 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrophages: central regulators of iron balance
Elena
Gammella
,
Paolo
Buratti
,
Gaetano
Cairo
* and
Stefania
Recalcati
Department of Biomedical Sciences for Health, University of Milan, Via Mangiagalli 31, 20133 Milano, Italy. E-mail: gaetano.cairo@unimi.it
First published on 29th May 2014
Abstract
Macrophages are important to immune function and also actively participate in iron homeostasis. The involvement of splenic and liver macrophages in the processing of effete erythrocytes and the subsequent return of iron to the circulation is well established, and the molecular details of iron recycling have been characterized recently. Another important aspect regarding iron handling by macrophages is their capacity to act as immune cells, which involves the inflammatory response, as well as other pathological conditions in which macrophages are central. This review discusses the latest advances in macrophage iron trafficking and the pathophysiological consequences of altered iron homeostasis in these cells.
 Elena Gammella | Elena Gammella is a researcher at the Department of Biomedical Sciences for Health of the University of Milan, Italy. She pursued her PhD in Experimental Pathology from the University of Milan in 2009. Her research is focused on the interaction of oxygen homeostasis, erythropoiesis, and iron metabolism. |
 Paolo Buratti | Paolo Buratti majored in Biotechnology at the University of Milan, Italy in 2013. After joining Prof. Cairo's group in 2012, he worked on the molecular basis of the interaction between iron metabolism and erythropoiesis for his thesis, and is presently involved in the analysis of the molecular regulation of proteins of iron metabolism. |
 Gaetano Cairo | Gaetano Cairo is full professor of General Pathology at the University of Milan School of Medicine, Italy. He obtained a university degree in Biological Sciences from the University of Pavia and in Agricultural Sciences from the University of Milan. His long standing research interest focuses on the molecular control of genes of iron homeostasis under pathophysiological conditions. |
 Stefania Recalcati | Stefania Recalcati earned a MD in 1995 from the University of Milan. In 1999 she completed her training in Gastroenterology at the University of Milan, and was licensed as a gastroenterology specialist. In 2003 she received a PhD in Gastroenterology from the same university. She is presently Assistant Professor at the Department of Biomedical Sciences for Health of the University of Milan, Italy, where she is in charge of a project investigating macrophage iron metabolism in health and disease. |
Introduction
Macrophages are differentiated cells of the mononuclear phagocyte system; they have a variety of functions in distinct locations.1 The major role of macrophages is associated with the innate immune response, as macrophages are essential for immune surveillance and the induction of the inflammatory response. However, macrophages also fulfill important homeostatic functions, such as the clearance of apoptotic cells and cellular debris, and iron recycling.2 This review focuses on the latter function; we consider the most recent and relevant insights into the molecular aspects of iron trafficking, and also examine the pathophysiological implications of altered iron homeostasis in the various subsets of macrophages.Role of macrophages in systemic iron trafficking
In humans, daily intestinal iron absorption (∼2 mg) is sufficient to counterbalance physiological loss, but it is clearly inadequate to provide sufficient iron to meet the ten-fold higher requirement for hemoglobin synthesis and erythropoietic cell proliferation. Erythroid cells acquire almost exclusively transferrin-bound iron, which is replenished by the recycling activity of macrophages. Macrophages in the reticuloendothelial system process old and/or defective red blood cells and return hemoglobin-derived iron to the circulation3 (Fig. 1). These specialized phagocytes are resident macrophages localized in the liver (Kupffer cells) and spleen red pulp, which (at least in the spleen) require the Spi-C transcription factor for their development and to exert their iron recycling function.4 Moreover, it has been shown that heme-regulated eIF2 kinase, which regulates protein synthesis to ensure that globin chains are synthesized in appropriate amounts, is also expressed in macrophages and is required for their maturation and erythrophagocytic activity.5 Notably, the phagocytosis of all types of blood cells by macrophages can be stimulated by interferon-γ under inflammatory conditions such as hemophagocytic lymphohistiocytosis, which can lead to cytopenias as well as severe and acute anemia.6 Such stimulation represents an additional mechanism at the basis of anemia of inflammation, based on increased red cell destruction (in the absence of hemolysis and hemorrhage), rather than impaired production.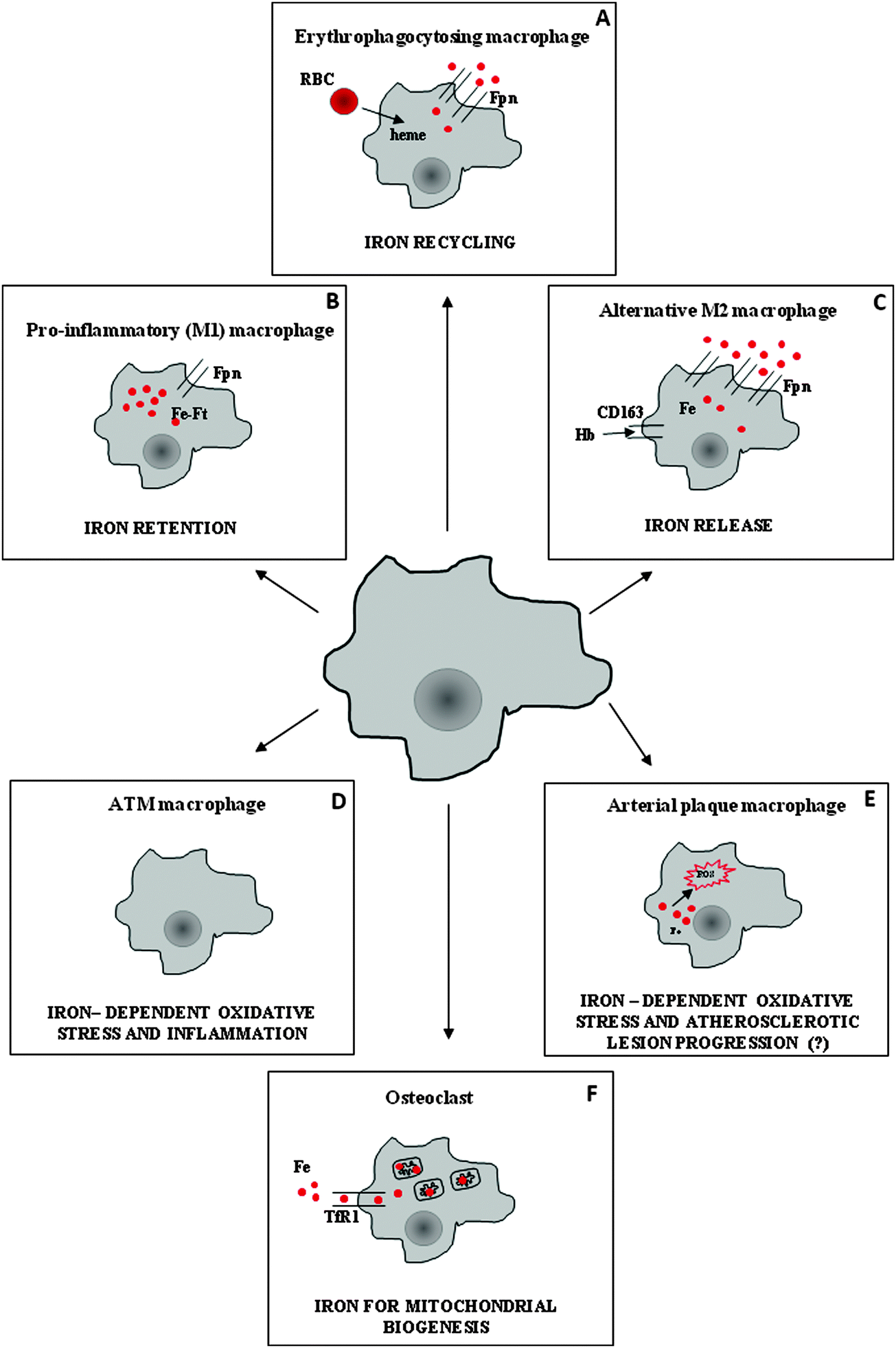 | ||
| Fig. 1 Iron handling in macrophage subsets performing distinct functions. (A) Clearance of senescent/damaged red blood cells (RBC) is the major function of macrophages involved in erythrophagocytosis in spleen red pulp and liver (Kupffer cells). These cells recycle iron derived from hemoglobin catabolism through the iron exporter ferroportin (Fpn). (B) During infection and inflammation, pro-inflammatory macrophages sequester iron into ferritin (Ft) to reduce iron availability to pathogens, at both the systemic and local levels. (C) A different population of macrophages (alternatively activated, M2), which is involved in inflammation resolution and tissue repair, actively internalizes heme iron through CD163, which is then exported by Fpn. (D) Obesity is associated with a switch of adipose tissue macrophages (ATM) from an M2-like phenotype to an M1-like phenotype; iron retention may enhance ROS production and pro-inflammatory activity. (E) High iron content in arterial plaque macrophages may lead to increased ROS production and favor the formation of foam cells and atherosclerotic plaque progression, but the role of iron in this setting is controversial. (F) Osteoclasts are multinucleated giant cells of the monocyte lineage that are responsible for bone resorption; for this activity they need high energy levels, and thus obtain a considerable amount of transferrin-bound iron for mitochondrial biogenesis. | ||
Ferroportin exports iron from macrophages (Fig. 2). To date, it has been the only known protein that transports ferrous iron from the cytoplasm across the plasma membrane (see Ward7 and Mitchell8 for a review). Transferrin, a circulating carrier that delivers iron for erythropoiesis and also satisfies the requirements of all other cells in which iron is used by proteins that perform essential functions, such as DNA synthesis (e.g., ribonucleotide reductase) or mitochondrial energy production (a number of proteins that contain heme groups and iron–sulfur clusters), binds only ferric iron efficiently (see Andrews9 for review). Therefore, iron exit from macrophages requires the ferroxidase activity of circulating ceruloplasmin. Iron recycling is negatively modulated by the interaction of ferroportin with hepcidin, a liver-derived peptide that induces the internalization and degradation of ferroportin, thus impairing its export activity (reviewed by Hentze10 and Pantopoulos11). Hepcidin also modulates intestinal iron absorption, and therefore it is the key regulator of systemic iron homeostasis: it is induced mainly by increased iron stores and inflammatory stimuli, and is decreased during conditions of high erythropoietic activity.12 Hepcidin levels are inappropriately low in iron-loaded hereditary hemochromatosis patients, which accounts for the observation that macrophages are paradoxically iron-deficient in individuals with body iron overload.13
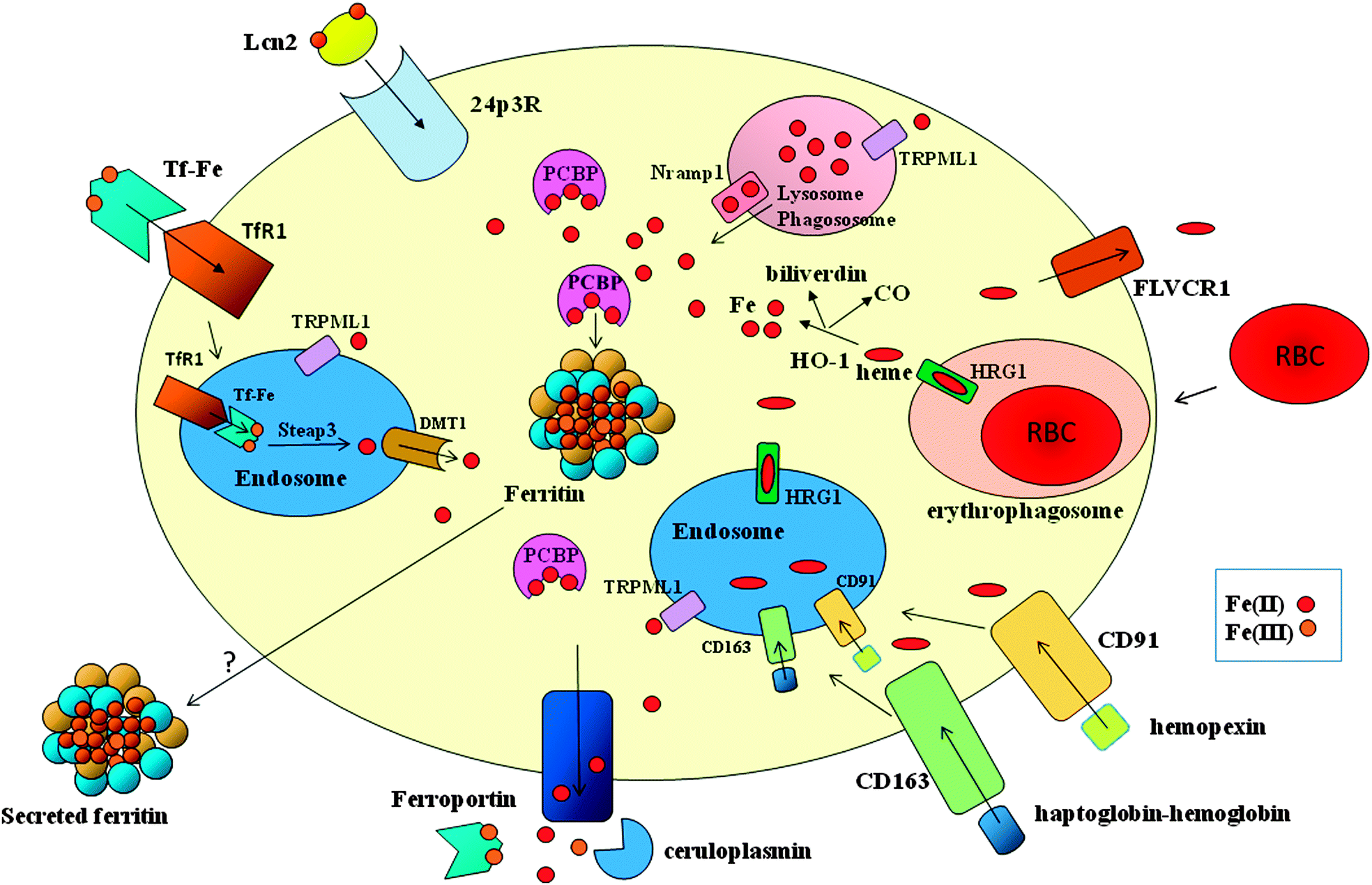 | ||
| Fig. 2 Major pathways of iron trafficking in macrophages. Macrophages acquire heme iron through ingestion of effete red blood cells and receptor-mediated internalization of proteins that bind extracellular free heme (interaction of hemopexin with CD91) or hemoglobin (interaction of haptoglobin with CD163). The heme moieties are degraded by heme oxygenase (HO-1) to release CO, biliverdin and iron, which is delivered by members of the PCBP family of chaperones to ferritin for storage. Part of heme can be exported through FLVCR1. Macrophages can also obtain iron by transferrin receptor (TfR1)-mediated endocytosis of diferric transferrin (Tf-Fe), followed by Steap3-dependent reduction and DMT1-mediated transport across the endosomal membrane. Iron can be transported across the endosomal (and lysosomal) membrane by means of the TRPML1 channel. Macrophages also express the lipocalin 2 (Lcn2) receptor 24p3R and can thus acquire siderophore-bound iron that has been complexed by Lcn2. Excess iron is mainly exported by ferroportin in ferrous form, and then oxidized by ceruloplasmin for efficient binding to transferrin, but secretion of iron-loaded ferritin may represent an alternative route. In macrophages, Nramp1 confers protection against intracellular microbes by exporting iron across the phagosomal membrane. | ||
Iron traffic inside macrophages
Macrophages in the red pulp of the spleen and Kupffer cells in the liver recognize alterations in the red cell surface that may include the modification of membrane determinants, such as proteins (e.g., Band 3), sialic acid, phosphatidylserine, or enhanced membrane rigidity that impairs deformability, and then capture and internalize red cells with a process similar to classic phagocytosis. Thus, the ingested erythrocytes are engulfed in phagolysosomes, in which oxidants and hydrolytic enzymes trigger the release of hemoglobin and then heme.14 Until recently, the successive step (the appearance of heme-derived iron in the cytosol) remained obscure because heme oxygenase 1 (HO-1), an inducible enzyme that cleaves heme to yield CO, iron, and biliverdin, is localized to the endoplasmic reticulum. The possibility that the endoplasmic reticulum contributes membrane constituents (and therefore also HO-1) to the formation of the phagocytic vacuole membrane was supported by observations that the iron transporters natural resistance associated macrophage protein (Nramp1) and divalent metal transporter 1 (DMT1) are recruited and localized at the erythrophagosomal membrane, where they promote efficient iron recycling.15 However, another report did not confirm the detection of DMT1 in the membrane of erythrophagosomes,16 and the hypothesis was in contrast with the topology of HO-1, whose catalytic domain faces the cytosol. This discrepancy has been resolved by a recent study that provided evidence showing how heme crosses the phagolysosomal membrane to be delivered to HO-1 for degradation. Heme-responsive gene 1 protein homolog (HRG1), which is highly expressed in macrophages and specifically localizes to the phagolysosomal membranes, transports heme from the phagolysosome and is essential for heme-iron recycling in macrophages17 (Fig. 2). Notably, lack of HO-1 caused the death of liver and spleen macrophages accompanied by abnormalities of iron homeostasis and kidney damage in mice;18 however, the phenotype was milder in the single human patient lacking HO-1.19 Excessive heme leads to tumor necrosis factor α-dependent and reactive oxygen species (ROS)-dependent programmed necrosis in macrophages, and cytotoxicity is exacerbated by the lack of HO-1.20 It has been demonstrated that feline leukemia virus, subgroup C, receptor (FLVCR) exports cytoplasmic heme from macrophages.21Cytosolic iron that is not used by phagocytic cells to synthesize proteins involved in vital functions can be stored in ferritin or exported by ferroportin (Fig. 2). The expression of both proteins is induced by heme and iron. Ferritin is a cytosolic iron storage protein composed of H and L subunits that are assembled in various proportions in different cells.22 Ferritin and ferroportin synthesis is controlled post-transcriptionally by the activity of iron regulatory proteins (IRPs), which regulate intracellular iron homeostasis by binding to conserved elements in target mRNAs.23 In the case of ferritin and ferroportin, IRP binding activity, which prevents mRNA translation, is decreased when iron levels are high. However, iron can also induce ferritin expression at the transcriptional level.24 Moreover, it has been demonstrated that the heme-activated transcription factor NRF2, which is key for HO-1 induction,25 is also involved in the increase of both ferritin H26 and ferroportin transcription.27 Therefore, the molecular basis of the partitioning between storage and release is presently not completely understood; however, this phenomenon may be relevant to account for the long-known biphasic nature of iron release from macrophages, which has been elegantly described in ferrokinetic experiments. In the initial phase, part of iron derived from hemoglobin promptly (within <1 h) returns to circulation, while the remaining iron is released over the course of days. Interestingly, the amount of iron released in these two distinct phases can be influenced by erythroid demand and is altered under pathological conditions.28
The fraction of iron stored in ferritin versus the fraction that returns to circulation is likely to depend on the still poorly understood mechanisms that control subcellular iron traffic, in which iron chaperon proteins (e.g., the poly(C) binding proteins) likely play a role. Although these proteins are important for the delivery of iron to ferritin and other iron-requiring proteins,29 their function has not been completely characterized.
It has been shown that iron can be transported across the endolysosomal membrane by the nonselective cation channel TRPML1,30 but its role in macrophages has not been characterized (Fig. 2).
Release by means of secreted ferritin is another possible fate that can be hypothesized for macrophage iron (Fig. 2). Although the source of serum ferritin remains unclear, recent studies suggest that macrophages contribute significantly to serum ferritin. The higher serum ferritin levels observed in mice with macrophage-specific ablation of IRP2 decreased dramatically after splenectomy;31 moreover, iron accumulation in spleen and liver macrophages and increased serum ferritin levels are present in mice with macrophage-specific ferroportin deletion.32 Thus, ferritin secretion might be an important mechanism of iron transport to circulation and/or within a tissue.
Because an average macrophage recycles one red cell per day, thus obtaining about 109 iron atoms, erythrophagocytosis represents the major route of iron uptake for macrophages; however, these cells can acquire iron by a number of different routes (Fig. 2). Macrophages express transferrin receptor 1 (TfR1), and therefore can internalize transferrin-bound iron, which is then reduced by Six-transmembrane epithelial antigen of the prostate 3 (Steap3) and transported in the cytoplasm by the divalent metal transporter 1 (DMT1), moreover, they acquire molecular iron via DMT1, and they also have other mechanisms to internalize heme iron by means of scavenger receptors. Haptoglobin binds cell-free hemoglobin (preventing the release of potentially toxic free heme), and hemopexin scavenges free heme. The macrophage transmembrane receptor CD163 binds the haptoglobin–hemoglobin complex, while hemopexin is recognized by low-density lipoprotein (LDL) receptor-related protein 1 (LRP1/CD9)33 (Fig. 2). Scavenger-mediated iron uptake can represent an important source of iron for macrophages under conditions of hemolysis or tissue damage. Since the storage and release of iron not obtained by erythrophagocytosis likely follows the same pathways described above, this issue has not been explored in depth.
Body iron metabolism under inflammatory conditions
As outlined at the beginning of this review, macrophages are components of innate immunity whose major function is host defense from pathogens. To this purpose, they possess a variety of strategies and biological weapons, including the capacity to modify the expression of proteins involved in iron metabolism in order to sequester iron, which represents a well known bacteriostatic mechanism (Fig. 1). In fact, microbes are strictly dependent on iron availability to grow, and thus are able to compete very efficiently for this essential metal.34 The importance of this issue has been emphasized recently by studies that link an increased risk of pathogen-dependent diseases to iron supplementation programs aimed at preventing iron-deficiency anemia.34 This competition for iron takes place at both the local and systemic levels. On one hand, given the large flux of iron that is processed daily during erythrophagocytosis, splenic and liver macrophages are primarily involved in the well-known hypoferremia that accompanies inflammatory conditions. However, as a side effect, iron accumulation in reticuloendothelial cells is one of the mechanisms that leads to the development of anemia associated with chronic inflammation (anemia of chronic disease, ACD).35 The molecular details of this response entail inflammatory stimulus-mediated regulation at the transcriptional and post-transcriptional levels of ferroportin (down-modulated) and ferritin (induced) in macrophages.36 However, the most important mechanism to reduce serum iron levels likely relies on the induction of hepcidin by pro-inflammatory cytokines like IL-6. Increased levels of circulating hepcidin block ferroportin-dependent iron release from macrophages, which in turn leads to hypoferremia and low transferrin saturation.3,9,10 Hepcidin induction and sustained hypoferremia require the Toll-like receptor 4 (TLR4)/MyD88 pathway.37 However, the response to inflammation is biphasic and the effect of hepcidin is preceded by an early phase (Myd88-independent) that mainly involves the direct suppression of ferroportin synthesis, which might be accompanied by the increased removal of circulatory iron through enhanced uptake via TfR1.38 The key role of elevated hepcidin levels in the pathogenesis of ACD has been demonstrated by studies showing that hepcidin overexpression in mice and humans is associated with iron retention in macrophages and hypoferremia, leading to functional iron deficiency and ACD.3,9,10,35 As a consequence, there has been great interest in developing hepcidin inhibitors as a new therapeutic strategy for treating ACD (reviewed in Sun39).Macrophage iron metabolism during infection
Iron sequestration by macrophages also occurs in the inflammatory microenvironment, where monocyte-derived macrophages recruited by inflammatory signals and activated resident macrophages face invading pathogens. In this context, the same machinery that operates to restrict systemic iron availability is at work; other specific mechanisms also act to sequester iron at the local level. Hepcidin is produced by macrophages at the site of infection by a lipopolysaccharide (LPS)-induced and TLR4-dependent pathway40,41 that also involves bone morphogenetic protein (BMP) signalling,42 and may represent an additional way to efficiently repress ferroportin-mediated iron export. On the other hand, recent studies shed light on the role of ferroportin in macrophages challenged by intracellular bacteria (e.g., Salmonella). In fact, ferroportin down-modulation with consequent intra-macrophage iron accumulation would spur intracellular pathogen growth.43 In agreement with this concept, hemochromatosis, which is characterized by hyperferremia and concomitant macrophage iron depletion, is associated with greater susceptibility to extracellular pathogens and resistance to intracellular pathogens.43 Therefore, it is notable that recent studies have shown that nitric oxide (NO) production stimulates Nrf2-dependent44 ferroportin expression and iron export in Salmonella-infected macrophages,45 possibly as a countermeasure to limit hepcidin-mediated ferroportin down-modulation. This finding represents a novel mechanism underlying the recognized antimicrobial role of NO. In the same vein, treatment with the calcium channel blocker nifedipine has been shown to induce ferroportin expression in the spleen, thereby mobilizing tissue iron and enhancing host resistance to intracellular pathogens.46 In this context, ferroportin polymorphisms identified in humans and resulting in impaired interaction with hepcidin can lead to decreased macrophage iron content, while iron accumulation is observed in individuals with alterations that lead to defective iron export. Evidence from in vitro experiments (and limited but interesting epidemiological studies) suggests that ferroportin polymorphisms can affect the host's response to intracellular and extracellular bacteria (reviewed in Kasvosve47).Pathogens utilize a variety of strategies to acquire iron from the environment despite its poor bioavailability; these mechanisms include extracellular iron mobilization, uptake and intracellular assimilation.48,49 Among the various strategies that a large number of bacteria employ to obtain iron, a significant role is played by the secretion of siderophores, small molecules that bind ferric iron from several sources with high affinity. Iron-siderophore complexes are then internalized, and specific and sophisticated iron release mechanisms are then used to make iron available for metabolic purposes.49
In this context, the relevant role of lipocalin2 (Lcn2, also named siderocalin, Neutrophil gelatinase-associated) is well known. Lcn2 is a protein that binds iron-loaded bacterial siderophores,50 and thus is essential in defending the host from extracellular pathogens.51 The Lcn2–siderophore complex can be transported across the cell-membrane by receptor-mediated endocytosis via the receptors 24p3R52 (Fig. 2). Recent studies have indicated that the protective action of Lcn2 is also important against intracellular pathogens. Experiments with macrophages obtained from Lcn2-deficient mice revealed that Lcn2 is required in order to limit the amount of iron available to Chlamydia pneumoniae and hence curb bacterial growth.53 Interestingly, in the absence of Lcn2, the host increases IL-10-dependent ferritin synthesis as an alternative protection mechanism.
Recent data suggest that the antimicrobial action of Nramp1, a cation transporter located in late phagolysosomes that confers resistance to infection with several intracellular pathogens,54 is caused by its recognized role in decreasing the availability of iron for intraphagosomal bacteria, and also results from interplay with Lcn2. In fact, Nramp1 increases Lcn2 expression through an NF-κB-mediated transcriptional mechanism that eventually confers protection against the intracellular bacterium S. typhimurium.55 A novel role, possibly not linked to its siderophore binding activity, was recently established for Lcn2 as a determinant of macrophage deactivation, a process that has been linked to the resolution phase of inflammation and is important for the outcome of bacterial infections. Lcn2, which is a marker of deactivated macrophages and is induced in patients with bacterial pneumonia, stimulated IL-10 production, thus impairing the immune response to Streptococcus pneumoniae.56 This role of Lcn2 apparently does not depend on its iron binding capacity, because S. pneumoniae does not use siderophores to acquire iron.
Metalloreductase Steap3, which is expressed at high levels in macrophages, is another protein that is important in regulating both iron homeostasis and TLR4-mediated inflammatory responses in macrophages (Fig. 2). Steap3 is a ferrireductase that transforms ferric iron derived from internalized transferrin to ferrous iron, which is the only form that can be transported to the cytoplasm.57 The down-regulation of Steap3 after LPS stimulation leads to iron accumulation in endosomes and lysosomes, accompanied by decreased cytosolic iron (a requisite for iron sequestration during inflammation). Furthermore, Steap3 appears to be important for the TLR4-mediated induction of inflammatory proteins.58
Iron metabolism and macrophage polarization
It is now increasingly recognized that macrophages can undergo classic pro-inflammatory (M1) or alternative (M2) polarization in response to various environmental and immune signals, and that these polarized cells have completely different biological properties.59 Classical activation (e.g., by microbial agents) is associated with the production of ROS and pro-inflammatory cytokines, and results in cytotoxicity and high microbicidal capacity; M2 polarisation is triggered by cytokines like IL-4 or IL-13,1,59 and results in cells that are important in the immune response to parasites and allergic stimuli. Moreover, M2 cells act in the regulation of adaptive immunity and in the control of cell growth and tissue remodelling during the resolution phase of the inflammatory process.Recent studies have shown that these two populations are also characterized by the divergent expression of the major proteins of iron homeostasis and very different iron handling.60,61 Contrary to the iron-sequestering activity of pro-inflammatory M1 cells, M2 macrophages express low levels of the iron storage protein ferritin and actively export iron via ferroportin (Fig. 1). This difference between M1 and M2 activity results in large differences in both intracellular and extracellular iron availability, which may in turn affect macrophage functions.36 The functional implications of this M2 macrophage activity regarding increasing iron availability in the microenvironment under specific pathophysiologic conditions have been recently discussed.48 The high capacity to internalize and detoxify heme iron (through CD163 and HO-1, respectively) is another characteristic of M2 macrophages62 that appears to be important under conditions characterized by bleeding and the accumulation of toxic heme iron.63
In addition to their distinct roles in the inflammatory response, differently polarized macrophage populations appear to have specific functions in other pathological settings, such as obesity and atherosclerosis.
Emerging role of iron in the functioning of particular types of macrophages
Atherosclerotic plaque macrophages
The macrophage is a key cell type in the formation and fate of atherosclerotic plaques, eventually evolving into a foam cell. A number of studies have recently elucidated the role of excess macrophage iron in atherosclerotic plaques (reviewed by Sullivan64) (Fig. 1). In fact, iron catalyzes ROS generation, and might therefore promote lipid oxidation and contribute to atherosclerotic plaque instability. Indeed, human atherosclerotic plaques contain approximately 10-fold more iron than healthy arteries, although probably with large variations in distribution.65Intraplaque hemorrhage, which promotes the progression and destabilization of atherosclerotic lesions, may be a source of iron. In this context, expression of CD16366 and HO-167 in macrophages exerts beneficial roles: CD163 internalizes potentially toxic heme from the microenvironment, which in turn stimulates cholesterol export; and HO-1 degrades the heme moiety. Recent reports have provided evidence in favor of the so-called “iron hypothesis”, which envisages a role for macrophage iron in the progression of atherosclerosis. In particular, a role has been suggested for hepcidin, which promotes plaque destabilization after erythrophagocytosis.68 Accordingly, reduction of macrophage iron content (obtained by inhibiting hepcidin, and thereby increasing ferroportin-mediated iron export) stimulated cholesterol efflux and prevented atherosclerosis in mice.69 Importantly, hepcidin levels and macrophage iron content correlated with vascular damage in monocytes of patients with a high risk of developing cardiovascular disease.70 Moreover, despite small divergences likely related to slightly different methodological procedures, a number of studies69,71,72 similarly reported that macrophages that exhibit an M2-like phenotype characterized by ferroportin-dependent iron release are atheroprotective (discussed in Hasty73). An interesting interplay between lipid and iron traffic in macrophages also emerges from these studies. Liver X receptors α71 and β,72 which are known to promote lipid efflux and decrease inflammation, induce both iron release and cholesterol export,69 possibly preventing lipid oxidation and foam cell formation; this mechanism may represent a novel atheroprotective strategy. In contrast to these reports, which suggest that iron accumulation in plaque macrophages is harmful, a recent work failed to demonstrate that hepcidin and macrophage iron act in atherosclerotic lesion formation,74 in agreement with a previous report showing a modest effect of iron chelation on atherosclerotic plaque development.75 These contrasting results may be related to differences in study design, such as the way of inducing iron overload (defective ferroportin-mediated iron export and/or parenteral iron injection vs. erythrophagocytosis associated with hemorrhage) and therefore the type of iron (non-heme vs. heme). In addition to modulating the properties of macrophages inside the atherosclerotic plaque, the finding that monocyte iron loading increases adhesion to the endothelium76 suggests that iron may also act during an earlier phase of atherosclerotic lesion progression.
Most tissues contain resident macrophages that are often involved in immune surveillance; however, their involvement in other functions is increasingly recognized.77 Recent studies have indicated that iron appears to be involved in the functions of at least two particular types of resident macrophages: adipose-tissue-associated macrophages (ATMs) and osteoclasts.
Osteoclasts
Osteoclasts, which derive from mononuclear precursors of the macrophage lineage, are multinucleate cells involved in bone resorption, and are centrally involved in skeletal growth and remodeling, as well as in calcium homeostasis.78 Recent work suggests that iron is important for bone formation, particularly for osteoclast differentiation and activation (Fig. 1). TfR1-mediated iron uptake79 and reduction of Fe3+ by the ferrireductase Steap family member 4 (ref. 80) are essential for osteoclastogenesis: iron is necessary for the biogenesis of mitochondria, which are required to sustain the high energy demand of osteoclasts. On one hand, iron serves for the synthesis of numerous heme and iron–sulfur-containing mitochondrial proteins; on the other hand, iron favors the production of nontoxic levels of ROS, which in turn act as signaling molecules to activate factors that stimulate mitochondrial biogenesis (e.g., CREB and PGC-1β). While iron is required to maintain correct bone remodeling, it is clear that, by leading to increased ROS production,81 iron excess may shift the balance between deposition and resorption in favor of the latter, thus explaining the well-known association between iron overload and osteoporosis.82 These studies provide new insights that correlate this bone defect that is typical of iron overload diseases with excessive osteoclastic activity, and open the way to possible therapeutic strategies based on iron chelators and/or antioxidants.Adipose tissue-associated macrophages
The contribution of the immune system to obesity has been increasingly appreciated. In obesity, adipocytes increase the recruitment of macrophages to adipose tissue (ATMs) with a concomitant shift in the activation state of macrophages from an anti-inflammatory M2 state towards an inflammatory M1 state. In turn, inflammatory M1 macrophages impair the function of adipose cells and trigger metabolic alterations, such as insulin resistance and liver pathology.83 The link between iron and energy homeostasis at the systemic level is also increasingly recognized (reviewed in Simcox84), as iron deficiency is associated with obesity, and iron overload increases diabetes risk by directly damaging pancreatic cells, as well as by increasing insulin resistance, possibly through adiponectin downregulation.85 Moreover, hepcidin was recently identified as a link between iron metabolism and glucose homeostasis,86 as well as nutrient status.87 At the cellular level, mammalian target of rapamycin (mTOR), a major sensor and controller of the metabolic state of the cell, has also been shown to regulate cellular iron homeostasis.88 In this context, a recent study analyzed iron metabolism in ATMs for the first time,89 and identified a population of iron-rich macrophages that also exhibit an M2-like expression profile (Fig. 1). Interestingly, the proportion of these cells with respect to iron-poor macrophages and their iron storage capacity were decreased in obese mice, concomitant with iron accumulation in adipocytes, which suggests complex iron trafficking among the cellular components of adipose tissue.Conclusion
It is becoming clear that macrophages are part of a continuum that spans a range of phenotypes and functions that are largely dictated by the microenvironment. This heterogeneity also concerns iron handling, which clearly differs among the various macrophage subpopulations. Defining the mechanisms of macrophage iron trafficking, which is a non-secondary function of these cells, will offer insights into the roles of distinct macrophage subtypes in a number of pathophysiological settings, and will also provide opportunities to explore possible therapeutic approaches by manipulating macrophage activity. For example, it will be possible to modulate ferroportin-mediated iron export from macrophages by targeting hepcidin with antagonists or agonists that have been characterized in pre-clinical models,39 or to use recent technologies90 to specifically deliver a macrophage-selective pharmacological effect.Acknowledgements
This work was supported by grants from Ministero dell'Istruzione, dell'Università e della Ricerca and Ministero della Salute.References
- F. O. Martinez, L. Helming and S. Gordon, Annu. Rev. Immunol., 2009, 27, 451–483 CrossRef CAS PubMed.
- T. Ganz, J. Innate Immun., 2012, 4, 446–453 CrossRef CAS PubMed.
- T. Ganz, Physiol. Rev., 2013, 93, 1721–1741 CrossRef CAS PubMed.
- M. Kohyama, W. Ise, B. T. Edelson, P. R. Wilker, K. Hildner, C. Mejia, W. A. Frazier, T. L. Murphy and K. M. Murphy, Nature, 2009, 457, 318–321 CrossRef CAS PubMed.
- S. Liu, R. N. Suragani, F. Wang, A. Han, W. Zhao, N. C. Andrews and J. J. Chen, J. Clin. Invest., 2007, 117, 3296–3305 CrossRef CAS PubMed.
- E. E. Zoller, J. E. Lykens, C. E. Terrell, J. Aliberti, A. H. Filipovich, P. M. Henson and M. B. Jordan, J. Exp. Med., 2011, 208, 1203–1214 CrossRef CAS PubMed.
- D. M. Ward and J. Kaplan, Biochim. Biophys. Acta, 2012, 1823, 1426–1433 CrossRef CAS PubMed.
- C. J. Mitchell, A. Shawki, T. Ganz, E. Nemeth and B. Mackenzie, Am. J. Physiol.: Cell. Physiol., 2014, 306, C450–C459 CrossRef CAS PubMed.
- N. C. Andrews, Blood, 2008, 112, 219–230 CrossRef CAS PubMed.
- M. W. Hentze, M. U. Muckenthaler, B. Galy and C. Camaschella, Cell, 2010, 142, 24–38 CrossRef CAS PubMed.
- K. Pantopoulos, S. K. Porwal, A. Tartakoff and L. Devireddy, Biochemistry, 2012, 51, 5705–5724 CrossRef CAS PubMed.
- C. Camaschella and A. Pagani, Int. J. Hematol., 2011, 93, 21–26 CrossRef CAS PubMed.
- G. Cairo, S. Recalcati, G. Montosi, E. Castrusini, D. Conte and A. Pietrangelo, Blood, 1997, 89, 2546–2553 CAS.
- A. D. Sheftel, A. B. Mason and P. Ponka, Biochim. Biophys. Acta, 2012, 1820, 161–187 CrossRef CAS PubMed.
- S. Soe-Lin, A. D. Sheftel, B. Wasyluk and P. Ponka, Exp. Hematol., 2008, 36, 929–937 CrossRef CAS PubMed.
- C. Delaby, C. Rondeau, C. Pouzet, A. Willemetz, N. Pilard, M. Desjardins and F. Canonne-Hergaux, PLoS One, 2012, 7, e42199 CAS.
- C. White, X. Yuan, P. J. Schmidt, E. Bresciani, T. K. Samuel, D. Campagna, C. Hall, K. Bishop, M. L. Calicchio, A. Lapierre, D. M. Ward, P. Liu, M. D. Fleming and I. Hamza, Cell Metab., 2013, 17, 261–270 CrossRef CAS PubMed.
- G. Kovtunovych, M. A. Eckhaus, M. C. Ghosh, H. Ollivierre-Wilson and T. A. Rouault, Blood, 2010, 116, 6054–6062 CrossRef CAS PubMed.
- A. Yachie, Y. Niida, T. Wada, N. Igarashi, H. Kaneda, T. Toma, K. Ohta, Y. Kasahara and S. Koizumi, J. Clin. Invest., 1999, 103, 129–135 CrossRef CAS PubMed.
- G. B. Fortes, L. S. Alves, R. de Oliveira, F. F. Dutra, D. Rodrigues, P. L. Fernandez, T. Souto-Padron, M. J. De Rosa, M. Kelliher, D. Golenbock, F. K. Chan and M. T. Bozza, Blood, 2012, 119, 2368–2375 CrossRef CAS PubMed.
- S. B. Keel, R. T. Doty, Z. Yang, J. G. Quigley, J. Chen, S. Knoblaugh, P. D. Kingsley, I. De Domenico, M. B. Vaughn, J. Kaplan, J. Palis and J. L. Abkowitz, Science, 2008, 319, 825–828 CrossRef CAS PubMed.
- P. Arosio, R. Ingrassia and P. Cavadini, Biochim. Biophys. Acta, 2009, 1790, 589–599 CrossRef CAS PubMed.
- S. Recalcati, G. Minotti and G. Cairo, Antioxid. Redox Signaling, 2010, 13, 1593–1616 CrossRef CAS PubMed.
- G. Cairo, L. Bardella, L. Schiaffonati, P. Arosio, S. Levi and A. Bernelli-Zazzera, Biochem. Biophys. Res. Commun., 1985, 133, 314–321 CrossRef CAS.
- R. Gozzelino and M. P. Soares, Antioxid. Redox Signaling, 2014, 20, 1754–1769 CrossRef CAS PubMed.
- E. C. Pietsch, J. Y. Chan, F. M. Torti and S. V. Torti, J. Biol. Chem., 2003, 278, 2361–2369 CrossRef CAS PubMed.
- C. Delaby, N. Pilard, H. Puy and F. Canonne-Hergaux, Biochem. J., 2008, 411, 123–131 CrossRef CAS PubMed.
- G. Fillet, Y. Beguin and L. Baldelli, Blood, 1989, 74, 844–851 CAS.
- C. C. Philpott, J. Biol. Chem., 2012, 287, 13518–13523 CrossRef CAS PubMed.
- X. P. Dong, X. Cheng, E. Mills, M. Delling, F. Wang, T. Kurz and H. Xu, Nature, 2008, 455, 992–996 CrossRef CAS PubMed.
- L. A. Cohen, L. Gutierrez, A. Weiss, Y. Leichtmann-Bardoogo, D. L. Zhang, D. R. Crooks, R. Sougrat, A. Morgenstern, B. Galy, M. W. Hentze, F. J. Lazaro, T. A. Rouault and E. G. Meyron-Holtz, Blood, 2010, 116, 1574–1584 CrossRef CAS PubMed.
- Z. Zhang, F. Zhang, P. An, X. Guo, Y. Shen, Y. Tao, Q. Wu, Y. Zhang, Y. Yu, B. Ning, G. Nie, M. D. Knutson, G. J. Anderson and F. Wang, Blood, 2011, 118, 1912–1922 CrossRef CAS PubMed.
- M. J. Nielsen, H. J. Møller and S. K. Moestrup, Antioxid. Redox Signaling, 2010, 12, 261–273 CrossRef CAS PubMed.
- H. Drakesmith and A. M. Prentice, Science, 2012, 338, 768–772 CrossRef CAS PubMed.
- G. Weiss and L. T. Goodnough, N. Engl. J. Med., 2005, 352, 1011–1023 CrossRef CAS PubMed.
- S. Recalcati, M. Locati, E. Gammella, P. Invernizzi and G. Cairo, Autoimmun. Rev., 2012, 11, 883–889 CrossRef CAS PubMed.
- A. Layoun, H. Huang, A. Calvé and M. M. Santos, Am. J. Pathol., 2012, 180, 2340–2350 CrossRef CAS PubMed.
- L. Tacchini, E. Gammella, C. De Ponti, S. Recalcati and G. Cairo, J. Biol. Chem., 2008, 283, 20674–20686 CrossRef CAS PubMed.
- C. C. Sun, V. Vaja, J. L. Babitt and H. Y. Lin, Am. J. Hematol., 2012, 87, 392–400 CrossRef CAS PubMed.
- X. B. Liu, N. B. Nguyen, K. D. Marquess, F. Yang and D. J. Haile, Blood Cells, Mol., Dis., 2005, 35, 47–56 CrossRef CAS PubMed.
- C. Peyssonnaux, A. S. Zinkernagel, V. Datta, X. Lauth, R. S. Johnson and V. Nizet, Blood, 2006, 107, 3727–3732 CrossRef CAS PubMed.
- X. Wu, L. M. Yung, W. H. Cheng, P. B. Yu, J. L. Babitt, H. Y. Lin and Y. Xia, PLoS One, 2012, 7, e44622 CAS.
- S. Recalcati, M. Locati and G. Cairo, Semin. Immunol., 2012, 24, 393–398 CrossRef CAS PubMed.
- N. Harada, M. Kanayama, A. Maruyama, A. Yoshida, K. Tazumi, T. Hosoya, J. Mimura, T. Toki, J. M. Maher, M. Yamamoto and K. Itoh, Arch. Biochem. Biophys., 2011, 508, 101–109 CrossRef CAS PubMed.
- M. Nairz, U. Schleicher, A. Schroll, T. Sonnweber, I. Theurl, S. Ludwiczek, H. Talasz, G. Brandacher, P. L. Moser, M. U. Muckenthaler, F. C. Fang, C. Bogdan and G. Weiss, J. Exp. Med., 2013, 210, 855–873 CrossRef CAS PubMed.
- S. M. Mair, M. Nairz, R. Bellmann-Weiler, T. Muehlbacher, A. Schroll, I. Theurl, P. L. Moser, H. Talasz, F. C. Fang and G. Weiss, J. Infect. Dis., 2011, 204, 685–694 CrossRef CAS PubMed.
- I. Kasvosve, Clin. Chim. Acta, 2013, 416, 20–25 CrossRef CAS PubMed.
- G. Cairo, S. Recalcati, A. Mantovani and M. Locati, Trends Immunol., 2011, 32, 241–247 CrossRef CAS PubMed.
- M. Miethke, Metallomics, 2013, 5, 15–28 RSC.
- S. Chakraborty, S. Kaur, S. Guha and S. K. Batra, Biochim. Biophys. Acta, 2012, 1826, 129–169 CAS.
- T. H. Flo, K. D. Smith, S. Sato, D. J. Rodriguez, M. A. Holmes, R. K. Strong, S. Akira and A. Aderem, Nature, 2004, 432, 917–921 CrossRef CAS PubMed.
- L. R. Devireddy, C. Gazin, X. Zhu and M. R. Green, Cell, 2005, 123, 1293–1305 CrossRef CAS PubMed.
- R. Bellmann-Weiler, A. Schroll, S. Engl, M. Nairz, H. Talasz, M. Seifert and G. Weiss, Immunobiology, 2013, 218, 969–978 CrossRef CAS PubMed.
- S. M. Vidal, D. Malo, J. F. Marquis and P. Gros, Annu. Rev. Immunol., 2008, 26, 81–132 CrossRef CAS PubMed.
- G. Fritsche, M. Nairz, S. J. Libby, F. C. Fang and G. Weiss, J. Leukocyte Biol., 2012, 92, 353–359 CrossRef CAS PubMed.
- J. M. Warszawska, R. Gawish, O. Sharif, S. Sigel, B. Doninger, K. Lakovits, I. Mesteri, M. Nairz, L. Boon, A. Spiel, V. Fuhrmann, B. Strobl, M. Müller, P. Schenk, G. Weiss and S. Knapp, J. Clin. Invest., 2013, 123, 3363–3372 CAS.
- R. S. Ohgami, D. R. Campagna, E. L. Greer, B. Antiochos, A. McDonald, J. Chen, J. J. Sharp, Y. Fujiwara, J. E. Barker and M. D. Fleming, Nat. Genet., 2005, 37, 1264–1269 CrossRef CAS PubMed.
- F. Zhang, Y. Tao, Z. Zhang, X. Guo, P. An, Y. Shen, Q. Wu, Y. Yu and F. Wang, Haematologica, 2012, 97, 1826–1835 CrossRef CAS PubMed.
- A. Sica and A. Mantovani, J. Clin. Invest., 2012, 122, 787–795 CAS.
- S. Recalcati, M. Locati, A. Marini, P. Santambrogio, F. Zaninotto, M. De Pizzol, L. Zammataro, D. Girelli and G. Cairo, Eur. J. Immunol., 2010, 40, 824–835 CrossRef CAS PubMed.
- G. Corna, L. Campana, E. Pignatti, A. Castiglioni, E. Tagliafico, L. Bosurgi, A. Campanella, S. Brunelli, A. A. Manfredi, P. Apostoli, L. Silvestri, C. Camaschella and P. Rovere-Querini, Haematologica, 2010, 95, 1814–1822 CrossRef CAS PubMed.
- F. Vallelian, C. A. Schaer, T. Kaempfer, P. Gehrig, E. Duerst, G. Schoedon and D. J. Schaer, Blood, 2010, 116, 5347–5356 CrossRef CAS PubMed.
- L. Nieuwenhuizen, R. E. Schutgens, K. Coeleveld, S. C. Mastbergen, G. Roosendaal, D. H. Biesma and F. P. Lafeber, Thromb. Res., 2014, 133, 390–395 CrossRef CAS PubMed.
- J. L. Sullivan, Biochim. Biophys. Acta, 2009, 1790, 718–723 CrossRef CAS PubMed.
- H. Gustafsson, M. Hallbeck, M. Norell, M. Lindgren, M. Engström, A. Rosén and H. Zachrisson, Magn. Reson. Med., 2014 DOI:10.1002/mrm.25267 [Epub ahead of print].
- C. A. Gleissner, I. Shaked, C. Erbel, D. Böckler, H. A. Katus and K. Ley, Circ. Res., 2010, 106, 203–211 CrossRef CAS PubMed.
- L. D. Orozco, M. H. Kapturczak, B. Barajas, X. Wang, M. M. Weinstein, J. Wong, J. Deshane, S. Bolisetty, Z. Shaposhnik, D. M. Shih, A. Agarwal, A. J. Lusis and J. A. Araujo, Circ. Res., 2007, 100, 1703–1711 CrossRef CAS PubMed.
- J. J. Li, X. Meng, H. P. Si, C. Zhang, H. X. Lv, Y. X. Zhao, J. M. Yang, M. Dong, K. Zhang, S. X. Liu, X. Q. Zhao, F. Gao, X. L. Liu, T. X. Cui and Y. Zhang, Arterioscler., Thromb., Vasc. Biol., 2012, 32, 1158–1166 CrossRef CAS PubMed.
- O. Saeed, F. Otsuka, R. Polavarapu, V. Karmali, D. Weiss, T. Davis, B. Rostad, K. Pachura, L. Adams, J. Elliott, W. R. Taylor, J. Narula, F. Kolodgie, R. Virmani, C. C. Hong and A. V. Finn, Arterioscler., Thromb., Vasc. Biol., 2012, 32, 299–307 CrossRef CAS PubMed.
- L. Valenti, P. Dongiovanni, B. M. Motta, D. W. Swinkels, P. Bonara, R. Rametta, L. Burdick, C. Frugoni, A. L. Fracanzani and S. Fargion, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 683–690 CrossRef CAS PubMed.
- G. Bories, S. Colin, J. Vanhoutte, B. Derudas, C. Copin, M. Fanchon, M. Daoudi, L. Belloy, S. Haulon, C. Zawadzki, B. Jude, B. Staels and G. Chinetti-Gbaguidi, Circ. Res., 2013, 113, 1196–1205 CrossRef CAS PubMed.
- J. J. Boyle, M. Johns, T. Kampfer, A. T. Nguyen, L. Game, D. J. Schaer, J. C. Mason and D. O. Haskard, Circ. Res., 2012, 110, 20–33 CrossRef CAS PubMed.
- A. H. Hasty and L. Yvan-Charvet, Circ. Res., 2013, 113, 1182–1185 CrossRef CAS PubMed.
- L. Kautz, V. Gabayan, X. Wang, J. Wu, J. Onwuzurike, G. Jung, B. Qiao, A. J. Lusis, T. Ganz and E. Nemeth, Cell Rep., 2013, 5, 1436–1442 CrossRef CAS PubMed.
- W. J. Zhang, H. Wei and B. Frei, Exp. Biol. Med., 2010, 235, 633–641 CrossRef CAS PubMed.
- A. E. Kartikasari, F. L. Visseren, J. J. Marx, S. van Mullekom, J. H. Kats-Renaud, B. S. Asbeck, L. H. Ulfman and N. A. Georgiou, Atherosclerosis, 2009, 205, 369–375 CrossRef CAS PubMed.
- L. C. Davies, S. J. Jenkins, J. E. Allen and P. R. Taylor, Nat. Immunol., 2013, 14, 986–995 CrossRef CAS PubMed.
- J. R. Edwards and G. R. Mundy, Nat. Rev. Rheumatol., 2011, 7, 235–243 CrossRef CAS PubMed.
- K. A. Ishii, T. Fumoto, K. Iwai, S. Takeshita, M. Ito, N. Shimohata, H. Aburatani, S. Taketani, C. J. Lelliott, A. Vidal-Puig and K. Ikeda, Nat. Med., 2009, 15, 259–266 CrossRef CAS PubMed.
- J. Zhou, S. Ye, T. Fujiwara, S. C. Manolagas and H. Zhao, J. Biol. Chem., 2013, 288, 30064–30074 CrossRef CAS PubMed.
- J. Tsay, Z. Yang, F. P. Ross, S. Cunningham-Rundles, H. Lin, R. Coleman, P. Mayer-Kuckuk, S. B. Doty, R. W. Grady, P. J. Giardina, A. L. Boskey and M. G. Vogiatzi, Blood, 2010, 116, 2582–2589 CrossRef CAS PubMed.
- G. F. Li, Y. Z. Pan, P. Sirois, K. Li and Y. J. Xu, Osteoporosis Int., 2012, 23, 2403–2408 CrossRef CAS PubMed.
- K. Sun, C. M. Kusminski and P. E. Scherer, J. Clin. Invest., 2011, 121, 2094–2101 CrossRef CAS PubMed.
- J. A. Simcox and D. A. McClain, Cell Metab., 2013, 17, 329–341 CrossRef CAS PubMed.
- J. S. Gabrielsen, Y. Gao, J. A. Simcox, J. Huang, D. Thorup, D. Jones, R. C. Cooksey, D. Gabrielsen, T. D. Adams, S. C. Hunt, P. N. Hopkins, W. T. Cefalu and D. A. McClain, J. Clin. Invest., 2012, 122, 3529–3540 CAS.
- C. Vecchi, G. Montosi, C. Garuti, E. Corradini, M. Sabelli, S. Canali and A. Pietrangelo, Gastroenterology, 2014, 146, 1060–1069 CrossRef CAS PubMed.
- K. Mleczko-Sanecka, F. Roche, A. R. da Silva, D. Call, F. D'Alessio, A. Ragab, P. E. Lapinski, R. Ummanni, U. Korf, C. Oakes, G. Damm, L. A. D'Alessandro, U. Klingmüller, P. D. King, M. Boutros, M. W. Hentze and M. U. Muckenthaler, Blood, 2014, 123, 1574–1585 CAS.
- M. Bayeva, A. Khechaduri, S. Puig, H. C. Chang, S. Patial, P. J. Blackshear and H. Ardehali, Cell Metab., 2012, 16, 645–657 CrossRef CAS PubMed.
- J. S. Orr, A. Kennedy, E. K. Anderson-Baucum, C. D. Webb, S. C. Fordahl, K. M. Erikson, Y. Zhang, A. Etzerodt, S. K. Moestrup and A. H. Hasty, Diabetes, 2014, 63, 421–432 CrossRef CAS PubMed.
- L. A. Needham, A. H. Davidson, L. J. Bawden, A. Belfield, E. A. Bone, D. H. Brotherton, S. Bryant, M. H. Charlton, V. L. Clark, S. J. Davies, A. Donald, F. A. Day, D. Krige, V. Legris, J. McDermott, Y. McGovern, J. Owen, S. R. Patel, S. Pintat, R. J. Testar, G. M. Wells, D. Moffat and A. H. Drummond, J. Pharmacol. Exp. Ther., 2011, 339, 132–142 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |