 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decreased serum zinc is an effect of ageing and not Alzheimer's disease†
Alan
Rembach‡
a,
Dominic J.
Hare‡
ab,
James D.
Doecke
cd,
Samantha C.
Burnham
c,
Irene
Volitakis
a,
Christopher J.
Fowler
a,
Robert A.
Cherny
a,
Catriona
McLean
e,
Rudolf
Grimm
f,
Ralph
Martins
g,
David
Ames
h,
Colin L.
Masters
a,
Ashley I.
Bush
a and
Blaine R.
Roberts
*a
aThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia. E-mail: blaine.roberts@florey.edu.au
bElemental Bio-imaging Centre, University of Technology, Sydney, Broadway, New South Wales, Australia
cCSIRO Preventative Health Flagship: Mathematics, Informatics and Statistics, Perth, Western Australia, Australia
dThe Australian e-Health Research Centre, Herston, Queensland, Australia
eAnatomical Pathology, The Alfred Hospital, Melbourne, Victoria, Australia
fAgilent Technologies, Santa Clara, California, USA
gJames McCusker Alzheimer's Disease Research Unit, Health Department of Western Australia, Western Australia, Australia
hNational Ageing Research Institute, Parkville, Victoria, Australia
First published on 20th March 2014
Abstract
We examined the distribution of zinc in the periphery (erythrocytes and serum) in a large, well-characterised cohort, the Australian Imaging, Biomarkers and Lifestyle (AIBL) study, in order to determine if there is systemic perturbation in zinc homeostasis in Alzheimer's disease (AD). We observed an age dependent decrease in serum zinc of approximately 0.4% per year. When correcting for the age dependent decline in serum zinc no significant difference between healthy controls (HC), mildly cognitively impaired (MCI) or AD subjects was observed.
Alzheimer's disease (AD) is the most prevalent cause of dementia in the aged community, and is a progressive and chronic neurodegeneration that is expected to afflict over 115 million individuals worldwide by 2050 if effective disease modifying treatments are not identified.1 AD is characterised by the accumulation of aggregated amyloid beta (Aβ) in extracellular plaques and neuronal tangles of phosphorylated tau,2 yet despite considerable knowledge concerning the underlying processes in AD, the precise toxic principal of this disorder has yet to be identified.
The concept of a loss of metallostasis,3 or a disturbance in metal homeostasis, has recently been proposed as a key element in the pathway leading to Aβ toxicity and AD.4–8 In particular, zinc has been the focus of intense research for its critical role in synaptic maintenance and neuronal transmission.9,10 In the brain, zinc is a redox-inert but essential biological metal involved in a range of catalytic activities11 and is implicated in AD for its role in amyloid precursor protein transcription,12–14 and in inducing soluble Aβ to aggregate and precipitate.15–18 Levels of peripheral zinc have been studied as putative disease biomarkers, though inconsistencies have emerged indicating that serum or plasma zinc may not reflect changes occurring in the central nervous system (CNS). A number of conflicting studies have shown a significant increase,19 a significant decrease,20–23 or no differences24,25 in peripheral zinc in AD when compared to healthy subjects. Furthermore, studies of the zinc concentration in cerebral spinal fluid (CSF) have produced varying results indicating either a decrease in CSF zinc in AD26 or no significant difference from control groups.27,28 Even within the AD brain itself, reported changes to total zinc levels are inconsistent,29,30 with decreased zinc reported in certain brain regions,31,32 contrasting an increase observed in other areas,33–36 These inconsistencies are most likely due to low statistical power from a limited number of samples.
AIBL is a unique resource of over 1000 enrolled AD, MCI and HCs. It is a longitudinal, prospective and multidisciplinary study that compares clinically characterised participants with amyloid imaging modalities,37 providing significant power to fully assess peripheral zinc status in a large, well-characterised AD cohort. Thus, the aim of this study was to examine the distribution of zinc in the periphery (erythrocytes and serum) in the AIBL study to determine if there is systemic perturbation in zinc homeostasis measurable in blood.
Serum zinc concentration was measured in the baseline AIBL cohort of 1084 subjects (complete, less 28 samples of the total 1112, which were deemed unsuitable for analysis). Comprehensive demographics, including age, sex, apolipoprotein-E type 4 (ApoEε4) allele status, Clinical Dementia Rating Scale Sum of Boxes score (CDR SOB; used to stage dementia) and mini-mental state examination (MMSE) questionnaire score are presented in Table 1. In the AIBL cohort, the HC group are significantly younger than the AD group (p < 0.0001), and contain a smaller number of ApoEε4 carriers (p < 0.0001). As expected, CDR SOB and MMSE score are consistently and significantly lower in the AD cohort (p < 0.0001). There was no difference in the proportion of males to females between the clinical classification groups (p = 0.335).
| Classification | HC | MCI | AD | p-value |
|---|---|---|---|---|
| N | 753 | 126 | 205 | |
| Age, years | 70.6 (7) | 76.2 (7.6) | 78.8 (8.6) | <0.001 |
| Sex, n female | 434 | 72 | 127 | 0.335 |
| ApoEε4 | 205 | 63 | 128 | <0.001 |
| CDR SOB | 0.0 (0.1) | 1.2 (0.8) | 5.8 (2.9) | <0.001 |
| MMSE | 28.9 (1.2) | 26.2 (2.7) | 18.9 (5.3) | <0.001 |
Analytical validity of our ICP-MS zinc assay was tested against a commercial standard serum (Seronorm™) and spike recovery from a characterised serum standard (Table S1, ESI†). 1084 individual serum samples were analysed over a 6-day period. Recovery of zinc from Seronorm™ standards was measured twice daily and was consistently within 10% of certified values. Similarly, spiked serum displayed good precision and accuracy in daily quality control checks. Twenty-five individual calibrations were performed, with acceptable background equivalent concentration (0.892 ± 0.65 μg L−1) and detection limits (0.511 ± 0.871 μg L−1) and linearity (R2 > 0.999) over the analysis period.
Serum zinc concentrations were subdivided according to clinical classification, sex and ApoEε4 allele distribution (Table 2). There was a subtle decrease in zinc concentration in AD subjects when compared to HCs (HC 12.730 ± 2.489 μM vs. AD 12.206 ± 2.808 μM). ANOVA of the marginal means across all three clinical groups showed a significant decrease in serum zinc levels (p = 0.003). Comparisons showed that this difference was driven by the HC vs. AD group difference (p = 0.001), with the HC-MCI group comparison not significantly different (p = 0.679).
| Group | [Zinc] (μM) | All three groups | HC vs. MCI | HC vs. AD | |||||
|---|---|---|---|---|---|---|---|---|---|
| HC | MCI | AD | Unadjusted p | Adjusted p | Unadjusted p | Adjusted p | Unadjusted p | Adjusted p | |
| Baseline all (n = 1084) | 12.73 (2.489) | 12.604 (2.082) | 12.206 (2.808) | 0.003 | 0.689 | 0.679 | 0.544 | 0.001 | 0.679 |
| Baseline males (n = 451) | 12.671 (2.178) | 12.76 (2.194) | 12.706 (3.332) | 0.896 | 0.230 | 0.758 | 0.469 | 0.747 | 0.223 |
| Baseline females (n = 633) | 12.774 (2.701) | 12.491 (2.006) | 11.91 (2.413) | <0.001 | 0.195 | 0.436 | 0.741 | <0.001 | 0.177 |
| ApoEε4 (+) (n = 396) | 12.919 (2.516) | 12.454 (1.925) | 12.013 (2.336) | <0.001 | 0.053 | 0.166 | 0.762 | <0.001 | 0.137 |
| ApoEε4 (−) (n = 688) | 12.658 (2.477) | 12.763 (2.244) | 12.524 (3.446) | 0.590 | 0.236 | 0.673 | 0.244 | 0.346 | 0.419 |
Independent of clinical classification we observed a small but significant decrease in serum zinc with age (Fig. 1a; see Fig. S1 (ESI†) for serum zinc levels in each classification), equating to a 0.051 ± 0.042 μM per year decrease in the AIBL cohort, or 0.40 ± 0.33% per year of mean serum zinc concentration. In AIBL, where AD subjects are, on average, 8.2 years older than their HC counterparts, this decrease in serum zinc with age represents a 0.418 ± 0.344 μM disparity between the two cohorts, which likely accounts for the 0.524 μM difference in serum zinc observed. Thus, when correcting for age as a covariate in the AIBL population,38 the previously observed decrease in serum zinc was abolished.
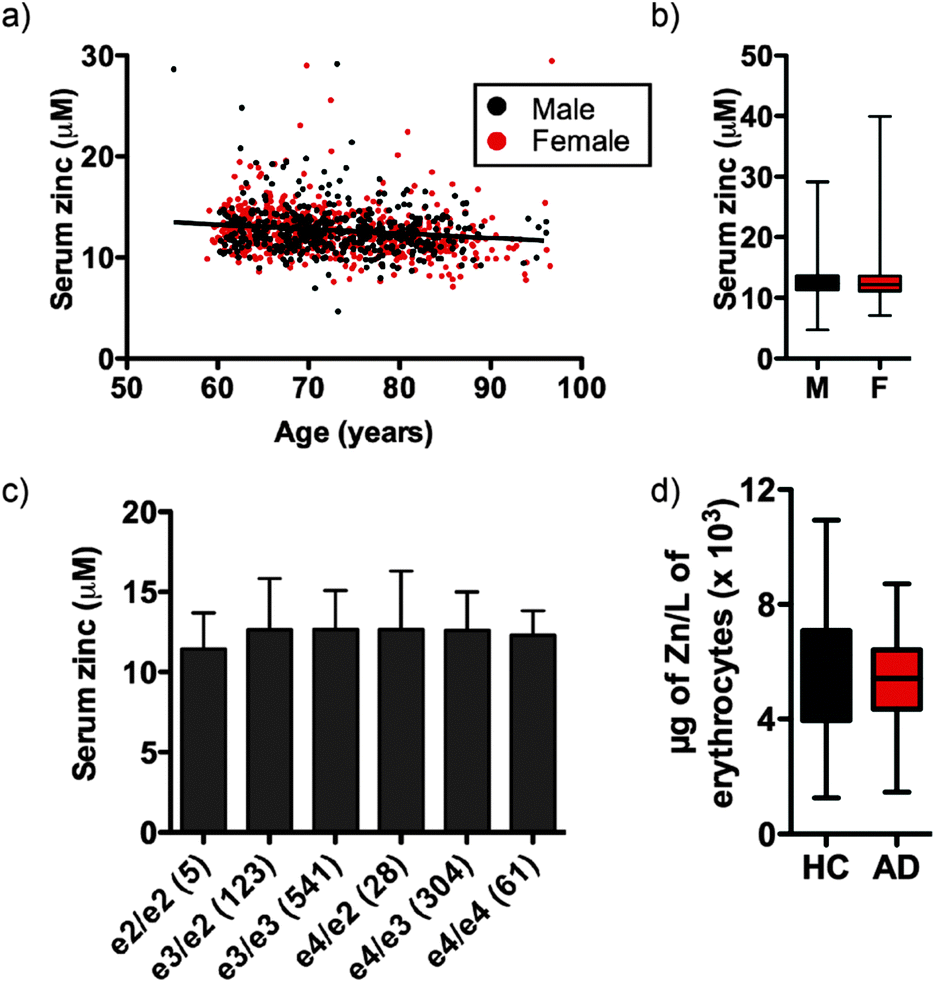 | ||
| Fig. 1 (a) Serum zinc decreases with age and is (b) consistent between sex, (c) ApoE allele status (n in parentheses) and (d) not significantly different in the erythrocyte fraction between HC and AD. | ||
Unlike previous reports,39 we did not observe a significant difference in serum zinc concentration according to sex (Fig. 1b). Serum zinc levels did appear lower in AD females as compared with AD males, however this did not reach statistical significance (female AD 11.910 ± 2.413 μM vs. male AD 12.706 ± 3.332 μM; p = 0.073). ANOVA of serum zinc levels according to ApoE allele status was also unchanged (Fig. 1c). As ApoEε4 carriers have increased risk of developing AD,40 we also examined the influence of ApoE allele status in each clinical classification. No statistically significant difference was observed between classes or within each classification (Table S2, ESI†), though there was a decreasing trend in ApoEε4 carriers across the three clinical classifications. Similarly, in a pilot study AD and HC participants (n = 40 per group) showed no significant difference in erythrocyte zinc concentration (Fig. 1d).
Understanding that subtle changes in serum zinc may not be reflected in bulk analysis, we used size exclusion inductively coupled plasma mass spectrometry (SEC-ICP-MS)41 to profile the zinc binding species present in plasma according to molecular size of protein species. Using SEC-ICP-MS we compared the zinc profile of 38 HC and 38 AD age and sex matched subjects. Reflecting the total serum zinc results, we observed no significant difference in zinc levels attributable to zinc-binding proteins in either clinical group (Fig. 2) (p > 0.05).
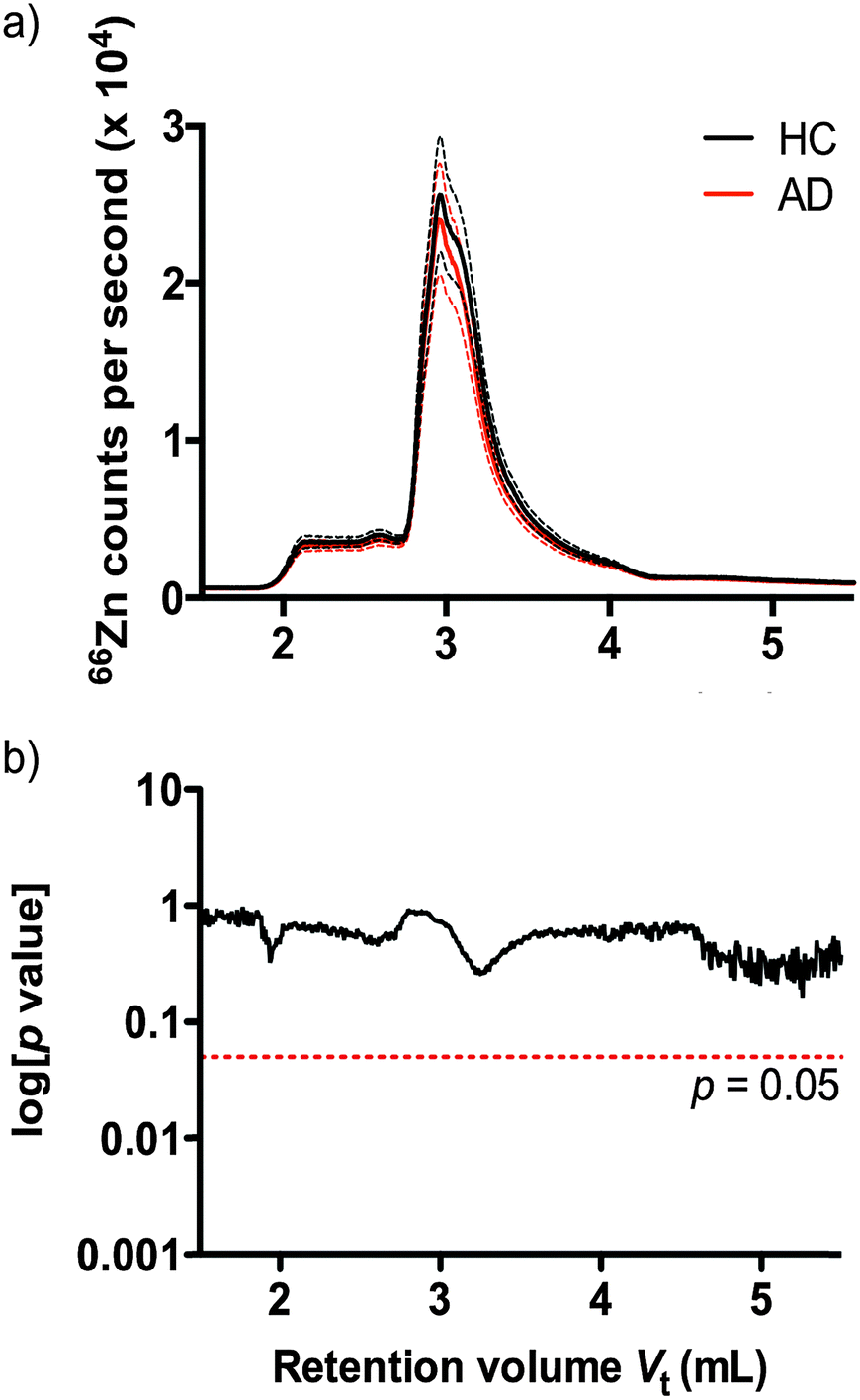 | ||
| Fig. 2 (a) SEC-ICP-MS chromatograms of mean 66Zn signal (solid line, ±1 SD dashed lines) in 38 AD and 38 HC subjects. (b) A two-tailed t-test on each point revealed no significant difference between groups. | ||
Peripheral zinc levels are highly regulated and are under the influence of homeostatic and environmental parameters such as diet or the concentration of other metals, including copper, and it is assumed that zinc homeostasis is compromised during the aging processes. In AIBL, the Cu/Zn ratio was slightly higher in the AD cohort (HC = 0.824; MCI = 0.806; AD = 0.798; p = 0.085) though again this was attributed to effects of age, as has been shown by others.42
We propose that previously reported decreases in serum zinc concentration may be attributed to either small data sets with low statistical power,20,21 or an effect of decreased zinc in the typically older AD population.22,23 The size of the AIBL study permits detailed study into the effects of age on serum zinc levels, which we found decreases inline with age. This does not, however, suggest that zinc is not implicated in AD pathology in the brain, where zinc enrichment in amyloid pathology is a robust feature of the disease.29,30,43 Regardless, we conclude that it is unlikely variations in zinc concentration due to changes in CNS metallostasis in AD can be detected in the periphery without the covariate influence of ageing masking subtle changes that are independent of disease.
Acknowledgements
We wish to thank the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (http://www.aibl.csiro.au/), including all scientists, participants and their families. This work was supported by the Cooperative Research Centre for Mental Health and by Operational Infrastructure Support from the Victorian State Government.Notes and references
- Alzheimer's Association, Alzheimer's Dementia, 2012, 8, 131–168 CrossRef PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- S. Ayton, P. Lei and A. I. Bush, Free Radical Biol. Med., 2013, 62, 76–89 CrossRef CAS PubMed.
- X. Huang, R. D. Moir, R. E. Tanzi, A. I. Bush and J. T. Rogers, Ann. N. Y. Acad. Sci., 2004, 1012, 153–163 CrossRef CAS PubMed.
- R. Squitti, Front. Biosci., 2012, 17, 451–472 CrossRef CAS.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, J. Neurochem., 2012, 120(suppl 1), 149–166 CrossRef CAS PubMed.
- I. Shcherbatykh and D. O. Carpenter, J. Alzheimer's Dis., 2007, 11, 191–205 CAS.
- A. I. Bush, J. Alzheimer's Dis., 2013, 33(suppl 1), S277–S281 Search PubMed.
- G. J. Brewer, BioFactors, 2012, 38, 107–113 CrossRef CAS PubMed.
- G. Lyubartseva and M. A. Lovell, BioFactors, 2012, 38, 98–106 CrossRef CAS PubMed.
- C. J. Frederickson, M. D. Hernandez and J. F. McGinty, Brain Res., 1989, 480, 317–321 CrossRef CAS PubMed.
- D. Bittel, T. Dalton, S. L. Samson, L. Gedamu and G. K. Andrews, J. Biol. Chem., 1998, 273, 7127–7133 CrossRef CAS PubMed.
- T. P. Dalton, D. Bittel and G. K. Andrews, Mol. Cell. Biol., 1997, 17, 2781–2789 CrossRef CAS PubMed.
- T. P. Dalton, Q. Li, D. Bittel, L. Liang and G. K. Andrews, J. Biol. Chem., 1996, 271, 26233–26241 CrossRef CAS PubMed.
- A. Bush, W. Pettingell, G. Multhaup, M. d Paradis, J. Vonsattel, J. Gusella, K. Beyreuther, C. Masters and R. Tanzi, Science, 1994, 265, 1464–1467 CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J. P. Vonsattel, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1997, 272, 26464–26470 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- C. Gonzalez, T. Martin, J. Cacho, M. T. Brenas, T. Arroyo, B. Garcia-Berrocal, J. A. Navajo and J. M. Gonzalez-Buitrago, Eur. J. Clin. Invest., 1999, 29, 637–642 CrossRef CAS PubMed.
- C. Jeandel, M. B. Nicolas, F. Dubois, F. Nabet-Belleville, F. Penin and G. Cuny, Gerontology, 1989, 35, 275–282 CrossRef CAS.
- L. Baum, I. H. S. Chan, S. K. K. Cheung, W. B. Goggins, V. Mok, L. Lam, V. Leung, E. Hui, C. Ng and J. Woo, BioMetals, 2010, 23, 173–179 CrossRef CAS PubMed.
- G. J. Brewer, S. H. Kanzer, E. A. Zimmerman, E. S. Molho, D. F. Celmins, S. M. Heckman and R. Dick, Am. J. Alzheimers Dis. Other Demen., 2010, 25, 572–575 CrossRef PubMed.
- H. Vural, H. Demirin, Y. Kara, I. Eren and N. Delibas, J. Trace Elem. Med. Biol., 2010, 24, 169–173 CAS.
- D. Shore, R. I. Henkin, N. R. Nelson, R. P. Agarwal and R. J. Wyatt, J. Am. Geriatr. Soc., 1984, 32, 892–895 CrossRef CAS PubMed.
- A. Haines, S. Iliffe, P. Morgan, T. Dormandy and B. Wood, Clin. Chim. Acta, 1991, 198, 261–266 CrossRef CAS.
- J. A. Molina, F. J. Jimenez-Jimenez, M. V. Aguilar, I. Meseguer, C. J. Mateos-Vega, M. J. Gonzalez-Munoz, F. de Bustos, J. Porta, M. Orti-Pareja, M. Zurdo, E. Barrios and M. C. Martinez-Para, J. Neural Transm.: Gen. Sect., 1998, 105, 479–488 CrossRef CAS.
- C. O. Hershey, L. A. Hershey, A. Varnes, S. D. Vibhakar, P. Lavin and W. H. Strain, Neurology, 1983, 33, 1350–1353 CrossRef CAS PubMed.
- R. N. Sahu, R. S. Pandey, M. N. Subhash, B. Y. Arya, T. S. Padmashree and K. N. Srinivas, Biol. Psychiatry, 1988, 24, 480–482 CrossRef CAS PubMed.
- J. R. Nuttall and P. I. Oteiza, Genes Nutr., 2014, 9, 379 CrossRef PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- F. M. Corrigan, G. P. Reynolds and N. I. Ward, BioMetals, 1993, 6, 149–154 CrossRef CAS PubMed.
- A. E. Panayi, N. M. Spyrou, B. S. Iversen, M. A. White and P. Part, J. Neurol. Sci., 2002, 195, 1–10 CrossRef CAS PubMed.
- G. Danscher, K. B. Jensen, C. J. Frederickson, K. Kemp, A. Andreasen, S. Juhl, M. Stoltenberg and R. Ravid, J. Neurosci. Methods, 1997, 76, 53–59 CrossRef CAS PubMed.
- D. Religa, D. Strozyk, R. A. Cherny, I. Volitakis, V. Haroutunian, B. Winblad, J. Naslund and A. I. Bush, Neurology, 2006, 67, 69–75 CrossRef CAS PubMed.
- D. L. Samudralwar, C. C. Diprete, B. F. Ni, W. D. Ehmann and W. R. Markesbery, J. Neurol. Sci., 1995, 130, 139–145 CrossRef CAS PubMed.
- C. M. Thompson, W. R. Markesbery, W. D. Ehmann, Y. X. Mao and D. E. Vance, Neurotoxicology, 1988, 9, 1–7 CAS.
- K. A. Ellis, A. I. Bush, D. Darby, D. De Fazio, J. Foster, P. Hudson, N. T. Lautenschlager, N. Lenzo, R. N. Martins, P. Maruff, C. Masters, A. Milner, K. Pike, C. Rowe, G. Savage, C. Szoeke, K. Taddei, V. Villemagne, M. Woodward, D. Ames and A. R. Group, Int. Psychogeriatr., 2009, 21, 672–687 CrossRef PubMed.
- Y. Hochberg and Y. Benjamini, Stat. Med., 1990, 9, 811–818 CrossRef CAS PubMed.
- I. M. Rea, Nutr. Res., 1989, 9, 121–125 CrossRef.
- E. Corder, A. Saunders, W. Strittmatter, D. Schmechel, P. Gaskell, G. Small, A. Roses, J. Haines and M. Pericak-Vance, Science, 1993, 261, 921–923 CAS.
- D. J. Hare, A. Grubman, T. M. Ryan, A. Lothian, J. R. Liddell, R. Grimm, T. Matsuda, P. A. Doble, R. A. Cherny, A. I. Bush, A. R. White, C. L. Masters and B. R. Roberts, Metallomics, 2013, 5, 1656–1662 RSC.
- A. Mezzetti, S. D. Pierdomenico, F. Costantini, F. Romano, D. De Cesare, F. Cuccurullo, T. Imbastaro, G. Riario-Sforza, F. Di Giacomo, G. Zuliani and R. Fellin, Free Radical Biol. Med., 1998, 25, 676–681 CrossRef CAS PubMed.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary methods, figure, tables and references. See DOI: 10.1039/c4mt00060a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |