Housecroft, M. N. Neuburger and J. A. Zampese, Eur. J. Inorg. Chem., 2010, 2000 CrossRef CAS.
- For instance, (a) G. Zucchi, V. Murugesan, D. Tondelier, D. Aldakov, T. Jeon, F. Yang, P. Thuéry, M. Ephritikhine and B. Geffroy, Inorg. Chem., 2011, 50, 4851 CrossRef CAS PubMed; (b) S. Petoud, S. M. Cohen, J.-C. G. Bünzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324 CrossRef CAS PubMed.
- (a) D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 3613 RSC; (b) S. Katagiri, Y. Tsukahara, Y. Hasegawa and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 8, 1492 CrossRef; (c) Y. Zheng, C. Tan, G. P. C. Drummen and Q. Wang, Spectrochim. Acta, Part A, 2012, 96, 387 CrossRef CAS PubMed; (d) Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134, 3979 CrossRef CAS PubMed; (e) J. Andres and A.-S. Chauvin, Inorg. Chem., 2011, 50, 10082 CrossRef CAS PubMed.
- (a) E. Brunet, O. Juanes, R. Sedano and J.-C. Rodríguez-Ubis, Photochem. Photobiol. Sci., 2002, 1, 613 RSC; (b) M. Starck, P. Kadjane, E. Bois, B. Darbouret, A. Incamps, R. Ziessel and L. J. Charbonnière, Chem.–Eur. J., 2011, 17, 9164 CrossRef CAS PubMed; (c) A. R. Ramya, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, Inorg. Chem., 2010, 49, 2407 CrossRef CAS PubMed; (d) S. Biju, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, J. Mater. Chem., 2009, 19, 5179 RSC; (e) A. P. S. Samuel, E. G. Moore, M. Melchior, J. Xu and K. N. Raymond, Inorg. Chem., 2008, 47, 7535 CrossRef CAS PubMed.
- D. Parker and J. A. G. Williams, Chem. Commun., 1998, 245 RSC ; and their in.
- F. R. Heirtzler, M. Neuburger, M. Zehnder and E. C. Constable, Liebigs Ann./Recl., 1997, 247 Search PubMed.
- (a) Y. Kawamura, H. Sasabe and C. Adachi, Jpn. J. Appl. Phys., 2004, 43, 7729 CrossRef CAS; (b) K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC; (c) A. Kobayashi, K. Suzuki, T. Yoshihara and S. Tobita, Chem. Lett., 2010, 39, 282 CrossRef CAS; (d) M. Hasegawa, A. Ishii, K. Furukawa and H. Ohtsu, J. Photopolym. Sci. Technol., 2008, 21, 333 CrossRef; (e) M. Hasegawa, S. Kunisaki, H. Ohtsu and W. Franz, Monatsh. Chem., 2009, 140, 751 CrossRef CAS PubMed.
- SMART, SAINT, XPREP and SADABS, Bruker AXS Inc., Madision, Wisconsin, 2004 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELXS-97, University of Göttingen, Göttingen, 1997 Search PubMed; (c) G. M. Sheldrick, SHELX-97, University of Göttingen, Göttingen, 1997 Search PubMed.
Footnotes |
| † Dedicated to Dr Toshihiko Hoshi, Professor Emeritus from Aoyama Gakuin University, on the occasion of his 77th birthday. |
| ‡ Electronic supplementary information (ESI) available: Detailed preparation and characterization of the complexes. CCDC 919427–919431 (a series of lanthanide complexes with L). Electronic absorption spectra of NdL and HoL, excitation spectra, luminescence decay curves, and representation of energy transfer mechanisms of the complexes. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00910f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |