 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc ions modulate protein tyrosine phosphatase 1B activity†
Elisa
Bellomo
*a,
Alberto
Massarotti
b,
Christer
Hogstrand
a and
Wolfgang
Maret
a
aKing's College London, Metal Metabolism Group, Division of Diabetes and Nutritional Sciences, School of Medicine, London, SE1 9NH, UK. E-mail: elisa.bellomo@kcl.ac.uk
bDipartimento di Scienze del Farmaco, Universitá degli Studi del Piemonte Orientale “A. Avogadro”, Largo Donegani, 2, 28100 Novara, Italy
First published on 25th April 2014
Abstract
Protein tyrosine phosphatases (PTPs) are key enzymes in cellular regulation. The 107 human PTPs are regulated by redox signalling, phosphorylation, dimerisation, and proteolysis. Recent findings of very strong inhibition of some PTPs by zinc ions at concentrations relevant in a cellular environment suggest yet another mechanism of regulation. One of the most extensively investigated PTPs is PTP1B (PTPN1). It regulates the insulin and leptin signalling pathway and is implicated in cancer and obesity/diabetes. The development of novel assay conditions to investigate zinc inhibition of PTP1B provides estimates of about 5.6 nM affinity for inhibitory zinc(II) ions. Analysis of three PTP1B 3D structures (PDB id: 2CM2, 3I80 and 1A5Y) identified putative zinc binding sites and supports the kinetic studies in suggesting an inhibitory zinc only in the closed and cysteinyl–phosphate intermediate forms of the enzyme. These observations gain significance with regard to recent findings of regulatory roles of zinc ions released from the endoplasmic reticulum.
Introduction
Protein tyrosine phosphatases (PTPs) do not simply counteract protein tyrosine kinases but have additional roles in regulating protein phosphorylation. The human genome contains over 100 PTPs that belong to the receptor-like and the non-transmembrane classes.1,2 They have been investigated in relation to their roles in normal physiology but also with regard to cancer, autoimmune and metabolic diseases, insulin resistance, obesity, and diabetes.3Among these enzymes, PTP1B (PTPN1) has attracted a lot of interest.‡ It is ubiquitously expressed and contains a hydrophobic domain at the C-terminus that targets it to the endoplasmic reticulum.4
PTP1B functions as a negative regulator of both the insulin and leptin receptor signalling pathways.5,6 The phenotype of PTP1B null mice confirmed the importance of this protein for metabolism.7 The mice are more insulin sensitive as they presented lower glucose levels with reduced insulinaemia when compared to littermate controls (WT); when exposed to a high fat diet, both PTP1B KO and heterozygotes were resistant to weight gain and insulin-resistance when compared to WT mice. Moreover, livers and skeletal muscles of KO mice had increased insulin receptor phosphorylation compared to WT.7 Leptin is also an important regulator of energy metabolism, and is directly linked to obesity.8 PTP1B KO mice revealed a role for PTP1B in the regulation of leptin signalling by dephosphorylating the down-stream target Janus Kinase 2 (JAK2),9 a known PTP1B substrate;10 mice deficient in both leptin (ob/ob mice) and PTP1B gained less weight, had less adipose tissue and an increased metabolic rate compared to controls.6
Since phosphatases oppose the actions of some oncogenic tyrosine kinases, they can also be tumour suppressors. This property was demonstrated for PTP1B. When overexpressed in NIH/3T3 fibroblasts, PTP1B protects the cells from transformation.11 More recent work has linked PTP1B to cancer. Investigations on breast cancer reported overexpression of PTP1B in >70% of human tumour sections.12 Studies on whole-body and mammary epithelium-specific deletion of PTP1B in mice revealed delays against HER2/Neu-induced tumourigenesis.13 However, deletion of PTP1B in mice with established breast tumours did not affect tumour growth,14 indicating that modulation of PTP1B activity might be effective in cancer prevention but not necessarily treatment.15
Given their central role in cellular functions, it is essential that PTPs are regulated tightly. At the protein level, the regulation can be achieved by mechanisms that include redox modulation, phosphorylation, sumoylation and dimerisation.16–19 Zinc ions are recognised as inhibitors of PTPs. At a concentration of 10 μM, Brautigan et al. showed that they inhibit a protein tyrosine phosphatase completely.20 However, it was then shown that zinc binds much tighter with an IC50 of 200 nM for human T-cell PTP (PTPN2).21 IC50 values of 17 nM for human PTP1B,22 93 nM for SHP-1 (PTPN6)22 and a Ki value of 21 pM for receptor protein tyrosine phosphate beta (PTPRB)23 were also determined. These tight inhibition constants are within the range of free cytosolic zinc(II) ion concentrations occurring during signalling events in cells, thus suggesting zinc modulation of PTP activity in vivo.24,25 However, detailed kinetic studies of zinc inhibition of PTP1B have not been reported. Measurement of zinc inhibition is based on enzymatic assays that are usually performed above the Km for substrate to obtain maximum velocities. This means that if zinc and the substrate compete for the same site, there will be more competition at high substrate concentrations and hence higher measured Ki values.
In this investigation, we developed and optimised enzymatic assays to measure zinc inhibition of PTP1B. We also discuss the issues that must be addressed when working with zinc in enzymatic assays of PTPs; the new assay allowed us to gain further insights into the zinc inhibition of PTP1B, which turns out to have specific features related to the mechanism of this enzyme. Analysis of three dimensional structures of PTP1B indicate a specific zinc site in the closed and phosphorylated intermediate and additional zinc-binding sites.
Experimental procedures
Reagents
Molecular biology-grade HEPES, ethylenediaminetetraacetic acid (EDTA), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), nitrilotriacetic acid (NTA), zinc sulphate (ZnSO4) and Triton X-100 were from Sigma-Aldrich; 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) and 6,8-difluoro-7-hydroxy-4-methylcoumarin (DiFMU) were from Invitrogen. Recombinant human PTP1B, residues 1–299, was from Millipore, supplied in 50 mM Hepes, pH 7.2, 1 mM DTT, 1 mM EDTA, and 0.05% (v/v) NP-40.Enzymatic assay
PTP1B was assessed fluorometrically for enzymatic activity at 25 °C in a freshly prepared buffer containing 50 mM Hepes/Na+, pH 7.4, 0.1 mM TCEP, 1 mM NTA and 0.01% (v/v) Triton X-100. The enzyme was added to the buffer to a final concentration of 2.5 nM. The reaction was initiated by adding the fluorogenic phosphatase substrate DiFMUP. Assays were performed in triplicates in a total volume of 100 μl in 96-well black optical bottom plates (Greiner Bio-One Ltd, Stonehouse, UK). Product formation (hydrolysis of DiFMUP to DiFMU) was monitored by fluorescence at 460 nm with 360 nm excitation using a fluorescence plate reader (Synergy HT, BioTek, Winooski, VT). The initial rate was determined from the linear portion of the progress curves. Concentrations were calculated from a DiFMUP–DiFMU mixture standard. Michaelis–Menten parameters and turnover number were obtained by fitting the initial rates to the Michaelis–Menten equation using SigmaPlot software 12.5 (SYSTAT Software Inc.).Zinc-inhibition assay
Free zinc ion concentrations were calculated with Maxchelator.26 1 mM NTA in the above solution was used to buffer zinc. The zinc-containing buffers were prepared fresh and equilibrated for 15 minutes before the enzyme was added. For incubation experiments, the enzyme was pre-incubated with the zinc-containing solutions for seven minutes at 25 °C in a closed Eppendorf tube; the substrate was then added and the reaction monitored. For competition experiments, the substrate was added to the reaction buffer containing zinc and the reaction was started by adding PTP1B. In the latter case, the enzyme was left at 25 °C for seven minutes before addition to the buffer to compensate for the possible loss of enzymatic activity upon pre-incubation with zinc. Inhibition constants (apparent Ki values) were obtained using SigmaPlot.ICP-MS
Inductively coupled plasma mass spectrometry (ICP-MS, Perkin Elmer Life Science, model Elan 610 DRC plus) analysis of metals was performed on all components of the assay. Samples were prepared in 5% (v/v) HNO3-washed polypropylene tubes (Elkay, Basingstoke, UK).Protein quantification
The protein concentration was determined spectrophotometrically (Synergy HT plate reader) using the Bio-Rad protein assay reagent and bovine serum albumin standards, following a modified Bradford assay.27Molecular modelling
The representation of protein structures was generated using PyMOL software.28 Three different X-ray structures of PTP1B (PDB id: 2CM2, 3I80 and 1A5Y) were considered. First, small organic compounds, ions and water molecules were removed. Polar hydrogens were added to the protein complexes and the resulting polar hydrogens were optimised with the use of the MolProbability server.29The docking study was performed considering the entire protein and using the software PLANTS 1.2.30 It generated 100 possible binding poses for Zn2+. The output was curated using an in house Perl script to select which Zn2+ ions were located near the donors of at least two of the Cys, His, Glu or Asp residues.31,32 In order to verify the molecular docking procedure, the X-ray structure of carbonic anhydrase II (PDB id: 1CAM), containing one zinc atom in the catalytic site, was selected as a positive control. One of the five Zn2+ docking poses selected was located within 2.0 Å of the Zn2+ coordinates in the crystal structure (ESI,† Table S1 and Fig. S2).
Data analysis and statistics
Data were analysed with Microsoft Excel 2010 and SigmaPlot 12.5. Values are reported as mean of at least three separate experiments ± SD. Statistical significance was assessed by one-way or two-way analysis of variance using SigmaStat (SYSTAT Software Inc.).Results
Enzymatic activity of PTP1B
Several parameters in the conventional PTP assays are not conducive for determining metal inhibition. Above all, this concerns the pH and the sensitivity of the assay. DiFMUP was used as a fluorogenic substrate as it allows assaying PTB1B even at sub-nanomolar concentrations (Fig. 1A). PTP1B activity is higher at acidic pH (Fig. 1B), a trait common to all PTPs. The fact that we were able to measure only residual activity below pH 5.0 is in accordance with published data.33 Because protons may compete with Zn2+ at zinc-binding sites, enzyme inhibition should not be measured at acidic pH. Therefore, reactions were recorded at pH 7.4, where enzymatic activity is lower. It is known that proteins can absorb on plastic surfaces as well as form oligomeric structures. Such processes can result in apparent inhibition of catalytic activity. To avoid this issue, Triton X-100 was added to the assay buffer.34 Relatively small concentrations proved to be sufficient to stabilise the activity (Fig. 1C). Accordingly, a concentration of 0.01% Triton X-100 in the assay buffer was chosen. Under these optimised conditions, the Km of PTP1B for DiFMUP was 3.9 ± 0.6 μM and the VMAX 129.9 ± 7.8 pmol min−1 (Fig. 1D), which resulted in a kcat of 8.66 s−1. Since the protein concentration of the commercial PTP1B varies, its concentration was measured to calculate this turnover number. In the inhibition experiments, the substrate concentration was set at 3 μM, i.e. below the Km, as higher concentrations might result in incorrect Ki determination.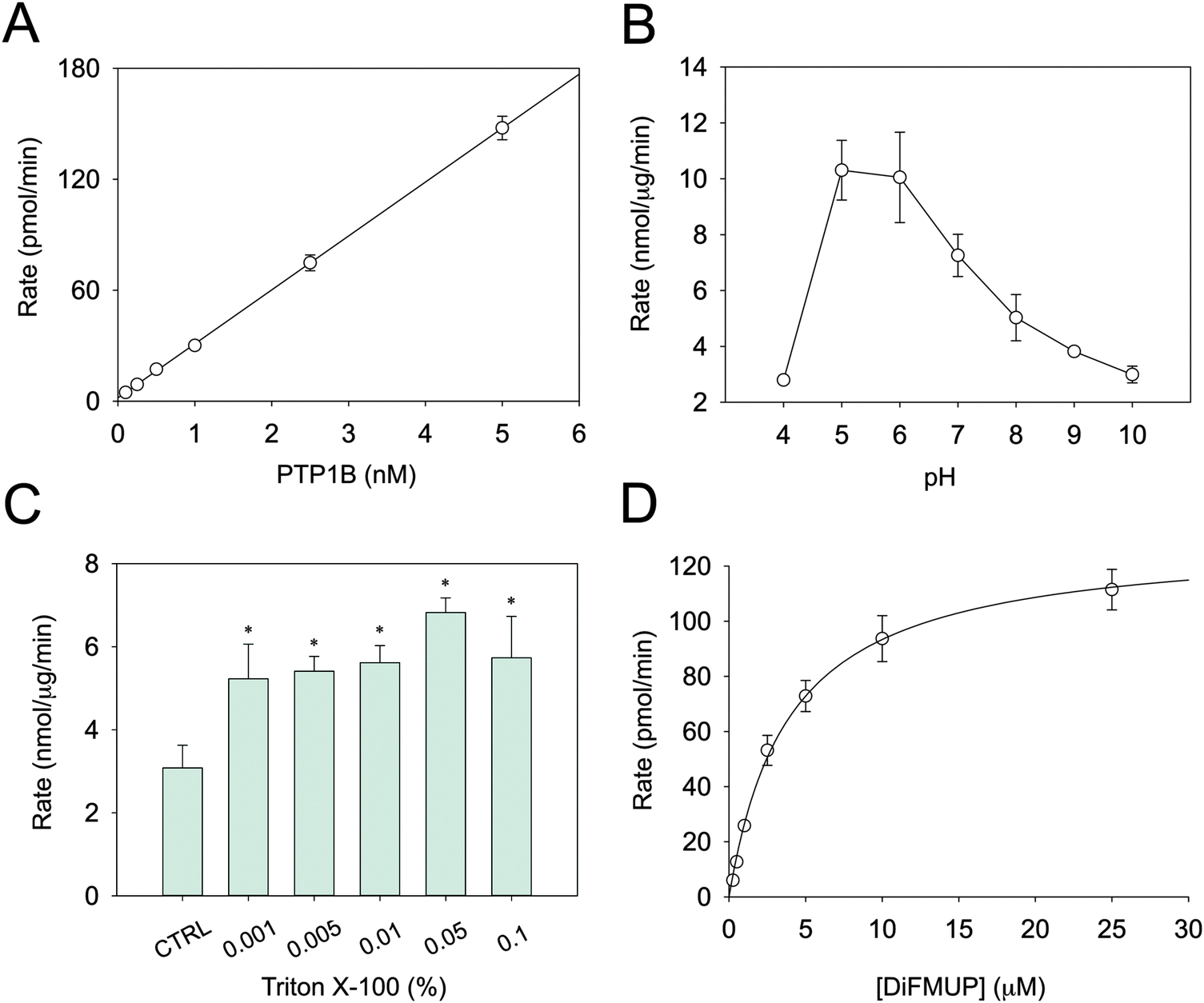 | ||
| Fig. 1 Optimisation of PTP1B enzymatic assay. (A) The rate of the reaction was measured at different enzyme concentrations in the presence of 10 μm DiFMUP. (B) PTP1B was assayed at different pH values and (C) Triton X-100 concentrations, using 2.5 nM PTP1B and 10 μM DiFMUP in 50 mM Hepes, 1 mM NTA and 0.1 mM TCEP. (D) In order to calculate Km, VMAX and kcat, enzymatic activity was measured in the presence of increasing concentrations of DiFMUP (0.25–25 μM), with 2.5 nM PTP1B in the above buffer at pH 7.4. *p < 0.001 compared to control. | ||
Determination of total and free Zn2+
A major issue when assessing PTP activity is the control of the concentration of metal ions. ICP-MS was used to measure the concentrations of Zn2+ in the solutions and buffers. All of them contained Zn2+ at various levels ranging from 0.1 to 3 μg L−1 (ppb), which correspond to nanomolar concentrations that are known to inhibit PTPs (Table 1).22,23,35 We used both Maxchelator26 and MINEQL+ 4.6 (Environmental Research Software, Hallowell, ME) to calculate the concentrations of free Zn2+ in the assay buffer. In the calculations, we also incorporated the total Zn2+ concentration measured with ICP-MS and the concentrations of EDTA and DTT present in the diluted commercial PTP1B in the assay. Metal buffering was provided by NTA, which has a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K of 8.3 for Zn2+ (ref. 36) and provides sufficiently high zinc buffering capacity at 1 mM. However, MINEQL+ 4.6 assigns a logK of 11.950 for Zn:[NTA]. Moreover, NTA forms 1:1 and 2:1 complexes with Zn2+.37 The 2:1 complex has a logK of 2.9. Modelling with MINEQL+ 4.6 indicates that the concentration of this complex compared to the 1:1 complex is negligible at pH 7.4 (ESI,† Fig. S1). Included in the calculations with MINEQL+ 4.6 were also Hepes (logK of 3.7 for Zn2+)38 and TCEP (logK of 2.1 for Zn2+).39 When we compared the free zinc concentrations calculated with Maxchelator to the ones obtained with MINEQL+ 4.6, we observed minor differences (Table 2), presumably due to differences in binding affinities of NTA for zinc as well as the fact that Maxchelator does not include other zinc species in its algorithm. We used the values obtained from Maxchelator for the estimated free zinc concentrations.
K of 8.3 for Zn2+ (ref. 36) and provides sufficiently high zinc buffering capacity at 1 mM. However, MINEQL+ 4.6 assigns a logK of 11.950 for Zn:[NTA]. Moreover, NTA forms 1:1 and 2:1 complexes with Zn2+.37 The 2:1 complex has a logK of 2.9. Modelling with MINEQL+ 4.6 indicates that the concentration of this complex compared to the 1:1 complex is negligible at pH 7.4 (ESI,† Fig. S1). Included in the calculations with MINEQL+ 4.6 were also Hepes (logK of 3.7 for Zn2+)38 and TCEP (logK of 2.1 for Zn2+).39 When we compared the free zinc concentrations calculated with Maxchelator to the ones obtained with MINEQL+ 4.6, we observed minor differences (Table 2), presumably due to differences in binding affinities of NTA for zinc as well as the fact that Maxchelator does not include other zinc species in its algorithm. We used the values obtained from Maxchelator for the estimated free zinc concentrations.
| Zn | Cu | Ca | Fe | |
|---|---|---|---|---|
| Water | 0.08 ± 0.02 | 0.06 ± 0.02 | 85.42 ± 2.56 | 4.41 ± 0.13 |
| 50 mM Hepes | 1.67 ± 0.45 | 0.29 ± 0.01 | 90.72 ± 1.67 | 2.72 ± 0.90 |
| 1 mM NTA | 2.81 ± 0.16 | 0.44 ± 0.01 | 89.97 ± 4.44 | 2.78 ± 0.25 |
| 1 mM TCEP | 0.27 ± 0.14 | 0.05 ± 0.01 | 57.43 ± 6.83 | 0.43 ± 0.21 |
| 0.01% Triton X-100 | 0.85 ± 0.13 | 0.12 ± 0.11 | 46.66 ± 4.63 | 0.99 ± 0.60 |
| 10 μM DiFMUP | 0.41 ± 0.20 | 0.13 ± 0.08 | 15.10 ± 2.80 | 0.27 ± 0.18 |
| 2.5 nM PTP1B | 0.93 ± 0.04 | 0.14 ± 0.02 | 25.71 ± 0.53 | 0.61 ± 0.11 |
| Zn (Tot) (M) | [Zn]free (M) Maxchelator | [Zn]free (M) MINEQL+ 4.6 |
|---|---|---|
| 10 × 10−6 | 2.659 × 10−11 | 2.93 × 10−11 |
| 30 × 10−6 | 8.145 × 10−11 | 8.98 × 10−11 |
| 100 × 10−6 | 2.926 × 10−10 | 3.22 × 10−10 |
| 300 × 10−6 | 1.128 × 10−9 | 1.24 × 10−9 |
| 500 × 10−6 | 2.633 × 10−9 | 2.9 × 10−9 |
| 600 × 10−6 | 3.951 × 10−9 | 4.34 × 10−9 |
| 700 × 10−6 | 6.146 × 10−9 | 6.72 × 10−9 |
| 800 × 10−6 | 1.053 × 10−8 | 1.14 × 10−8 |
| 900 × 10−6 | 2.371 × 10−8 | 2.45 × 10−8 |
| 950 × 10−6 | 5.007 × 10−8 | 4.45 × 10−8 |
| 990 × 10−6 | 2.565 × 10−7 | 8.87 × 10−8 |
| 1 × 10−3 | 1.7 × 10−6 | 1.06 × 10−7 |
Low nanomolar Zn2+ concentrations inhibit PTB1B
Enzymatic activity was determined in the presence of different zinc concentrations. The substrate was added to the zinc-containing buffer and the reaction was started by the addition of the enzyme (Fig. 2A). Remarkably, zinc was capable of inhibiting the enzyme by 55% only. By fitting the data to a non-linear regression curve with SigmaPlot, we calculated an inhibition constant of 5.6 nM. This value is lower than the one previously observed when the enzyme was pre-incubated with zinc-containing solutions before assessing the enzymatic activity.35 We therefore also incubated PTP1B with increasing concentrations of zinc for up to seven minutes and measured enzymatic activity. When we fitted the data to a non-linear regression curve, we calculated an inhibition constant of 13.8 nM (Fig. 2B). Under these conditions we noted a loss of enzymatic activity at high free zinc concentrations (>250 nM). In order to achieve such a high free zinc concentration, a total concentration of 990 μM zinc needs to be added to the buffer (Table 2). These levels are at the limit of NTA's zinc buffering capacity and therefore free zinc concentrations are poorly controlled. Because of this effect we re-calculated the inhibition constant without the data points at high zinc concentrations. We observed a shift in apparent Ki of almost one order of magnitude (from 13.8 nM to 2.2 nM). A complete loss of enzymatic activity was never observed in the absence of pre-incubation. We also noted that the percentage of enzyme inhibition was higher when pre-incubating the enzyme at a given zinc concentration for a longer period of time. We hypothesised that this effect was due to the incubation itself and therefore equilibrated the enzyme with 1.7 μM zinc at different times. Without incubation, zinc inhibited the enzyme 50%. The percentage of inhibition, however, increased to 58%, 62% and 72% when the enzyme was incubated with zinc for 1, 3, or 10 minutes, respectively. Thus, when pre-incubating the enzyme with zinc, additional inhibition of the enzyme occurs, presumably through zinc binding at lower affinity sites.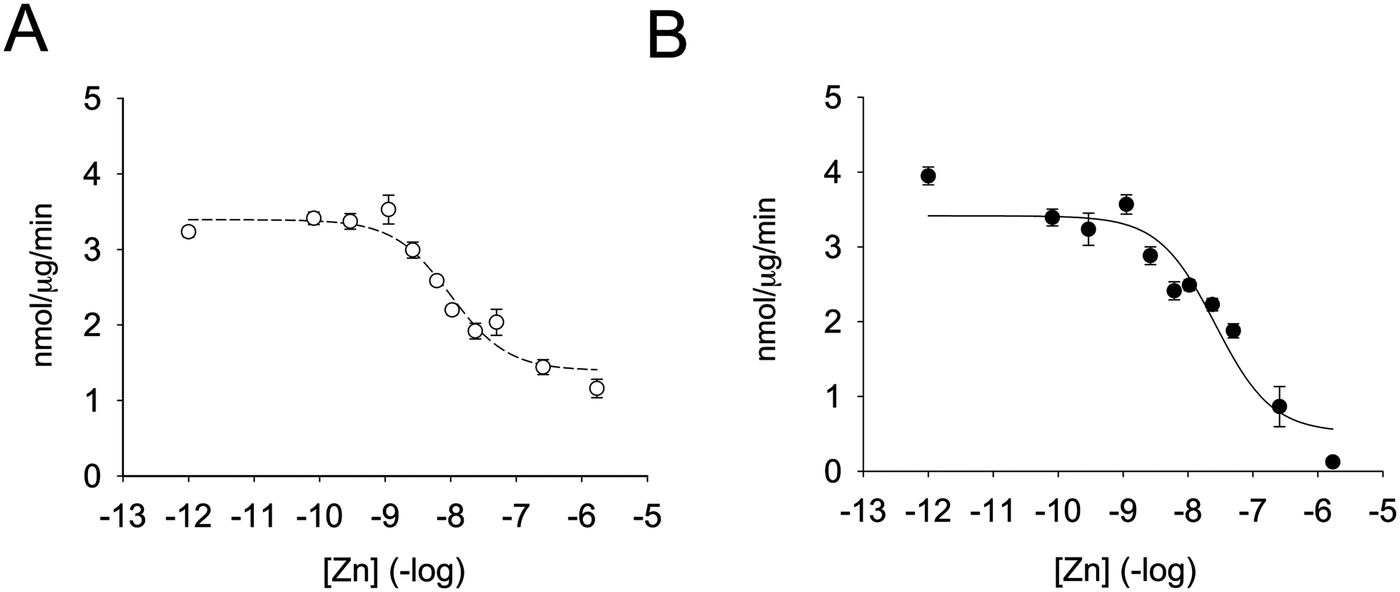 | ||
| Fig. 2 Zinc inhibition of PTP1B. The enzyme was assayed at increasing concentrations of free zinc, as calculated using Maxchelator (Table 2) in 50 mM Hepes, 1 mM NTA, 0.1 mM TCEP and 0.01% Triton X-100, pH 7.4. The enzyme was either added to the buffer containing zinc and 3 μM DiFMUP (A) or pre-incubated for seven minutes with zinc before addition 3 μM DiFMUP (B). | ||
Zn2+ inhibition of the pre-steady state
At the beginning of the catalytic reaction, PTP1B is in an open conformation, corresponding to the dephosphorylated state. The enzyme then binds the substrate, becomes phosphorylated, and releases the product. These pre-steady state kinetics precede the slower steps that determine the steady state kinetics, as the rate-limiting step is the dephosphorylation of the enzyme before a new catalytic cycle can occur (Fig. 3A). When assessing product formation with the fluorescence plate reader, a delay of about 30 seconds occurs between the start of the enzymatic reaction (when the substrate is added to the enzyme) and the beginning of the recording. Once the background fluorescence counts were subtracted and the 30 seconds delay taken into account, we noticed that the progress curve does not intersect with the abscissa at time zero (Fig. 3B). This indicates that a burst kinetic step is observed even at low enzyme concentrations (2.5 nM). Such a burst is typical for the ping–pong mechanism of this enzyme. We therefore estimated the amplitude of the burst for each zinc concentration and compared it to the control. Increasing the concentration of zinc has a minor effect on the amplitude of fluorescence, indicating that the burst is still present even at high zinc concentrations (Fig. 3C, white bars): 1.7 μM of zinc inhibits the burst by 36%. Interestingly, the percentage of inhibition observed is smaller than the one calculated for the steady state kinetics (55%, Fig. 2A). When we analysed the burst amplitude upon pre-incubation of the enzyme with zinc, we also noticed a higher percentage of inhibition compared to the same experiments performed without incubation (Fig. 3C, black bars).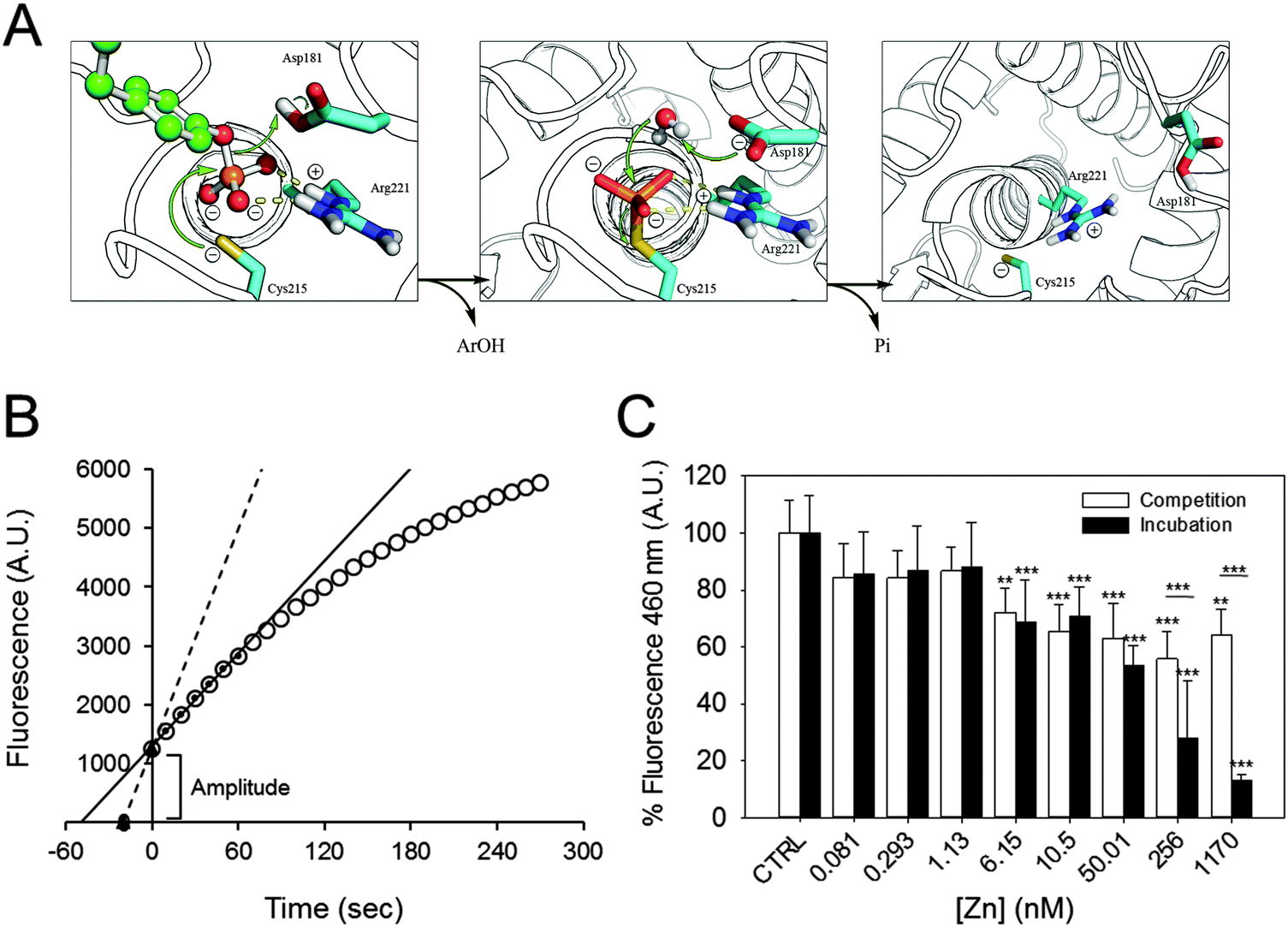 | ||
| Fig. 3 Zinc inhibition of PTP1B at different catalytic steps. (A) PTPs catalyse dephosphorylation of the substrate by a ping–pong mechanism. The substrate (green) binds to the enzyme, which rapidly catalyses the dephosphorylation and the release of the product Ar(OH). A water molecule subsequently attacks the phospho–enzyme intermediate to release inorganic phosphate (Pi) and to regenerate the enzyme. (Modified from Brandao et al.49) (B) Schematic of how the steady state rate and extrapolated pre-steady state amplitude were calculated. For steady state kinetics, the first 60 seconds of the reaction (small black circles) were used to draw a linear regression line (black), from which the slope was calculated. The lag-time between the start of the reaction and the first acquired datapoint was 30 seconds; a regression line was drawn between the extrapolated zero and the first measured point (black triangles and dotted line). (C) The amplitude in fluorescence observed at the beginning of the experiment was calculated in incubation (black) as well as in competition experiments (white) and is presented as percentage of control, in the absence of zinc. ***p < 0.001. **p < 0.01. | ||
Effect of EDTA on Zn2+-inhibited PTP1B activity
We examined next whether the effect of zinc inhibition is reversible by adding EDTA. In these sets of experiments, the enzyme was added to the buffer containing 256 nM free zinc and the reaction was initiated by adding the substrate. EDTA (2.5 mM) was added either before (EDTA + S) or after (S + EDTA) the substrate (S) DiFMUP (Fig. 4). We noticed that EDTA restored full enzymatic activity when added to the reaction buffer (94% of total activity). However, when EDTA was added after the substrate, the recovery of activity, although noticeable, was significantly lower than the one observed when EDTA was added prior to DiFMUP (84% versus 94%, Fig. 4A). Similar responses were observed when the enzyme was pre-incubated with zinc. Although addition of EDTA to the reaction buffer promoted a recovery in activity, this was not as complete (from 3% activity in the presence of zinc to 23% after EDTA addition) as observed without pre-incubation in the time period investigated. Moreover, addition of EDTA after DiFMUP promoted a small recovery of activity that was however not statistically significant (Fig. 4B).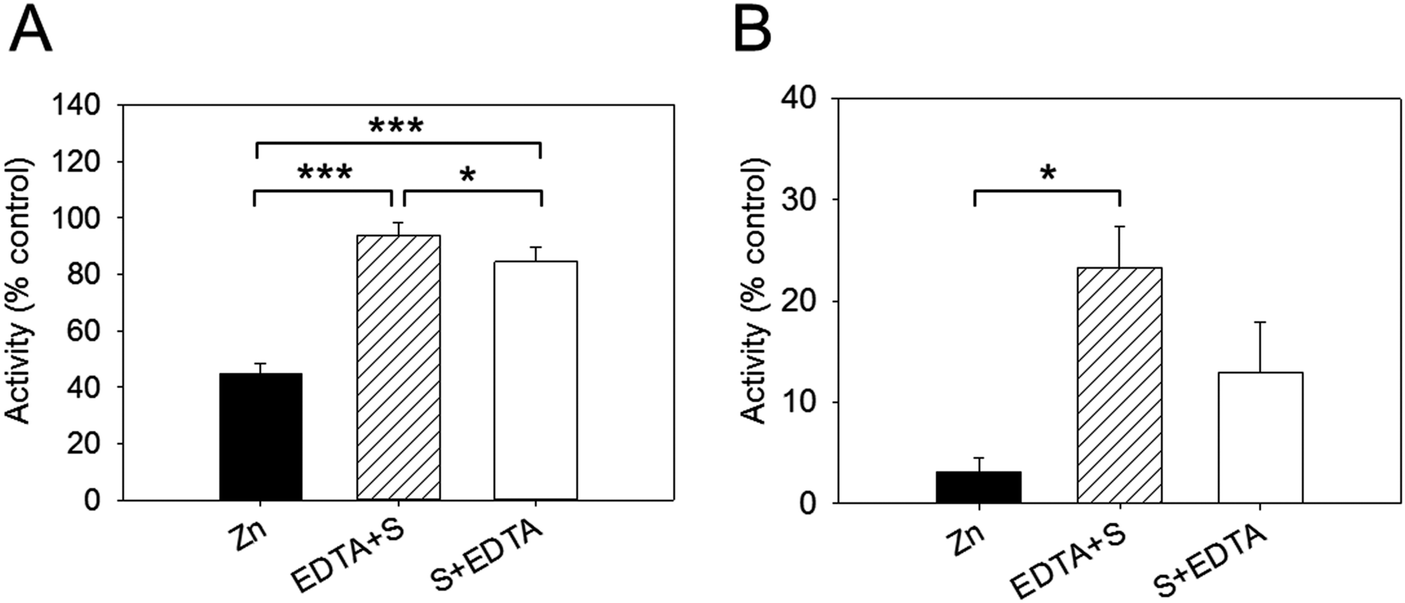 | ||
| Fig. 4 Effect of EDTA on zinc-inhibited PTP1B activity. The buffer contained 256 nM free zinc in these experiments. (A) 2.5 nM PTP1B was added directly to the buffer before 2.5 mM EDTA was added either before (EDTA + S) or after (S + EDTA) the addition of 3 μm DiFMUP. The initial rate was determined and is given as percentage of control in the absence of zinc. (B) The same experiment was performed but PTP1B was pre-incubated with 256 nM free zinc for seven minutes before further additions were made. ***p < 0.001, *p < 0.05. | ||
Zn2+ binding to PTP1B
We examined whether PTP1B has Zn2+ binding sites. Since PTP1B crystal structures in the presence of zinc have not been reported, we performed a knowledge-based molecular docking approach to identify putative binding pockets. The approach employs rigid protein structures taken form the Protein Data Bank.40 First, the whole protein was examined using a docking algorithm to predict zinc-binding sites; second, the suggested sites were filtered and only sites where zinc could coordinate at least two conserved residues (see Experimental procedures) were retained. The resulting zinc poses were ranked based on a docking scoring function (Table 3). This method was applied to three PTP1B X-ray crystal structures that represent the different conformations during the catalytic cycle of the enzyme. The results show zinc binding to the surface of the open (Fig. 5A) and closed (Fig. 5B) conformation as well as to the cysteinyl–phosphate intermediate of PTP1B (Fig. 5C). We noted that these sites were not conserved among the three structures examined, possibly because of the different orientation of the amino acid side chains (Table 3). Significantly, the catalytic site can accommodate zinc ions only in the closed conformation (Fig. 5B) and when the catalytic cysteine is phosphorylated (Fig. 5C). In the closed conformation (PDB id: 3I8O) the binding occurs between Asp181, Arg221, Gln262 and the catalytic Cys215. In the cysteinyl–phosphate intermediate (PDB id: 1A5Y), the binding occurs between phospho–Cys215, Asp181 and Gln266. We noted that in the latter PDB file there is a mutation, Q262A, explaining why we failed to detect Gln262 as a binding residue in the catalytic site. The open conformation failed to accommodate zinc in the catalytic domain as a consequence of the increased distance between the above residues in the catalytic pocket.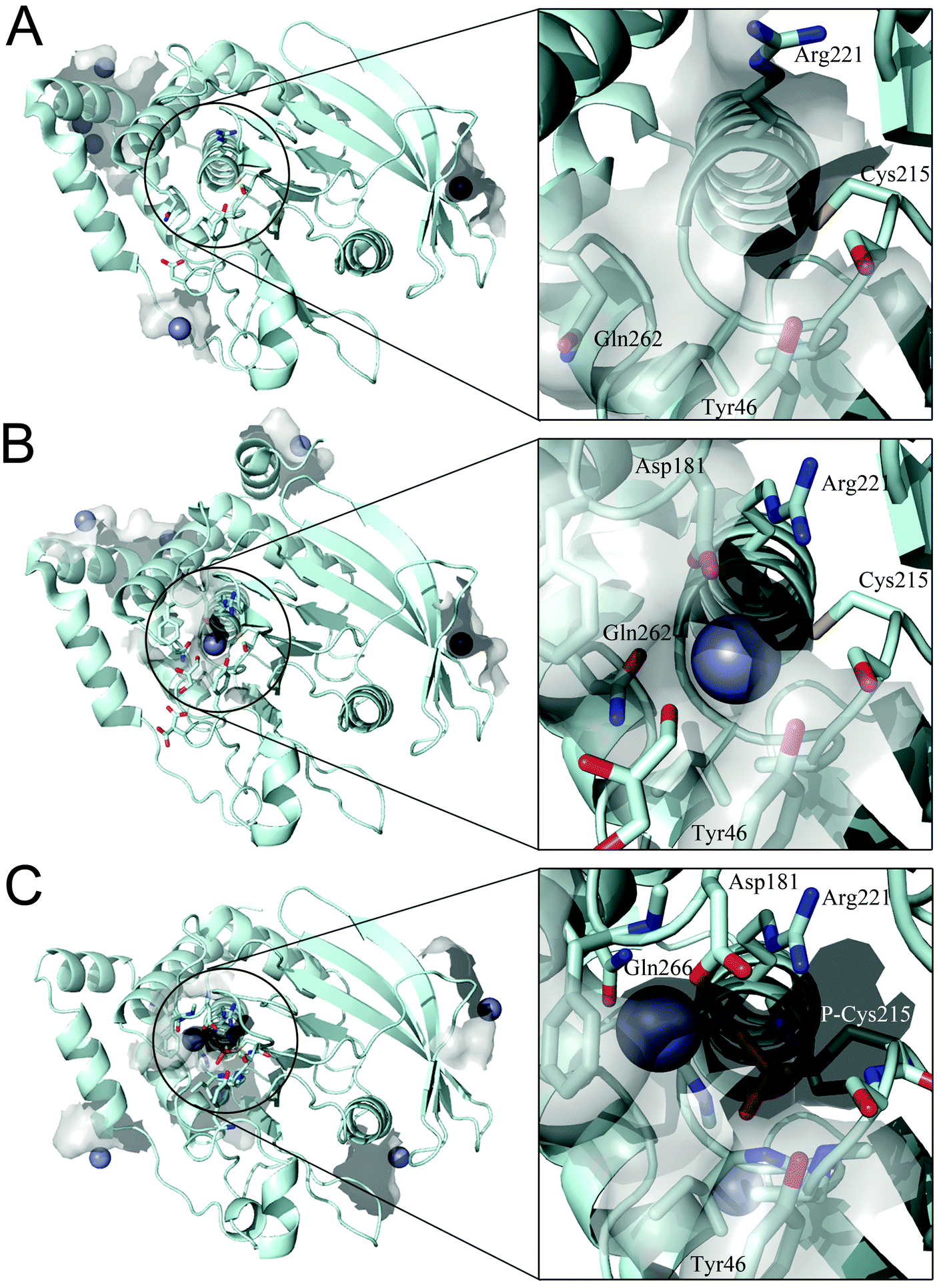 | ||
| Fig. 5 Docking simulation performed on different PTP1B crystal structures. (A) Open form (PDB id: 2CM2), (B) closed form (PDB id: 3I8O) and (C) cysteinyl–phosphate intermediate (PDB id: 1A5Y). Details of the catalytic site are shown in the square frame. Protein structure is represented in pale cyan, zinc ions as grey spheres and relevant amino acids as sticks. | ||
| Pose 1 | Pose 2 | Pose 3 | Pose 4 | Pose 5 | |
|---|---|---|---|---|---|
| PTP1B open form (PDB id: 2CM2, res.: 1.50 Å) | |||||
| Estimate binding energy (kcal mol−1) | −26.4817 | −25.1668 | −24.1878 | −23.8156 | −23.4430 |
| Amino acids within 4 Å |
Lys141
Thr143 Glu159 Glu161 |
Glu2
Glu4 Glu6 |
Glu4
Lys5 Glu8 |
His0
His1 Glu2 |
Asp29
Pro31 Cys32 Met258 |
| PTP1B closed form (PDB id: 3I80, res.: 2.25 Å) | |||||
| Estimate binding energy (kcal mol−1) | −27.3148 | −24.4115 | −24.353 | −24.1576 | −23.8244 |
| Amino acids within 4 Å |
Lys141
Thr143 Glu159 Glu161 |
Glu2
Met3 Glu4 Ile275 Ala278 |
Glu8
Asp11 |
Asp181
Cys215 Arg221 Gln262 |
Asp289
Lys292 Glu293 |
| PTP1B cysteinyl–phosphate intermediate (Q262A) (PDB id: 1A5Y, res.: 2.50 Å) | |||||
| Estimate binding energy (kcal mol−1) | −25.9348 | −23.3821 | −23.1203 | −22.8746 | −22.8391 |
| Amino acids within 4 Å |
Met74
Glu75 Glu76 Asp252 |
His60
Glu101 |
Asp181
P–Cys215 Gln266 |
His25
Glu26 |
Glu129
Glu132 |
Discussion
PTP1B is involved in metabolic diseases (e.g. diabetes and obesity) and cancer3 and has over 30 known substrates.41 This enzyme is therefore considered to be a major potential pharmaceutical target.42 Inhibition of protein tyrosine phosphatases with experimental drugs has been studied for many years but has suffered from issues of specificity of inhibitors with regard to other phosphatases. Vanadium compounds are one class of inhibitors. They have insulin-like and anti-diabetic effects in a variety of animal models.43 The mode of action of vanadium is through inhibition of PTP1B, as it mimics the phosphate group in the transition state of the enzyme,44 therefore competing with the substrate for the binding site. Remarkably, there is much less work on zinc, which is also a known PTP inhibitor45 and binds with much higher affinity than vanadium compounds. Zinc, in contrast to vanadium, is a nutritionally essential element. Therefore its effect on PTB1B is physiologically much more relevant and the above observations on the biological effects of vanadium compounds give an indication of what effects zinc might have. The total cellular zinc concentration is in the range of a few hundred micromolar while its free cytosolic concentration is in the high picomolar range.24,46,47In this study we investigated zinc inhibition in more detail as there is no comprehensive study of how zinc inhibits the enzyme. Using DiFMUP as a substrate,48 we have found that the very strong inhibition is dependent on the conformation of the enzyme. By performing competition experiments, we found an inhibition constant of 5.6 nM for zinc. The reaction catalysed by protein tyrosine phosphatases is explained very elegantly in the work of Brandao et al.49 Briefly, the enzyme initially is present in an open conformation. The binding of substrate triggers a conformational change in the phosphatase that culminates in the closure of the catalytic pocket by a conserved WPD loop. The general kinetic mechanism is known as ping–pong: in the first step the thiol of Cys215 attacks the phosphate ester group of the substrate, which generates a phospho–enzyme intermediate and the product, which is immediately released. In the second and rate limiting step, a water molecule attacks the phospho–enzyme, releasing the inorganic phosphate and regenerating the enzyme49 (Fig. 3A). We estimated the burst of product formation by measuring the amplitude in fluorescence observed at the beginning of the experiment. We noted that the effect of zinc on the burst was minor and much less than the effect on the steady state kinetics. These data suggest that zinc binds and inhibits the enzyme after the burst has occurred. When adding EDTA to the reaction in order to rescue activity, we observed that EDTA restores enzymatic activity. However, the extent to which this is achieved depends on whether the chelator is added before or after the substrate. Addition of the substrate seemed to decrease the ability of EDTA to restore full enzymatic activity, and this supports the hypothesis that zinc inhibits the enzyme after the first catalytic step. In fact, our examination of the 3D structures of PTP1B revealed that zinc binds to the catalytic pocket only when the enzyme is in a closed conformation or when it is in the phospho-intermediate form. The transition from the open to the closed conformation involves a movement of the so-called WPD loop. WPD loop closure and release of the product occur concomitantly. Thus, the possibility of finding PTP1B in a closed, dephosphorylated conformation is very low,50 supporting the hypothesis that zinc binds predominantly to the phospho-intermediate form of PTP1B. Indeed, the aspartic acid (Asp181) of the WPD loop is close to Gln262 and to the phospho–Cys215 in the closed conformation and zinc binding in this position could trigger inhibition of the second catalytic step (phosphate release) in the ping–pong mechanism. Infact, the Gln262 and Asp181 act in concert to hydrolyse the cysteinyl–phosphate intermediate through coordinating a water molecule. Moreover, Arg221 is also close to this putative zinc-binding pocket. This residue is important for the catalytic activity as it plays a role in substrate binding and first transition state stability. In the absence of substrate, the enzyme is in an open conformation: the WPD loop is far from the catalytic cysteine and the binding site for zinc is not present. When we pre-incubated PTP1B with zinc, we noticed that the activity was inhibited to a higher percentage, although we failed to rescue the enzymatic activity completely with EDTA, at the time points tested. We hypothesise that zinc might have additional effects on PTP1B. Indeed, we found that PTP1B possesses many acidic residues on its surface, which are potential binding sites for zinc (Fig. 5). How such binding inhibits the enzyme up to 100% is not clear but may involve aggregation; it has been reported that PTP1B dimerises, a known mechanism to inhibit protein tyrosine phosphatases.17,51 The identification of a putative site for zinc binding and PTP1B activity modulation could be important for the development of pharmaceutical zinc-containing drugs. However, the design of such compounds would need to be very specific as the putative site does not allow accessibility to a large molecule. A zinc-delivering system might be more appropriate. In this way it would be possible to address specificity, e.g. in docking a zinc-containing drug to a specific PTP.
Key questions resulting from this work are under which conditions zinc inhibition is physiologically relevant and why virtually no attention was given to the significance of zinc inhibition for the biology of this important enzyme. One issue certainly is that PTPs are always supplied with relatively high concentrations of chelating agents to keep them active. As this study shows, it is experimentally extremely challenging to have the correct conditions to investigate zinc inhibition. The second critical issue is that only in the last couple of years it became evident that free zinc concentrations are in the range of the apparent Ki of PTP1B inhibition and that they undergo controlled fluctuations. The concentration of zinc in the cytosol is tightly regulated by a plethora of proteins: two families of zinc transporters (ZnT and Zip), metallothioneins (MT), and other binding proteins.52 The resting concentration of free zinc in the cytosol is in the high picomolar range although it fluctuates in response to extracellular stimuli.24,25,46 Zinc ions are transported from the extracellular space or released from intracellular compartments. The endoplasmic reticulum (ER) is a storage compartment for zinc. Growth factor stimulation triggers zinc release from the ER through the zinc transporter Zip7 in the TamR breast cancer cell line.53 PTP1B is an ER-bound protein. Its catalytic pocket faces the cytosol where it dephosphorylates the insulin receptor as well as other substrates.4,41 Zip7-mediated zinc release from the ER could be a specific pathway to inhibit ER-bound PTP1B. Because of the co-localisation of Zip7 and PTP1B, there is no need for increasing cytosolic zinc globally in order to inhibit the enzyme. However, from the data presented here, we hypothesise that inhibition will occur only in the presence of a PTP1B substrate when PTP1B is enzymatically active. Zinc is considered to be an insulin-mimetic. The inhibition of PTP1B can prolong the insulin signal through the insulin receptor.54 Prolonged exposure of pancreatic beta cells to high glucose concentrations resulted in increased cytosolic zinc levels.55 Recently, Fernandez-Ruiz et al. investigated the involvement of PTP1B in pancreatic beta cell mass and found that ablating the enzyme in mice as well as silencing it in a mouse pancreatic beta cell line (MIN6) results in beta cell expansion.56 As beta cell mass increases in the early stage of diabetes, when pancreatic islets start to compensate for the insulin insensitivity, we hypothesise that the increase in cytosolic zinc observed at prolonged high glucose concentration could potentially be a signal for PTP1B inhibition and beta cell proliferation. While this discussion focuses only on the possible effects on insulin signalling, from the involvement of the enzyme in leptin signalling and cancer processes it seems obvious that there are many additional implications.
In summary, a specific mechanism for the modulation of PTP1B by zinc ions suggests that the phosphorylated enzyme intermediate is required in addition to inhibitory zinc concentrations, which can be achieved locally at the ER.
Since both the catalytic mechanism described for PTP1B and the amino acids participating in catalysis are conserved among different members of the PTP family,57 the proposed mechanism responsible for the modulation of PTP1B activity by zinc could be general and applicable to other PTPs. We have previously described the inhibition by zinc (Ki = 21 pM) of another member of the PTP family, the receptor PTP beta.23 Even in that case a mixed type inhibition was noted, supporting the view that modulation by zinc ions is dependent on the enzyme being active and catalysing the dephosphorylation of a substrate.
Other enzymes are inhibited by very low concentrations of zinc.58 Among those, caspases have been shown to be strongly inhibited by zinc ions.21 An investigation of the mode of inhibition of caspase-3 by zinc revealed that zinc is a mixed type inhibitor.59 The binding site for zinc, which inhibits enzymatic activity, does not prevent binding of the substrate, and does not involve the catalytic cysteine. These data together indicate that zinc modulates the activity of different enzymes in a new way that might not necessarily involve the catalytic thiol.
Conclusions
We have optimised a new assay for the study of zinc inhibition of protein tyrosine phosphatases. By using this tool as well as molecular docking techniques, we have identified a novel mechanism of PTP1B inhibition by zinc ions. We show here that zinc binds in the catalytic pocket of the phospho-intermediate form of PTP1B, therefore only after activation in the presence of a substrate, with an apparent Ki of 5.6 nM. We also show that enzyme inhibition can also occur through a second mechanism that is believed to involve binding to surface residues. The modulation of PTP1B in the range of available cellular zinc ion concentrations and the localisation of PTP1B and Zip7 on the ER membrane strongly suggest physiological significance. These data will need further confirmation by structural studies employing NMR spectroscopy and X-ray diffraction experiments, which we have currently begun, as well as cell biological work to investigate PTP1B activity towards physiological substrates in response to varied cellular zinc concentrations.Acknowledgements
This work was supported by a grant from the Biotechnology and Biological Sciences Research Council (BB/K001442/1). We acknowledge Mr Andrew Cakebread for metal analysis by inductively coupled plasma mass spectrometry and the Genomics Centre, Franklin-Wilkins Building, King's College London. We also thank Ms Bettina Schuhn.References
- E. H. Fischer, Cell signaling by protein tyrosine phosphorylation, Adv. Enzyme Regul., 1999, 39, 359–369 CrossRef CAS.
- N. K. Tonks, Protein tyrosine phosphatases: from genes, to function, to disease, Nat. Rev. Mol. Cell Biol., 2006, 7, 833–846, DOI:10.1038/nrm2039.
- M. Feldhammer, N. Uetani, D. Miranda-Saavedra and M. L. Tremblay, PTP1B: a simple enzyme for a complex world, Crit. Rev. Biochem. Mol. Biol., 2013, 48, 430–445, DOI:10.3109/10409238.2013.819830.
- J. V. Frangioni, P. H. Beahm, V. Shifrin, C. A. Jost and B. G. Neel, The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence, Cell, 1992, 68, 545–560 CrossRef CAS.
- K. A. Kenner, E. Anyanwu, J. M. Olefsky and J. Kusari, Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling, J. Biol. Chem., 1996, 271, 19810–19816 CrossRef CAS PubMed.
- A. Cheng, N. Uetani, P. D. Simoncic, V. P. Chaubey, A. Lee-Loy, C. J. McGlade, B. P. Kennedy and M. L. Tremblay, Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B, Dev. Cell, 2002, 2, 497–503 CrossRef CAS.
- M. Elchebly, P. Payette, E. Michaliszyn, W. Cromlish, S. Collins, A. L. Loy, D. Normandin, A. Cheng, J. Himms-Hagen, C. C. Chan, C. Ramachandran, M. J. Gresser, M. L. Tremblay and B. P. Kennedy, Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene, Science, 1999, 283, 1544–1548 CrossRef CAS.
- J. M. Friedman, Leptin, leptin receptors, and the control of body weight, Nutr. Rev., 1998, 56, s38–s46 CrossRef CAS ; discussion s54–s75.
- J. M. Zabolotny, K. K. Bence-Hanulec, A. Stricker-Krongrad, F. Haj, Y. Wang, Y. Minokoshi, Y. B. Kim, J. K. Elmquist, L. A. Tartaglia, B. B. Kahn and B. G. Neel, PTP1B regulates leptin signal transduction in vivo, Dev. Cell, 2002, 2, 489–495 CrossRef CAS.
- M. P. Myers, J. N. Andersen, A. Cheng, M. L. Tremblay, C. M. Horvath, J. P. Parisien, A. Salmeen, D. Barford and N. K. Tonks, TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B, J. Biol. Chem., 2001, 276, 47771–47774, DOI:10.1074/jbc.C100583200.
- T. A. Woodford-Thomas, J. D. Rhodes and J. E. Dixon, Expression of a protein tyrosine phosphatase in normal and v-src-transformed mouse 3T3 fibroblasts, J. Cell Biol., 1992, 117, 401–414 CrossRef CAS.
- J. R. Wiener, B. J. Kerns, E. L. Harvey, M. R. Conaway, J. D. Iglehart, A. Berchuck and R. C. Bast, Jr., Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression, J. Natl. Cancer Inst., 1994, 86, 372–378 CrossRef CAS PubMed.
- S. G. Julien, N. Dube, M. Read, J. Penney, M. Paquet, Y. Han, B. P. Kennedy, W. J. Muller and M. L. Tremblay, Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis, Nat. Genet., 2007, 39, 338–346, DOI:10.1038/ng1963.
- K. K. Balavenkatraman, N. Aceto, A. Britschgi, U. Mueller, K. K. Bence, B. G. Neel and M. Bentires-Alj, Epithelial protein-tyrosine phosphatase 1B contributes to the induction of mammary tumors by HER2/Neu but is not essential for tumor maintenance, Mol. Cancer Res., 2011, 9, 1377–1384, DOI:10.1158/1541-7786.MCR-11-0198.
- N. Aceto and M. Bentires-Alj, Targeting protein-tyrosine phosphatases in breast cancer, OncoTargets Ther., 2012, 3, 514–515 Search PubMed.
- D. L. Lohse, J. M. Denu, N. Santoro and J. E. Dixon, Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1, Biochemistry, 1997, 36, 4568–4575, DOI:10.1021/bi963094r.
- D. Bandyopadhyay, A. Kusari, K. A. Kenner, F. Liu, J. Chernoff, T. A. Gustafson and J. Kusari, Protein-tyrosine phosphatase 1B complexes with the insulin receptor in vivo and is tyrosine-phosphorylated in the presence of insulin, J. Biol. Chem., 1997, 272, 1639–1645 CrossRef CAS PubMed.
- S. Dadke, S. Cotteret, S. C. Yip, Z. M. Jaffer, F. Haj, A. Ivanov, F. Rauscher, 3rd, K. Shuai, T. Ng, B. G. Neel and J. Chernoff, Regulation of protein tyrosine phosphatase 1B by sumoylation, Nat. Cell Biol., 2007, 9, 80–85, DOI:10.1038/ncb1522.
- A. M. Bilwes, J. den Hertog, T. Hunter and J. P. Noel, Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization, Nature, 1996, 382, 555–559, DOI:10.1038/382555a0.
- D. L. Brautigan, P. Bornstein and B. Gallis, Phosphotyrosyl-protein phosphatase. Specific inhibition by Zn, J. Biol. Chem., 1981, 256, 6519–6522 CAS.
- W. Maret, C. Jacob, B. L. Vallee and E. H. Fischer, Inhibitory sites in enzymes: zinc removal and reactivation by thionein, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1936–1940 CrossRef CAS.
- H. Haase and W. Maret, Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling, Exp. Cell Res., 2003, 291, 289–298, DOI:10.1016/S0014-4827(03)00406-3.
- M. Wilson, C. Hogstrand and W. Maret, Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase beta activity, J. Biol. Chem., 2012, 287, 9322–9326, DOI:10.1074/jbc.C111.320796.
- A. Krezel and W. Maret, Zinc-buffering capacity of a eukaryotic cell at physiological pZn, JBIC, J. Biol. Inorg. Chem., 2006, 11, 1049–1062, DOI:10.1007/s00775-006-0150-5.
- J. L. Vinkenborg, T. J. Nicolson, E. A. Bellomo, M. S. Koay, G. A. Rutter and M. Merkx, Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis, Nat. Methods, 2009, 6, 737–740, DOI:10.1038/nmeth.1368.
- D. M. Bers, C. W. Patton and R. Nuccitelli, A practical guide to the preparation of Ca(2+) buffers, Methods Cell Biol., 2010, 99, 1–26, DOI:10.1016/B978-0-12-374841-6.00001-3.
- T. Zor and Z. Selinger, Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies, Anal. Biochem., 1996, 236, 302–308, DOI:10.1006/abio.1996.0171.
- L. Shrodinger, 1.3r1 edn, 2010.
- I. W. Davis, A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall, 3rd, J. Snoeyink, J. S. Richardson and D. C. Richardson, MolProbity: all-atom contacts and structure validation for proteins and nucleic acids, Nucleic Acids Res., 2007, 35, W375–W383, DOI:10.1093/nar/gkm216.
- O. Korb, T. Stutzle and T. E. Exner, Empirical scoring functions for advanced protein-ligand docking with PLANTS, J. Chem. Inf. Model., 2009, 49, 84–96, DOI:10.1021/ci800298z.
- M. Laitaoja, J. Valjakka and J. Janis, Zinc coordination spheres in protein structures, Inorg. Chem., 2013, 52, 10983–10991, DOI:10.1021/ic401072d.
- C. Andreini, I. Bertini and G. Cavallaro, Minimal functional sites allow a classification of zinc sites in proteins, PLoS One, 2011, 6, e26325, DOI:10.1371/journal.pone.0026325.
- G. H. Peters, S. Branner, K. B. Moller, J. N. Andersen and N. P. Moller, Enzyme kinetic characterization of protein tyrosine phosphatases, Biochimie, 2003, 85, 527–534 CrossRef CAS.
- J. Montalibet, K. I. Skorey and B. P. Kennedy, Protein tyrosine phosphatase: enzymatic assays, Methods, 2005, 35, 2–8, DOI:10.1016/j.ymeth.2004.07.002.
- A. Krezel and W. Maret, Thionein/metallothionein control Zn(II) availability and the activity of enzymes, JBIC, J. Biol. Inorg. Chem., 2008, 13, 401–409, DOI:10.1007/s00775-007-0330-y.
- A. Krezel and W. Maret, Dual nanomolar and picomolar Zn(II) binding properties of metallothionein, J. Am. Chem. Soc., 2007, 129, 10911–10921, DOI:10.1021/ja071979s.
- D. L. Rabenstein and R. J. Kula, Ligand-exchange kinetics and solution equilibriums of cadmium, zinc, and lead nitrilotriacetate complexes, J. Am. Chem. Soc., 1969, 91, 2492–2503, DOI:10.1021/ja01038a017.
- E. Chekmeneva, R. Prohens, J. M. Diaz-Cruz, C. Arino and M. Esteban, Thermodynamics of Cd2+ and Zn2+ binding by the phytochelatin (gamma-Glu-Cys)4-Gly and its precursor glutathione, Anal. Biochem., 2008, 375, 82–89, DOI:10.1016/j.ab.2008.01.008.
- A. Krezel, R. Latajka, G. D. Bujacz and W. Bal, Coordination properties of tris(2-carboxyethyl)phosphine, a newly introduced thiol reductant, and its oxide, Inorg. Chem., 2003, 42, 1994–2003, DOI:10.1021/ic025969y.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- E. Ferrari, M. Tinti, S. Costa, S. Corallino, A. P. Nardozza, A. Chatraryamontri, A. Ceol, G. Cesareni and L. Castagnoli, Identification of new substrates of the protein-tyrosine phosphatase PTP1B by Bayesian integration of proteome evidence, J. Biol. Chem., 2011, 286, 4173–4185, DOI:10.1074/jbc.M110.157420.
- A. J. Barr, Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development, Future Med. Chem., 2010, 2, 1563–1576, DOI:10.4155/fmc.10.241.
- R. L. Khandelwal and S. Pugazhenthi, In vivo effects of vanadate on hepatic glycogen metabolizing and lipogenic enzymes in insulin-dependent and insulin-resistant diabetic animals, Mol. Cell. Biochem., 1995, 153, 87–94 CrossRef CAS.
- M. J. Gresser and A. S. Tracey, Vanadium in Biological Systems, Kluwer Academic Publishers, Netherlands, 1990 Search PubMed.
- J. M. Samet, R. Silbajoris, W. Wu and L. M. Graves, Tyrosine phosphatases as targets in metal-induced signaling in human airway epithelial cells, Am. J. Respir. Cell Mol. Biol., 1999, 21, 357–364, DOI:10.1165/ajrcmb.21.3.3656.
- R. A. Bozym, R. B. Thompson, A. K. Stoddard and C. A. Fierke, Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor, ACS Chem. Biol., 2006, 1, 103–111, DOI:10.1021/cb500043a.
- J. L. Vinkenborg, M. S. Koay and M. Merkx, Fluorescent imaging of transition metal homeostasis using genetically encoded sensors, Curr. Opin. Chem. Biol., 2010, 14, 231–237, DOI:10.1016/j.cbpa.2009.11.022.
- S. Welte, K. H. Baringhaus, W. Schmider, G. Muller, S. Petry and N. Tennagels, 6,8-Difluoro-4-methylumbiliferyl phosphate: a fluorogenic substrate for protein tyrosine phosphatases, Anal. Biochem., 2005, 338, 32–38, DOI:10.1016/j.ab.2004.11.047.
- T. A. Brandao, A. C. Hengge and S. J. Johnson, Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps, J. Biol. Chem., 2010, 285, 15874–15883, DOI:10.1074/jbc.M109.066951.
- S. K. Whittier, A. C. Hengge and J. P. Loria, Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases, Science, 2013, 341, 899–903, DOI:10.1126/science.1241735.
- I. Anderie, I. Schulz and A. Schmid, Characterization of the C-terminal ER membrane anchor of PTP1B, Exp. Cell Res., 2007, 313, 3189–3197, DOI:10.1016/j.yexcr.2007.05.025.
- T. Fukada and T. Kambe, Molecular and genetic features of zinc transporters in physiology and pathogenesis, Metallomics, 2011, 3, 662–674, 10.1039/c1mt00011j.
- K. M. Taylor, S. Hiscox, R. I. Nicholson, C. Hogstrand and P. Kille, Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7, Sci. Signaling, 2012, 5, ra11, DOI:10.1126/scisignal.2002585.
- H. Haase and W. Maret, Fluctuations of cellular, available zinc modulate insulin signaling via inhibition of protein tyrosine phosphatases, J. Trace Elem. Med. Biol., 2005, 19, 37–42, DOI:10.1016/j.jtemb.2005.02.004.
- E. A. Bellomo, G. Meur and G. A. Rutter, Glucose regulates free cytosolic Zn(2)(+) concentration, Slc39 (ZiP), and metallothionein gene expression in primary pancreatic islet beta-cells, J. Biol. Chem., 2011, 286, 25778–25789, DOI:10.1074/jbc.M111.246082.
- R. Fernandez-Ruiz, E. Vieira, P. M. Garcia-Roves and R. Gomis, Protein Tyrosine Phosphatase-1B Modulates Pancreatic beta-cell Mass, PLoS One, 2014, 9, e90344, DOI:10.1371/journal.pone.0090344.
- A. J. Barr, E. Ugochukwu, W. H. Lee, O. N. King, P. Filippakopoulos, I. Alfano, P. Savitsky, N. A. Burgess-Brown, S. Muller and S. Knapp, Large-scale structural analysis of the classical human protein tyrosine phosphatome, Cell, 2009, 136, 352–363, DOI:10.1016/j.cell.2008.11.038.
- W. Maret, Inhibitory zinc sites in enzymes, BioMetals, 2013, 26, 197–204, DOI:10.1007/s10534-013-9613-7.
- A. G. Daniel, E. J. Peterson and N. P. Farrell, The bioinorganic chemistry of apoptosis: potential inhibitory zinc binding sites in caspase-3, Angew. Chem., 2014, 53, 4098–4101, DOI:10.1002/anie.201311114.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mt00086b |
| ‡ The nomenclature used for protein tyrosine phosphatases in this manuscript is the most common. The updated nomenclature for the proteins mentioned here is as follow: PTPN1 (protein tyrosine phosphatase, non-receptor type 1) for PTP1B, PTPN6 (protein tyrosine phosphatase, non-receptor type 6) for SHP-1, PTPN2 (protein tyrosine phosphatase, non-receptor type 2) for TC-PTP and PTPRB (protein tyrosine phosphatase, receptor type B) for R-PTP-beta (http://www.genenames.org/genefamilies/PTP). |
| This journal is © The Royal Society of Chemistry 2014 |