
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neuroprotective peptide–macrocycle conjugates reveal complex structure–activity relationships in their interactions with amyloid β†
Mingfeng
Yu
a,
Timothy M.
Ryan
b,
Samantha
Ellis
c,
Ashley I.
Bush
 b,
James A.
Triccas
c,
Peter J.
Rutledge
*a and
Matthew H.
Todd
*a
b,
James A.
Triccas
c,
Peter J.
Rutledge
*a and
Matthew H.
Todd
*a
aSchool of Chemistry, The University of Sydney, Sydney, New South Wales 2006, Australia. E-mail: matthew.todd@sydney.edu.au; peter.rutledge@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 2180 Tel: +61 2 9351 5020
bThe Florey Institute of Neuroscience and Mental Health, Parkville, Victoria 3010, Australia
cSydney Medical School, The University of Sydney, Sydney, New South Wales 2006, Australia
First published on 11th August 2014
Abstract
Interactions between amyloid β (Aβ) and metal ions are thought to mediate the neuropathogenic effects of Aβ in Alzheimer's disease. The construction of small molecules capable of synergistically chelating metal ions and recognizing Aβ would allow new insights into the biology of this disease and provide a possible therapeutic approach. We report herein the synthesis and biological evaluation of tetraazamacrocycle–(G)KLVFF hybrids and their metal complexes. The results obtained from ThT and bis-ANS extrinsic fluorescence assays, tyrosine intrinsic fluorescence assay and proteolytic assay imply complex, multifaceted structure–activity relationships in the interaction of these conjugates with Aβ. Many of the compounds tested rescued cells from Aβ-induced cytotoxicity. The attendant simplicity and ready diversification of the synthesis of these conjugates makes them attractive for further investigation.
Introduction
Alzheimer's disease (AD) is a progressive and multifactorial neurodegenerative disorder leading to the most common form of dementia in the elderly.1,2 The disease imposes a huge and growing burden on society.3 Progress towards understanding the underlying cause of AD has been made across a number of disciplines, yet its etiology and pathogenesis remain to be fully and precisely elucidated.4 Though it is not without its critics,5–8 the widely-supported amyloid hypothesis posits that aggregation of amyloid β (Aβ) and subsequent deposition into senile plaques (SPs) are involved in the progression of AD.9Metal ions play an important role in the assembly of Aβ.1,10 Zinc(II)11 and copper(II),12 even at the trace (nanomolar) concentrations found in commonly-used laboratory buffers and culture media,13 induced marked Aβ aggregation in vitro. Elevated concentrations of copper, zinc and iron have been observed in SPs of AD patients.14 Redox-active copper(I/II) and iron(II/III) bound to Aβ can undergo Fenton-type chemistry to generate reactive oxygen species such as hydrogen peroxide (H2O2) and the hydroxyl radical (˙OH),15,16 which can cause considerable oxidative damage to biological molecules and trigger neurodegeneration. These findings suggest that metal chelators could offer interesting new therapeutic benefits for AD by disrupting metal–Aβ interactions.
Current research efforts in this area centre on the construction of small molecules capable of synergistically chelating metal ions and recognizing Aβ.17,18 Such molecules consist of a metal chelator (e.g. clioquinol) and a known Aβ recognition group (e.g. curcumin, thioflavin-T). A particularly intriguing example of such a bifunctional molecule is the cyclen–pentapeptide hybrid 1 (Fig. 1A) in which cyclen is the metal chelator and the KLVFF peptide the Aβ recognition moiety.19 This hybrid was found to capture copper(II) bound to Aβ, become proteolytically active, interfere with Aβ oligomerization and aggregation, cleave Aβ into fragments, and prevent H2O2 formation and toxicity in neuronal cell culture. This precedent raises two interesting questions: (1) Is sequestration of the metal ion pivotal? (2) What structural elements are required for proteolysis to occur? Answering these questions may help to explain, among other things, why molecule 1 does not self-cleave. We became interested in exploring this structure for its possible therapeutic potential but also more broadly for the insights it might give to the behaviour of Aβ. We report herein the synthesis and biological evaluation of ten novel tetraazamacrocycle–(G)KLVFF hybrids: the amide- or triazole-linked derivatives 2 and 4–6 (Fig. 1A) and their metal complexes 20, 21 and 23–26 (Scheme 1).
 | ||
| Fig. 1 (A) Structures of tetraazamacrocycle–(G)KLVFF hybrids 1–6; (B) electronic similarities between a Z-amide bond and a 1,4-disubstituted triazole ring.20 | ||
 | ||
Scheme 1 Synthesis of tetraazamacrocycle–(G)KLVFF hybrids 1–6 and their metal complexes 19–26. Reagents and conditions: (a) appropriate carboxylic acid (9, 10, 11 or 12), PyBOP, NMM, DMF, rt, 1 h; (b) TFA/TIS/H2O (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5), rt, 2 h, followed by RP-HPLC purification, 1: 53%, 2: 63%, 3: 52%, 4: 60%, 5: 60%, 6: 58%; (c) CuCl2·2H2O or ZnCl2, EtOH, reflux, 6 h, 19: 94%, 20: 81%, 21: 54%, 22: 85%, 23: 88%, 24: 67%, 25: 53%, 26: 69%. :5:5), rt, 2 h, followed by RP-HPLC purification, 1: 53%, 2: 63%, 3: 52%, 4: 60%, 5: 60%, 6: 58%; (c) CuCl2·2H2O or ZnCl2, EtOH, reflux, 6 h, 19: 94%, 20: 81%, 21: 54%, 22: 85%, 23: 88%, 24: 67%, 25: 53%, 26: 69%. | ||
These molecules were designed to provide insight into the structure–activity relationships that might be operating. Cyclam has a stronger binding affinity for copper(II) than does cyclen (logK: 26.5 for copper(II)–cyclam vs. 23.3 for copper(II)–cyclen),21 suggesting that copper(II)–cyclam complexes are more likely to be formed in vivo than the corresponding copper(II)–cyclen complexes.22 Furthermore, the metal complexes of cyclen and cyclam tend to have different hydrolytic abilities, thus cyclam–KLVFF hybrid 2 was chosen to evaluate the role of the azamacrocyclic metal complex in any observed peptide cleavage. Synthetic pentapeptide KLVFF, a short Aβ fragment (Aβ16–20), has been shown to bind full-length Aβ and disrupt its assembly into Aβ fibrils.23,24 To examine the importance of the length and nature of the recognition sequence, and its position relative to the metal centre, additional glycine spacers were introduced (hybrids 3 and 4). The triazole moiety was incorporated into tetraazamacrocycle–KLVFF hybrids to explore whether coordinative saturation of copper(II) with the additional triazole ligand affects proteolytic activity (hybrids 5 and 6). Given that hybrids 5vs.3 and 6vs.4 are structurally identical except for the linker (triazole vs. amide), the biological potential of 1,4-disubstituted triazole as a Z-amide bioisostere20 (Fig. 1B) could be assessed by comparing triazole-linked hybrids 5 and 6 with amide-tethered hybrids 3 and 4. All six tetraazamacrocycle–(G)KLVFF hybrids 1–6 and their corresponding copper(II) and zinc(II) complexes were synthesized and evaluated; the change in metal ion is of obvious interest to determine whether the specific nature of the metal complex is important in the biological potency of these conjugates.
Results and discussion
(a) Synthesis of tetraazamacrocycle–(G)KLVFF hybrids and their metal complexes
The resin-bound oligopeptides KLVFF 7 and GKLVFF 8 were assembled via Fmoc-strategy solid phase peptide synthesis (SPPS) on Wang resin (Scheme S1, ESI†), and further coupled with tri-Boc-tetraazamacrocycle–acetic acid 9 or 10 to give resin-bound Boc-protected tetraazamacrocycle–(G)KLVFF compounds 13–16 (Scheme 1). Removal of Boc groups and detachment from the solid support were achieved in one pot by treatment of resin-bound compounds 13–16 with a cleavage/scavenger cocktail of TFA/TIS/H2O (90:5:5). The crude products were purified by RP-HPLC and lyophilized to give the desired amide-tethered hybrids 1–4 as fluffy white solids in good overall yields. This solid-phase based synthetic procedure was successfully applied to the preparation of triazole-linked hybrids 5 and 6 by introducing tri-Boc-tetraazamacrocycle–triazole–acetic acid 11 and 12 respectively to the resin-bound oligopeptide 7. Elemental analysis data indicated that each of the isolated compounds 1–6 incorporated four equivalents of constitutive TFA (presumably three associated with secondary amino groups of the tetraazamacrocycle and one with the ε-amino group of the lysine residue).
The trifluoroacetate salts of compounds 1–6 were complexed directly with copper(II) and zinc(II) as reported previously for related systems.25–27 Reaction with CuCl2·2H2O was carried out in EtOH at reflux for 6 hours to afford copper(II)–tetraazamacrocycle complexes 19, 20 and 22–25, which were isolated by centrifugation. The copper(II)–cyclen complexes appear blue whereas the copper(II)–cyclam complexes are purple powders (Fig. S1, ESI†). Zinc(II) complexes of hybrids 2 and 6 were also prepared by reacting the ligands with ZnCl2 under similar conditions. Other metal cyclam–amino acid/peptide complexes that we have reported previously all exhibited characteristic singly charged cations in the high resolution mass spectra.25 In contrast, all the metal complexes of tetraazamacrocycle–(G)KLVFF hybrids gave rise exclusively to a cluster of [M–2Cl]2+ peaks with the correct isotope patterns. The purity of these metal complexes was confirmed by elemental analysis (ESI†). The UV-Vis spectra of the copper(II) complexes showed that λmax and ε values associated with the cyclen complexes (λmax = 582–591 nm, ε = 211–258) were larger than those for the corresponding cyclam complexes (λmax = 552–555 nm, ε = 110–138). Absolute values of λmax and ε are expected to be solvent-, pH- and substituent-dependent, but the same relationship of these values (cyclen > cyclam) has been observed previously,28 as has the reverse relationship (cyclam > cyclen).29 No significant differences in λmax and ε values were observed between the triazole-linked complexes and the amide-tethered complexes.
(b) In vitro biological evaluation
With tetraazamacrocycle–(G)KLVFF hybrids 1–6 and their metal complexes 19–26 in hand, a series of in vitro biological assays was conducted using Aβ42 that included thioflavin-T (ThT) extrinsic fluorescence, tyrosine intrinsic fluorescence, bis-ANS extrinsic fluorescence and neurotoxicity assays, along with MALDI-TOF-MS analysis. Azide-capped pentapeptide 27 (see Scheme S1, ESI† for the synthetic procedure) and the simple, non-peptidic metal–cyclam complexes 28 and 2925 were also evaluated as controls (Fig. 2). The fresh Aβ stock solution used for these studies was prepared by a modified literature procedure (ESI†).19 Pretreatment of Aβ with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) has been previously considered to be of benefit with respect to solubilizing the peptide and monomerizing β-sheet peptide aggregates, however this step was omitted because the alcohol has recently been shown to increase Aβ aggregation in solution.30 | ||
| Fig. 2 Structures of three control compounds 27–29. | ||
A continuous ThT extrinsic fluorescence assay33,34 was employed to determine the effects of compounds 1–6 and 19–29 on Aβ aggregation. These compounds (10 and 50 μM) were incubated with monomeric Aβ (5 μM) and ThT (20 μM) in PBS buffer (pH 7.4) for 23 hours, and the fluorescence of ThT was measured continuously throughout the incubation (excitation at 444 nm and emission at 485 nm). It was found that addition of the test compounds gave rise to (1) an increase (1, 3, 27 and 29), (2) a decrease (2, 6, 20, 21, 23, 24 and 26) or (3) little change (4, 5, 19, 22, 25 and 28) in the fluorescence intensity with comparison to that obtained from the case where monomeric Aβ alone was incubated with ThT (Fig. 3 and Fig. S2, ESI†), demonstrating that these compounds could (1) promote, (2) inhibit or (3) hardly affect the Aβ fibril formation respectively (Table 1).
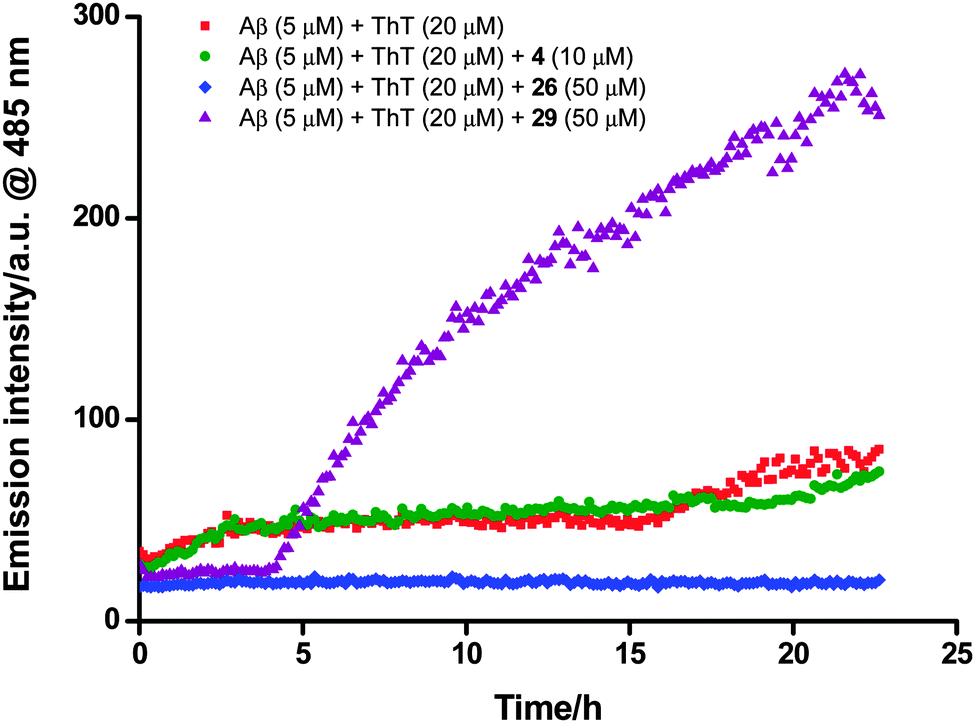 | ||
| Fig. 3 Selected data from ThT extrinsic fluorescence assay: addition of the peptidic zinc(II) complex 26 (50 μM) quenched the ThT fluorescence (blue diamond) with comparison to that obtained from an incubated solution of Aβ (5 μM) and ThT (20 μM) alone (red square), but the simple, non-peptidic zinc(II) complex 29 (50 μM) enhanced the ThT fluorescence (purple up-pointing triangle). The free ligand 4 (10 μM) exerted little influence on the ThT fluorescence (green circle). | ||
| Compound | Effect on Aβ aggregationa | |
|---|---|---|
| 10 μM | 50 μM | |
| a (1) Inhibition: weak (–), moderate (––), strong (–––); (2) no effect: 0; (3) promotion: weak (+), moderate (++), strong (+++). | ||
| 1 | 0 | +++ |
| 2 | ––– | 0 |
| 3 | +++ | 0 |
| 4 | 0 | 0 |
| 5 | 0 | 0 |
| 6 | – | 0 |
| 19 | – | – |
| 20 | 0 | ––– |
| 21 | – | ––– |
| 22 | 0 | 0 |
| 23 | 0 | –– |
| 24 | 0 | –– |
| 25 | 0 | 0 |
| 26 | – | ––– |
| 27 | 0 | ++ |
| 28 | 0 | 0 |
| 29 | + | +++ |
Zinc(II) complexes 21 and 26 strongly inhibited formation of Aβ fibrils at both concentrations. The equivalent copper(II) complexes 20 and 25 respectively showed significantly reduced inhibitory activity: some activity was observed for 20 but only at high concentration, while no activity was observed for 25 at both concentrations. This result implies that the nature of the metal ion in such conjugates is an important factor in Aβ fibril inhibitory activity. The corresponding free ligands 2 and 6 had little effect on Aβ fibril formation at 50 μM, but surprisingly exhibited inhibitory activity at 10 μM. Copper(II) complexes 23 and 24 displayed modest activity against Aβ fibril formation at high concentration, while the corresponding free ligands 4 and 5 exerted little influence on Aβ aggregation. As ThT fluorescence is a spectroscopic measure of aggregation, and since it is known that the addition of compounds that are spectroscopically active can skew the results,35 we confirmed these results using a pelleting assay, where the amount of aggregate formed over 24 hours was measured by the proportion of Aβ peptide that pelleted at 100000 × g (Fig. S3, ESI†). The results showed that the ThT assay was measuring the proportion of aggregated Aβ accurately, confirming the results presented in Table 1. In addition, aggregation could be influenced by an interaction between the phosphate buffer and metal ions. We find no difference in the results when the PBS buffer is substituted for Tris-HCl (20 mM, pH 7.5), suggesting that the PBS buffer is not interfering with the aggregation assay.
The copper(II)–cyclen complexes 19 and 22 had been reported to inhibit markedly the formation of ThT-positive Aβ aggregates.19 However, in the present study, these two complexes were found to have little effect on Aβ aggregation at either 10 or 50 μM. The corresponding free amine ligands 1 and 3, which differ only in the length of peptide sequences (KLVFF in 1vs. GKLVFF in 3), showed contrasting results: hybrid 1 significantly promoted the formation of Aβ fibrils at high concentration (but did not affect Aβ aggregation at low concentration), whereas compound 3 strongly activated Aβ fibril generation at low concentration (but had little effect on Aβ fibril formation at high concentration).
The peptide control 27 moderately accelerated Aβ fibril formation at high concentration. The simple (non-peptidic) zinc(II) complex 29 promoted the generation of Aβ fibril. In contrast, the equivalent copper(II) complex 28 showed no activity against Aβ fibril formation, providing further evidence for the importance of the nature of the metal ion.
The promotion of Aβ aggregation by compounds 1, 3, 27 and 29 was signalled by a dramatic increase in ThT fluorescence intensity during the incubation. It is conceivable that these compounds may somehow themselves aggregate to give a false positive, however incubation of each of compounds 1, 3, 27 and 29 with ThT in the absence of Aβ did not trigger any measurable fluorescence change (this control was performed for every compound used in this study, including the zinc(II) and copper(II) complexes, with the same result). This result confirmed that these compounds on their own do not form ThT-positive aggregates. There remains the possibility that ThT-positive heterofibrils are being formed between the added compounds and Aβ, which from a biochemical standpoint warrants further investigation.
These data reveal few if any obvious trends, making it difficult to develop a unified rationale for the effects observed. Overall, the peptidic zinc(II) complexes 21 and 26 exhibited the strongest Aβ fibril inhibitory activity among the test compounds whereas the simple, non-peptidic zinc(II) complex 29 was the only compound to accelerate Aβ fibril formation at both concentrations. None of the copper(II) complexes (19, 20, 22–25 and 28) promoted Aβ aggregation. Two of the unmetallated ligands (2 and 6) showed inhibitory activity against Aβ fibril formation at low concentration but hardly affected Aβ aggregation at high concentration, whereas the rest of them (1 and 3–5) either exerted little influence on or even strongly accelerated the formation of Aβ fibrils. There are no obvious patterns when comparing the different chelators (cyclam vs. cyclen), different spacers (N-benzylamide vs. KLVFF vs. GKLVFF), and different linkers (triazole vs. amide). The conclusion from this assay is that, rather than a simple Aβ:(G)KLVFF interaction giving a pre-defined outcome, there must be a complex combination of factors that control the interaction between these compounds and Aβ. It is possible that heterofibril assemblies36,37 form between the compounds and Aβ under the control of complex noncovalent interactions; while this is not equivalent to inhibiting fibril formation, such assemblies could still be important from a medicinal chemistry perspective. Further work is required to investigate this possibility.
Excitation of Aβ (10 μM) in Tris buffer (50 mM, pH 7.5) at 275 nm gave rise to the expected time-invariant maximum emission intensity at ca. 314 nm (Fig. 4A). This tyrosine fluorescence was quenched upon addition of copper(II) chloride (16 μM) as previously reported,40,42 and subsequent incubation for 1 hour did not cause any further decrease (Fig. S5, ESI†). After the 1 hour incubation of Aβ with copper(II), the test compound (40 μM) was added and co-incubated for a further hour. The total fluorescence of these three-component mixtures was measured, from which any intrinsic fluorescence of the test compounds themselves (measured separately – Fig. S5, ESI†) was subtracted. The resulting fluorescence intensity was compared with that obtained from the incubated mixture of Aβ and copper(II) alone to determine whether the test compounds could extract and capture copper(II) bound to Aβ (Fig. 4). Alternate additions of these compounds, either preloading the compounds with copper(II), or pre-incubating the Aβ with the compounds did not alter the results.
 | ||
| Fig. 4 (A) Selected data from tyrosine intrinsic fluorescence assay: tyrosine emission intensity of Aβ (10 μM) in Tris buffer (50 mM, pH 7.5) at ca. 314 nm (blue) was quenched upon addition of copper(II) chloride (16 μM) (red); subsequent incubation of the resulting mixture with compound 4, 24, or 27 (40 μM) had little effect on (green), partially restored (orange), or further reduced (purple) the tyrosine intrinsic fluorescence respectively; (B) summary of the effects of compounds 1–6 and 19–29 on the copper(II)-induced quenching of Aβ tyrosine fluorescence. | ||
Hybrid 1 had previously been reported to reverse the copper(II)-induced quenching of Aβ tyrosine fluorescence due to sequestration of the metal ion from Aβ.19 However, in the present study, this free ligand exerted little effect on the tyrosine fluorescence of copper(II)-bound Aβ (Fig. 4B). Similar results were observed for the ligands 3, 4 and 6. In contrast, addition of ligands 2 and 5 resulted in the partial recovery of the tyrosine fluorescence, indicating a low to moderate ability to sequester copper(II) from Aβ.
Surprisingly, the tyrosine intrinsic fluorescence was partially restored upon incubation with several of the metal complexes (21–24 and 26). These compounds lack any obvious metal-chelating ability: the azamacrocycle already holds a metal ion, and the peptide portion shows no demonstrable ability to sequester copper(II) under these conditions when tested as compound 27. These results therefore imply that beyond (or perhaps instead of) the chelation effect, another, different interaction contributes to the revival of tyrosine fluorescence. The other compounds tested exert little or no influence on the tyrosine fluorescence. Compound 27 in fact further reduced the tyrosine fluorescence, which may be due to the oxidation of the tyrosine residue by the azido group.43 As many of these compounds are coloured we cannot rule out some indirect influence on the tyrosine fluorescence through inner-filtering effects. Three points in our results suggest that this effect is not a significant factor: (i) the concentration of the compound, while fairly high, does not give rise to an OD greater than 0.1 arbitrary units at the excitation wavelength of tyrosine; (ii) only one uncoloured compound decreased the tyrosine fluorescence further, which, if inner filtering were significant, should have happened with more of the coloured compounds; and (iii) mixtures of Aβ and the compounds in the absence of copper(II) did not indicate any significant effect of either molecule on the fluorescence signal of the other, i.e. the total tyrosine fluorescence was simply the sum of the respective signals from the compound and Aβ.
Overall, more of the metal complexes were capable of restoring the tyrosine fluorescence than the free ligands, suggesting that alternative interaction(s) between metal complexes and copper(II)-bound Aβ (i.e. other than the chelation and sequestration of copper), are responsible for this process. Thus we propose that the fluorescence is regained not through the chelation of copper, but possibly via the displacement of copper(II) from Aβ by the binding of these compounds. Previous deployment of this assay has used either glycine or histidine in large excess to bind copper40,42 rather than a discrete macrocyclic chelator to compete with Aβ for copper(II) as used here; it is plausible that in those cases too a binding interaction or displacement is occurring, rather than simple metal chelation as previously proposed. While the tyrosine fluorescence results observed in the present study would be explained by a more complex interaction between Aβ and the compounds tested, more work is required to validate this hypothesis. Such work is clearly important to confirm the meaning of this assay more generally.
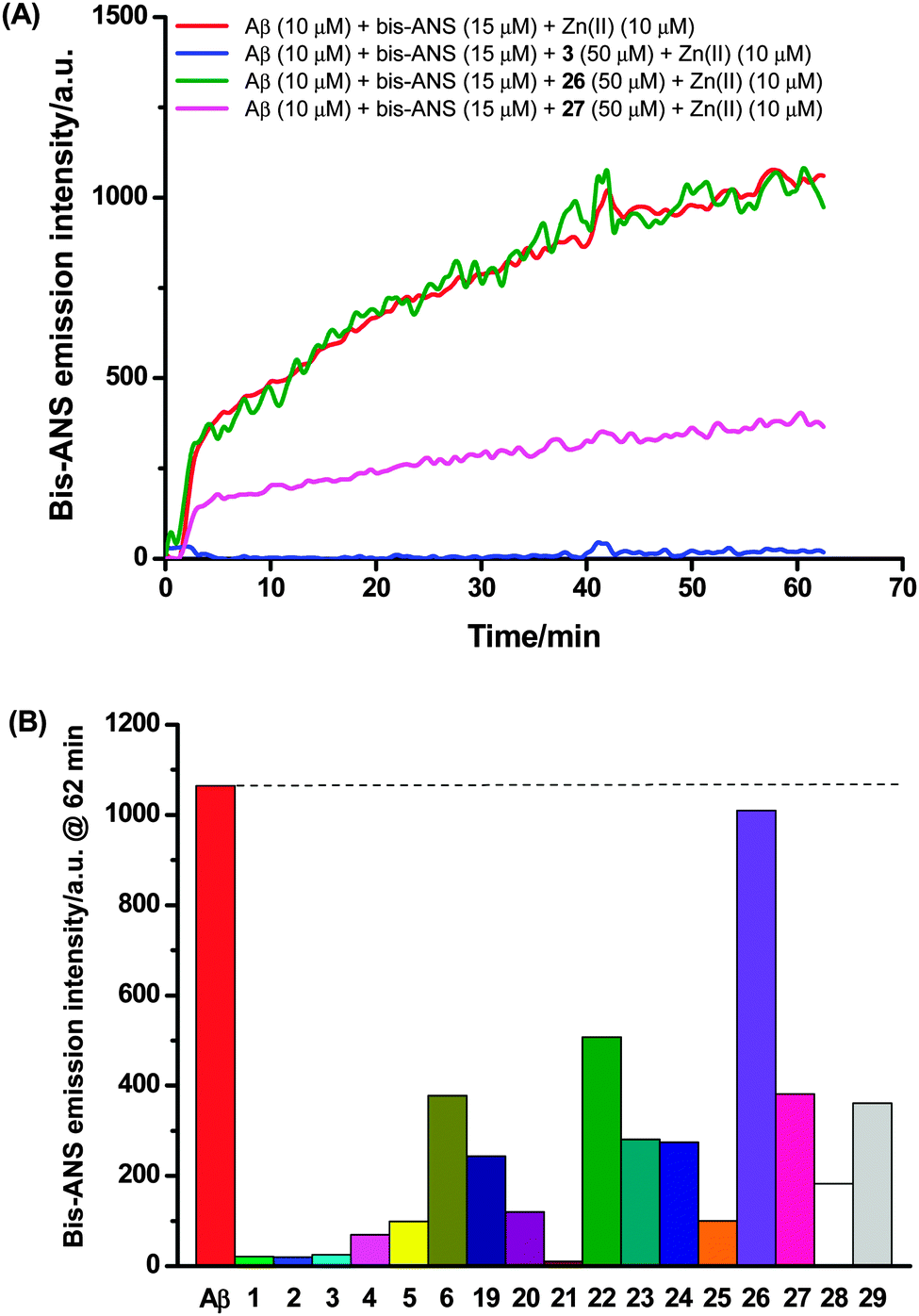 | ||
| Fig. 5 (A) Selected data from bis-ANS extrinsic fluorescence assay: incubation of compound 3, 26, or 27 (50 μM) with Aβ (10 μM), bis-ANS (15 μM) and zinc(II) (10 μM) completely quenched (blue), exerted little influence on (green), or reduced (pink) the bis-ANS fluorescence respectively with comparison to that obtained from an incubated solution of Aβ (10 μM), bis-ANS (15 μM) and zinc(II) (10 μM) alone (red); (B) summary of the effects of compounds 1–6 and 19–29 on the bis-ANS fluorescence intensity at the end of the assay. | ||
SH-SY5Y neuronal cells were incubated with Aβ (10 μM) and the test compounds (50 μM) for 4 days; neuronal viability was then assessed using the same fluorescence assay (Fig. 6A). It was found that (1) most of these compounds (1, 3, 5, 6, 20–24 and 27) rescued SH-SY5Y neuronal cells from Aβ-induced neurotoxicity; (2) three compounds (2, 4 and 26) exerted little protective effect on neurons; (3) four compounds marginally (19, 25) or clearly (28 and 29) exacerbated the total neurotoxicity. Incubation of SH-SY5Y neuronal cells with the test compounds (50 μM) in the absence of Aβ suggested that only compounds 19, 25 and 28 themselves were cytotoxic (Fig. 6B), which correlates well with the heightened neurotoxicity observed with these compounds in the Aβ neurotoxicity assay.
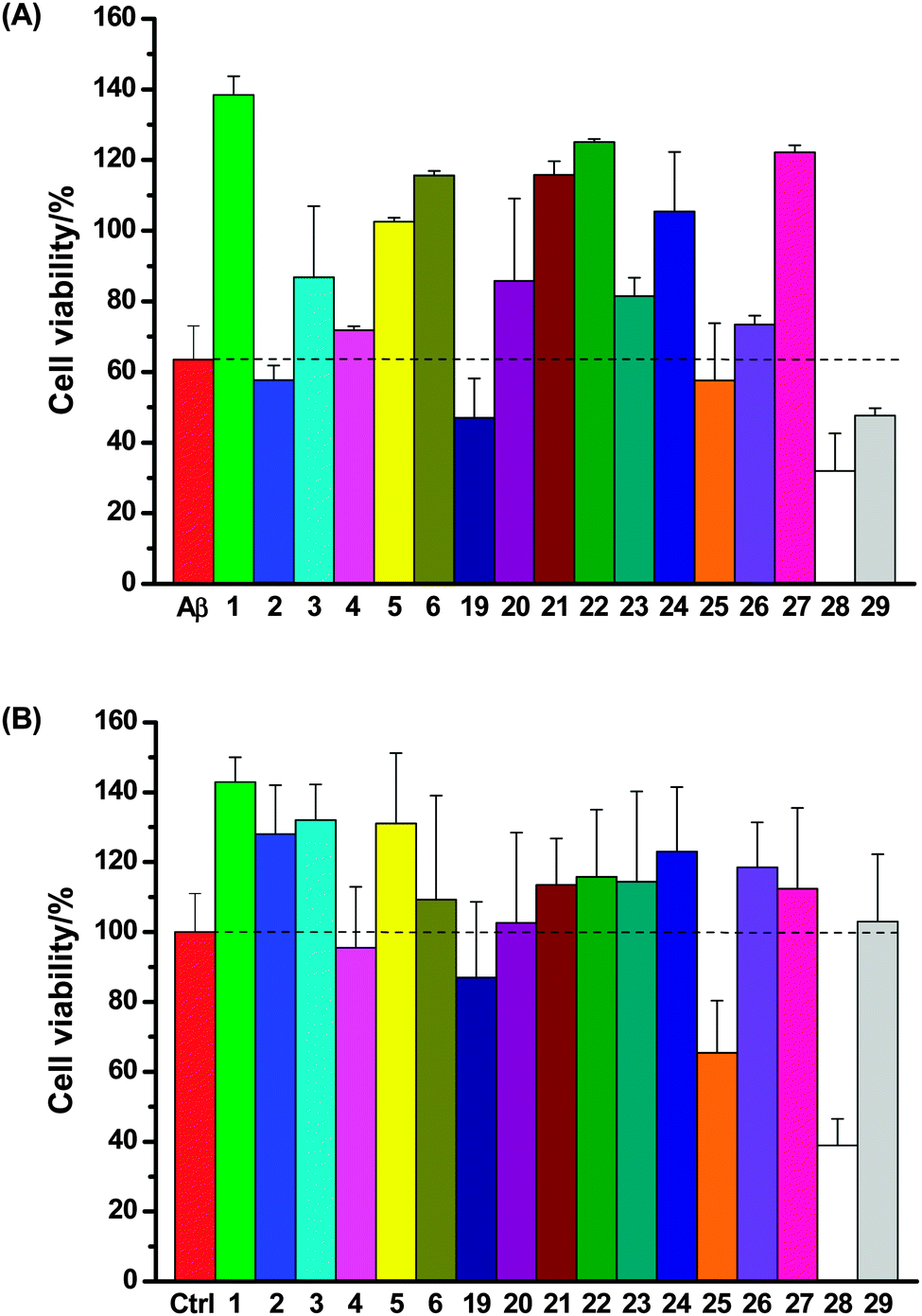 | ||
| Fig. 6 (A) Effects of compounds 1–6 and 19–29 on Aβ-mediated toxicity in neuronal cell culture; (B) cytotoxicity of compounds 1–6 and 19–29 in neuronal cell culture in the absence of Aβ (Ctrl = SH-SY5Y neuron cells only (no test compound was added)). | ||
Experimental
Synthesis and characterization of tetraazamcrocycle–(G)KLVFF hybrids and their metal complexes
See ESI† for complete experimental procedures and spectroscopic data.Aβ42 stock solution preparation
Aβ42 (a lyophilized powder, the W. M. Keck Foundation Biotechnology Resource Laboratory, Yale University) (1 mg) was pre-treated with ammonium hydroxide as described previously.30 The treated peptides were suspended in 60 mM NaOH (200 μL) and incubated for 5 min at room temperature. The resulting solution was diluted with Milli-Q water (700 μL) and sonicated at room temperature for a further 5 min in a water bath. The sonicated solution was neutralized with 10 × PBS (PBS is defined as 50 mM sodium phosphate, 2.7 mM KCl, 137 mM NaCl, pH 7.4, 100 μL) and centrifuged for 10 min at 14000 × g in a benchtop centrifuge. The optical density at 214 nm of the supernatant, containing the resolubilized Aβ42, was determined using a quartz microplate and a Flexstation 3 microplate reader (Molecular Devices) equipped with absorbance optics. The concentration was calculated from the 214 nm absorbance value using the molar extinction coefficient for Aβ42 of 94530 M−1 cm−1.34 Recovery of the peptide was typically 70–80%. The Aβ42 stock solution was immediately used for ThT extrinsic fluorescence assay, MALDI-TOF-MS, tyrosine intrinsic fluorescence assay, bis-ANS extrinsic fluorescence assay and neurotoxicity assay.
ThT extrinsic fluorescence assay
The effects of compounds 1–6 and 19–29 on the Aβ42 aggregation was evaluated using a continuous ThT fluorescence assay described previously.33,34 Compounds were dissolved in DMSO (Sigma Aldrich) to yield 1 mM stocks. These solutions were added into a 96-well microtitre plate (Wallac) containing ThT (Sigma Aldrich) and PBS (pH 7.4) to give final compound concentrations of 10 and 50 μM with a ThT level of 20 μM; Aβ42 was added to give a final concentration of 5 μM. The plate was sealed with acetate adhesive seals (MP Biomedicals) to minimize evaporative loss, and incubated at 37 °C for 23 h. During incubation, the plate was shaken every 7 min for 3 s prior to the measurement of the ThT fluorescence intensity (excitation at 444 nm and emission at 485 nm) using a Flexstation 3 microplate reader (Molecular Devices).Aβ42 pelleting assay
The results of the ThT assay were confirmed using a pelleting assay. Samples were prepared and treated as described for the ThT assay, and incubated at 37 °C for 24 h with shaking every 7 min for 3 s using an orbital plate shaker (350 opm). 100 μL of the sample was centrifuged at 100000 × g (Beckman Coulter, TL-100 benchtop ultracentrifuge), and the supernatant, pellet and starting material were assayed for protein concentration using a microBCA assay (Pierce) according to the manufacturer's instructions. The results were confirmed using the Direct Detect protein quantitation instrument from Millipore.
MALDI-TOF-MS
The proteolytic effects of compounds 1–6 and 19–29 on Aβ42 were investigated using MALDI-TOF-MS as described previously.19 The test compound (50 μM) was incubated with or without Aβ42 (10 μM) in PBS buffer (pH 7.4) for 4 and 7 days at 37 °C under sterile conditions. Each sample was desalted by a reverse phase C18 Zip-tip, and the resulting solution (1 μL) was mixed 1:1 with matrix solution (10 mg mL−1 α-cyano-4-hydroxycinnamic acid in a mixture of acetonitrile/water/TFA (50:49.9:0.1)) and spotted onto a ground steel target (MTP 384, Bruker Daltonics). Spotted samples were analyzed using a Bruker Daltonics Ultraflex III MALDI-TOF in reflector mode, with a detection range of 900–5000 Da, using appropriate peptide calibrants (Bruker Daltonics). Collected data were baseline corrected and smoothed using the Flexanalysis software module (Bruker Daltonics).
Tyrosine intrinsic fluorescence assay
Tyrosine intrinsic fluorescence assay19,40,42 was employed to investigate the effects of compounds 1–6 and 19–29 on the interaction between Aβ42 and copper(II). The tyrosine fluorescence spectra (excitation at 275 nm and emission at 290–305 nm) of Aβ42 (10 μM) in Tris buffer (50 mM, pH 7.5) in a 96-well microtitre plate (Wallac) were acquired using a Flexstation 3 microplate reader (Molecular Devices) immediately after dissolution. After incubation for 1 h at 25 °C, fluorescence was measured again. Copper(II) chloride was added to give a final concentration of 16 μM, and the fluorescence spectra of the resulting mixtures were acquired immediately. After incubation for 1 h at 25 °C, fluorescence was measured again. The test compounds (1 mM stocks in DMSO) were added to give a final concentration of 40 μM, and the fluorescence spectra of the resulting mixtures were acquired immediately. After incubation for 1 h at 25 °C, fluorescence was measured again.Bis-ANS extrinsic fluorescence assay
The ability of compounds 1–6 and 19–29 to interfere with zinc(II) related enhancement of Aβ self-association was analyzed using the dye 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (bis-ANS). The assay was conducted by adding bis-ANS, Aβ42 and compound to a microtiter plate containing 1 × PBS (200 μL) to final concentrations of 15, 10 and 50 μM respectively at 25 °C. Upon mixing, the fluorescence emission was recorded at 485 nm with excitation at 390 nm using a Flexstation 3 Plate reader (Molecular Devices, Sunnyvale, California) at 1 minute intervals. After 5 minutes, zinc(II) was added to a final concentration of 10 μM, and the fluorescence intensity was monitored for a further hour. All data were corrected for the intensity of changes of samples containing no Aβ42. Data reduction was done by taking the final point of the 60 minute time course.Neurotoxicity assay
Neuronal cell line SH-SY5Y was cultured in RPMI (Invitrogen) supplemented with 10% heat inactivated FBS (Invitrogen), HEPES (25 mM; GIBCO), L-glutamine (100 mM; GIBCO) and β-mercaptoethanol. Cells were seeded at 1 × 105 cells per well of a 96-well microtitre plate (Falcon, BD Biosciences) in 100 μL of culture media and allowed to adhere overnight under standard cell culture conditions (37 °C, 5% CO2 and 95% humidity). Compounds 1–6 and 19–29 at a final concentration of 50 μM in 0.1% DMSO (Sigma-Aldrich) were combined with Aβ42 at final concentrations of 1, 5 and 10 μM in triplicate in appropriate wells. Cells, media and Aβ42 alone were used as controls. The plates were incubated for 96 h (37 °C, 5% CO2 and 95% humidity). Resazurin (Sigma-Aldrich) was added at a final concentration of 0.05% (w/v) per well and incubated for 4 h before a measurement of fluorescent intensity was recorded on a FLUOstar OMEGA at 540/595 nm. Addition of compounds incorporating zinc(II) or copper(II) directly to resazurin did not alter the output fluorescence after 4 days incubation at 37 °C in 5% CO2. The percentage cell viability was calculated in relation to the maximum and minimum measurement of fluorescence caused by cells only (100% survival) or media alone (100% inhibition). All experiments were performed in duplicate, n = 2.Conclusions
Tetraazamacrocycle–(G)KLVFF hybrids 1–6 were prepared efficiently in good overall yields using a solid-phase based synthetic procedure. Complexation of these six conjugates with zinc(II) and copper(II) salts proceeded smoothly to give the corresponding metal complexes 19–26 in good to excellent yields. The biological activities of these tetraazamacrocycle–(G)KLVFF hybrids and their metal complexes were evaluated in vitro using several assays. Both the extrinsic fluorescence assays using ThT and bis-ANS, and the tyrosine intrinsic fluorescence assay imply complex, multifaceted structure–activity relationships in the interaction of these conjugates and Aβ. None of the compounds tested degrade Aβ into fragments. However most of these compounds protect neurons from Aβ toxicity.The previously reported ability of compound 1 to capture copper(II) ions and become proteolytically active could not be replicated in the present study. The differences might be explained by the different sources of Aβ or their slightly different methods of preparation. In the previous report, the Aβ was prepared in HFIP, but this was not used here as it has been found that HFIP can increase Aβ aggregation on its own.30 While it is known that Aβ prepared with alternative pretreatments to HFIP, such as ammonium hydroxide (NH4OH), differs in its aggregation speed, NH4OH pretreatment was used in the present work since our previous work shows that this treatment results in an almost monomeric starting solution.30 The methods of preparation of the metal complexes also differed: in the present work the metal complexes were isolated and purified, whereas in the previous work the complexes were prepared in situ with a slight excess of the ligand over the metal (1.2:1). The association constants for ligands/metal ions of this general class are high,48–51 meaning one would expect a vanishingly small free metal ion concentration from a 1:1 mixture of metal and ligand, but the slight excess of the free ligand used in the previous work could mean it is possible there was some free ligand present in the sample when it was biologically evaluated.
More generally for the overall set of compounds described in this paper, the results do not clearly articulate any consistent SARs and instead the data – particularly for the pre-formed metal complexes – suggest that interactions between compounds of this class and Aβ are complex. This is not unexpected given the a priori complexity of any binding interaction and what has been learned from the recent use of simple non-metallated peptides in the inhibition of amyloid formation.52–58 The results from the tyrosine intrinsic fluorescence assay in particular should caution that the changes in optical output observed using this assay may be caused by more complex changes than simple metal sequestration (which certainly should be more likely with the free ligands tested than the pre-formed metal complexes, in contrast with the outcomes discussed above). Further work is needed to verify whether other peptide- or chelator–Aβ interactions can give rise to the observed fluorescence quenching. Despite the lack of clear patterns and SARs from the fluorescence and proteolytic assays with isolated protein, many of the compounds screened demonstrated an impressive ability to rescue cells from Aβ-induced cytotoxicity. The attendant simplicity and ready diversification of their synthesis makes this a promising class of compounds for further investigation.
Acknowledgements
We thank the National Breast Cancer Fund for a Novel Concept Grant (NC-10-69) and the Australian Research Council for funding (DP120104035). M. Yu was supported by a University of Sydney International Scholarship (USydIS). T. M. Ryan is on a fellowship with the Australia Alzheimer's Dementia Research Foundation (AADRF).References
- P. A. Adlard and A. I. Bush, J. Alzheimer's Dis., 2006, 10, 145–163 Search PubMed.
- A. Bush and R. Tanzi, Neurotherapeutics, 2008, 5, 421–432 CrossRef CAS PubMed.
- M. D. Hurd, P. Martorell, A. Delavande, K. J. Mullen and K. M. Langa, N. Engl. J. Med., 2013, 368, 1326–1334 CrossRef CAS PubMed.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed.
- A. D. Korczyn, Alzheimer's Dementia, 2008, 4, 176–178 CrossRef CAS PubMed.
- C. Reitz, Int. J. Alzheimer's Dis., 2012, 369808, DOI:10.1155/2012/369808.
- R. A. Armstrong, Int. J. Alzheimer's Dis., 2011, 630865, DOI:10.4061/2011/630865.
- S. W. Pimplikar, Int. J. Biochem. Cell Biol., 2009, 41, 1261–1268 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CAS.
- P. Faller, ChemBioChem, 2009, 10, 2837–2845 CrossRef CAS PubMed.
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. d. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Science, 1994, 265, 1464–1467 CAS.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. E. Bacarra, D. M. Romano, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1998, 273, 12817–12826 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Inorg. Chem., 2004, 9, 954–960 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS PubMed.
- X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. A. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS PubMed.
- C. Hureau, I. Sasaki, E. Gras and P. Faller, ChemBioChem, 2010, 11, 950–953 CrossRef CAS PubMed.
- J. J. Braymer, A. S. DeToma, J. S. Choi, K. S. Ko and M. H. Lim, Int. J. Alzheimer's Dis., 2011, 623051, DOI:10.4061/2011/623051.
- W. Wu, P. Lei, Q. Liu, J. Hu, A. P. Gunn, M. Chen, Y. Rui, X. Su, Z. Xie, Y. Zhao, A. I. Bush and Y. Li, J. Biol. Chem., 2008, 283, 31657–31664 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- V. J. Thom, G. D. Hosken and R. D. Hancock, Inorg. Chem., 1985, 24, 3378–3381 CrossRef CAS.
- S. J. Paisey and P. J. Sadler, Chem. Commun., 2004, 306–307 RSC.
- M. M. Pallitto, J. Ghanta, P. Heinzelman, L. L. Kiessling and R. M. Murphy, Biochemistry, 1999, 38, 3570–3578 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS PubMed.
- M. Yu, J. R. Price, P. Jensen, C. J. Lovitt, T. Shelper, S. Duffy, L. C. Windus, V. M. Avery, P. J. Rutledge and M. H. Todd, Inorg. Chem., 2011, 50, 12823–12835 CrossRef CAS PubMed.
- M. Yu, Q. Yu, P. J. Rutledge and M. H. Todd, ChemBioChem, 2013, 14, 224–229 CrossRef CAS PubMed.
- M. Yu, N. H. Lim, S. Ellis, H. Nagase, J. A. Triccas, P. J. Rutledge and M. H. Todd, ChemistryOpen, 2013, 2, 99–105 CrossRef CAS PubMed.
- L. M. P. Lima, D. Esteban-Gómez, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2012, 51, 6916–6927 CrossRef CAS PubMed.
- H. Ozay, Y. Baran and Y. Ishii, Spectrochim. Acta, Part A, 2011, 83, 525–531 CrossRef CAS PubMed.
- T. M. Ryan, J. Caine, H. D. T. Mertens, N. Kirby, J. Nigro, K. Breheney, L. J. Waddington, V. A. Streltsov, C. Curtain, C. L. Masters and B. R. Roberts, PeerJ, 2013, 1, e73 Search PubMed.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS.
- T. M. Ryan, A. Friedhuber, M. Lind, G. J. Howlett, C. Masters and B. R. Roberts, J. Biol. Chem., 2012, 287, 16947–16954 CrossRef CAS PubMed.
- G. McColl, B. R. Roberts, A. P. Gunn, K. A. Perez, D. J. Tew, C. L. Masters, K. J. Barnham, R. A. Cherny and A. I. Bush, J. Biol. Chem., 2009, 284, 22697–22702 CrossRef CAS PubMed.
- S. A. Hudson, H. Ecroyd, T. W. Kee and J. A. Carver, FEBS J., 2009, 276, 5960–5972 CrossRef CAS PubMed.
- B. O'Nuallain, A. D. Williams, P. Westermark and R. Wetzel, J. Biol. Chem., 2004, 279, 17490–17499 CrossRef PubMed.
- C. J. Sarell, P. G. Stockley and S. E. Radford, Prion, 2013, 7, 359–368 CrossRef CAS PubMed.
- J. Suh, S. H. Yoo, M. G. Kim, K. Jeong, J. Y. Ahn, M. S. Kim, P. S. Chae, T. Y. Lee, J. Lee, J. Lee, Y. A. Jang and E. H. Ko, Angew. Chem., Int. Ed., 2007, 46, 7064–7067 CrossRef CAS PubMed.
- J. Suh, Acc. Chem. Res., 2003, 36, 562–570 CrossRef CAS PubMed.
- C. D. Syme, R. C. Nadal, S. E. J. Rigby and J. H. Viles, J. Biol. Chem., 2004, 279, 18169–18177 CrossRef CAS PubMed.
- W. Garzon-Rodriguez, A. K. Yatsimirsky and C. G. Glabe, Bioorg. Med. Chem. Lett., 1999, 9, 2243–2248 CrossRef CAS.
- Q. F. Ma, J. Hu, W. H. Wu, H. D. Liu, J. T. Du, Y. Fu, Y. W. Wu, P. Lei, Y. F. Zhao and Y. M. Li, Biopolymers, 2006, 83, 20–31 CrossRef CAS PubMed.
- V. Kadlcik, C. Sicard-Roselli, T. A. Mattioli, M. Kodicek and C. Houée-Levin, Free Radical Biol. Med., 2004, 37, 881–891 CrossRef CAS PubMed.
- W.-T. Chen, Y.-H. Liao, H.-M. Yu, I. H. Cheng and Y.-R. Chen, J. Biol. Chem., 2011, 286, 9646–9656 CrossRef CAS PubMed.
- W.-T. Chen, C.-J. Hong, Y.-T. Lin, W.-H. Chang, H.-T. Huang, J.-Y. Liao, Y.-J. Chang, Y.-F. Hsieh, C.-Y. Cheng, H.-C. Liu, Y.-R. Chen and I. H. Cheng, PLoS One, 2012, 7, e35807 CAS.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, J. Neurochem., 2012, 120, 149–166 CrossRef CAS PubMed.
- I. I. I. H. LeVine, Arch. Biochem. Biophys., 2002, 404, 106–115 CrossRef.
- Y. H. Lau, J. R. Price, M. H. Todd and P. J. Rutledge, Chem. – Eur. J., 2011, 17, 2850–2858 CrossRef CAS PubMed.
- E. Tamanini, A. Katewa, L. M. Sedger, M. H. Todd and M. Watkinson, Inorg. Chem., 2009, 48, 319–324 CrossRef CAS PubMed.
- E. Tamanini, K. Flavin, M. Motevalli, S. Piperno, L. A. Gheber, M. H. Todd and M. Watkinson, Inorg. Chem., 2010, 49, 3789–3800 CrossRef CAS PubMed.
- K. Jobe, C. H. Brennan, M. Motevalli, S. M. Goldup and M. Watkinson, Chem. Commun., 2011, 47, 6036–6038 RSC.
- M. A. Findeis, G. M. Musso, C. C. Arico-Muendel, H. W. Benjamin, A. M. Hundal, J.-J. Lee, J. Chin, M. Kelley, J. Wakefield, N. J. Hayward and S. M. Molineaux, Biochemistry, 1999, 38, 6791–6800 CrossRef CAS PubMed.
- T. L. Lowe, A. Strzelec, L. L. Kiessling and R. M. Murphy, Biochemistry, 2001, 40, 7882–7889 CrossRef CAS PubMed.
- S. M. Chafekar, H. Malda, M. Merkx, E. W. Meijer, D. Viertl, H. A. Lashuel, F. Baas and W. Scheper, ChemBioChem, 2007, 8, 1857–1864 CrossRef CAS PubMed.
- M. Convertino, A. Vitalis and A. Caflisch, J. Biol. Chem., 2011, 286, 41578–41588 CrossRef CAS PubMed.
- T. Härd and C. Lendel, J. Mol. Biol., 2012, 421, 441–465 CrossRef PubMed.
- S. A. Sievers, J. Karanicolas, H. W. Chang, A. Zhao, L. Jiang, O. Zirafi, J. T. Stevens, J. Munch, D. Baker and D. Eisenberg, Nature, 2011, 475, 96–100 CrossRef CAS PubMed.
- C. T. Middleton, P. Marek, P. Cao, C.-c. Chiu, S. Singh, A. M. Woys, J. J. de Pablo, D. P. Raleigh and M. T. Zanni, Nat. Chem., 2012, 4, 355–360 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mt00122b |
| This journal is © The Royal Society of Chemistry 2014 |