 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photophysical and electrochemical properties and temperature dependent geometrical isomerism in alkyl quinacridonediimines†
Iqbal
Javed
*abc,
Ayub
Khurshid
*de,
Muhammad Nadeem
Arshad
f and
Yue
Wang
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: javedkhattak79@gmail.com; yuewang@jlu.edu.cn; Tel: +46-704763801
bDepartment of Chemistry, Umeå University, SE-901 87 Umeå, Sweden
cDepartment of Chemistry & Biochemistry, University of Agriculture, Faisalabad, Pakistan
dDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Kingdom of Saudi Arabia. E-mail: kayub@kfu.edu.sa
eDepartment of Chemistry, COMSATS Institute of Information Technology, Abbottabad, KPK, Pakistan 22060
fCentre of Excellence for Advanced Materials Research (CEAMR), King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia
First published on 27th November 2013
Abstract
Quinacridone diimines 1–10 were synthesized by the condensation of anilines with alkyl substituted quinacridones (QA). Photophysical and electrochemical properties of the compounds were investigated. Unconventional behavior of absorption spectra suggested a decrease in π_conjugation within the QA skeleton as well as lack of extended π_conjugation between the QA skeleton and the N-phenyl rings. A computational study of compounds 1–10, a variable temperature 1H NMR study of compounds 2, 7 and 10 (for instance), and single crystal X-ray analysis of 2, 3, 6, 7, 8 and 10 indicated that the anomalous behavior is due to the buckled, non-planar structure of the quinacridones. Moreover the molecules existed in two interconvertible geometric isomeric forms at different temperatures. Molecular orbital calculations were performed at B3LYP/6-31+G(d), B3PW91/6-31G(d) and PBEPBE/6-31G(d) levels of theory at B3PW91/6-31G(d) optimized structures for both isomers of all compounds (1–10); the results obtained are in close agreement with the experimentally determined values.
Introduction
Quinacridone (QA) and its derivatives are widely used organic pigments that display excellent fastness properties as well as pronounced photovoltaic and photoconductive activities.1–3 Many investigations on QA derivatives have been performed to explore the effects of different structural parameters on their physical properties. For example, Müllen and co-workers have carefully studied the phase-formation behavior and aggregation properties of some soluble QAs.4,5 Nakahara and co-workers have applied alkyl QA derivatives in Langmuir–Blodgett films to control the orientation and packing of the chromophores.6 Jones and co-workers have reported the crystal structure of QA.7 Schmidt and co-workers have reported different polymorphic forms of QA that differ significantly in terms of their coloristic properties.8 Our group has explored the polymorphs and pseudopolymorphs of N,N-di (n-butyl) quinacridone.9The literature reveals that a great deal of effort has been devoted to the synthesis and characterization of various QA derivatives; however little attention has been paid to functionalization of the carbonyl group.10 To fully exploit the potential applications of such functional compounds in organic material based devices, it is necessary to design and synthesize some new types of QA derivatives with different molecular structures. A careful understanding of the relationship between molecular structures, conformations and optical and electronic properties in solution and condensed states will lead to the development of new strategies for the preparation of high-performance organic optical and electronic materials.
A series of QA diimines were synthesized by the condensation of alkyl substituted quinacridones with aromatic amines. The introduction of the aniline base backbone generally not only enhances the conducting and optoelectronic properties of the derivatives but also broadens the absorption band due to extended π-conjugation but this requires coplanarity, at least to within 15°.11 Physical properties are closely related to structure but after measuring the photophysical properties of the synthesized compounds the coplanarity was questionable as they show anomalous behavior in their physical properties.
For in-depth structural exploration of these compounds, single crystals have been grown at different temperatures for some compounds and single crystal X-ray analysis is carried out. Density Functional Theory (DFT) calculations are also performed in an attempt to attain a certain level of predictability in these systems. To get an unclouded picture we embarked on the study of the variable temperature 1H NMR spectrum of compounds 2, 7 and 10 for instance. We also evaluated the relationship between the structure, conformation and optoelectronic properties of the QA diimines.
On the basis of single crystal X-ray analysis, molecular modeling calculations and the variable temperature 1H NMR spectrum study, herein we report that these QA diimines exist in different geometrical isomeric forms at different temperatures with the buckled QA skeleton in a non-planar fashion.
Computational methods
DFT calculations were performed using the Gaussian 03 suite of programs.12 Geometries of both isomers of QA diimines, 1–10, were optimized using the hybrid functional B3PW91 method, which consists of Becke's three-parameter (B3) hybrid exchange functional in conjunction with the correlation functional of the Perdew and Wang (PW91) method. Frequency calculations were performed in order to confirm these structures as true minima (absence of an imaginary frequency). Molecular orbital calculations were performed at B3LYP/6-31+G(d), B3PW91/6-31G(d) and PBEPBE/6-31G(d) levels of theory at B3PW91/6-31G(d) optimized structures.Experimental section
Instrumentation
1H NMR spectra were recorded on a Bruker AVANCE 500 MHz spectrometer with tetramethylsilane as the internal standard. Elemental analysis and mass spectroscopy were carried out on Flash EA 1112 and GC/MS mass spectrometers, respectively. UV-vis absorption spectra were recorded using a PE UV-Vis lambdazo spectrometer.Electrochemical measurements
Cyclic voltammetry was performed on a BAS 100 W instrument with a scan rate of 100 mV s−1. A three-electrode configuration was used for the measurement: a platinum electrode as the working electrode, a platinum wire as the counter electrode, and an Ag/Ag+ electrode as the reference electrode. A 0.1 M solution of tetrabutylammonium perchlorate (TBAP) in CH2Cl2 was used as the supporting electrolyte.X-ray crystallography
Single crystals suited for X-ray structural analysis were obtained by slow diffusion of petroleum ether into the chloroform solution of compounds. Diffraction data were collected on a Rigaku R-AXIS RAPID diffractometer (Mo Kα radiation, graphite monochromator) in the ω rotation scan mode. The structure determination was done using direct methods by using SHELXTL 5.01v and refinements with full-matrix least-squares on F2. The positions of hydrogen atoms were calculated and refined isotropically.Synthesis
Quinacridone was purchased from Tokyo Kasei Kogyo Company. 3,5-Dimethylaniline, 1-bromobutane and 1-bromooctane were obtained from Acros Organics. Diethyl-2, 5-dihydroxy-1, 4-dicarboxylate was purchased from Aldrich. The chemicals were used directly without further purification.The CnQA (n = 4,8), N,N-di(n-butyl)-1,3,8,10-tetramethylquinacridone (TMQA) and N,N-di(n-butyl)-2,9-difluoroquinacridone (DFDBQA) were synthesized according to the similar method reported in the literature.5
Synthesis of N,N-diphenyl-N,N′-di(n-butyl)quinacridonediimine (1)
Aniline (4 mmol) and triethylamine (TEA) 1.2 mL (8.5 mmol) were dissolved in 20 mL of toluene. 0.6 mL (5.4 mmol) of titanium chloride (TiCl4) in 5 mL of toluene was added dropwise over 15 min using an addition funnel. The addition funnel was rinsed with 3 mL of toluene. DBQA (1 mmol) was added using a powder addition funnel at once. The funnel was rinsed with 3 mL toluene. The reaction mixture was refluxed for about 8 hours. The reaction mixture was cooled and filtered, the filtrate obtained was dried and loaded on the silica gel column and eluted with CH2Cl2: diethyl ether (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The desired product was obtained as a red solid in 50% yield.
:1). The desired product was obtained as a red solid in 50% yield.
Compound 1
1H NMR (CDCl3, ppm) (500 MHz): δ 8.50 (s, 2H), 7.43 (d, 2H), 7.34 (t, 6H), 7.11 (m, 2H), 7.01 (t, 4H), 6.90 (d, 4H), 4.32 (t, 2H), 3.40 (m, 2H), 1.99 (m, 2H), 1.55 (m, 4H), 1.01 (m, 6H), 0.89 (t, 2H), MS: m/z 574.31 [M]+. Anal. Calcd for C40H38N4; C, 83.49; H, 6.66; N, 9.75. Found: C, 83.78; H, 6.46; N, 9.70.The same procedure was adopted for compounds 2–10 and reaction conditions are tabulated in Table S1 (ESI†).
N,N-Bis(4-fluorophenyl)-N,N′-di(n-butyl)quinacridonediimine (2)
1H NMR (CDCl3, ppm) (500 MHz): δ 8.43 (s, 2H), 7.54 (d, 4H), 7.34 (m, 2H), 7.03 (t, 6H), 6.80 (m, 4H), 4.32 (t, 2H), 3.43 (t, 2H), 1.90 (m, 2H), 1.35–1.65 (m, 6H), 1.01–1.01 (m, 6H), MS: m/z 612.29 [M]+. Anal. Calcd for C40H36 F2N4; C, 78.66; H, 5.94; N, 9.17. Found: C, 78.66; H, 5.95; N, 9.17.N,N-Diphenyl-N,N′-di(n-butyl)-2,9-difluoroquinacridonediimine (3)
1H NMR (CDCl3, ppm) (500 MHz): δ 8.19 (s, 2H), 7.59 (s, 2H), 7.37 (t, 4H), 7.21 (d, 2H), 7.04 (t, 4H), 6.90 (d, 4H), 4.30 (t, 2H), 3.37 (d, 2H), 1.97 (m, 2H), 1.59 (m, 2H), 1.31 (m, 4H), 1.00 (m, 6H), MS: m/z 612.30 [M]+. Anal. Calcd for C40H36 F2N4 C, 78.66; H, 5.94; N, 9.17. Found: C, 78.74; H, 5.95; N, 8.73.N,N-Bis(4-fluorophenyl)-N,N′-di(n-butyl)-2,9-difluoroquinacridonediimine (4)
1H NMR (CDCl3, ppm) (500 MHz): δ 8.16 (s, 2H), 7.56 (s, 2H), 7.22 (m, 2H), 7.07 (t, 6H), 6.85 (m, 4H), 4.31 (t, 2H), 3.47 (d, 2H), 1.94 (m, 2H), 1.94 (m, 2H), 1.54 (m, 2H), 1.32 (m, 4H), 1.00 (m, 6H), MS: m/z 646.30 [M]+. Anal. Calcd for C40H34F4N4; C, 74.29; H, 5.30; N, 8.66.N,N-Diphenyl-N,N′-di(n-butyl)-1,3,8,10-tetramethylquinacridonediimine (5)
1H NMR (CDCl3, ppm) (500 MHz): δ 7.28 (t, 4H), 6.96 (t, 2H), 6.75 (d, 8H), 3.41–3.44 (m, 4H), 2.75–2.84 (m, 4H), 2.36 (s, 6H), 1.34–1.55 (m, 6H), 1.02 (t, 6H), MS: m/z 630.30 [M]+. Anal. Calcd for C44H46N4; C, 83.77; H, 7.35; N, 8.88. Found: C, 83.44; H, 7.31; N, 8.76.N,N-Bis(4-fluorophenyl)-N,N′-di(n-butyl)-1,3,8,10-tetramethylquinacridonediimine (6)
1H NMR (CDCl3, ppm) (500 MHz): δ 7.13 (s, 2H), 6.98 (m, 4H), 6.72 (m, 8H), 3.46 (m, 4H), 2.83 (m, 4H), 2.30 (s, 6H), 1.33–1.57 (m, 10H), 1.03 (s, 6H), MS: m/z 666.40 [M]+. Anal. Calcd for C44H44 F2N4; C, 79.25; H, 6.65; N, 8.40. Found: C, 79.44; H, 6.41; N, 8.52.N,N-Bis(2,3,4,5,6-pentafluorophenyl)-N,N′-di(n-butyl)quinacridonediimine (7)
1H NMR (CDCl3, ppm) (500 MHz): δ 8.55 (s, 2H), 7.61 (t, 4H), 7.41 (d, 2H), 7.04 (m, 2H), 4.25 (mt, 4H), 1.88 (m, 4H), 1.56 (m, 4H), 1.11 (t, 6H), MS: m/z 754.8 [M]+. Anal. Calcd for C40H28 F10N4; C, 63.66; H, 3.74; N, 7.42. Found: C, 63.99; H, 3.73; N, 7.43.N,N-Bis(3,5-dimethylphenyl)-N,N′-di(n-butyl)-2,9-difluoroquinacridonediimine (8)
1H NMR (CDCl3, ppm) (500 MHz): δ 8.17 (s, 2H), 7.73 (s, 2H), 7.13 (d, 4H), 6.68 (s, 2H), 6.52 (s, 4H), 6.90 (d, 4H), 4.16–4.36 (m, 2H), 3.46 (s, 2H), 2.29 (s, 12H), 1.23–1.41 (m, 8H), 0.98 (t, 6H), MS: m/z 666.31 [M]+. Anal. Calcd for C44H44F2N4; C, 79.25; H, 6.65; N, 8.40. Found: C, 79.76; H, 6.31; N, 8.46.N,N-Dinaphthyl-N,N′-di(n-butyl)quinacridonediimine (9)
1H NMR (CDCl3, ppm) (500 MHz): δ 8.55–8.75 (m, 2H), 8.15–8.18 (m, 2H), 7.89 (d, 2H), 7.54 (t, 6H), 7.38–7.41 (m, 6H), 7.04–7.10 (m, 4H), 6.72 (s, 2H), 3.39–3.59 (m, 2H), 1.54 (d, 2H), 1.25–1.31 (m, 4H), 0.74–0.90 (m, 10H), MS: m/z 674.30 [M]+. Anal. Calcd for C48H42N4; C, 85.43; H, 6.27; N, 8.30. Found: C, 85.10; H, 6.14; N, 8.10.N,N-Bis(4-cyanophenyl)-N,N′-di(n-octyl)quinacridonediimine (10)
1H NMR (CDCl3, ppm) (500 MHz): δ 7.83–8.04 (m, 4H), 7.62 (d, 8H), 7.53 (t, 2H), 7.28 (s, 2H), 6.95 (t, 6H), 3.89–4.38 (m, 4H), 1.73–2.00 (m, 4H), 1.31–1.42 (m, 22H), 0.90 (t, 4H), MS: m/z 736.20 [M]+. Anal. Calcd for C50H52N6; C, 81.49; H, 7.11; N, 11.40. Found: C, 81.15; H, 6.99; N, 11.41.Results and discussion
Synthesis
The synthetic procedure is outlined in Scheme 1. Alkyl substituted QAs were synthesized according to the standard procedure.5 Introduction of the alkyl chain at the N–H position prevents the hydrogen bonding between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H, therefore the solubility of QA is increased. The soluble alkyl substituted QAs were treated with anilines to form a new series of QA derivatives. These compounds are soluble in common organic solvents, such as toluene, chloroform and tetrahydrofuran (THF) etc. The chemical structures were verified by 1H NMR, mass spectrometry and elemental analysis. Structures of some of the compounds were further confirmed by single crystal X-ray analysis.
O and N–H, therefore the solubility of QA is increased. The soluble alkyl substituted QAs were treated with anilines to form a new series of QA derivatives. These compounds are soluble in common organic solvents, such as toluene, chloroform and tetrahydrofuran (THF) etc. The chemical structures were verified by 1H NMR, mass spectrometry and elemental analysis. Structures of some of the compounds were further confirmed by single crystal X-ray analysis.
 | ||
| Scheme 1 Synthesis procedure for quinacridone derivatives (1–10) from alkyl substituted QAs. | ||
Photophysical properties
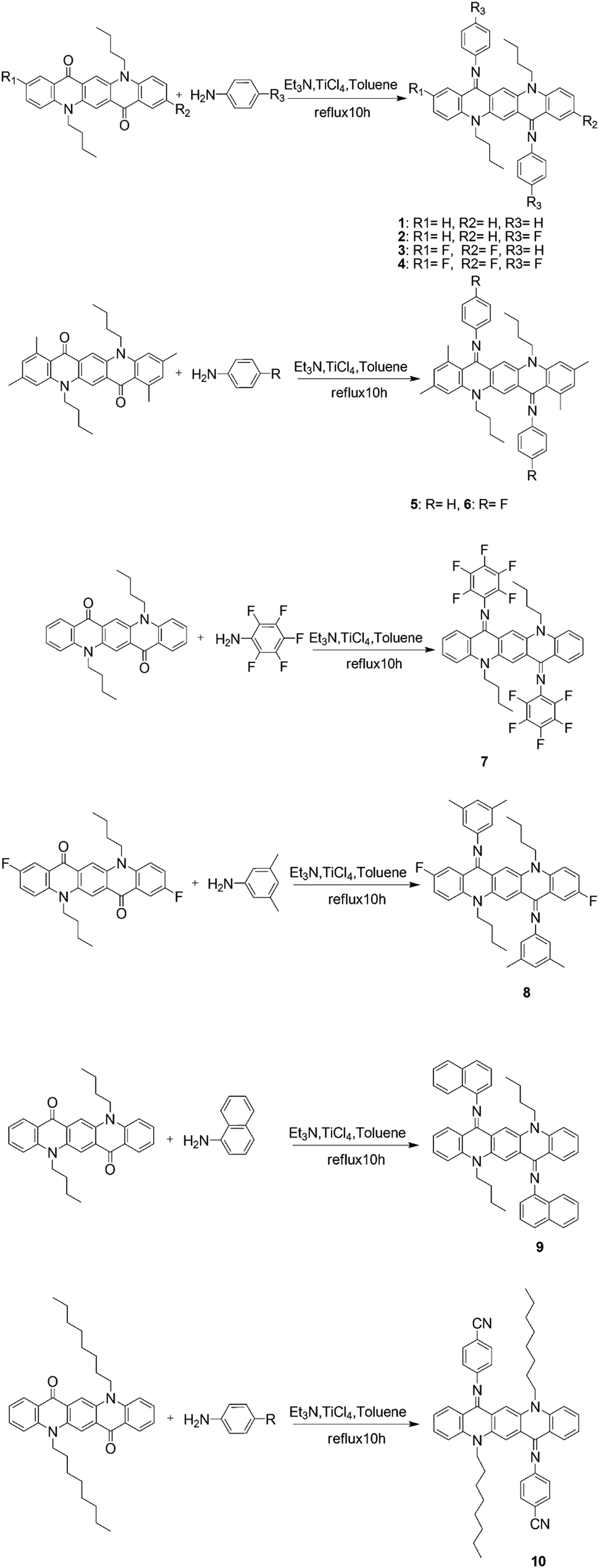
Absorption properties of all the derivatives were measured both as thin films on quartz and in CHCl3 as a solution (Fig. 1). In the solution state, these compounds cover the absorption range from 450 nm to 600 nm and their maximum absorption wavelength λmax values are summarized in Table S2 (ESI†).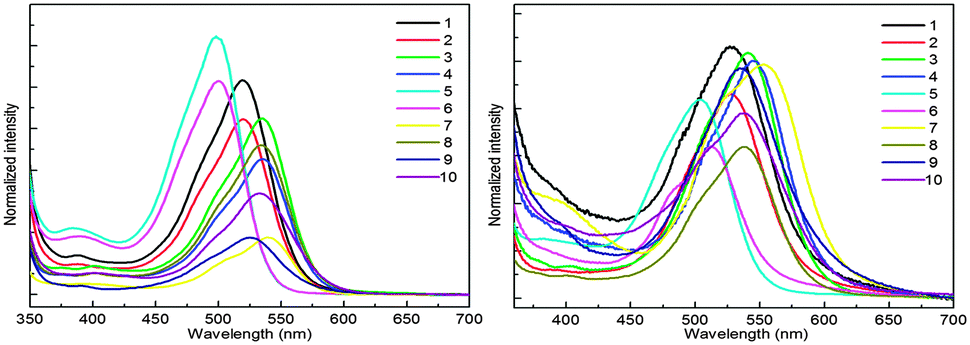 | ||
| Fig. 1 Absorption spectra in the CHCl3 solution state (left) and in the thin film state (right). | ||
It is worth noting that λmax of the majority of these compounds is blue shifted with respect to their parent non-iminated compounds (alkyl substituted QA), which is quite opposite to the expected red shifting as a result of extended π-conjugation. A generally accepted concept, for the relationship between structure and absorption, requires maximum planarity for producing the red shift because of increase of π-conjugation. From the observed absorption spectra it can be deduced that the N-phenyl ring might not be in the plane of the quinacridone nucleus, so it is not involved in extended π-conjugation. This conjecture was confirmed by single crystal X-ray analysis. Single crystal structure analysis of six compounds reveals that the QA nucleus is not planar but slightly buckles to a “butterfly” conformation. In addition, the N-phenyl rings are twisted out of the plane of the central ring system and prefer to be in conjugation with the nitrogen lone pair and not in extended π-conjugation. The QA skeleton is buckled to a large extent in compounds 5 and 6, which results in large blue shifts in the UV-Vis spectrum and this is confirmed by the crystal structure of 6 where the QA skeleton is buckled by 31.68° (Fig. 8). Compound 7 is the only molecule which exhibits a slight red shift in λmax by 17 nm as each N-phenyl ring is substituted by five fluorine atoms, which enhances intermolecular interactions.
Absorption spectra were also recorded in acetone and toluene in order to observe the effect of the polarity of solvents on absorption wavelengths (Fig. 2). Generally, QAs form more stable aggregates in less polar solvents and, as a result, the absorption maxima are red shifted. On the other hand, the absorption maxima are blue shifted in more polar solvents, probably due to the loss of aggregation. The solvent effect, i.e., the maximum absorption wavelength shifts to a longer wavelength with decreasing polarity, was not observed in regularity. This means that different substituents obviously affect the absorption of substituted QA.
 | ||
| Fig. 2 Absorption in acetone (a) and toluene (b). | ||
Absorption spectra of these derivatives as thin films on quartz from CHCl3 solution were also recorded (Fig. 1). The λmax is slightly red shifted and this spectral displacement on going from solution to the solid state is in accordance with the molecular exciton theory of Kasha.13 It is also important to note that the spectral shape in the solid state is quite similar to that in solution and the numbers of absorption bands are the same indicating that the molecular nature is not changed.
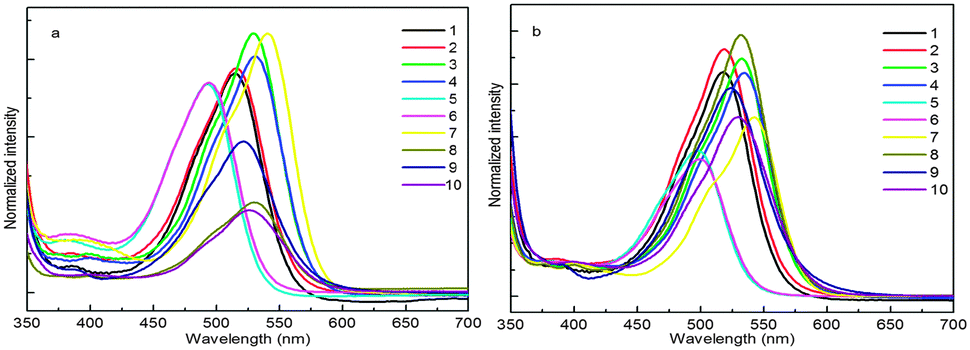
Twisting of the N-phenyl ring and buckling of the QA skeleton
From the analysis of absorption spectra it is deduced that the N-phenyl ring is not in the plane of the central ring system and therefore not in conjugation with the π-system of the QA skeleton. The twisting of the N-phenyl ring out of the plane might be due to the steric hindrance of the peri-hydrogen of the central ring system with ortho hydrogen of the N-phenyl ring14 (Fig. 3a). | ||
| Fig. 3 Quinacridone diimines in (a) sterically hindered form, (b) the “(A)” isomer, (c) the “(B)” isomer and (d) the monosubstituted product. | ||
QA carbonyl groups are less reactive towards nucleophilic attack than aliphatic carbonyl groups as these are the part of a conjugation system. The second carbonyl group left after mono-substitution was found to be more reactive than the first one. This is inferred from the fact that the mono-substituted product (Fig. 3d) was very difficult to obtain and the reaction generally yielded a mixture of starting material and the disubstituted product with very small or negligible amounts of mono-substituted product. A proposed explanation is that when the first imine is formed after mono-substitution, the QA system buckles and moves the second carbonyl group out of conjugation, making it more reactive than the first one. After disubstitution the QA nucleus is buckled and the N-phenyl ring is out of the plane of the QA nucleus. This buckling and non-planarity of the N-phenyl ring are evident in X-ray structures of molecules.
Variable temperature NMR studies
The 1H NMR spectrum of the synthesized compounds was complicated and difficult to interpret in full detail. The aromatic region contains several overlapping peaks due to the QA skeleton and the N-phenyl protons. This is not what would be expected if only one isomeric form were present as in the crystal structure. We postulate that complex spectra of the compounds may be due to different conformational and geometrical isomers being present in solution which can be differentiated at different temperatures (Fig. 4).15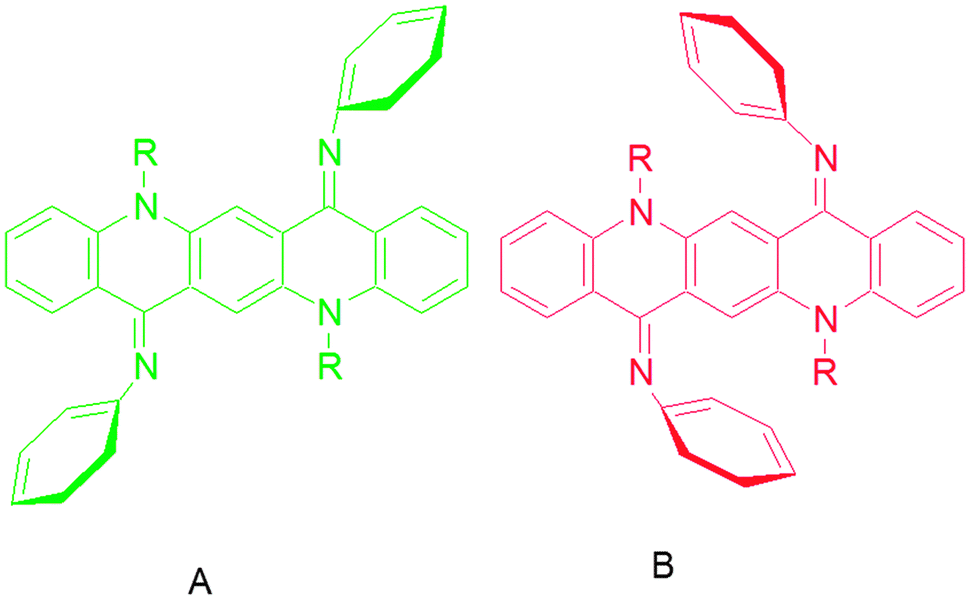 | ||
| Fig. 4 Geometrical isomers “(A)” and “(B)” of quinacridone diimines. | ||
A variable temperature 1H NMR study of 2, 7 and 10 gives an evidence of different geometrical isomers (ESI†).
A variable temperature 1H NMR study of compound 2 revealed interesting molecular dynamics, one at lower temperature (200–230 K) and the other at higher temperatures (298–335 K). The 1H NMR spectrum of 2 (in chloroform-d3) at ambient temperature shows two broad singlets at δ 3.50 and 4.30, with a relative ratio of 1.0:0.58 and are attributed to ZZ(2B) (major) and EE(2A) (minor) isomers, respectively (vide infra). The broad signals become sharp when the temperature is reduced however at further lower temperatures, broadening of the signals is also observed (Fig. 5 and ESI†). The signal at δ 3.43 is broadened at 233 K and it splits at 223 K into two signals at δ 3.08 and 3.72 in a relative ratio of 1:1. The coalescence temperature is expected to be somewhere between 233 and 223 K. These new signals cannot be attributed to the EZ isomer because at a further lower temperature (213 K) the signal at δ 4.33 also splits in a similar fashion into two signals at δ 4.24 and 4.48 in a relative ratio of 1:1. The splitting of both peaks at lower temperatures indicates some process other than E–Z interconversion, mainly because of the fact that both isomers show a similar splitting pattern but different peaks appear in the NMR spectrum. The energy of the dynamic process is calculated to be 10.2 kcal mol−1 which is much lower than expected for the E–Z isomerization around a double bond.15 Since the signals due to N–CH2 are affected at lower temperature, we believe that the variable NMR behavior may be associated with some conformational process involved around N–CH2. At room temperature, the N-alkyl chains are free rotating however at lower temperature, free rotation of N-alkyl chains may be restricted due to interaction with the peri-hydrogens. Due to the restricted rotation, both hydrogens on N–CH2 (Ha and Hb) become diastereotopic due to the absence of the symmetry plane in the molecule, and behave differently in NMR measurements. Due to the restricted rotation, one alkyl chain is restricted above the plane of quinacridone whereas the other is restricted below the plane of the quinacridone. Some signals in the aromatic region (δ 7.5 and 8.4) are also broadened which may be assigned to the peri-hydrogens. We realized that the NMR signals at δ 3.46 and 4.34 are quite sharp at a temperature range of 263–283; however broadening is also observed at 303 (vide supra), which clearly points to some dynamic process just above room temperature. Since chloroform-d3 boils at 333 K, we have carried out high temperature NMR measurements in benzene. The 1H NMR spectrum of 2 (in benzene-C6D6) at ambient temperature shows two sharp singlets at δ 3.26 and 3.89, with a relative ratio of 1.0:0.58, which are attributed to ZZ(2B) (major) and EE(2A) (minor) isomers, respectively. However the dynamic processes in benzene proceed at different rates compared to chloroform. For example, the signals at δ 3.26 and 3.89 start broadening at 310 K and coalesced at 330 K. This indicates that the dynamic process is quite fast in benzene compared to the rate in chloroform. Since the EZ isomer is quite stable over the temperature range of ca. 320–335 K (the temperature range under study), it is a quite stable species on the potential energy surface. These findings are consistent with the results from the computational analysis where predicted relative energy of the EZ isomer is not very different from the EE isomer. The energy difference between ZZ(2B) and ZE is relatively large whereas the difference in relative energies between EZ and EE(2A) isomers is small (vida infra).
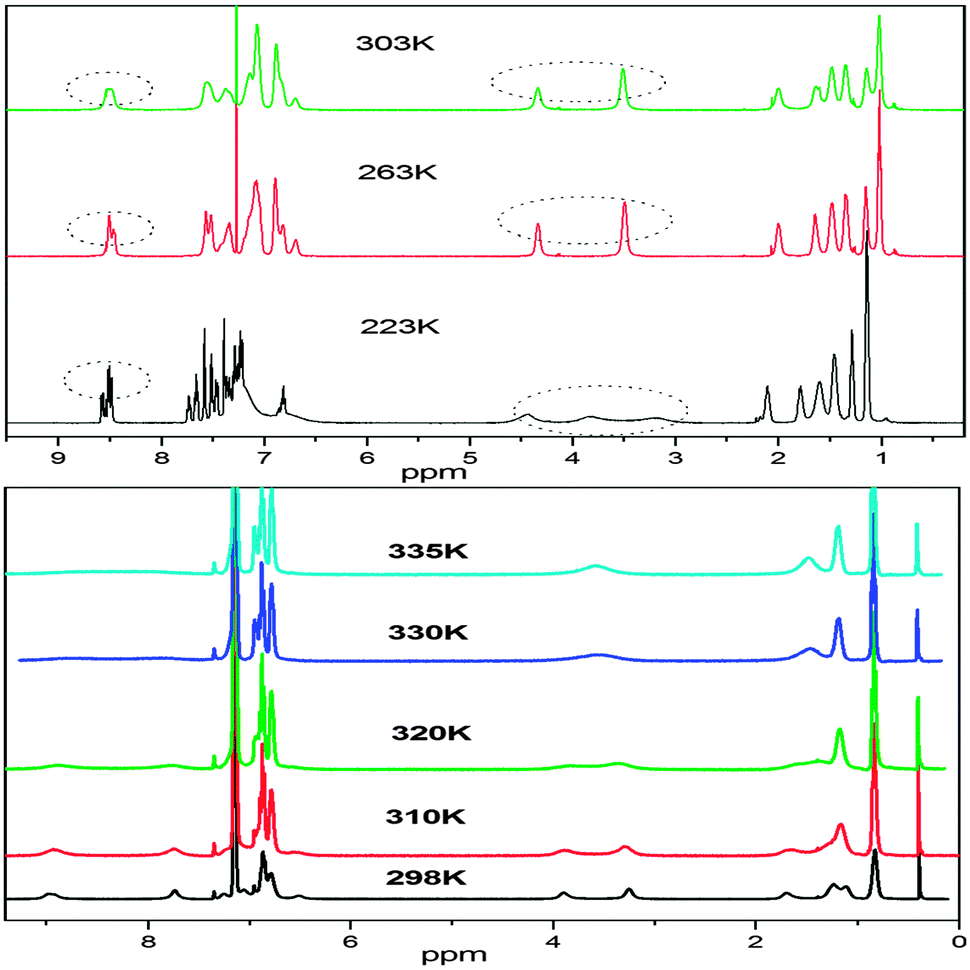 | ||
| Fig. 5 Variable temperature 1H NMR study of 2 in CDCl3 (upper) and C6D6 (lower). | ||
The variable temperature NMR behavior of compounds 7 and 10 is also analyzed (ESI†). The room temperature NMR spectrum (in benzene) of 7 shows a single peak at δ 4.38 which is a composite signal from the coalesced NCH2 groups of the isomer mixture due to rapid interconversion on the NMR time-scale at room temperature. These observations point towards the fact is that the ZZ–EE isomerization is much faster in 7 compared to 2. At ambient temperature the interconversion rate is fast on the NMR time scale. Occurrence of the ZZ(7B) isomer of compound 7 cannot be observed in the variable temperature NMR spectrum at any temperature under study. Not only the signal at δ 4.38 is broad at 298 K, but also the signals at δ 7.8–8.8. At 260 K, two broad singlets at δ 3.70 and 4.40 are also observed which coalesce at 275 K. The signals at δ 3.70 and 4.40 may be attributed to the EZ isomer because two signals are expected for the unsymmetrical isomer and they both coalesce at the same temperature (275 K) and are converted into a broad singlet at δ 4.38. The coalescence temperature is believed to be around 275 K. At further lower temperature (245 K), the peaks at δ 3.70 and 4.40 start splitting into two peaks with equal intensity. This process is very similar to the restricted conformation process described above for 2. However the splitting of the peak at δ 4.38 is not observed at temperatures as low as 230 K, which indicates that the restriction in conformational flexibility is achieved earlier for the Z component of the EZ isomer or for the ZZ(7B) isomer. This is very much consistent with the structural analysis. For the ZZ(7B) isomer (or the Z component of the EZ isomer), the phenyl ring of the imine part is bent towards the alkyl chain which pushes the latter towards the peri-hydrogens and therefore causes more steric hindrance for rotation. Therefore based on the structural analysis, the order of restricted conformational flexibility should be ZZ(B) > EZ > EE(A) and this is also observed experimentally. The high temperature NMR spectrum of compound 7 (in benzene) from 298–338 K is also shown in the ESI.† No significant change in the NMR of 7 could be observed in this temperature range, which indicates that the dynamic (E–Z) isomerization processes complete at 298 K (vide supra). Quite contrary to variable temperature of 2, no significant difference in the rates of dynamic processes is observed with the change of solvent from chloroform to benzene.
The variable temperature NMR spectrum of compound 10 is more complicated probably because in this case a number of conformational and geometrical isomerization processes are taking place simultaneously (ESI†). Compound 10 is structurally different than compounds 2 and 7 in two ways; it contains a long more flexible alkyl chain and a cyano group on the phenyl ring. The electron withdrawing effect of cyano probably speeds up the imino nitrogen inversion process and lowers the barrier to EZ isomerisation. The proton NMR spectral analysis of compound 10 reveals some flips of the chemical shifts. At room temperature, the signal at δ 3.54 is attributed to the EE(10A) isomer. EE–EZ isomerization is observed at 260 K, however the EZ isomer has two peaks, one at 3.04 and the other at 3.50, which is quite opposite to the behavior of 2 and 7 in variable temperature NMR. The protons due to the Z-component of the EZ isomer appear downfield compared to the E fragment, whereas in 2 and 7 a reverse phenomenon is observed. At lower temperature, each of the peaks split into two, which is attributed to restricted conformation (vide supra).
The rate constant, k, for the interconversion of the isomers at the coalescence temperature (Tc) 330 K for the broad NCH2 signals in benzene-d6 was calculated from the Gutowsky–Holm equation, and the imine interconversion barrier (ΔG‡) was calculated to be 15.42 kcal mol−1 by using the Eyring equation.
To further confirm the “(A)” and “(B)” isomers of QA diimines we grew crystals of these compounds at ambient and low temperatures. Importantly, we got single crystals of 3, 6, 8 and 10 at low temperature with “(B)” conformation and of 2 and 7 with “(A)” conformation at ambient temperature, which shows that QA diimines exist in two isomeric forms and more likely interconvert through intermediates.
Computational study
A lack of the expected enhanced properties of QA derivatives 1–10 stimulated an in-depth structural exploration through single crystals X-ray analysis and simulation models. Geometries of the QA derivatives were optimized using the hybrid functional B3PW91 and the structural analysis reveals close resemblance to the X-ray structure. The phenyl ring of the imine moiety makes an average angle of ca. 50–51° with the QA skeleton (B) for the internal isomers compared with 53–54° for the external analogue. Another striking difference is the orientation of the butyl group: the butyl group is bent towards inside in the case (A) (external), whereas in (B) (internal) it is bent towards outside due to steric interaction with the phenyl group of the imine group, which is bent inside (Fig. 8–13).In order to evaluate the stability of the EZ isomer relative to EE(A) and ZZ(B) isomers, we have calculated relative energies of all these isomers for compounds 7 and 10. For both these molecules, the EE isomer is the most stable isomer however the EZ isomer is unstable relative to the EE isomer by 2.55 and 1.54 kcal mol−1 for 7 and 10, respectively. The ZZ isomers for 7 and 10 lie relative to the EE isomer at 5.9 and 5.33 kcal mol−1, respectively (see Tables S6 and S7, ESI†).
Molecular orbitals of both isomers ((A) and (B)) are analyzed and MO energies and isodensity plots of HOMO − 2 to LUMO + 2 are shown in Fig. 6. Molecular orbitals of both isomers have strong resemblance among themselves regarding isodensity however their energies are quite different (see Table S3, ESI†). For 1a, the HOMO (−5.0 eV at B3PW91) is located primarily on the QA skeleton. The next highest occupied MOs (HOMO − 1 and HOMO − 2) are 0.43 and 0.53 eV below the HOMO and are centered mainly on the phenyl ring of the imine group. The lowest unoccupied orbital (LUMO) lies 2.99 eV above the HOMO. The LUMO and the next higher orbitals (LUMO + 1) are centered mainly on the QA core, however LUMO + 2 is exclusively based on the QA core. The internal orientation of the phenyl ring of imines has little effect on the isodensity of all of these orbitals.
 | ||
| Fig. 6 MO energies and isodensity plots of HOMO − 2 to LUMO + 2. | ||
A comparison of eigenvalues of HOMOs and LUMOs of QAs 1–10 (A) and (B) is shown in Table S3 (ESI†) at three different levels of theory. B3PW91/6-31G(d) and B3LYP/6-31+G(d) methods are quite good at predicting the energy of HOMOs (see Table S4, ESI†) in comparison with the experimental methods, which is consistent with the fact that hybrid functionals are better at predicting the HOMO eigenvalues than those without hybrid functionals (PBEPBE);16,17 although the former (B3PW91) slightly underestimates the energies, the latter (B3LYP) overestimates them. The simulated LUMO eigenvalues are much higher (for both hybrid and non-hybrid functionals) than the experimental values and this discrepancy is because of the poor description of virtual orbitals by time independent DFT methods. Hybrid functionals although significantly increase the accuracy of the HOMO eigenvalues, however they are less accurate in computing the HOMO–LUMO gap when compared with non-hybrid functionals PBEPBE (see Table S3, ESI†). The difference between theoretical and experimental HOMO–LUMO gaps decreases with increasing size of hydrocarbons.18 QAs are big enough to show the accurate HOMO–LUMO gap through calculations, however we think that the shape of the molecule and planarity are also key features. The QA skeleton is not even planar and it shows buckling effect.
X-ray single crystal structure determination
Suitable single crystals of one geometrical isomer were obtained for 2 and 7 at ambient temperature and for the other geometrical isomer were obtained at low temperature (−10 °C for 3, 6, 8 and 10). It is worth noting that except 10, which has CN groups as electron withdrawing groups, all other crystals obtained have fluorine atoms in the skeleton, which betokens that halogens may induce crystallization.N,N-Bis(4-fluorophenyl)-N,N′-di(n-butyl)quinacridonediimine (2)
The crystal structure for 2 is shown in Fig. 8 and the molecules in the unit cell lie at the inversion centre. In the crystal form both phenyl groups at N are out of the plane of the QA body by 53.68(5)° indicating that phenyl rings prefer to be in conjugation with a lone pair of nitrogen and both N-phenyl rings are oriented away from the alkyl chain, which confirmed the ‘EE(2A)’ form of conformation at ambient temperature. A small contortion is observed in the N-phenyl ring and is mainly due to the electron withdrawing character of fluorine. The angle between carbons a, b and c (122.73(15)°) is slightly larger than expected for pure sp2 hybridization at C b (Fig. 7a). A C2–H2⋯F1 type weak hydrogen bonding interaction has been observed with dH⋯F = 2.53 Å and symmetry operation is 2 − x, −y, −z.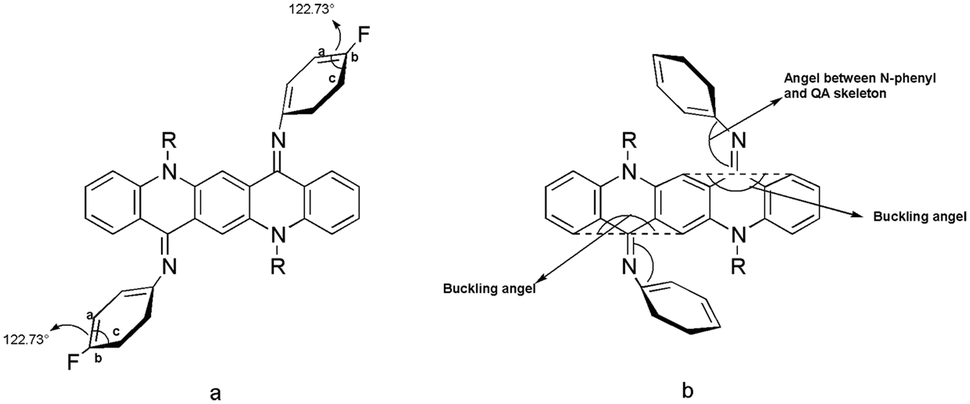 | ||
| Fig. 7 Different kinds of angles in QA diimine structure. | ||
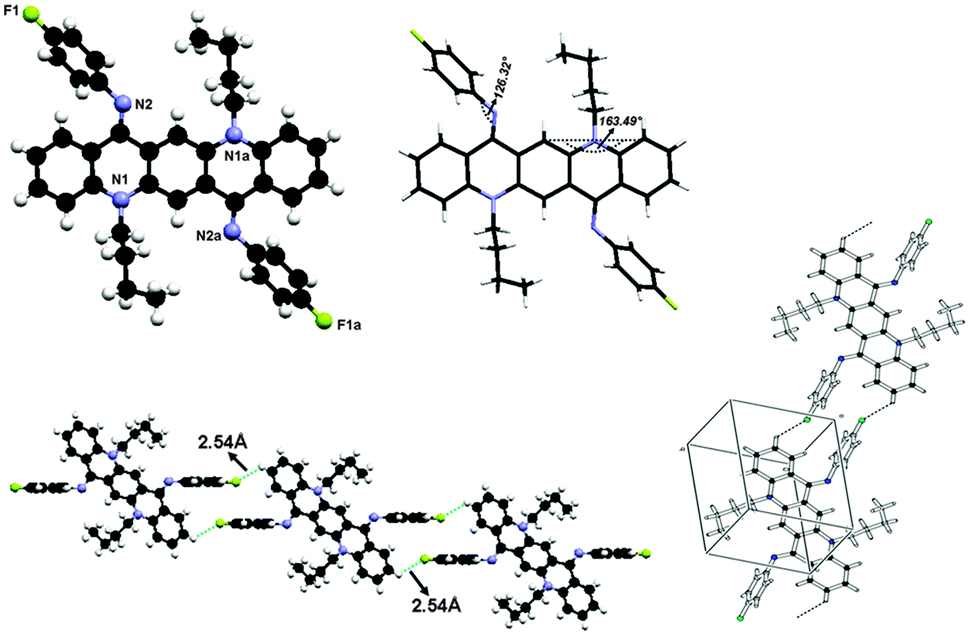 | ||
| Fig. 8 Crystal structure of compound 2 where the additional “a” letters in the atom labels indicate that these atoms are at equivalent positions (−x, −y, 1 − z), showing C–H⋯F hydrogen bonding interaction and unit cell packing. | ||
The bond distances in this aromatic ring are shorter than the rg value for benzene (1.399 ± 0.001 Å) and this is also attributed to the fluorine substituent at the para position of the aromatic ring. It is also worth noting that the QA skeleton is not planar, but is buckled by 16.51(2)° and the angle between the QA body and the N-phenyl ring is 126.32(7)°.
As far as molecular packing is concerned molecules are connected to each other by N-phenyl rings through hydrogen bonding in such a way that the QA skeleton seems to be embedded perpendicularly between N-phenyl rings of different molecules.
N,N-Diphenyl-N,N′-di(n-butyl)-2,9-difluoroquinacridonediimine (3)
The crystal structure of 3 is shown in Fig. 9 and crystallographic parameters are given in the ESI.† This crystal was grown at low temperature and therefore it exists in ‘ZZ(3B)’ form of the isomer as N-phenyl rings are perpendicular to the QA skeleton pointing towards alkyl chains. The end-on-view of the crystal structure in Fig. 9 reveals that the QA skeleton is buckled to a different extent (7.72° and 11.18°) at the points of substitution. This might be due to the fact that at the temperature of crystallization the molecule has not fully flipped to ‘ZZ(3B)’ form of conformation. It is also worth noting that the buckling in this case is smaller than 2, this is due to the substituent effect at the N-phenyl ring in 2. In addition N-phenyl rings are rotated out of the central plane at different angles (128.90(9)° and 130.35(7)°). Atoms C(38) and C(2) in the molecule at (x, y, z) act as hydrogen-bond donors, via atoms H(38) and H(2) to atoms F(1) and F(2) at (x − 1, y − 1, z) and (1 + x, 1 + y, z) respectively, though the side rings of the QA skeleton are the only interactions which join the molecules together in such a way that the QA skeleton is framed linearly with N-phenyl rings perpendicular to it on both sides, where dH⋯F = 2.48 Å and 2.50 Å for both interactions, respectively.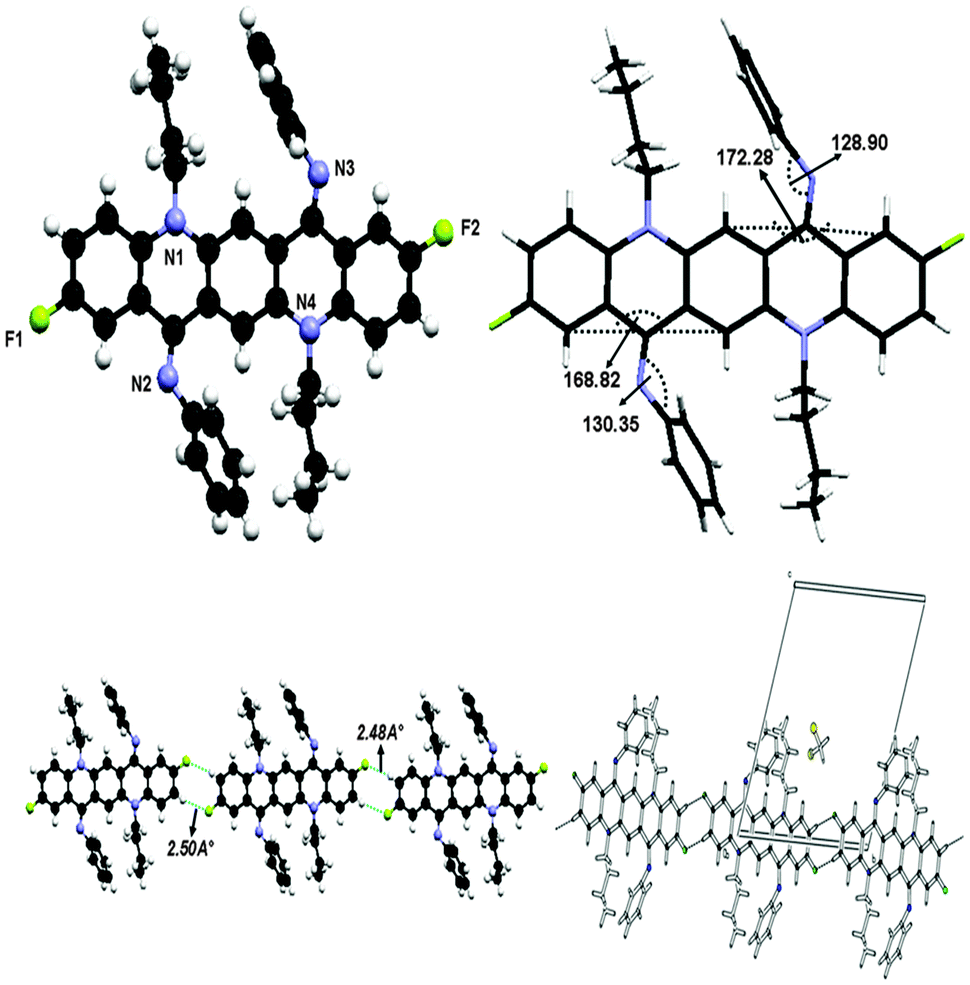 | ||
| Fig. 9 Crystal structure of compound 3 showing C–H⋯F hydrogen bonding interaction and unit cell packing. | ||
N,N-Bis(4-fluorophenyl)-N,N′-di(n-butyl)-1,3,8,10-tetramethylquinacridonediimine (6)
This crystal was grown at low temperature and it also exists in ‘ZZ(6B)’ form at low temperature as shown in Fig. 10. The molecule has line of symmetry and lies at the inversion centre in its crystal structure. In this molecule the substituent factor is of more concern as QA skeleton side rings are substituted with four methyl groups while N-phenyl rings have fluorine at the para position so the central QA skeleton is buckled by 31.68(3)°, which is 15.17(2)° larger than 2. The N-phenyl rings are out of the plane of the central QA skeleton by 53.41(8)° and are rotated out of the central plane at an angle of 126.59(7)°. The crystal structure in Fig. 10 shows that each molecule is connected to four molecules with two types of hydrogen bonding between C22–H22A⋯F1 and C19–H19A⋯N1 having dH⋯X (F/N) bond distances of 2.51 Å and 2.72 Å and symmetry operations are 1 + x, y, z − 1 and 1 − x, 2 − y, −z respectively.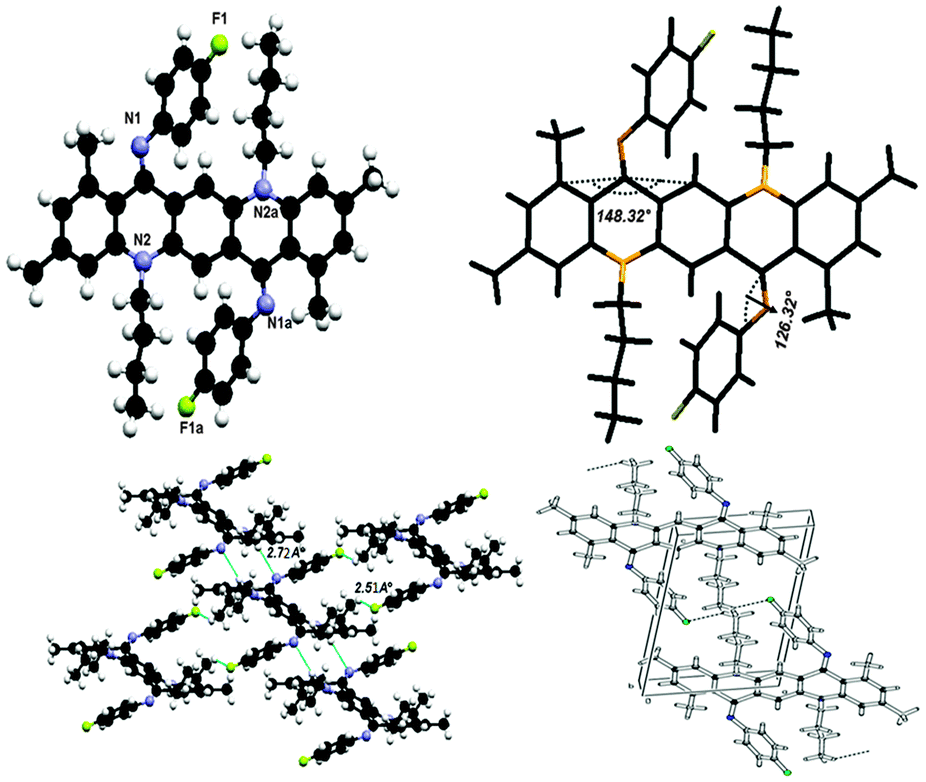 | ||
| Fig. 10 Crystal structure of compound 6 where the additional “a” letters in the atom labels indicate that these atoms are at equivalent positions (−x, 2 − y, −z), showing C–H⋯F and C–H⋯N hydrogen bonding interactions and unit cell packing. | ||
N,N-Bis(2,3,4,5,6-pentafluorophenyl)-N,N′-di(n-butyl)quinacridonediimine (7)
In the crystal structure of 7, shown in Fig. 11, molecules lie at the inversion centre. In the crystal form both N-phenyl groups at N are out of the plane of the QA body by 54.29(9)° indicating that phenyl rings prefer to be in conjugation with the lone pair of nitrogen and both N-phenyl rings are oriented away from the alkyl chain which confirmed the ‘EE(7A)’ form of conformation at ambient temperature.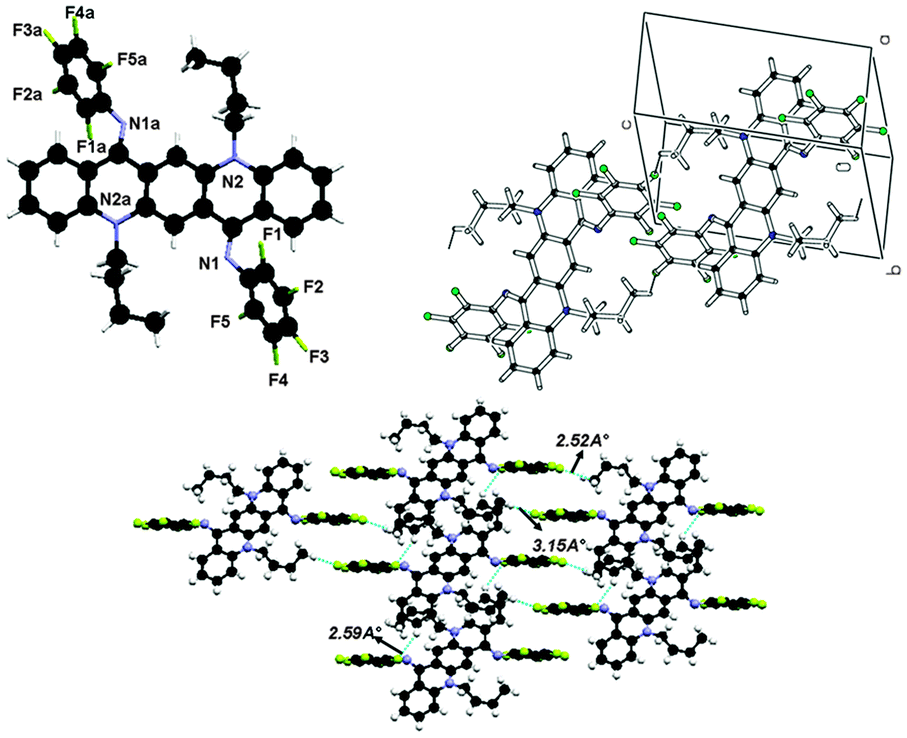 | ||
| Fig. 11 Crystal structure of compound 7 where the additional “a” letters in the atom labels indicate that these atoms are at equivalent positions (−x, 1 − y, 1 − z), showing C–H⋯F hydrogen bonding interaction and unit cell packing. | ||
A contortion is observed in the N-phenyl ring, which is quite larger than the previous compounds due to the electron withdrawing character of the five fluorine atoms. The angles are slightly larger than expected for pure sp2 hybridization. The bond distances in this aromatic ring are shorter than the rg value of benzene (1.399 ± 0.001 Å) and this may be attributed to fluorine atoms substituted at the aromatic ring. In this molecule, the QA skeleton is also not planar, and is buckled by 20.52(2)° and the angle between the QA body and the N-phenyl ring is 125.71(8)°.
As far as molecular interactions are concerned each molecule is connected to four other molecules through two types of hydrogen bonds. Atoms C(14) and C(20) in the molecule at (x, y, z) act as hydrogen-bond donors, via atoms H(14) and H(20b) to atoms F(1) and F(3) at (1 + x, y, z) and (x, 1 + y, 1 + z) respectively to form the dimers (Fig. 12). The dH⋯A distance in C14–H14⋯F1 and C2–H20b⋯F3 is 2.59 Å and 2.53 Å respectively.
 | ||
| Fig. 12 Crystal structure of compound 8 where the additional “a” letters in the atom labels indicate that these atoms are at equivalent positions (1 − x, 1 − y, −z), showing C–H⋯F hydrogen bonding interaction and unit cell packing. | ||
As far as molecular packing is concerned molecules are connected to each other by N-phenyl rings through hydrogen bonding in such a way that the QA skeleton seems to be embedded perpendicularly between N-phenyl rings of different molecules.
N,N-Bis(3,5-dimethylphenyl)-N,N′-di(n-butyl)-2,9-difluoroquinacridonediimine (8)
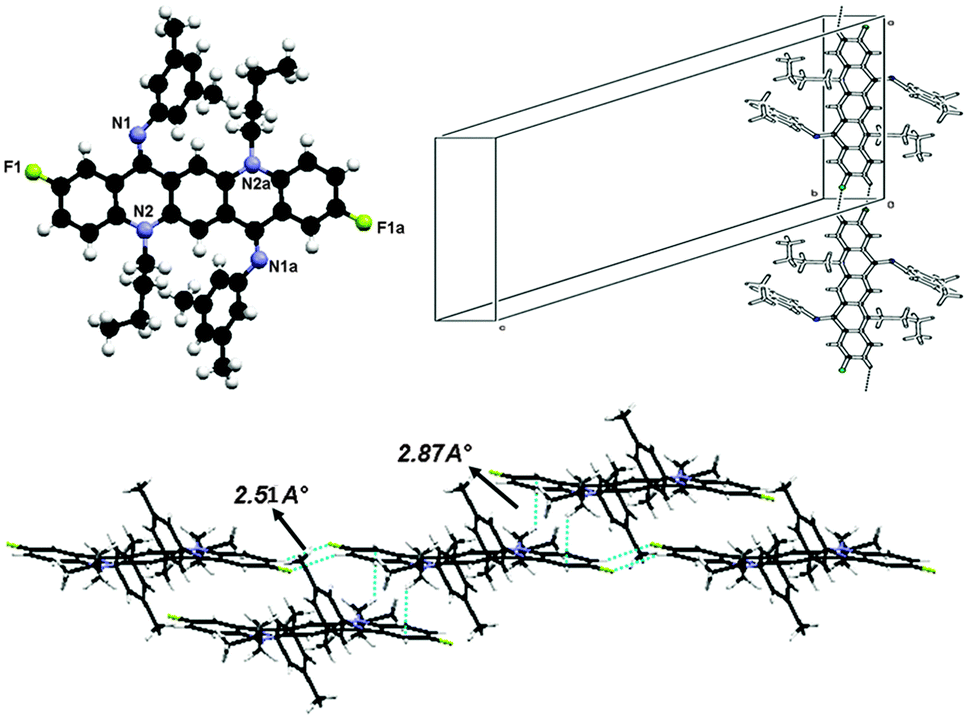
X-ray crystal structure determination of 8 shown in Fig. 12 reveals the ZZ(8B) form of conformation as this crystal is also grown at low temperature. The molecule lies at the inversion centre in the crystal structure of 8. The central QA skeleton is not planar like other crystals and is buckled by 27.64(2)° in a butterfly fashion. Also the N-phenyl rings do not lie in the plane of the central ring system but are twisted out of the plane by approximately 51.43(9)°. As far as molecular packing is concerned this crystal has different behavior than rest of the crystals as the central QA skeleton of one molecule stacks with other molecule's QA skeleton by CH2⋯π interactions. Molecules are commingled linearly by hydrogen bonding between the side rings of the QA skeleton. The interesting feature of this crystal is that the molecules are stacked together in a staircase in such a way that large cavities are generated among the molecules. The weak hydrogen bonding interaction through C2–H2⋯F1, where the flourine atom is situated at (−x, 1 − y, −z) with respect to the donor carbon atom at (x, y, z) via H2, helps to stabilize the crystal structure of the molecule, where dH⋯F = 2.51.N,N-Bis(4-cyanophenyl)-N,N′-di(n-octyl)quinacridonediimine (10)
This molecule is also non-planar, lies at the inversion centre as it has a plane of symmetry and is triclinic, belonging to space group p1.As this single crystal was grown at low temperature, it exists in the ‘ZZ(10B)’ form of conformation (Fig. 13). There is no π⋯π stacking or intermolecular hydrogen bonding involved between molecules. The only interactions which might be involved are van der Waals forces. The molecule is buckled by 22.74(1)° and the N-phenyl ring is twisted out of the plane of the central core by 51.5(9)°. This large buckling and twisting effect is due to the presence of the electron withdrawing ‘CN’ group at the N-phenyl ring. As far as molecular packing is concerned the central core of the molecules is located at the edges of the unit cell while the N-phenyl ring is embedded at the center of the unit cell.
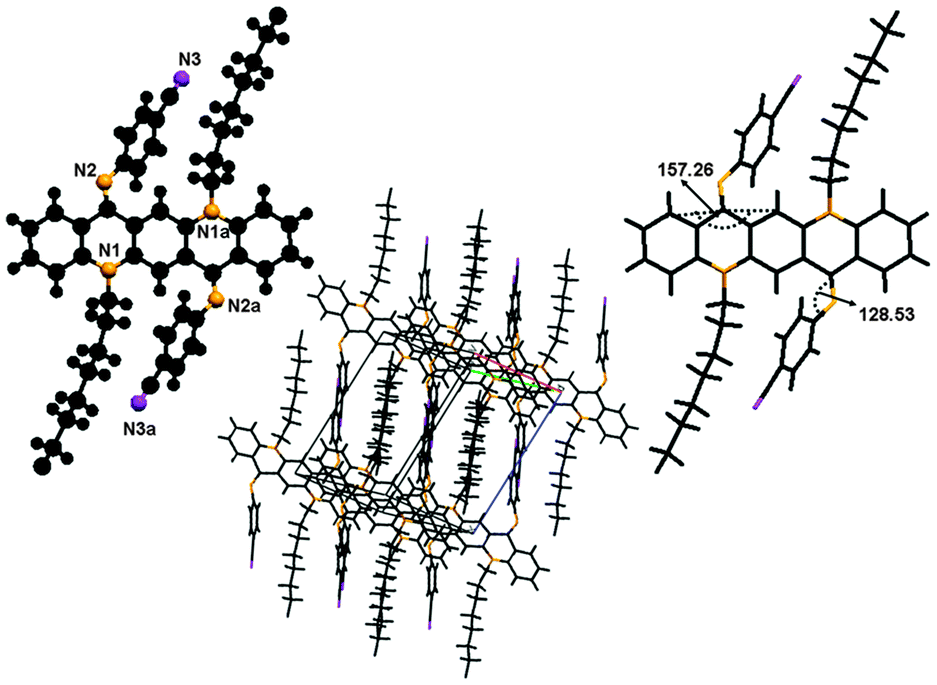 | ||
| Fig. 13 Crystal structure of compound 10, where the additional “a” letters in the atom labels indicate that these atoms are at equivalent positions (2 − x, 1 − y, −z), showing C–H⋯F hydrogen bonding interaction and unit cell packing. | ||
Redox properties
To study the effect of different groups on the electrochemical properties of these QA derivatives, cyclic voltammetry (CV) was performed in anhydrous CH2Cl2 solution with 0.1 M TBAP as a supporting electrolyte. The potentials relative to standard ferrocene (Fc/Fc+) are summarized in Table S5 (ESI†). The cyclic voltammograms of all the derivatives are shown in S-2 (ESI†). Most of the derivatives exhibit a reversible reduction and oxidation wave potential except (5 and 8) which display irreversible oxidation and (1) which displays irreversible reduction waves. The first half wave reduction and oxidation potentials of the compounds are (E1/2red = −1.98 V–1.20 V) and (E1/2ox = 0.22 V–0.68 V), respectively. The electrochemical data give an unclouded picture of the effect of electron donating and electron withdrawing groups on electrochemistry of the compounds. Compounds 7 and 10 with strong electron withdrawing groups on N-phenyl rings have higher E1/2red (−1.45 V, −1.20 V) and E1/2ox (0.67 V, 0.62 V) while compound 5 with four methyl groups as electron donating groups on the QA skeleton has a relatively low E1/2red (1.98 V) and E1/2ox (0.40 V). This effect is also translated in the LUMO and HOMO levels, compounds having electron withdrawing groups have relatively low LUMO and HOMO levels.Conclusions
A series of QA diimines 1–10 were synthesized by the condensation of anilines with alkyl substituted QAs (QA) and their photophysical and electrochemical properties were investigated. Unconventional behavior of absorption spectra suggested the lack of π-conjugation within the QA skeleton as well as the lack of extended π-conjugation between the QA skeleton and N-phenyl rings. A computational study, a variable temperature 1H NMR study of compounds 2, 7 and 10 (for instance) and single crystal X-ray analysis of 2, 3, 6, 7, 8 and 10 gave an unclouded picture of anomalous behavior, indicating a buckled, non-planar structure and the existence of molecules in two different isomeric forms at different temperatures. A theoretical study was carried out for both isomers of all compounds using a computational approach; results obtained are in close agreement with the experimentally determined values.Acknowledgements
This work was supported by the National Natural Science Foundation of China (50773027 and 50733002), the Major State Basic Research Development Program (2009CB939700), and 111 Project (B06009). We acknowledge Tobias Sparrman, Department of chemistry,Umeå University, SE-901 87 Umeå, Sweden for VT NMR facilities. K.A acknowledges Queen's University, Canada, for the computational facility.Notes and references
- H. Masahiro, K. Shuji and Y. Masaaki, Jpn. J. Appl. Phys., 1996, 35, L349 Search PubMed.
- T. Shichiri, M. Suezaki and T. Inoue, Chem. Lett., 1992, 1717 CrossRef CAS.
- H. Nakahara, K. Kitahara, H. Nishi and K. Fukuda, Chem. Lett., 1992, 711 CrossRef CAS.
- S. De Feyter, A. Gesquière, F. C. De Schryver, U. Keller and K. Müllen, Chem. Mater., 2002, 14, 989 CrossRef CAS.
- U. Keller, K. Müllen, S. De Feyter and F. C. De Schryver, Adv. Mater., 1996, 8, 490 CrossRef CAS.
- H. Nakahara, K. Fukuda, M. Ikeda, K. Kitahara and H. Nishi, Thin Solid Films, 1992, 210/211, 555 CrossRef.
- G. D. Potts, W. Jones, J. F. Bullock, S. J. Andrews and S. J. Maginn, Chem. Commun., 1994, 2565 RSC.
- E. F. Paulus, F. J. J. Leusen and M. U. Schmidt, CrystEngComm, 2007, 9, 131 RSC.
- Y. Fan, Y. Zhao, L. Ye, B. Li, G. Yang and Y. Wang, Cryst. Growth Des., 2009, 9(3), 1421 CAS.
- (a) I. Javed, T. Zhou, F. Muhammad, J. Guo, H. Zhang and Y. Wang, Langmuir, 2012, 28(2), 1439 CrossRef CAS PubMed; (b) I. Javed, Z. Zhang, T. Peng, T. Zhou, H. Zhang, M. I. Khan, Y. Liu and Y. Wang, Solar Energy Mater. Solar Cells, 2011, 95(9), 2670 CrossRef CAS PubMed.
- P. A. Tucker, A. Hoekstra, J. M. Ten Cate and A. Vos, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 733 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.04 E.01, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- M. Kasha, in NATO Advanced Study Institute Series, Series B, ed. B. D. Bartolo, Plenum Press, New York, 1976, vol. 12, p. 337 Search PubMed.
- H. W. Boone, M. A. Bruck, R. B. Bates, A. B. Padias and H. K. Hall, Jr., J. Org. Chem., 1995, 60, 5279 CrossRef CAS.
- C. Gahl, D. Brete, F. Leyssner, M. Koch, E. R. McNellis, J. Mielke, R. Carley, L. Grill, K. Reuter, P. Tegeder and M. Weinelt, J. Am. Chem. Soc., 2013, 135, 4273 CrossRef CAS PubMed.
- U. Salzner, J. B. Lagowski, P. G. Pickup and R. A. Poirier, J. Comput. Chem., 1997, 18, 1943 CrossRef CAS.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554 CrossRef CAS PubMed.
- S. S. Naghavi, T. Gruhn, V. Alijani, G. H. Fecher, C. Felser, K. Medjanik, D. Kutnyakhov, S. A. Nepijko, G. Schonhense, R. Rieger, M. Baumgarten and K. Mullen, J. Mol. Spectrosc., 2011, 265, 95 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, cyclic voltammetric and crystallographic data. CCDC 923157 (2), 923158 (3), 923159 (6), 923160 (7), 923161 (8) and 923162 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00477e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |