
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new insight into the zinc-dependent DNA-cleavage by the colicin E7 nuclease: a crystallographic and computational study†
Anikó
Czene
a,
Eszter
Tóth
b,
Eszter
Németh
b,
Harm
Otten
c,
Jens-Christian N.
Poulsen
c,
Hans E. M.
Christensen
d,
Lubomír
Rulíšek
e,
Kyosuke
Nagata
f,
Sine
Larsen
*c and
Béla
Gyurcsik
*ab
aMTA-SZTE Bioinorganic Chemistry Research Group, Dóm tér 7, H-6720 Szeged, Hungary
bDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: gyurcsik@chem.u-szeged.hu; Fax: +36 62544340
cDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: sine@chem.ku.dk; Fax: +45 35320322
dDepartment of Chemistry, Technical University of Denmark, Kemitorvet, Building 207, 2800 Kgs. Lyngby, Denmark
eInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo namesti 2, 166 10 Prague 6, Czech Republic
fNagata Special Laboratory, Faculty of Medicine, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8575, Japan
First published on 21st August 2014
Abstract
The nuclease domain of colicin E7 metallonuclease (NColE7) contains its active centre at the C-terminus. The mutant ΔN4-NColE7-C* – where the four N-terminal residues including the positively charged K446, R447 and K449 are replaced with eight residues from the GST tag – is catalytically inactive. The crystal structure of this mutant demonstrates that its overall fold is very similar to that of the native NColE7 structure. This implicates the stabilizing effect of the remaining N-terminal sequence on the structure of the C-terminal catalytic site and the essential role of the deleted residues in the mechanism of the catalyzed reaction. Complementary QM/MM calculations on the protein–DNA complexes support the less favourable cleavage by the mutant protein than by NColE7. Furthermore, a water molecule as a possible ligand for the Zn2+-ion is proposed to play a role in the catalytic process. These results suggest that the mechanism of the Zn2+-containing HNH nucleases needs to be further studied and discussed.
Introduction
The colicin E7 bacterial toxin of Escherichia coli belongs to the HNH family of metallonucleases.1,2 It is expressed in E. coli under environmental stress to protect the host cell from related bacteria and bacteriophages. Only the nuclease domain (NColE7) of the protein enters the target cell, killing it through non-specific digestion of the nucleic acids.3 The host cell is protected by the Im7 immunity protein, which when co-expressed with the nuclease prevents substrate binding.4 Crystal structures of NColE7 variants have been published in the presence or absence of its immunity protein, its metal ion cofactor, and the substrate DNA.3–11 The HNH motif in the active centre at the C-terminus of NColE7 contains conserved H and N amino acids in the HHX14NX8HX3H (amino acids 544–573) pattern.12 In this ββα – metal ion binding stretch H544, H569 and H573 act as ligands to the Zn2+-ion,7 while H545 is supposed to generate the nucleophilic agent by deprotonating a water molecule.9,13,14 The catalytic mechanism is well established, with the exception of the protonation of the leaving group. The hypothesized pathways of this step are dependent on the quality of the metal ion in the active centre. The proton may be donated by a water molecule coordinated to a Ni2+-ion in NColE915 and to a Mg2+-ion in Vvn,16 related HNH nucleases. But for NColE7 the question remains open due to the absence of the corresponding inner-sphere water molecule around the Zn2+-ion.7,17 A putative proton channel was suggested in the related colicin E9 nuclease18–20 involving the amino acids that correspond to R538, E542 and H569 in colicin E7.4 However, H569 is bound to the Zn2+-ion and the change in its protonation state would be unfavourable.Recently we have shown that the deletion of the N-terminal KRNK (446–449) sequence in NColE7 cancelled the catalytic activity,21 in agreement with the previously demonstrated importance of the R447 residue.13 The requirement of cooperation of the N- and C-termini in NColE7 to exert its catalytic action may be developed into an allosteric activation control mechanism in a new artificial nuclease.22 This property would be an advantage over the artificial chimeric nucleases created by fusion of e.g. zinc-finger or TALE proteins and the FokI nuclease domain,23–25 where the allosteric control present in the native FokI26 is lost by exchanging its DNA binding domain with zinc-finger motifs. This may e.g. account for the known moderate cytotoxicity of zinc-finger nucleases designed for gene therapy.27 Therefore, it is important to understand the regulatory elements in artificial nucleases intended for in vivo applications.28
To better understand the role of the N-terminal sequence crystallization was undertaken for the ΔN4-NColE7-C* protein – where the KRNK (446–449) sequence is replaced by a part of the GST purification tag lacking positively charged side chains. Previously its biochemical and biophysical properties were studied.21,29 Here we present the structure determination to 1.7 Å resolution and a comparison to the previously published structures of NColE7. The possible influence of the mutation at the C-terminus was also elucidated by recloning the ΔN4-NColE7 gene. The NColE7–DNA complexes can only be crystallized with an inactive form of the protein. To obtain information on the local geometries of the active site, we performed also QM/MM calculations.
Materials and methods
Construction of the genes of the mutant proteins
The gene encoding the ΔN4-NColE7 mutant was cloned from the pQE70 plasmid (a generous gift of Prof. K.-F. Chak, Institute of Biochemistry and Molecular Biology, National Yang Ming University, Taipei, Taiwan12) and inserted into the pGEX-6P-1 (GE Healthcare) vector between the EcoRI/XhoI restriction enzyme sites (E/X) as described earlier.21 The same fragment was also built in the pET-21a vector (Novagen) with the same restriction enzymes – (E/X)ΔN4-NColE7 gene – and was recloned into the NdeI/XhoI sites (N/X) of the same vector: the (N/X)ΔN4-NColE7 gene. Since the insert DNA sequence contained a C-terminal stop codon, no C-terminal tags were expected to form. To check the effect of the C-terminal mutation, we have also cloned the gene of the NColE7-C* mutant into the pGEX-6P-1 vector.Protein expression and purification
The expression and purification of the GST-ΔN4-NColE7-C* protein (for the protein sequences see Fig. 1.) and the cleavage of the glutathione S-transferase (GST) affinity tag at the N-terminus by the human rhinovirus C3 protease30 was performed as described previously.29 The expression of the GST-ΔN4-NColE7, GST-NColE7-C* and (E/X)ΔN4-NColE7 proteins could not be carried out because of their toxicity to the host cell. On the other hand, the (N/X)ΔN4-NColE7 protein was successfully overexpressed. The purification of this mutant was carried out using a Sepharose Fast Flow (GE Healthcare) ion exchange column equilibrated with phosphate-buffer (PBS: 0.14 M NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH = 7.7). The elution was carried out with a NaCl concentration gradient up to 1.0 M through 15 column volume in the same buffer. The protein was loaded onto a Source 30S (GE Healthcare) column under the same conditions as mentioned above. The molecular mass of the purified protein was determined by ESI-MS as described by Czene et al.21 The theoretical average mass of the holo protein (N/X)ΔN4-NColE7 with its first Met removed and in complex with one Zn2+-ion is 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 563.3 Da, in excellent agreement with the ESI-MS results of 14563.0 Da. This indicates that the purified protein was in its Zn2+-bound form.
563.3 Da, in excellent agreement with the ESI-MS results of 14563.0 Da. This indicates that the purified protein was in its Zn2+-bound form.
 | ||
| Fig. 1 Multiple alignment of the amino acid sequences of the five designed mutant proteins. Rows 1: (GST)ΔN4-NColE7-C*, 2: (GST)ΔN4-NColE7, 3: (E/X)ΔN4-NColE7, 4: (N/X)ΔN4-NColE7 and 5: (GST)NColE7-C*, respectively. The amino acids in red are those originating from the expression vectors at the N-termini, and the result of the unexpected mutation at the C-terminus of the ΔN4-NColE7-C* mutant. In the remaining 8 rows the amino acid sequences of nuclease domains of colicins and related toxins is shown for identification of the conserved N-terminal amino acids. Highly conserved residues are in bold, while bold and italic means conserved amino acids. | ||
Structure determination and refinement
Details of the crystallization of ΔN4-NColE7-C* and measurement of X-ray diffraction data have been provided earlier in ref. 29. The overall symmetry of the diffraction pattern corresponds to the Laue class 3m and the systematically absent reflections are consistent with the space groups P3121 or P3221. The volume-to-mass ratio VM of 1.99 Å3 Da−1 and the corresponding solvent content of 35% match well a structure with one molecule per asymmetric unit (Mr ∼16.2 kDa). It was possible to solve the structure by the Molecular Replacement method in the space group P3221 using MOLREP31 with the native NColE7 structure (PDB entry: 1M085) as a search model.In the subsequent refinement of the structure, difficulties in modelling of the polypeptide chain were encountered in tracing the tagged N-terminus of the protein. The refined model in the space group P3221, which had the lowest acceptable penalty score in XDS32 contained density close to the crystallographic twofold axis that could not be resolved into a meaningful chemical model and thus the space group published in ref. 29 was reconsidered. The N-terminus of the ΔN4-NColE7-C* protein (Fig. 1) contains a GPLGSPEF additional sequence remaining after the cleavage of the GST tag during the protein expression and purification procedure. It was possible to trace the SPEF residues in this segment, found in a part of the crystal structure adjacent to the twofold axis. In the space group P3221 the first four (GPLG) residues would run into density of the same four residues of the symmetry related molecule (Fig. S1, ESI†). Refinements in other possible space groups were therefore attempted. The structure could be refined in P32 with a pseudo twofold axis replacing the crystallographic twofold axis in P3221. The refined model had a significantly lower Rfree and a comparable Rwork to the model obtained in P3221. In the space group C2, which would correspond to three molecules per asymmetric unit one of them is related to another molecule in the unit cell by a crystallographic twofold axis giving rise to similar problems as in the P3221 space group. Due to the low redundancy refinement was not attempted in P1. The results are summarized in Table 1.
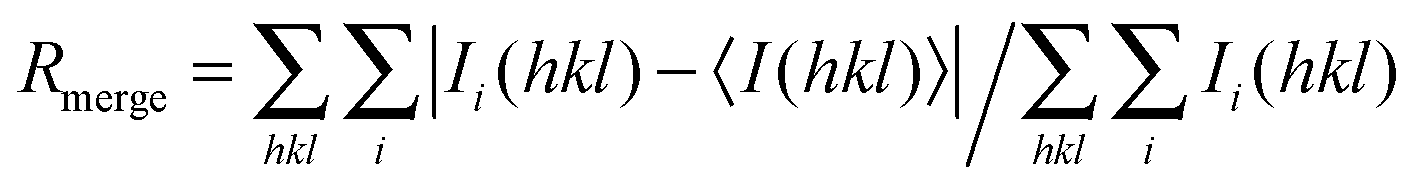 , where Ii(hkl) is the intensity measurement for a given reflection and Ii(hkl)i is the average intensity for multiple measurements of this reflection. The values in parentheses correspond to the highest resolution shell
, where Ii(hkl) is the intensity measurement for a given reflection and Ii(hkl)i is the average intensity for multiple measurements of this reflection. The values in parentheses correspond to the highest resolution shell
| Space group | P32 | P3221 | C2 |
|---|---|---|---|
| a With respect to Engh and Huber parameters.33 b The three Ramachandran outliers34,35 N461, L465 and D471 had well defined backbone density for the modelled conformations in both molecules. | |||
| Resolution range/Å | 40–1.7(1.8–1.7) | 40–1.7(1.8–1.7) | 40–1.7(1.8–1.7) |
| Unit-cell parameters a, b, c (Å) | 55.4, 55.4, 73.2 | 55.4, 55.4, 73.2 | 96.0, 55.4, 73.2 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90.0, 90 |
| Total reflections | 154648(24128) |
154714(24240) |
153962(24038) |
| Unique reflections | 53902(8489) |
27529(4395) |
78323(12217) |
| Average multiplicity/redundancy | 2.9(2.8) | 5.6(5.5) | 2.0(2.0) |
| Completeness (%) | 97.3(93.0) | 99.4(97.6) | 94.2(90.6) |
| 〈I/σ(I)〉 | 15.7(2.7) | 22.8(3.9) | 15.2(2.4) |
| R merge (%) | 4.1(39.3) | 4.4(43.6) | 3.2(36.8) |
| Number of molecules per asymmetric unit | 2 | 1 | 3 |
| Refinement Rwork/Rfree | 0.200(0.290)/0.234(0.331) | 0.191(0.260)/0.277(0.320) | 0.210(0.230)/0.250(0.300) |
| R.m.s. deviation from idealitya | |||
| Bonds/Å | 0.01 | ||
| Angles/° | 1.28 | ||
| Number of atoms (protein) | 4179 | ||
| Number of water molecules | 104 | ||
| Number of bound Zn2+-ions | 2 | ||
| Number of atoms in ions (2 acetates, 9 sulfates, 2 chlorides) | 55 | ||
| Ramachandran plot outliersb/% | 0.8 | ||
After refinement with Refmac536 and manual rebuilding in COOT,37 it was possible to obtain a chemically sound model that contains all residues of the sequence from 446S (KRNK of WT NColE7 replaced by SPEF) to H573. Four N-terminal (442G, 443P, 444L, 445G, which is part of the GST tag) and 12 C-terminal residues (Q574–D585, this part of the sequence is indicated as the C* mutations) were not modelled due to a disorder. The residual fragmented density close to the non-crystallographic twofold axis was modelled as water molecules. It was possible to trace the amino acids in the electron density up to those introduced by random mutation in the C-terminus. Thus, the final structural model displays an intact ββα type metal binding site. In the refinement noncrystallographic symmetry (NCS) was not applied to allow for disruption of the twofold symmetry that relates the two crystallographical independent molecules. The difference and omit maps revealed the positions of the Zn2+-ion. Residual density was modelled as water molecules if they fulfilled the expected geometrical conditions. Furthermore, nine sulfate, two chloride and two acetate ions were modelled in the difference electron density. The almost perfect twofold symmetry that relates the two molecules also with respect to the overall B-values, 27.3 Å2 and 27.1 Å2 for the A and B molecule, respectively, is not completely maintained in the position of the ions vide infra.
QM/MM calculations on protein–DNA complexes
The protein setup was performed based on the crystal structures 3FBD10 and 1M085 as well as the construction of the QM/MM system, following the recommended protocol38,39 that is almost identical to those used in our previous QM/MM studies.40–42 The quantum system (System 1) consisted of 169 atoms for NColE7 and 156 for ΔN4-NColE7 (Fig. S2, ESI†) including the Zn2+-ion, H544, H545, H569, H573, V555 and the side chain of R447 (only for NColE7). The MM part was divided into System 2 (surrounding of the quantum region) that is allowed to move in the QM/MM minimizations and System 3 (the rest of the protein) that is kept fixed.The QM/MM calculations were carried out by employing a modified version of the ComQum program.38,39,43 The Turbomole 6.3 program44 was used for the quantum chemical calculations carried out using DFT method and using Perdew–Burke–Ernzerhof (PBE) functional45 and Ahlrich's def2-SVP basis set.46 Resolution of the identity (density-fitting) was used to expedite the DFT calculations. MM calculations were carried out in the AMBER 8 program package (sander module) and the ff0347,48 force field (parm99 set in Amber).
Further details are deposited in the ESI† (see chapters S1–S3 and references therein).
PDB code
The coordinates of the ΔN4-NColE7-C* refined in space group P32 have been deposited in the PDB with the reference: 3ZFK.Results
Description of the structure of the ΔN4-NColE7-C* protein
The two protein chains in the final model are virtually identical, the superimposition of their backbones gives an rmsd (root mean square deviation) of 0.05 Å (Fig. S3, ESI†). It is noteworthy that the four residues of fused GST-tag that have replaced the four deleted residues (KRNK) in the intact NColE7 sequence are visible in the electron density. The close interactions between the N-termini of the A and B molecules illustrated in Fig. 2a involve only the first four amino acids of the tag. Mass spectra and N-terminal analysis have verified that the protein is intact. However, in the C-terminus only the residues that are part of the NColE7 sequence are visible in the electron density. Inspection of the structure shows that there is sufficient space in the crystal packing to accommodate the last 12 residues in disordered conformations.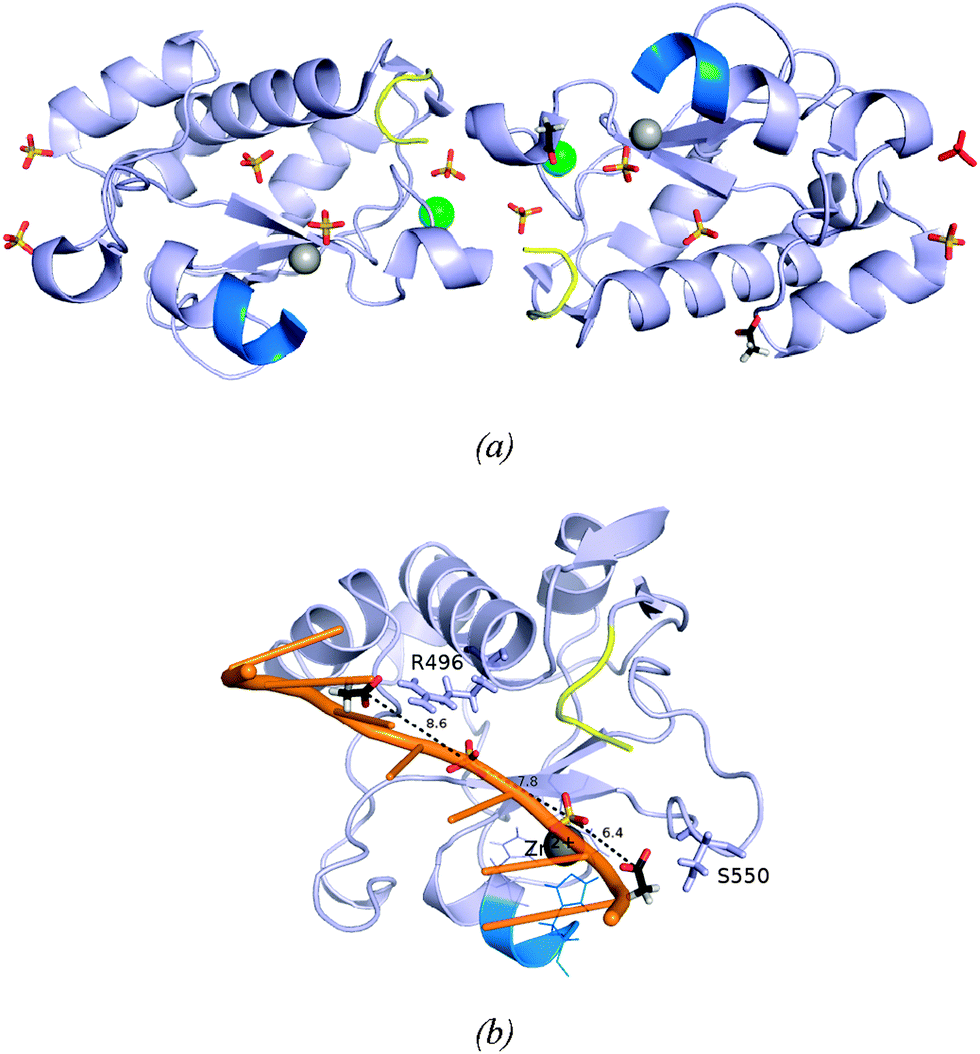 | ||
| Fig. 2 (a) The two independent molecules A and B of ΔN4-NColE7-C* with bound ions. Molecule A is in the left of the figure, and molecule B to the right. Their N-termini are shown in yellow and the C-termini in bright blue. Zn2+-ions are illustrated as grey spheres, chloride ions as green spheres; sulfate and acetate ions are shown in a stick representation. The sulfate ion drawn in red bound to molecule B is a crystallographically related match of the sulfate ion in the top left bound to molecule A. (b) Molecule B with its bound anions. The side chains of the hydrogen bond partners of the acetate ions are shown in a stick representation. The two acetate ions (bound to the labeled residues) and two sulfate ions close to the active site of molecule B are bound similarly to the phosphodiester groups of one of the DNA chains in the structure 2IVH.8 | ||
Electron densities interpreted as chloride ions are located in similar positions on the surface of the two molecules, their neighbors are G455 and three water molecules, all in the expected hydrogen bond distances. Fig. 2a shows how the molecules related by the non-crystallographic twofold axes are linked by sulfate ions that neutralize positively charged side chains. One of these (marked in red) is located on the non-crystallographic twofold axis. Two of the sulfate ions serve as ligands to the bound Zn2+-ions. The sulfate ions associated with the molecules A and B conform with the overall twofold symmetry that relates the two molecules. This is not the case for the acetate ions. The two acetate ions that were identified in the electron density are both bound to molecule B, where they form hydrogen bonds with R496 and S550. It is worth mentioning that the anions bound to the B molecule nicely mimic the predicted binding sites of the DNA phosphodiester groups (Fig. 2b).
Some distinct interactions involving the amino acids of the truncated N-terminus could be identified in the crystal structure of ΔN4-NColE7-C*. One of these involve the highly conserved W464 located in a hydrophobic pocket formed by the residues of the N-terminal loop (L465, P475, V476, P477), the central part of the protein (F499, W500, L509, F513) and the HNH motif (V563) as shown in Fig. 3a and b. Residues from the N-terminus interact with the HNH loop and with one of the β-sheets within the C-terminal HNH motif as shown in Fig. 3c. These interactions make the N-terminal loop fill the space (Fig. 3d) between the HNH motif and the central part of the protein including nonspecific DNA and Im7 binding helices.
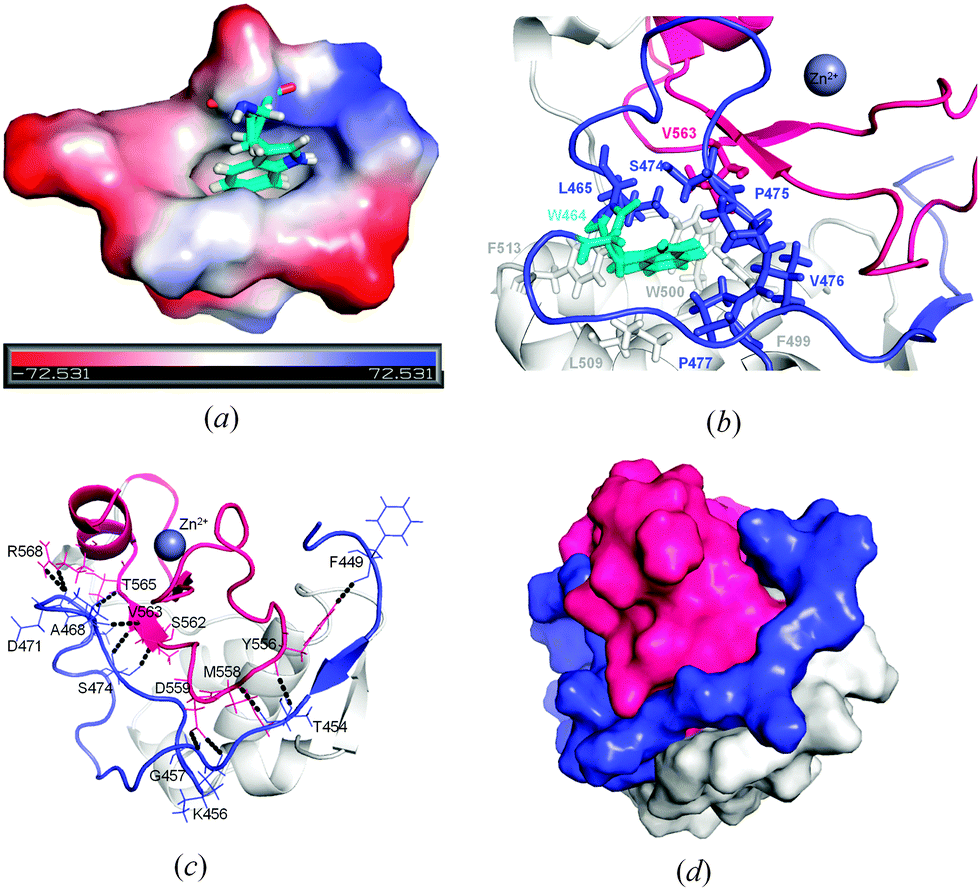 | ||
| Fig. 3 Interactions of the N-terminal loop. (a) The vacuum electrostatic surface of the surroundings of W464. (b) The hydrophobic environment around W464. (c) The identification of possible hydrogen bonds formed between the amino acids of the N-terminal loop and those of the HNH motif. (d) Shape complementarity of the N-terminal loop between the C-terminal HNH motif and the central parts of the protein (grey). In (b), (c) and (d) the N-terminus is colored in blue, while the HNH motif is red. | ||
These interactions – most of them found in the other known NColE7 structures – may stabilize the active site. This is supported by our recent studies on the HNH motif itself,49 on the ΔN25-NColE7 mutant lacking the major part of the N-terminal loop,21 as well as on the NColE7 triple mutant including T454A, K458A and W464A mutations.50
Fig. 4 shows the environment of the Zn2+-ions bound in the two molecules, A and B. Both are coordinated by three histidine residues and a sulfate ion. The C-terminal H573 is poorly defined in both molecules. The refined B-value for the Zn2+-ion is significantly higher in molecule A than in molecule B, 40.54 and 24.57, respectively. The excess residual density at the position of the Zn2+-ion in molecule A could indicate a disorder that has not been sufficiently well modeled. The closest water molecule to Zn2+ is W2072, in a distance of 4.0 Å. The B-value of the Zn2+-ion in molecule B is significantly lower and residual density is found 1.25 Å away from the Zn2+-ion. Furthermore a water molecule (W2065) is located in the second coordination sphere of Zn2+, at a distance of 3.9 Å.
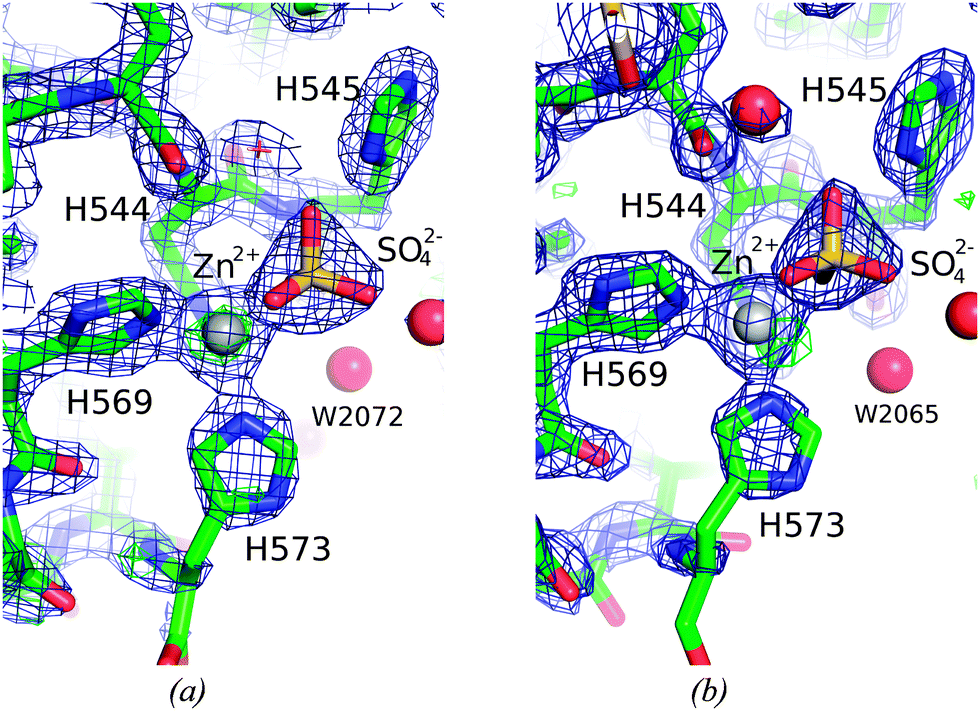 | ||
| Fig. 4 The surroundings of Zn2+-ion in the active site of the two molecules. A Sigma-A weighted 2Fobs–Fcalc electron density map at 1.5 sigma (0.48 e− Å−3) is shown in blue and Fobs–Fcalc difference map electron density at 3.0 sigma cut-off (0.37 e− Å−3) in green and red, respectively for the (a) A and (b) B molecule in the crystal structure of ΔN4-NColE7-C*. The peak in the difference density 1.25 Å away from the Zn2+-ion was not modelled. | ||
Computational results: QM/MM calculations – comparison of the catalytic centre from the calculations and crystal structures
QM/MM calculations provide a valuable complementary method for the local structural details of proteins e.g., in ref. 51 and 52, QM/MM calculations were performed for the ΔN4-NColE7-Zn2+–DNA and the active NColE7-Zn2+–DNA model structures. We expected that these calculations should predict the active state of the protein complexed with Zn2+ and DNA, and give important atomic details (position of hydrogen atoms, protonation states, etc.).As can be seen in Fig. 5a, the overall optimized structure of the two proteins is similar whereas small structural differences are confined to the active centre. These differences are seen in the orientations of the metal coordinating histidines, notably H544 and the position of the metal ion in ΔN4-NColE7. The bond angles to ligands in the coordination sphere of the metal ion are more significantly influenced than the bond lengths. Surprisingly, in both QM/MM optimized structures the Zn2+-ion in the active centre became pentacoordinated in a slightly distorted trigonal bipyramidal geometry (Fig. 5b).
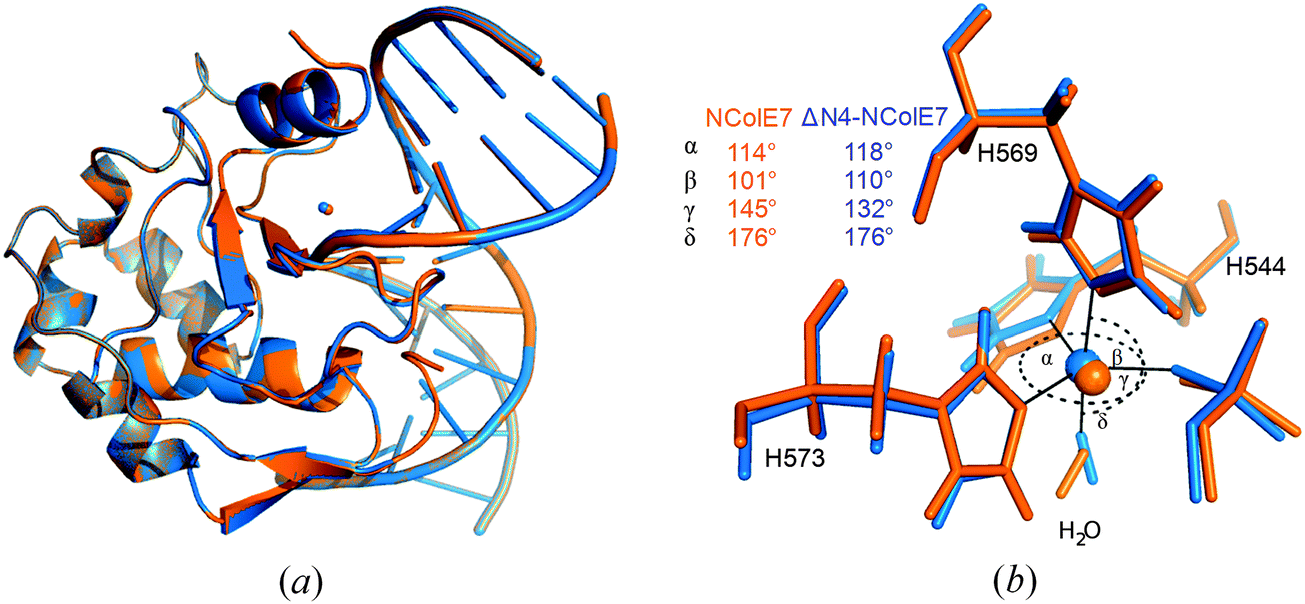 | ||
| Fig. 5 (a) The superimposed structures of NColE7–DNA (orange) and ΔN4-NColE7–DNA (blue) complexes, both optimized using the QM/MM method. (b) The active centre of the corresponding structures: the Zn2+-ion is pentacoordinated with three His residues, one water molecule and an oxygen atom of the scissile phosphate in its coordination sphere. | ||
The deletion of the N-terminal KRNK residues likely causes changes also in key steps of the catalysis. The supposed general base H545 is tilted in the modelled ΔN4-NColE7 structure (Fig. 6a), as compared to the optimized NColE7–DNA complex structure. The catalytic water molecule activated by this residue is in different orientation in the two optimized structures. When R447 is present, it serves as a hydrogen bond donor to this water molecule in the optimized structure.
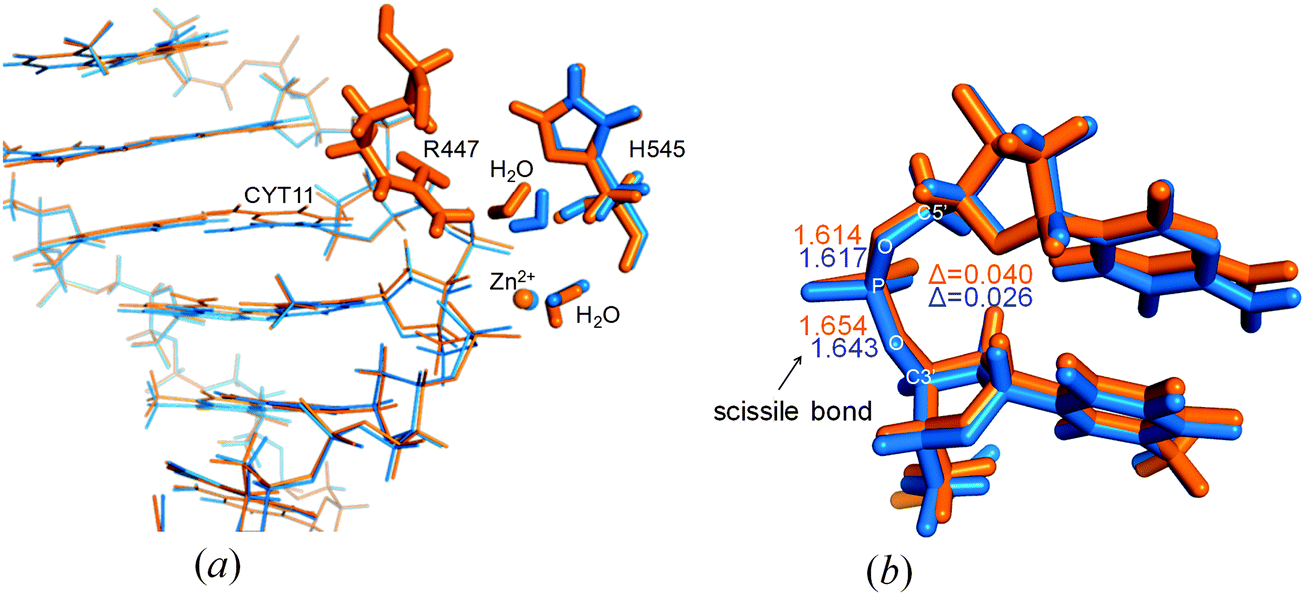 | ||
| Fig. 6 (a) DNA binding of NColE7 (orange) and ΔN4-NColE7 (blue) in the QM/MM optimized structures. Major differences appear in the position of the H545 and its hydrogen-bonded water molecule and in the position of the plane of CYT11. (b) P–O bond lengths within the scissile phosphodiester bond in the same structures. | ||
Changes in the active site of NColE7 affected the substrate DNA chain. One of the two bases at the scissile phosphate (CYT11) is tilted in the NColE7–DNA complex (Fig. 6a) and is no longer coplanar with its guanine pair. This strain in DNA may facilitate its hydrolytic cleavage. In contrast, in the ΔN4-NColE7–DNA optimized structure the CYT11 is nearly coplanar to its pair on the opposite strand. We also noted a difference in the P–O3′ scissile bond lengths in the optimized structures (Fig. 6b): the increased length in the presence of NColE7 suggests higher probability of the cleavage by the WT enzyme compared to the ΔN4-NColE7 mutant.
Discussion
Comparison of the ΔN4-NColE7-C* crystal structure with other available NColE7 structures
Fig. 7a shows that the mutations at the N- and C-termini did not influence significantly the ΔN4-NColE7 fragment (residues 450–573) of the structure compared to NColE7 (PDB entry: 1MZ86) the all-atom rmsd being 0.23 Å. Accordingly, the lack of DNase activity is not due to conformational changes but it is directly related to the mutations. The only clear difference was observed in the electrostatics: ΔN4-NColE7-C* is significantly less positive than NColE7 (Fig. S4, ESI†).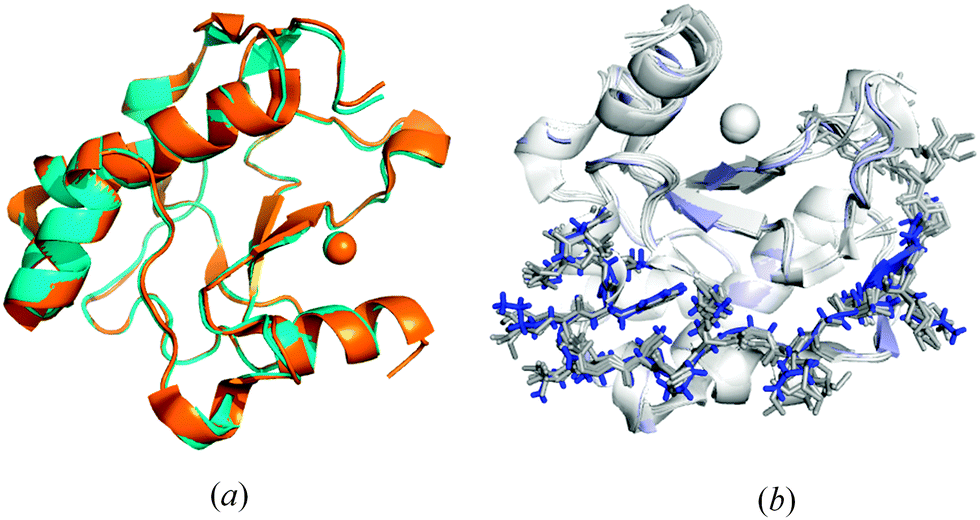 | ||
| Fig. 7 (a) Structural alignment based on the identical amino acids of the ΔN4-NColE7-C* (blue; PDB code: 3ZFK) and WT NColE7 (orange; PDB code: 1MZ86) shows good agreement between the structures of the two proteins. (b) Structural alignment of the N-terminal loops in selected NColE7 domains including the PDB structures 1M08,51MZ8,61PT3,31ZNS,71ZNV,77CEI,42IVH,82JAZ,92JB0,92JBG9 and 3FBD10 all in light grey and the Δ4-NColE7-C* in blue. | ||
The structure and relative position of the N-terminus are not affected by DNA or Im7 binding in NColE7 variant crystal structures.3–10 It also remained unchanged in the ΔN4-NColE7-C* protein upon the mutation of the KRNK sequence at the N-terminus as Fig. 7b shows. This confirms that additional interactions exist between the N-terminal loop and the rest of the protein, which are responsible for keeping the positive charges at the N-terminus close to the catalytic centre.
In ΔN4-NColE7-C* it was also possible to identify all the hydrogen bonds formed by the conserved N560 with the backbone atoms of E546, K547, G557 and D557 in the flexible HNH loop (Fig. S5, ESI†).9 N560 has a high impact on the catalytic activity, since it fixes the position of the general base H545.9,13 The position of the four active site histidines is well preserved in most of the NColE7 structures. The only exceptions are the DNA complexes of the H545 mutated inactive enzyme (1ZNS,72IVH8) – where one or more His side-chains are tilted (Fig. 8a). Similarly, the rotation of the H573 side-chain imidazole ring by ∼17 degrees is observed in the active site of ΔN4-NColE7-C* relative to the corresponding imidazole in the 1M08 NColE7 structure.
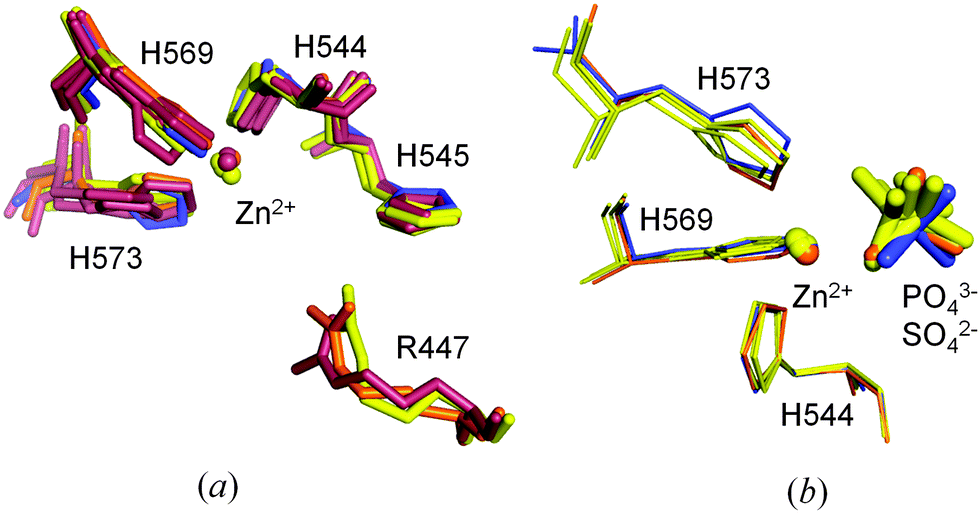 | ||
| Fig. 8 (a) Comparison of the arrangement of the metal ion and coordinating histidine residues in the active site of ΔN4-NColE7-C* (in blue) with other NColE7 structures (NColE7 in orange: 1M08;5 NColE7/Im7 in yellow: 1MZ8,61ZNV,77CEI,42JAZ,92JB0,92JBG;9 NColE7–DNA in brown: 1PT3,32IVH,81ZNS,73FBD10). (b) Orientation of the phosphate ions (1M08, 1MZ8, 2JAZ) and the sulfate ions (2JBG and the ΔN4-NColE7-C*) applying the same color coding. | ||
A slight shift in the position is seen for the Zn2+-ion and the substrate mimicking sulfate ion in ΔN4-NColE7-C* relative to the Zn2+ containing NColE7 structures in Fig. 8b. This did not affect the strong metal ion binding of ΔN4-NColE7-C*.21 By contrast ΔN25-NColE7 and T454A/K458A/W464A-NColE7 mutants with modified N-terminal sequences showed impaired Zn2+-binding ability due to the structural changes of the active centre.21,50 Consequently, we suggest that the interactions between the N-terminus and the HNH motif are essential for the mutual stabilization of their structure and for the proper folding of the catalytic centre.
The comparison of the ΔN4-NColE7-C* structure and other published crystal structures to the QM/MM computational models of NColE7–DNA and ΔN4-NColE7–DNA can give a deeper insight into the catalytic differences in the active centre. While the Zn2+-coordination is similar in the compared structures, as shown by Zn2+-donor atom distances listed in Table 2, the difference between the P–O(3′) and P–O(5′) bond lengths of the scissile phosphate is only significant in the optimized active structure, but not in the crystal structures. It must be emphasized that all the experimental results were obtained for inactive forms of the protein, i.e. either the metal ion was absent or a functional residue was mutated. This difference between the computed NColE7–DNA and ΔN4-NColE7–DNA complexes may indicate the slight changes in the active centre that finally contribute to the loss of nuclease activity upon deletion of the N-terminal positively charged residues.
| Structures | Zn2+–O(DNA) | Zn2+–H544 | Zn2+–H569 | Zn2+–H573 | P–O (3′) | P–O (5′) | ΔP–O (3′–5′) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1PT3 (without Zn2+) | — | — | — | — | 1.607 | 1.588 | 0.019 | 3 |
| 3FBD (without Zn2+) | — | — | — | — | 1.616 | 1.594 | 0.022 | 10 |
| 2IVH (H545Q) | 1.769 | 2.039 | 2.136 | 1.837 | 1.604 | 1.596 | 0.008 | 8 |
| 1ZNS (H545E) | 2.017 | 1.798 | 1.941 | 2.047 | 1.591 | 1.579 | 0.012 | 7 |
| 3ZFK (without DNA) A | — | 1.977 | 2.187 | 2.287 | — | — | — | This work |
| 3ZFK (without DNA) B | — | 1.672 | 2.073 | 2.090 | — | — | — | This work |
| ΔN4-NColE7–DNA calc. | 2.003 | 2.081 | 2.147 | 2.044 | 1.643 | 1.617 | 0.026 | This work |
| NColE7–DNA calc. | 2.067 | 2.095 | 2.148 | 2.117 | 1.654 | 1.614 | 0.040 | This work |
The Zn2+-ion and the positively charged N-terminal amino acids in the active site – new possible roles in the catalytic function
In the crystal structure of ΔN4-NColE7-C* the B molecule had a Zn2+-binding site with significant residual density in the vicinity, at a difference map peak of 1.25 Å, as shown in Fig. 2. The major difference lies not in the protein monomers, but in the metal ions: Zn2+-ion B402 is less rigid and with a significantly lower (62%) atomic displacement parameter than that of the Zn2+-ion in the A molecule. The metal–ligand coordination is essentially the same in both molecules, but our modelling studies hinted at the coordination of an additional water molecule (Fig. 4). This is in agreement with the results of the QM/MM calculations. In the optimized structures (Fig. 6a) a water molecule that was weakly bound (R(Zn–O) = 3.7 Å) in the starting structure approached the metal ion to 1.9 Å and 2.0 Å for ΔN4-NColE7 and NColE7, respectively. Such water molecules may be activated during the catalytic process to serve either as a general acid or base. This model is consistent with an artificial nuclease system53,54 and a similar type of coordination was observed experimentally in an NColE9 crystal structure (1FSJ55). It is worth mentioning that in the crystal structure of ΔN4-NColE7-C* H2O(2072) in the A molecule and H2O(2065) in the B molecule are located ∼4.0 Å from the Zn2+-ion.Sequence comparison of related colicin and pyocin bacterial toxins showed that the arginine corresponding to R447 in colicin E7 is highly conserved (Fig. 1). Crystal structures of HNH nucleases showed that an arginine stretches into the active site e.g. R447 in NColE7,5,6,10 R5 in NColE9,56 R99 in Vvn,16 R57 in Sm endonuclease57 and R93 in nuclease A.58 The distance from the side chain of this arginine to the metal ion located in the active centre is ∼6–7 Å in the presence of DNA or a phosphate/sulfate ion situated between the positively charged residues, and >10 Å in the absence of it. Based on Vvn crystal structures it was hypothesized that the arginine side chain binds and stabilizes the cleaved DNA to decelerate the reverse reaction.16,17 In NColE920 and in Sm endonuclease59 related HNH nucleases the arginine corresponding to R447 in NColE7 was proposed to stabilize the pentavalent transition state. The R447A mutation in NColE7 reduced the in vitro DNase activity to ∼15% of the initial value13 supposed to be due to the decreased DNA binding affinity. In contrast, we have recently shown that the mutations of the positive charges within the KRNK (446–449) sequence do not significantly affect the strength of the DNA binding.60 This study also demonstrated that the positively charged lysines can partially replace the missing arginine in its function.
Surprisingly, precise location of R447 in the presence of the DNA has only been determined in the 18bp DNA–D493Q NColE7 mutant complex,10 and it was not possible to locate it in the three other crystal structures of DNA complexes.3,7,8 In this complex R447 clearly interacts with the phosphodiester group of the DNA molecule. In spite of the close contacts, this residue has unusually high temperature factors 60–70 Å2 compared to those of the rest of the molecule (Fig. S6, ESI†) indicating its high flexibility. The experiments on the cytotoxic behavior of the mutants (see later) also demonstrated that even the location of the positively charged amino acid residue in the amino acid sequence close to the N-terminus is not strictly determined.
Considering the above observations we can not exclude that the flexible arginine side-chain mediates the proton transfer in NColE7. After protonating the leaving group it may become instantly re-protonated by H545. These two residues get close (Fig. 9) to each other and a water-mediated hydrogen bond is formed between them during the catalytic cycle. The initial source of the proton on H545 is the water molecule, which has to become deprotonated to perform the nucleophilic attack at the partially positively charged phosphorus atom. This would be in agreement with the recently proposed shuttle mechanism according to which the leaving group is protonated by the hydrogen ion originating from the same water molecule that initiated the nucleophilic attack.61
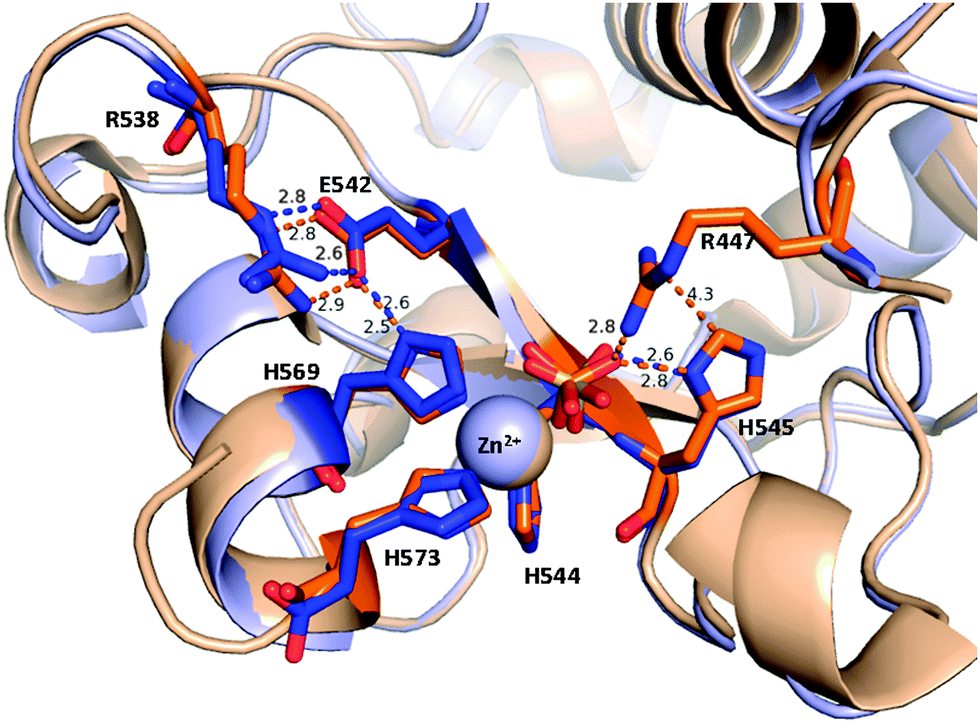 | ||
| Fig. 9 Short interatomic distances reflecting possible hydrogen bonds in the active site of ΔN4-NColE7-C* molecule A (blue) in comparison with NColE7 (PDB: 1M08, in orange). The hydrogen bonds among R538, E542 and H569 (the H569 residue also binds the Zn2+-ion) form a putative proton channel. | ||
The above mechanism would be similar to the one found in a serine recombinase-mediated DNA cleavage.62 Although arginine is rarely mentioned in the literature to behave as an acid but the environment of an enzyme active site can significantly shift the pKa values of critical residues. It can be considered plausible in biological systems,63 especially in the presence of a basic leaving group as it is the 3′-alcoholate ion. This would provide a reasonable answer to the still unsolved but intriguing question about the identity of the general acid that protonates the leaving group in NColE7. It shall also be mentioned that the multiple roles of R447 can be partially replaced by other positive charges, such as lysine side-chains or the N-terminal amino group in a suitable position.
The effect of the N- and C-terminal mutations on the cytotoxicity
Toxic variants of NColE7 kill the bacterial cell through their nuclease activity already during the cloning process due to the minor level of expression in the applied cloning systems.22,64 Taking advantage of this we can obtain information on the catalytic activity of an NColE7 mutant already in the early stage of the experiments. At the same time, the minuscule probability to obtain a gene of erroneous sequence, resulting in an inactive mutant, is increased. These mutations may reveal the necessity of each amino acid residue and may affect the protein structure, metal ion or DNA binding or the catalytic process itself. This event led to a C-terminal modification resulting in extra 9 amino acids within the inactive (GST)ΔN4-NColE7-C* protein. This points to the fact that the expected GST-ΔN4-NColE7 protein was cytotoxic for the E. coli cells.To understand this and to investigate the impact of the mutations at the two termini of the GST-ΔN4-NColE7-C* protein we have recloned the ΔN4-NColE7 gene into the pET-21a vector as described in Materials and methods (the expected sequences are depicted in Fig. 1). While the (E/X)ΔN4-NColE7 variant was cytotoxic, the (N/X)ΔN4-NColE7 protein of correct sequence was successfully overexpressed in E. coli as a Zn2+-bound protein (the ESI-MS proof of the metal ion binding is shown in Fig. S7, ESI†) similarly to the GST-ΔN4-NColE7-C* mutant. The latter protein was proven to bind both Zn2+-ions and DNA in vitro strongly,21 thus it is the lack of another feature that allows for expression without cytotoxic effects.
The data collected in Table 3 show that the ΔN4-NColE7 with an arginine close to the N-terminus in a similar position to the WT NColE7 is cytotoxic. The only exception is the active GST-ΔN4-NColE7 nuclease. However, in this protein the GST fusion tag carries several positive charges both on the surface and in the linker region (Fig. S8, ESI†). One of these may allow for the catalytic process in GST-ΔN4-NColE7, but not in GST-ΔN4-NColE7-C*. The access of the bulky GST tag to the active site is most probably prohibited by the newly formed C-terminus in the latter protein. In parallel to this the same C* mutation can not prevent the GST-NColE7-C* to be cytotoxic, providing further proof of the importance of the KRNK sequence.
| Position of R near the N-terminus | C-terminal modification | Cytotoxicity | |
|---|---|---|---|
| a There is no positively charged residue within a distance of 10 amino acids of the original position of the R447. b The gene of ΔN4-NColE7 was inserted into a plasmid that added a short sequence containing the arginine at position R445. | |||
| NColE7 | 447 | − | + |
| GST-ΔN4-NColE7-C* | —a | + | − |
| GST-ΔN4-NColE7 | —a | − | + |
| (E/X)ΔN4-NColE7 | 445b | − | + |
| (N/X)ΔN4-NColE7 | — | − | − |
| GST-NColE7-C* | 447 | + | + |
Conclusions
The truncated mutant of NColE7 nuclease, lacking the positively charged KRNK amino acid string at the end of the N-terminal sequence proved to be catalytically inactive. The presence of the N-terminal loop – distant in the sequence from the catalytic centre – playing an intramolecular allosteric role in the enzymatic process, as found in NColE7, is a fascinating phenomenon to study. This feature of NColE7 may be applied in the engineering of controlled artificial nucleases. The crystal structure of the ΔN4-NColE7-C* mutant, determined at 1.7 Å resolution revealed direct information about the structural role of the remaining N-terminal sequence, and it also provided indirect information on the role of the deleted KRNK (446–449) string. The additional C-terminal modification was shown to control the enzymatic activity by preventing the distal positively charged amino acids on bulky chains from approaching the catalytic site.As the structure of ΔN4-NColE7-C* does not significantly differ from the structures determined so far for NColE7 variants we can conclude that the lack of its catalytic activity is not due to a change in the overall structure. The extensive interactions between the unchanged part of the N-terminal loop and the rest of the protein are not influenced by the deletion of the KRNK amino acid sequence. These interactions may contribute to the mutual stabilization of the N-terminal loop and the catalytic centre.
The cytotoxic activity of the designed mutant proteins indicated that it can be associated with a positively charged residue close to the N-terminus. R447 could not be modeled in the majority of NColE7 variant crystal structures, which indicates a great flexibility. It leads us to propose that R447 beside the previously suggested functions may also behave as a flexible general acid that assist in the transfer of the proton from the general base to the leaving group. According to the QM/MM calculations the presence of R447 also facilitates the distortion of the substrate DNA chain promoting the hydrolysis of the scissile phosphodiester group. QM/MM calculations pointed out that in the active NColE7–DNA complex – that cannot be studied by crystallographic methods due to the catalytic reaction – the bond length of the scissile bond in contrast to the crystal structures of inactive variants is elongated, and the plane of the nucleobase at the cleavage site is tilted. These phenomena were not observed in the optimized ΔN4-NColE7 structure.
Both the QM/MM calculations and the careful inspection of the crystal structure revealed the possibility of the coordination of a water molecule to the Zn2+-ion as the fifth ligand. Such a metal ion activated water molecule may participate in the catalytic process as a nucleophilic reagent after passing its proton to an amino acid side-chain of the protein, or as a general acid providing its proton in the final step of the reaction.
Our results showed that the mechanism of action of NColE7 requires some revisions. The essential catalytic role of the positively charged amino acids (such as R447 in the WT enzyme), as well as the presence of a water molecule as the fifth ligand in the Zn2+ coordination sphere has to be considered. The detailed description of the particular role of these residues will be possible upon further combined experimental and computational studies.
Acknowledgements
Financial support from the Hungarian Scientific Research Found (OTKA-NKTH CK80850), TÁMOP-4.2.2/B-10/1-2010-0012, TÁMOP 4.2.4.A/2-11-1-2012-0001 ‘National Excellence Program’ and JSPS is greatly acknowledged. E.T. and A.C. thank Danish Ministry of Science, Innovation & Higher Education for the fellowships provided. L.R. acknowledges the support from the Grant Agency of the Czech Republic (grant 1431419S).References
- R. D. Finn, J. Tate, J. Mistry, P. C. Coggill, S. J. Sammut, H. R. Hotz, G. Ceric, K. Forslund, S. R. Eddy, E. L. Sonnhammer and A. Bateman, Nucleic Acids Res., 2008, 36, D281–D288 CrossRef CAS.
- A. Veluchamy, S. Mary, V. Acharya, P. Mehta, T. Deva and S. Krishnaswamy, Bioinformation, 2009, 4, 80–83 CrossRef.
- K. Hsia, K. Chak, P. Liang, Y. Cheng, W. Ku and H. S. Yuan, Structure, 2004, 12, 205–214 CrossRef CAS.
- T. Ko, C. Liao, W. Ku, K. Chak and H. S. Yuan, Structure, 1999, 7, 91–102 CrossRef CAS.
- Y. Cheng, K. Hsia, L. G. Doudeva, K. Chak and H. S. Yuan, J. Mol. Biol., 2002, 324, 227–236 CrossRef CAS.
- M. Sui, L. Tsai, K. Hsia, L. Doudeva, W. Ku, G. Han and H. Yuan, Protein Sci., 2002, 11, 2947–2957 CrossRef CAS.
- L. Doudeva, D. Huang, K. Hsia, Z. Shi, C. Li, Y. Shen, Y. Cheng and H. Yuan, Protein Sci., 2006, 15, 269–280 CrossRef CAS.
- Y. Wang, W. Yang, C. Li, L. G. Doudeva and H. S. Yuan, Nucleic Acids Res., 2007, 35, 584–594 CrossRef CAS.
- H. Huang and H. S. Yuan, J. Mol. Biol., 2007, 368, 812–821 CrossRef CAS.
- Y.-T. Wang, J. D. Wright, L. G. Doudeva, H.-C. Jhang, C. Lim and H. S. Yuan, J. Am. Chem. Soc., 2009, 131, 17345–17353 CrossRef CAS.
- K. B. Levin, O. Dym, S. Albeck, S. Magdassi, A. H. Keeble, C. Kleanthous and D. S. Tawfik, Nat. Struct. Mol. Biol., 2009, 16, 1049–1055 CAS.
- K. Chak, W. Kuo, F. Lu and R. James, J. Gen. Microbiol., 1991, 137, 91–100 CrossRef CAS.
- Z. Shi, K. Chak and H. Yuan, J. Biol. Chem., 2005, 280, 24663–24668 CrossRef CAS.
- J. H. Eastberg, J. Eklund, R. Monnat Jr. and B. L. Stoddard, Biochemistry, 2007, 46, 7215–7225 CrossRef CAS.
- R. H. van den Heuvel, S. Gato, C. Versluis, P. Gerbaux, C. Kleanthous and A. J. Heck, Nucleic Acids Res., 2005, 33, e96 CrossRef.
- C. L. Li, L. I. Hor, Z. F. Chang, L. C. Tsai, W. Z. Yang and H. S. Yuan, EMBO J., 2003, 22, 4014–4025 CrossRef CAS.
- K. Hsia, C. Li and H. Yuan, Curr. Opin. Struct. Biol., 2005, 15, 126–134 CrossRef CAS.
- C. Garinot-Schneider, A. J. Pommer, G. R. Moore, C. Kleanthous and R. James, J. Mol. Biol., 1996, 260, 731–742 CrossRef CAS.
- A. J. Pommer, S. Cal, A. H. Keeble, D. Walker, S. J. Evans, U. C. Kühlmann, A. Cooper, B. A. Connolly, A. M. Hemmings, G. R. Moore, R. James and C. Kleanthous, J. Mol. Biol., 2001, 314, 735–749 CrossRef CAS.
- D. C. Walker, T. Georgiou, A. J. Pommer, D. Walker, G. R. Moore, C. Kleanthous and R. James, Nucleic Acids Res., 2002, 30, 3225–3234 CrossRef CAS.
- A. Czene, E. Németh, I. G. Zóka, N. I. Jakab-Simon, T. Körtvélyesi, K. Nagata, H. E. M. Christensen and B. Gyurcsik, J. Biol. Inorg. Chem., 2013, 18, 309–321 CrossRef CAS.
- E. Németh, G. K. Schilli, G. Nagy, C. Hasenhindl, B. Gyurcsik and C. Oostenbrink, J. Comput.-Aided Mol. Des., 2014, 28, 841–850 CrossRef.
- Y. G. Kim, J. Cha and S. Chandrasegaran, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1150–1160 Search PubMed.
- J. K. Joung and J. D. Sander, Nat. Rev. Mol. Cell Biol., 2013, 14, 49–55 CrossRef CAS.
- M. Christian, T. Cermak, E. L. Doyle, C. Schmidt, F. Zhang, A. Hummel, A. J. Bogdanove and D. F. Voytas, Genetics, 2010, 186, 757–761 CrossRef CAS.
- D. A. Wah, J. A. Hirsch, L. F. Dorner, I. Schildkraut and A. K. Aggarwal, Nature, 1997, 388, 97–100 CrossRef CAS.
- T. I. Cornu and T. Cathomen, Methods Mol. Biol., 2010, 649, 237–245 CAS.
- B. Gyurcsik and A. Czene, Future Med. Chem., 2011, 3, 1935–1966 CrossRef CAS.
- A. Czene, E. Tóth, B. Gyurcsik, H. Otten, J. N. Poulsen, L. Lo Leggio, S. Larsen, H. E. M. Christensen and K. Nagata, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2013, 69, 551–554 CrossRef CAS.
- P. Walker, L. Leong, P. NG, S. Tan, S. Waller, D. Murphy and A. Porter, Bio/Technology, 1994, 12, 601–605 CrossRef CAS.
- A. Vagin and A. Teplyakov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 22–25 CrossRef CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS.
- R. A. Engh and R. Huber, Acta Crystallogr., Sect. A: Found. Crystallogr., 1991, 47, 392–400 CrossRef.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- V. B. Chen, W. B. Arendall 3rd, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- U. Ryde, J. Comput.-Aided Mol. Des., 1996, 10, 153–164 CrossRef CAS.
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335–347 CrossRef CAS.
- L. Rulisek, E. I. Solomon and U. Ryde, Inorg. Chem., 2005, 44, 5612–5628 CrossRef CAS PubMed.
- M. Srnec, F. Aquilante, U. Ryde and L. Rulisek, J. Phys. Chem. B, 2009, 113, 6074–6086 CrossRef CAS.
- V. Klusak, C. Barinka, A. Plechanovova, P. Mlcochova, J. Konvalinka, L. Rulisek and J. Lubkowski, Biochemistry, 2009, 48, 4126–4138 CrossRef CAS PubMed.
- T. A. Rokob and L. Rulísek, J. Comput. Chem., 2012, 33, 1197–1206 CAS.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- M. C. Lee and Y. Duan, Proteins, 2004, 55, 620–634 CrossRef CAS PubMed.
- B. Gyurcsik, A. Czene, H. Jankovics, N. I. Jakab-Simon, K. Ślaska-Kiss, A. Kiss and Z. Kele, Protein Expression Purif., 2013, 89, 210–218 CrossRef CAS PubMed.
- E. Németh, T. Körtvélyesi, M. Kožíšek, P. W. Thulstrup, H. E. M. Christensen, M. N. Asaka, K. Nagata and B. Gyurcsik, J. Biol. Inorg. Chem., 2014 DOI:10.1007/s00775-014-1186-6.
- U. Ryde and K. Nilsson, J. Am. Chem. Soc., 2003, 125, 14232–14233 CrossRef CAS PubMed.
- L. Rulisek and U. Ryde, J. Phys. Chem. B, 2006, 110, 11511–11518 CrossRef CAS PubMed.
- X. Sheng, X. Guo, X. M. Lu, G. Y. Lu, Y. Shao, F. Liu and Q. Xu, Bioconjugate Chem., 2008, 19, 490–498 CrossRef CAS PubMed.
- X. Sheng, X. M. Lu, J. J. Zhang, Y. T. Chen, G. Y. Lu, Y. Shao, F. Liu and Q. Xu, J. Org. Chem., 2007, 72, 1799–1802 CrossRef CAS PubMed.
- U. C. Kühlmann, A. J. Pommer, G. R. Moore, R. James and C. Kleanthous, J. Mol. Biol., 2000, 301, 1163–1178 CrossRef PubMed.
- M. J. Maté and C. Kleanthous, J. Biol. Chem., 2004, 279, 34763–34769 CrossRef PubMed.
- S. V. Shlyapnikov, V. V. Lunin, M. Perbandt, K. M. Polyakov, V. Y. Lunin, V. M. Levdikov, C. Betzel and A. M. Mikhailov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 567–572 CrossRef CAS.
- M. Ghosh, G. Meiss, A. Pingoud, R. E. London and L. C. Pedersen, J. Biol. Chem., 2005, 280, 27990–27997 CrossRef CAS PubMed.
- P. Friedhoff, I. Franke, G. Meiss, W. Wende, K. L. Krause and A. Pingoud, Nat. Struct. Biol., 1999, 6, 112–113 CrossRef CAS PubMed.
- E. Németh, T. Körtvélyesi, P. W. Thulstrup, H. E. M. Christensen, M. Kozísek, K. Nagata, A. Czene and B. Gyurcsik, Protein Sci., 2014, 23, 1113–1122 CrossRef PubMed.
- J. A. Bueren-Calabuig, C. Coderch, E. Rico, A. Jimenez-Ruiz and F. Gago, ChemBioChem, 2011, 12, 2615–2622 CrossRef CAS PubMed.
- R. A. Keenholtz, K. W. Mouw, M. R. Boocock, N. S. Li, J. A. Piccirilli and P. A. Rice, J. Biol. Chem., 2013, 288, 29206–29214 CrossRef CAS PubMed.
- P. J. Silva, C. Schulz, D. Jahn, M. Jahn and M. J. Ramos, J. Phys. Chem. B, 2010, 114, 8994–9001 CrossRef CAS PubMed.
- L. C. Anthony, H. Suzuki and M. Filutowicz, J. Microbiol. Methods, 2004, 58, 243–250 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mt00195h |
| This journal is © The Royal Society of Chemistry 2014 |