 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox sulfur chemistry of the copper chaperone Atox1 is regulated by the enzyme glutaredoxin 1, the reduction potential of the glutathione couple GSSG/2GSH and the availability of Cu(I)†
Jens
Brose
a,
Sharon
La Fontaine
b,
Anthony G.
Wedd
a and
Zhiguang
Xiao
*a
aSchool of Chemistry and The Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: z.xiao@unimelb.edu.au; Fax: +61 3 9347 5180; Tel: +61 3 9035 6072
bSchool of Life and Environmental Sciences, Deakin University, Burwood, Victoria 3125, Australia
First published on 5th February 2014
Abstract
Glutaredoxins have been characterised as enzymes regulating the redox status of protein thiols via cofactors GSSG/GSH. However, such a function has not been demonstrated with physiologically relevant protein substrates in in vitro experiments. Their active sites frequently feature a Cys–xx–Cys motif that is predicted not to bind metal ions. Such motifs are also present in copper-transporting proteins such as Atox1, a human cytosolic copper metallo-chaperone. In this work, we present the first demonstration that: (i) human glutaredoxin 1 (hGrx1) efficiently catalyses interchange of the dithiol and disulfide forms of the Cys12–xx–Cys15 fragment in Atox1 but does not act upon the isolated single residue Cys41; (ii) the direction of catalysis is regulated by the GSSG/2GSH ratio and the availability of Cu(I); (iii) the active site Cys23–xx–Cys26 in hGrx1 can bind Cu(I) tightly with femtomolar affinity (KD = 10−15.5 M) and possesses a reduction potential of Eo′ = −118 mV at pH 7.0. In contrast, the Cys12–xx–Cys15 motif in Atox1 has a higher affinity for Cu(I) (KD = 10−17.4 M) and a more negative potential (Eo′ = −188 mV). These differences may be attributed primarily to the very low pKa of Cys23 in hGrx1 and allow rationalisation of conclusion (ii) above: hGrx1 may catalyse the oxidation of Atox1(dithiol) by GSSG, but not the complementary reduction of the oxidised Atox1(disulfide) by GSH unless Cuaq+ is present at a concentration that allows binding of Cu(I) to reduced Atox1 but not to hGrx1. In fact, in the latter case, the catalytic preferences are reversed. Both Cys residues in the active site of hGrx1 are essential for the high affinity Cu(I) binding but the single Cys23 residue only is required for the redox catalytic function. The molecular properties of both Atox1 and hGrx1 are consistent with a correlation between copper homeostasis and redox sulfur chemistry, as suggested by recent cell experiments. These proteins appear to have evolved the features necessary to fill multiple roles in redox regulation, Cu(I) buffering and Cu(I) transport.
Introduction
Much of the copper in mammalian cells is imported by the high-affinity Cu(I) transporter Ctr1 and is then distributed by metallo-chaperones to intra-cellular destinations.1 A major pathway involves delivery of copper by Atox1 to the Type P1B ATPase transporters ATP7A/7B in the trans-Golgi network (TGN) for incorporation into enzymes.1,2 Atox1 and the six N-terminal metal binding domains (MBDs) of ATP7A/7B all feature the high affinity Cu(I)-binding motif Cys–xx–Cys incorporated into a ferredoxin-like βαββαβ fold.3 Both in vivo and in vitro experiments suggest that copper delivery by Atox1 involves direct protein–protein interactions mediated by Cu(I) binding and exchange between Cys–xx–Cys motifs of the protein partners.4–6 In addition, recent studies also indicate that Cu(I)-mediated dimerisation of Atox1 is involved in its function as a transcription factor.7,8Cellular copper levels are maintained by regulation of Ctr1 levels at the plasma membrane and by reversible trafficking of ATP7A/7B from the TGN to vesicles and/or the plasma membrane for copper export. The latter process is controlled at the stage of copper transfer from Atox1 to ATP7A/7B via kinase phosphorylation at multiple sites on the ATPases.9–11
Glutaredoxins (Grxs) are thiol–disulfide oxido-reductase enzymes proposed to catalyse (de)glutathionylation of protein thiols (P-SH) via cofactors glutathione GSH and its oxidised form GSSG (see recent reviews12,13). However, such proposals have not been documented by in vitro experiments with physiologically relevant protein substrates. Model substrates including mercaptoethanol disulfide14 have been used as well as glutathionylated proteins generated by pure GSSG under non-physiological conditions.15
Many Grxs such as human glutaredoxin 1 (hGrx1) adopt the thioredoxin fold and feature a solvent-exposed Cys–xx–Cys motif that is the enzyme active site (Cys23–xx–Cys26 in Fig. 1).16,17 The equivalent Cys–xx–Cys motifs in Atox1 and the ATP7A/B domains MBD1–6 have evolved to bind CuI with high affinity (KD ∼ 10−17.5 M).18 However, it has been proposed that the hGrx1 active site (Cys–Pro–Tyr–Cys) is adapted to prevent binding of metal ions due to the presence of distorting Pro residues both within and near the active site.19
 | ||
| Fig. 1 Protein molecular structures: (a) hGrx1 (PDB code: 1JHB; thioredoxin fold) (b) hGrx1 (C8,26,79,83S)-GSH complex (1B4Q) and (c) CuI-Atox1 (1TL4; ferredoxin fold). Labelled amino acid residues and the GSH fragment are shown as sticks while the Cu(I) atom in CuI-Atox1 represented as a black sphere. Protein N- and C-termini are indicated by N and C, respectively. | ||
Yeast two-hybrid and mammalian co-immunoprecipitation experiments provide evidence that both the metallo-chaperone Atox1 and the hGrx1 interact with the ATPases ATP7A/7B and that these interactions require copper and the MBDs of ATP7A/7B.20,21 Although other interpretations are possible, the observations have been rationalised in terms of direct regulation of the Cu-ATPases by hGrx1 with a consequent influence on copper homeostasis. Over-expression of hGrx1 in neuronal cells perturbs copper metabolism.22 However, complementary experiments on hGrx1-null cells are needed for definitive assessment.
GSH (0.1–10 mM in the cytosol)23 regulates the redox status of cells via inter-conversion with GSSG.12,13 It can also act as a buffer of Cuaq+. The nature of Cu(I)–GSH interactions seems to vary with conditions and the specific complexes involved remain to be defined, together with the corresponding formation constants. However, at its typical cellular concentrations of >1 mM, GSH appears to buffer free Cuaq+ concentrations towards femtomolar levels.18 Although such affinities are too weak for GSH to compete effectively with copper chaperone proteins in the cytosol, recent evidence indicates that GSH may play a key role in copper uptake by Ctr1, possibly by mediating transfer of Cu(I) from Ctr1 to the chaperones.24
A recent report links copper homeostasis (via copper transporters/chaperones) with the GSH/GSSG couple in the cytosol.25 Redox enzymes of the thioredoxin family such as hGrx1 and glutathione reductase may participate by connecting the abundant thiol pool of GSH to the Cu(I)-binding thiol pool of proteins. It is also possible that the exposed Cys–xx–Cys motifs in Atox1 and MBDs may be protected from oxidation by glutathionylation (i.e., formation of Cys–S–SG bonds) mediated by hGrx1. Such processes are emerging as mechanisms of redox regulation and signalling, comparable to phosphorylation.26,27
The present work has examined redox sulfur chemistry involving Atox1 and the couple GSSG/2GSH mediated by hGrx1 with the aim of defining a potential molecular role for hGrx1 in copper metabolism. It provides the first demonstration that hGrx1 actually binds Cu(I) with femtomolar affinity and catalyses the redox sulfur chemistry of Atox1 (a physiologically relevant substrate) in a way regulated by both the reduction potential of the couple GSSG/2GSH and the availability of Cu(I). The evidence suggests possible roles for hGrx1 in copper metabolism including redox regulation of copper trafficking and delivery of copper to the redox partners of copper transporters. The yeast homologue of Atox1 (Atx1) was identified originally as an antioxidant (Atx) in yeast cells lacking a functional superoxide dismutase 1 (Sod1).28 This work identifies a plausible molecular mechanism for such action that involves both hGrx1 and copper.
Results and discussion
Protein generation and quantification
hGrx1 features five Cys residues (at positions 8, 23, 26, 79, 83). Two of them constitute the enzyme active site defined by Cys23–xx–Cys26 (Fig. 1a). To evaluate the contributions of these residues towards Cu(I)-binding and thiol–disulfide oxido-reductase activity, wild type hGrx1 and four variants (C23S, C26S, C23,26S and C8,79,83S) were generated and isolated. The identity of each protein was confirmed by DNA sequencing of the expression plasmids and electrospray ionization mass spectrometry (ESI-MS) analysis of the isolated proteins (Table S1, ESI†). Each sample as isolated was converted to its fully reduced apo-form by incubation with excess dithiothreitol (Dtt) or tris(2-carboxyethyl)phosphine (Tcep), followed by buffer change to remove excess reductant under anaerobic conditions in a glove-box ([O2] < 1 ppm). Elman analysis29 confirmed the expected thiol content for each sample based on concentrations estimated from the solution absorbance at 280 nm using a calculated molar absorptivity of ε = 2980 M−1 cm−1.Atox1 was expressed and isolated using reported protocols.18,30 As indicated previously, purified samples were a mixture of two components (7401.7 and 7270.5 Da) corresponding to molecules with and without the first methionine residue.30 The relative contents of these two components varied from batch to batch. Three equivalents of cysteine thiols were detected in all fully reduced samples.
hGrx1 binds Cu(I) with femtomolar affinity
hGrx1 possesses five Cys residues including two in the active site motif Cys23–xx–Cys26. The latter sequence is commonly present in high affinity Cu(I)-binding proteins such as Atox1 or in protein domains such as the MBDs of ATP7A/7B. Consequently, hGrx1 is likely to bind Cu(I) with high affinity. Two Cu(I) probes bicinchoninic anion (Bca) and bathocuproine disulfonate anion (Bcs) (Fig. 2) were employed to evaluate the Cu(I) binding properties of hGrx1 and its protein variants. Atox1 was used for comparison.18 | ||
| Fig. 2 Structures of Cu(I) probe ligands. | ||
Both Bca and Bcs bind Cu(I) specifically to yield 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 chromophoric complexes [CuIL2]3− (L = Bca or Bcs) with high affinity (β2 = 1017.2 M−2 for Bca and 1019.8 M−2 for Bcs).18 Consequently, depending on the total concentrations of the ligands L relative to Cu(I), they can buffer free Cuaq+ concentrations (hereafter expressed as p[Cu+] = −log[Cuaq+]) in the range p[Cu+] = 12–16 and 15–19, respectively.31
:2 chromophoric complexes [CuIL2]3− (L = Bca or Bcs) with high affinity (β2 = 1017.2 M−2 for Bca and 1019.8 M−2 for Bcs).18 Consequently, depending on the total concentrations of the ligands L relative to Cu(I), they can buffer free Cuaq+ concentrations (hereafter expressed as p[Cu+] = −log[Cuaq+]) in the range p[Cu+] = 12–16 and 15–19, respectively.31
A solution with p[Cu+] = 12.7 was prepared by generating the anion [CuI(Bca)2]3− (34 μM) in KPi buffer (50 mM, pH 7.0) (see caption of Fig. 3). Increasing amounts of either wild type hGrx1 (hGrx1-wt) or the triple variant hGrx1-C8,79,83S (hGrx1-tm, in which the three Cys residues outside the active site Cys23–xx–Cys26 are replaced with Ser) were added to samples of this probe solution. This induced decreases in the concentration of [CuI(Bca)2]3−, signalling transfer of Cu(I) from the probe complex to the proteins. The data in Fig. 3a indicate that, when free Cuaq+ concentration was buffered at p[Cu+] = 12.7, hGrx1-wt bound more than one equivalent of Cu(I) while hGrx1-tm bound one equivalent only. When free Cuaq+ concentration was constrained further to p[Cu+] ∼ 14, both hGrx1-wt and hGrx1-tm bound one equivalent of Cu(I) only (Fig. 3b). Therefore, hGrx1-wt can bind at least two equivalents of Cu(I) with different affinities: KD < 10−14 M for the highest affinity binding site and KD = 10−13–10−14 M for lower affinity binding of a second equivalent of Cu(I). On the other hand, the removal of the three Cys residues in hGrx1-tm outside the active site also removed the lower affinity binding.
 | ||
| Fig. 3 Determination of the Cu(I) binding stoichiometry of the highest affinity site in hGrx1. Removal of Cu(I) from the probe complex [CuI(Bca)2]3− by hGrx1 proteins was detected by the decrease in its concentration (monitored by the absorbance at 562 nm) with increasing concentration of the added protein (solid circles for hGrx1-wt; empty circles for hGrx1-tm). The probe complex was prepared with the following compositions: [Cu]tot = ∼34 μM; [NH2OH]tot = 1.0 mM, [Bca]tot = 100 μM for (a) and 200 μM for (b) in KPi buffer (25 mM, pH 7.0). Note: p[Cuaq+] in the initial probe complex solution was calculated to be 12.7 in (a) and 13.9 in (b). | ||
Variants C23S and C26S (with one Cys residue in the active site Cys23–xx–Cys26 motif in hGrx1-wt replaced) displayed complete loss of the Cu(I) site of highest affinity but retained the weaker affinity (Table 1; Fig. S1, ESI†). This weaker Cu(I) binding induced a tendency for the proteins to precipitate, most likely due to inter-protein linkage.
| Protein | logKD |
Competing L (μM) | ||
|---|---|---|---|---|
| pH 7.0a | pH 5.7b | Bca | Bcs | |
| a In KPi buffer. b In Mes buffer. c With 7 M urea in the solution. d Less reliable due to slow changes in spectra. | ||||
| hGrx1-wt | −15.5(1) | 500/250 | ||
| −15.6(1) | 80 | |||
| −17.5(1)c | 250/125 | |||
| −14.3(1) | 400/200 | |||
| hGrx1-tm | −15.5(1) | 500/250 | ||
| −15.6(1) | 80 | |||
| −16.6(1)c | 250/125 | |||
| −14.4(1) | 400/200 | |||
| Atox1 | −17.4(1) | 500/250 | ||
| −16.5(1)c | 250/125 | |||
| −14.6(1) | 400/200 | |||
| hGrx1-C23S | −13.5(3)d | 200 | ||
| hGrx1-C26S | −13.3(3)d | 200 | ||
The above experiments demonstrate that both Cys23 and Cys26 are required for the highest affinity for Cu(I) (KD < 10−14 M). Equivalent experiments with the free Cuaq+ concentration at the more stringent constraint of p[Cu+] = ∼15 (increased total ligand concentration; see caption of Fig. 4) led to an effective competition for Cu(I) between ligands Bca and P = hGrx1-wt or hGrx1-tm (Fig. 4, data sets (ii)). This allowed an estimation of their Cu(I) affinities viaeqn (1) and (2) (Fig. 4):31
| [CuIL2]3− + P ⇌ CuI–P + 2L2− (L = Bca) | (1) |
 | (2) |
 | ||
| Fig. 4 Determination of KD for the site with highest affinity for Cu(I) in hGrx1. The experiments are similar to those of Fig. 3. (a) hGrx1-wt; (b) hGrx1-tm. The data in a(i) and b(i) are for hGrx1-C23S and hGrx1-C26S, respectively. The probe complex was prepared as following: [Cu]tot = 38 μM; [Bca]tot = 500 μM and [NH2OH]tot = 1.0 mM in KPi buffer (25 mM, pH 7.0). p[Cu+] was calculated to be 14.9. The experimental data points shown with empty circles in (iii) were obtained from a 1:1 dilution of each solution in (ii). The solid traces in (ii, iii) are the fitting curves of the experimental data points to eqn (3). The dashed traces in (iii) are the simple 1:1 dilution of data sets (ii) and demonstrate that the equilibrium position in (iii) has adjusted from that present in (ii). | ||
Curve fitting of each of the two sets of experimental data (ii and iii) for each protein to eqn (2) based on the known β2 for [CuI(Bca)2]3− (1017.2 M−2)18 yielded the same KD = 10−15.5 M at pH 7.0 for both hGrx1-wt and hGrx1-tm. The individual estimates were indistinguishable within experimental error (Table 1). This affinity falls within the effective buffer range of probe Bcs (Fig. 2), as well as that of Bca. Equivalent experiments with Bcs generated similar competition for Cu(I) and provided the same estimate (KD = 10−15.6 M) based on β2 = 1019.8 M−2 for [CuI(Bcs)2]3− (Fig. S2, ESI;†Table 1). On the other hand, variants C23S and C26S were unable to compete for Cu(I) under the same conditions (Fig. 4; data sets (i)). These experiments demonstrated that the two Cys residues in the active site of hGrx1 contribute solely to its femtomolar affinity for Cu(I). The other three Cys residues may contribute to the weaker binding sites.
Interestingly, a conserved cis-Pro residue adjacent to the β3 strand and another within the Cys–xx–Cys motif occur in the thioredoxin family of enzymes. They have been proposed to play a role in preventing assembly of Fe–S clusters in this site and possibly in preventing binding of other metal ions as well.19 Two such Pro residues are present in hGrx1 studied in this work (Pro71 and motif Cys–Pro–Tyr–Cys). However, the present work demonstrates that the enzyme is able to bind one equivalent of Cu(I) with femtomolar affinity. The thioredoxin from E. coli can do the same.32
The protein local environment fine tunes the affinities of hGrx1 and Atox1 for Cu(I)
Copper chaperone Atox1 is one of the key proteins involved in copper nutrition in human cells.4 Its solvent-exposed Cys–xx–Cys motif binds Cu(I) with higher affinity (KD = 10−17.4 M at pH 7.0) than does hGrx1 (KD = 10−15.5 M).18 This is likely associated with differences in protein local environments that fine-tune properties (ligand pKa, reduction potential, Cu(I) binding affinity) for their specific biological functions. To explore a possible correlation of these properties, the affinities of hGrx1-tm and Atox1 for Cu(I) were compared as a function of solution pH.The affinities of hGrx1-tm and Atox1 for Cu(I) both decreased in the pH range 7.0 to ∼5.0. The decrease per pH unit is smaller for hGrx1-tm than for Atox1 and the affinities are the same at about pH 5.5 (Fig. 5a; Table 1). This is consistent with the very low pKa (∼3.5)33 estimated for Cys-23 in hGrx1. This is the key to its catalytic role as an oxido-reductase and is responsible for the weaker Cu(I) affinity of hGrx1. This also correlates with the fact that the reduction potential of the Cys–xx–Cys motif in hGrx1 is more positive than that of Atox1 (vide infra). Interestingly, upon unfolding the proteins with urea (7.0 M; pH 7.0), the affinity of hGrx1-tm for Cu(I) increased (KD decreased) by about an order of magnitude (KD = 10−16.6 M) whereas that of Atox1 decreased to an essentially identical value (KD = 10−16.5 M; Fig. 5b; Table 1). Within experimental error, these values are indistinguishable from the Cu(I) affinities reported recently for several proteins containing a Cys–xx–Cys motif (including Atox1) under the same unfolding condition.32
 | ||
| Fig. 5 Comparison of affinities for Cu(I) under different conditions: (a) variation of logKD for CuI-Grx1-tm (red circles) and CuI-Atox1 (blue circles) with pH; (b) determination of logKD for CuI-Grx1-tm (red triangles) and CuI-Atox1 (blue triangles) under denaturing conditions. Equilibrium transfer of Cu(I) from the probe complex [CuI(Bcs)2]3− (composition: [Cu]tot = 38 μM; [Bcs]tot = 250 μM) to protein hGrx1-tm or Atox1 was monitored in KPi buffer (50 mM, pH 7.0) containing urea (7.0 M). The experimental data points in b(ii) were obtained from a 1:1 dilution of each solution in b(i); the traces in (i, ii) are the fitting curves of the experimental data points for hGrx1-tm to eqn (3) which generate a consistent logKD = −16.6 ± 0.1 for CuI-Grx1-tm. An essential identical logKD = −16.5 was determined for CuI-Atox1 under the same conditions. | ||
Clearly, it is the protein local environment that fine-tunes the properties of these Cys–xx–Cys motifs for their specific functions. The unusually low pKa of Cys23 in hGrx1 is attributable to several factors including its location at the N-terminus of an α helix (that may stabilize the thiolate anion via partial positive charge associated with the helix dipole) and electrostatic interaction with positively charged residues near the active site (such as Lys20) and perhaps the adjacent residue Pro24 which may impose a more favourable positioning of Cys23 relative to the helix; (Fig. 1a).12,16,34 Upon protein unfolding, these secondary structural impacts on the Cys–xx–Cys motif are removed and the properties of the Cys–xx–Cys motifs from different sources become similar. Notably, under denaturing conditions, the affinity of hGrx1-wt for Cu(I) is still higher by an order of magnitude than those of Atox1, hGrx1-tm and other proteins (Table 1 and ref. 32), apparently due to an involvement of its other Cys residues in Cu(I) binding under the conditions.
The reduction potential of Atox1 is more negative than that of hGrx1
Preliminary experiments suggested that the extent of oxidation of the Cys–xx–Cys motifs of both Atox1 and hGrx1 could be controlled by the glutathione disulphide/glutathione redox couple GSSG/GSH that can buffer the effective reduction potential of the solution. This allowed estimation of the protein reduction potentials. The experiments were performed in a series of redox buffers containing varying molar ratios of GSSG/GSH at total concentrations in the range [GSH + 2GSSG] = 1.0–4.0 mM in KPi buffer (50 mM, pH 7.0). Each protein (10 μM) was incubated overnight in the redox buffers under anaerobic conditions to ensure attainment of equilibrium. The systems were quenched by alkylation with excess iodoacetamide, allowing estimation of the ratio of the fully reduced to fully oxidized protein forms (i.e., P-(SH)2/P-(SS)) by ESI-MS analysis (see Fig. 9 below and Fig. S4 and S5, ESI†).35 At a fixed pH, the reduction potentials of the redox buffers and of the target proteins, respectively, may be calculated via the Nernst relationships of eqn (3) and (4):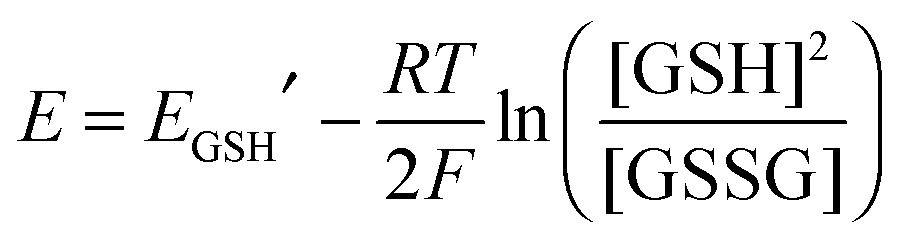 | (3) |
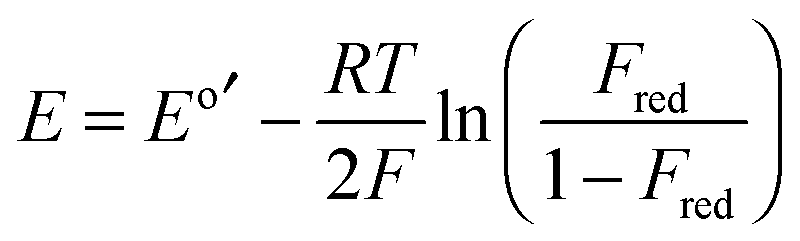 | (4) |
E GSH′ is the conditional standard reduction potential of the couple GSSG/2GSH which is pH-dependant and is equal to −240 mV at pH 7.0;36Eo′ is the conditional standard reduction potential of the couple P-(SS)/P-(SH)2 which is also pH-dependant; Fred is the fraction of the fully reduced protein to the sum of the fully reduced and fully oxidized protein forms (i.e., Fred = [P-(SH)2]/[P-(SH)2 + P-(SS)]). The minor form P-(SH)(SSG) (see Fig. 9 below and Fig. S4, ESI†) was not included in eqn (4) since it was a redox intermediate in equilibrium with both fully reduced and fully oxidized forms and cancels out in the calculation.37 This redox intermediate reached a maximal fractional level of ∼10% at ∼50% oxidation–reduction (Fig. 6, empty circles).
 | ||
| Fig. 6 Determination of reduction potentials of the CxxC motif in Atox1 (shown in green) and hGrx1-tm (shown in red). Each protein (∼10 μM) was incubated overnight under anaerobic conditions in a series of redox buffers GSSG/GSH (total [GSH + 2 GSSG] = 1.0–4.0 mM in KPi (50 mM, pH 7.0)) with reduction potentials defined by eqn (3). The free thiols were then alkylated with excess iodoacetamide (∼10 mM). ESI-MS analysis of the fractions of the reduced and oxidized forms was carried out as shown in Fig. 8 below and Fig. S3 (ESI†). The reduction potential window of cells under typical cellular conditions (∼1% GSSG with total [GSH] varying in the range 1–10 mM) is estimated, viaeqn (3), to be between −210 mV to −240 mV (shaded in yellow-green), consistent with a recent in-cell determination in yeast cells.44 The fractions of P-(SH)(SSG) detected for Atox1 and hGrx1-tm are shown in green and red empty circles, respectively. | ||
The fully reduced fraction Fred for both Atox1 and hGrx1-tm changed sensitively within different reduction potential windows (Fig. 6). Curve-fitting of the experimental data to eqn (4) led to the estimates Eo′ = −188 mV and −118 mV at pH 7.0 for Atox1 and hGrx1-tm, respectively. The reduction potential of Atox1 is 70 mV more negative than that of hGrx1-tm. This correlates with the lower pKa of Cys23 in hGrx1 and its lower affinity for Cu(I). These differences are attributed mainly to the positively charged environment surrounding the catalytic residue Cys23 in hGrx1.12,16,34
The reduction potentials at pH 7.0 determined in this work for Atox1 (−188 mV) and hGrx1 (−118 mV) are, respectively, 40 mV and 100 mV more positive than recent literature values of −229 mV and −220 mV.25 A slightly more negative value for hGrx1 (−232 mV) was reported earlier.37 The reduction potential of a thiol group is pH-dependant.36 The two literature estimates and the present work were conducted in the same buffer using the same couple as redox buffer. The differences lie in the more complex approaches used previously to quench the reaction and quantify the protein oxidation states. The method of alkylation and ESI-MS analysis employed here is justified by the following experiments: (i) controls in which a solution of iodoacetamide with extra GSSG was added as quenching reagent provided the same results as those using iodoacetamide alone; this demonstrates that the rate of alkylation was rapid compared the rate of change of the redox equilibrium; (ii) both the alkylation/ESI-MS probe and the solution probe [CuI(Bca)2]3− provided equivalent results in the kinetics experiments of Fig. 8c given below (red crosses versus blue dots; see also Fig. 9 below). The fact that the reduction potential of hGrx1 is 70 mV more positive than that of Atox1 (Fig. 6) is consistent with all the experimental data presented below.
The active site variant hGrx1-C26S is more active in disulfide reduction than the native enzyme
A classic assay based on coupled enzyme reactions was employed (Fig. 7a).33 The first reaction is reduction of mercaptoethanol disulfide (HOC2H4S)2 to mercaptoethanol HOC2H4SH via oxidation of GSH to GSSG with hGrx1 as catalyst. The second reaction reduces product GSSG back to GSH with glutathione reductase (Grase) as catalyst. The oxidized product nicotinamide adenine dinucleotide phosphate (NADP+) of reducing agent NADPH was detected directly by the absorbance change for NADPH at 340 nm (ε = 6319 M−1 cm−1).14,38 The assay conditions were set to ensure that the overall oxidation rate of NADPH was proportional to hGrx1 concentration (Table 2). Under these conditions (Fig. 7), the catalytic turnover rate for NADPH was ∼280 min−1 for hGrx1. Conversion of the N-terminal Cys23 to Ser to form variant hGrx1-C23S led to ∼90% loss of activity while substitution of both Cys23 and Cys26 to Ser in hGrx1-C23,26S led to complete loss (Fig. 7b(iv)). In contrast, substitution of the C-terminal Cys26 to Ser in hGrx1-C26S increased the activity by >100% (Fig. 7b(i, ii); Table 2). Addition of three equivalents of Cu(I) (relative to hGrx1) into the same assay mixture for hGrx1-wt decreased the activity by ∼70% (Fig. 7b(ii, iii); Table 2). The femtomolar affinity of the hGrx1 active site for Cu(I) ensures that it will compete strongly with GSH for the added Cu(I). These observations demonstrate that: (i) binding of Cu(I) to hGrx1 inhibits its catalytic activity for thiol–disulfide exchange; (ii) a mono-thiol mechanism via Cys23 only is more efficient than a dithiol mechanism involving both Cys23 and Cys26. In fact, the presence of Cys26 inhibits activity in the current assay (compare Fig. 7b(i, ii)), as reported previously with a similar assay.17 | ||
| Fig. 7 hGrx1 catalytic activity. (a) Reaction scheme: hGrx1 catalyses reduction of mercaptoethanol disulfide by GSH to yield oxidation product GSSG which is reduced back to GSH by NADPH catalysed by the second enzyme glutathione reductase (Grase). The overall catalytic rates were detected by the decreasing absorbance of NADPH at 340 nm. (b) Relative activity: (i) hGrx1-C26S; (ii) hGrx1-wt; (iii) hGrx1-wt + 3 equiv. of Cu(I); (iv) hGrx1-C23,26S. | ||
| Protein | Turnover rate for NADPH (min−1) | Turnover rate for (HOC2H4S)2 (min−1) | Relative to WT |
|---|---|---|---|
| hGrx1 | 562 | 281 | 1.0 |
| C23S | 58 | 29 | 0.1 |
| C26S | 1280 | 640 | 2.3 |
| C23,26S | 0 | 0 | 0 |
| hGrx1 + 3Cu(I) | 176 | 88 | 0.3 |
It has been suggested that (HOC2H4S)2 is not a direct substrate. Incubation with GSH is proposed to spontaneously generate the mixed disulfide HOC2H4S–SG that is the real substrate. A ping-pong mechanism has been proposed (Scheme 1).39,40 The presence of Cys26 close to the crucial Cys23 residue allows formation of an internal disulfide bond that inhibits the enzyme activity. Likewise, high affinity Cu(I) binding to the active site Cys23–xx–Cys26 motif may also suppress the nucleophilic attack of the Cys23 thiolate on the mixed disulfide substrate and so inhibit catalytic activity.
 | ||
| Scheme 1 Mechanism for reduction of (HOC2H4S)2 by GSH catalysed by hGrx1.39,40 The catalysis is inhibited by either Cu(I) binding to the active site motif Cys23–xx–Cys26 or by formation of a disulfide bond between Cys23 and Cys26. It is eliminated by a removal of the critical residue Cys23. The S atoms of Cys23 and Cys26 are represented, respectively, by a bold red S and an unbold red S. | ||
The unique catalytic role of Cys23 is attributed primarily to its very low pKa (∼3.5)33 that ensures its existence at physiological pH as a thiolate ready for nucleophilic attack on disulfide bonds. Its solvent-exposed location over a surface cleft may also be important to allow it to act as a GSH binding site to facilitate formation of the mixed Cys23S–SG (Fig. 1b; Scheme 1).
hGrx1 catalyses oxidation of Atox1 by GSSG
One of the primary functions proposed for hGrx1 is to prevent irreversible oxidation of protein thiols by catalysing their reversible (de)glutathionylation employing cofactors GSSG/GSH.41 However, the reported activities of hGrx1 and similar enzymes have been dominated by assays based on model substrates such as (HOC2H4S)2, as presented above.A new assay was developed here to evaluate how hGrx1 may mediate the sulfur redox chemistry of protein thiols involved in copper nutrition. It mimics cellular reactions and conditions based on catalytic oxidation of the fully reduced Cu(I)-binding protein Atox1 by GSSG with hGrx1 as catalyst. Its design is shown schematically in the inset of Fig. 8. The catalytic oxidation reaction (framed by the green dotted line) was monitored and regulated by a binding/buffering reaction that provided competition for Cu(I) between reduced Atox1 and a chromophoric Cu(I) probe ligand L (L = Bca or Bcs; framed by the red dotted line). The availability of Cu(I) to Atox1 was controlled by ligand L and the oxidation rate was followed in real time by the loss of high affinity for Cu(I) upon conversion of Atox1 to oxidised forms (internal disulfide or glutathionylated). The released Cu(I) binds rapidly to the probe ligand L to yield a chromophoric complex [CuIL2]3−.
 | ||
| Fig. 8 Comparison of the oxidation rate of Atox1 (35 μM) by GSSG (400 μM) catalysed by hGrx1-wt (blue trace), hGrx1-C26S (green trace) or no catalyst (red trace) in KPi buffer (20 mM; pH 7.0; NH2OH, 1.0 mM). Concentration of hGrx1 = 2.0 μM. p[Cuaq+] was buffered at (a) ≤16 by buffer: [Cu(I)]tot = 35 μM, [Bca]tot = 500 μM; (b) ≤17 M by buffer [Cu(I)]tot = 35 μM, [Bcs]tot = 140 μM; (c) no buffer. The rates in (a, b) were followed in real time by increase in concentration of the Cu(I) probe [CuI(Bca)2]3− and the rate in (c) by quenching the oxidation at various reaction time points with the same probe [CuI(Bca)2]3− but lower [Bca]tot = 100 μM (see Experimental section for details). The red crosses shown in (c) document an alternative quantification by quenching the oxidation via alkylation and ESI-MS analysis (see Fig. 9 below). Inset: scheme for the assay that consists of the catalysis (framed in green dots) and the associated detection of Cuaq+-buffering and Cu(I)-transfer (framed in red dots). | ||
The assay was conducted in KPi buffer at pH 7.0. Substrate Atox1 was provided as a mixture of apo-Atox1 and CuI-Atox1 in a ratio controlled by the concentration of ligand L. When ligand Bca at 500 μM was used (buffering free Cuaq+ concentration at p[Cu+] = ∼16), Atox1 was expected to be present predominantly (>95%) as CuI-Atox1 (as predicted from its known KD = 10−17.4 M).18 Under such conditions, Atox1 (35 μM) was oxidised slowly by GSSG (400 μM) when hGrx1-C26S (its most active form; 2.0 μM) was used as catalyst (Fig. 8a; initial turnover rate, ∼0.1 min−1). hGrx1-wt exhibited no catalytic activity under these conditions (Fig. 8a and Fig. S3a, ESI†).
When the higher affinity ligand Bcs at 140 μM was used (imposing p[Cu+] = ∼17), ∼80% of Atox1 was expected to be present as CuI-Atox1. hGrx1-wt then exhibited an initial turnover rate of ∼0.2 min−1 while that of hGrx1-C26S was enhanced 20-fold to an initial turnover rate of ∼2 min−1 (Fig. 8b and Fig. S3b, ESI†).
On the other hand, when substrate Atox1 was presented in apo form only (no added Cu(I)), both hGrx1-wt and hGrx1-C26S catalysed oxidation of Atox1 by GSSG much more efficiently (Fig. 8c and Fig. S3c, ESI†). hGrx1-C26S was again the more active enzyme than hGrx1-wt (initial rate ∼7.7 min−1vs. ∼2.3 min−1). The rates in these experiments were determined by transferring aliquots of reaction mixture at different reaction times into solutions containing Cu(I) probe [CuI(Bca)2]3− to quench and quantify the oxidation. Alternatively, the oxidation rates could be determined by transferring aliquots of the same reaction mixture into solutions of excess iodoacetamide to quench and quantify by alkylation of the Cys thiols and ESI-MS analysis.35 The outcomes of the two approaches were the same, within the experimental error (blue dots versus red crosses in Fig. 8c).
Equivalent experiments with hGrx1 variants as catalysts showed that the catalytic activity of hGrx1-tm was always lower than that of hGrx1-C26 but indistinguishable from that of hGrx1-wt. On the other hand, hGrx1-C23S and hGrx1-C23,26S exhibited no catalytic activity. It must be noted that, under the different conditions tested in Fig. 8, free Cuaq+ was buffered at such low concentrations (p[Cu+] ≥ 16) that all forms of hGrx1 could not compete for Cu(I). It can be concluded that the Cuaq+ buffer itself had little impact on the enzyme activity and the observed catalytic activities may be attributed to the Cu-free forms of the hGrx1 and its variants.
Three conclusions may be drawn (the nature of the overall processes is depicted in Scheme 2): (i) Cys23 in hGrx1 plays a central role in the catalytic oxidation of Atox1 by GSSG. The adjacent residue Cys26 may suppress the catalysis via formation of an internal disulfide bond with Cys23. The other non-active site Cys residues (Cys8,79,83) have little impact on the catalysis; (ii) apo-Atox1 is thermodynamically vulnerable to oxidation but the process is slow (even under oxidising conditions: initial concentration of GSSG, 400 μM) unless catalysed by hGrx1; (iii) binding of Cu(I) to Atox1 protects it from oxidation.
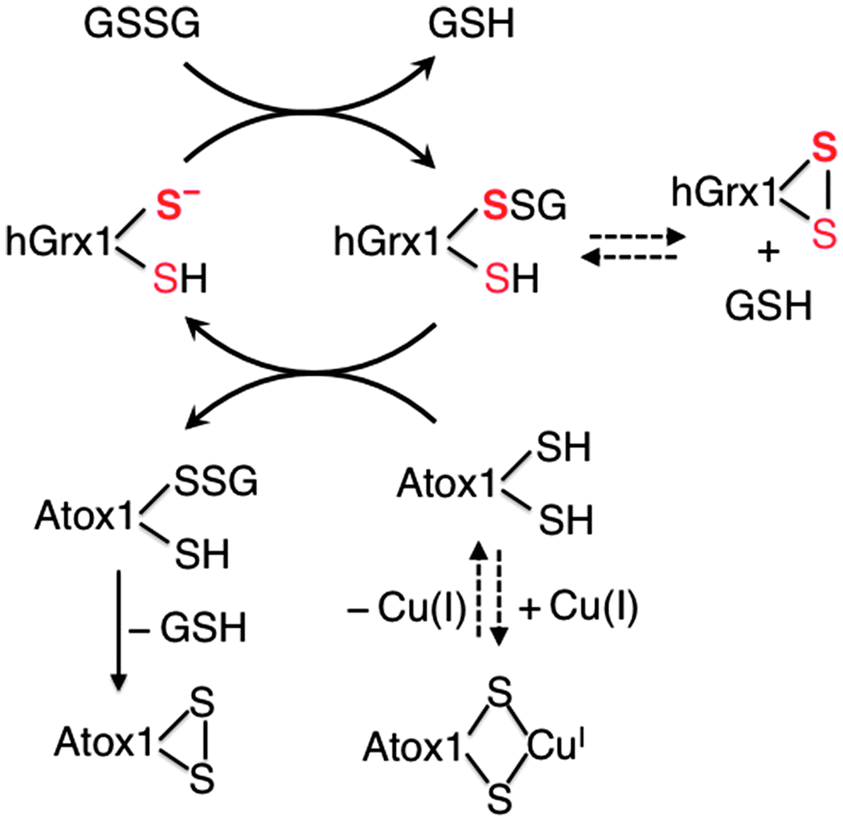 | ||
| Scheme 2 Proposed reaction mechanism for oxidation of Atox1 by GSSG catalysed by hGrx1. The catalysis is inhibited either by formation of a disulfide bond in the active site of hGrx1 or by Cu(I) binding to Atox1. It is promoted by formation of an internal disulfide bond in Atox1. | ||
The internal disulfide forms of Atox1 and hGrx1 are thermodynamically favoured relative to glutathionylated forms
A number of different oxidation processes promoted by GSSG may lead to loss of high affinity binding of Cu(I) to the dithiol Cys–xx–Cys motif in Atox1. They include formation of an internal disulfide bond leading to Atox1-(SS) and/or mixed disulfide forms such as Atox1-(SH)(SSG) and Atox1-(SSG)2. To address this issue, the reaction product(s) formed during the course of catalytic oxidation of Atox1 by hGrx1-wt (Fig. 8c; red crosses) were characterised. The reactions were quenched by alkylation of protein thiols, as described above. The products were characterised directly by ESI-MS analysis (Fig. 9).35 Two ions are expected for each of the reactant and products as the Atox1 samples were a mixture of molecules with and without an N-terminal methionine residue. Atox1 possesses three Cys residues (Cys12 and Cys15 in the Cys–xx–Cys motif and Cys41, Fig. 1c) and the reduced substrate form was detected as the fully alkylated species Atox1-(SA)3 (Fig. 9). The two Cys residues in the Cys–xx–Cys motif were oxidised to an internal disulfide bond: Atox1-(SA)(SS) (>96%) after 15 min with minimal production of glutathionylated forms: Atox1-(SSG)(SS) (<2%); Atox1-(SA)2(SSG) (<2%). | ||
| Fig. 9 ESI-MS analysis (deconvoluted spectra) of the oxidation of Atox1 by GSSG catalyzed by hGrx-wt (under the conditions of Fig. 8c). The reaction was quenched by alkylation of unreacted thiols by iodoacetamide to label residues indicated as –SA. The two ions identified by parentheses differ in the mass of an N-terminal methionine as the Atox1 reactant was a mixture of two forms. | ||
To assess the oxidation products formed under non-catalytic conditions, Atox1 was incubated overnight in redox buffers containing different ratios of GSSG and GSH. The products were again alkylated and detected by ESI-MS. Under physiologically relevant conditions within the reduction potential window of normal cells (E′, −210 to −240 mV),12 Atox1 remained predominantly in its fully reduced form (Fig. 10a). When the buffer was made more oxidising at E′ ∼ −190 mV, about 50% of the protein appeared as a mixture of several oxidised forms including Atox1-(SA)(SS) (∼40%), Atox1-(SSG)(SS) (<1%) and Atox1-(SA)2(SSG) (∼10%) (Fig. 10b). When the GSSG:GSH ratio was increased ∼100 fold to the non-physiological value of E′ > −100 mV, essentially quantitative conversion to the disulfide form ensued (Fig. 10c; ions detected: Atox1-(SA)(SS), >98%; Atox1-(SSG)(SS), <1%; Atox1-(SA)2(SSG), <1%). Even under the extreme conditions of a very high concentration of pure GSSG (25 mM), the oxidation forms of the Cys–xx–Cys motif in Atox1 were still dominated by the disulfide bond (Atox1-(SSG)(SS), ∼75%; Atox1-(SSG)3, ∼23%) while the isolated single Cys41 was trapped largely in glutathionylated form (∼98%) (Fig. 10d).
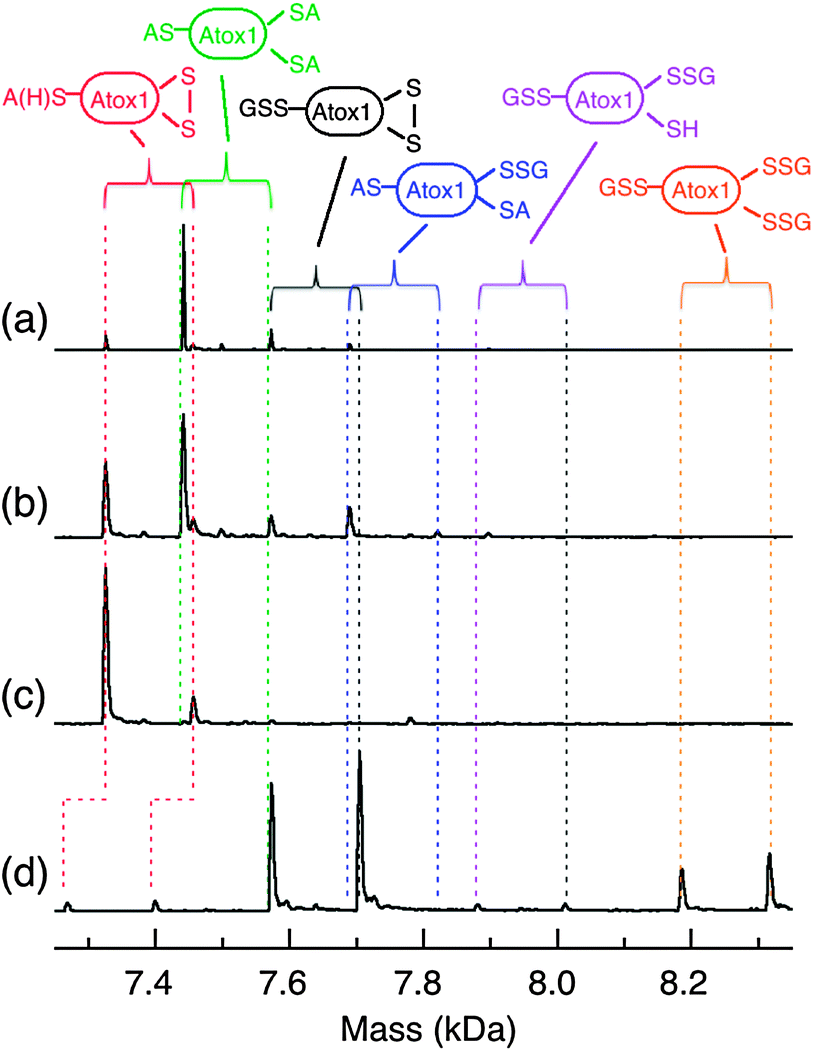 | ||
| Fig. 10 ESI-MS analysis of the oxidized forms of Atox1 (10 μM) upon oxidation by GSSG under non-catalytic condition in the following redox buffer (potential E): (a) GSSG (0.06 mM)/GSH (3.88 mM) (−222 mV); (b) GSSG (0.36 mM)/GSH (3.28 mM) (−195 mV); (c) GSSG (0.39 mM)/GSH (0.03 mM) (−69 mV); (d) GSSG (25 mM). Each reaction mixture was incubated overnight under anaerobic condition in KPi (50 mM, pH 7.0), followed by either alkylation with iodoacetamide (∼10 mM) and ESI-MS analysis (a–c) or by direct ESI-MS analysis without alkylation (d). Note: the relative content of the two components of Atox1 protein in (a–c) is different from that in (d) due to different batches of isolation. | ||
Equivalent experiments with hGrx1-tm showed that the oxidised forms were dominated by hGrx1-(SS) (>90%) under all physiologically relevant conditions. The content of the minor component hGrx1-(SH)(SSG) varied with conditions and reached up to ∼10% of the total oxidation products after the protein was about 50% oxidised (Fig. S4, ESI†).
The above experiments demonstrated that: (i) only the protein thiols in the Cys–xx–Cys motifs are thermodynamically susceptible to oxidation by GSSG and isolated single Cys residues (such as Cys41 in Atox1) are susceptible only at very high GSSG concentrations (Fig. 10). This is consistent with previous observations that single Cys residues in proteins are not easily glutathionylated under physiological conditions;25,42 (ii) the dominating oxidised form of the Cys–xx–Cys motif for P = Atox1 or hGrx1 (and likely for similar proteins) is P-(SS) under all conditions; (iii) under physiologically relevant conditions, the doubly glutathionylated form P-(SSG)2 was not observed and the singly glutathionylated form P-(SH)(SSG) was detected as a minor product. The content of the latter was minimal (<2%) under catalytic conditions, but increased slightly under non-catalytic conditions. Apparently, it is the formation of the internal disulfide bond in Atox1 that provides the driving force for the oxidation of the Cys–xx–Cys motif by GSSG.
However, species P-(SH)(SSG) are proposed to appear as intermediates of both substrate Atox1 and catalyst hGrx1 in the overall oxidation (Scheme 2). Their concentrations are kept minimal under catalytic conditions but may accumulate slightly under non-catalytic conditions. Consequently, previous conclusions of glutathionylation of ATP7A based on detection by immunoprecipitation may represent a small fraction only of the total ATP7A present.21
hGrx1 can catalyse reduction of Atox1 by GSH but only in the presence of Cu(I)
Oxidation of the Cys–xx–Cys motif in Atox1 under physiologically relevant conditions results in Atox1-(SS) (Fig. 9 and 10 and Fig. S3, ESI;†Scheme 2). Consequently, this internal disulfide form is a potential substrate for reduction by GSH with hGrx1 as catalyst. It was prepared readily by oxidation of reduced Atox1 with 2–3 equivalents of K3[FeIII(CN)6], followed by separation over a desalting column. The assay was similar to that employed in Fig. 8 but operated in the opposite direction (inset, Fig. 11). The assay was conducted in a metal buffer controlled by [CuI(Bcs)2]3−/Bcs2− (p[Cu+] = 15.5). Atox1-(SS) has little affinity for Cu(I) but reduction by GSH generates the reduced form that binds Cu(I) with high affinity (KD = 10−17.4 M at pH 7.0).18 This form scavenges Cu(I) from the chromophoric probe [CuI(Bcs)2]3− at a reaction rate much fast than the Atox1-(SS) reduction rate. Consequently, the reduction may be followed in real time via the decrease in probe concentration.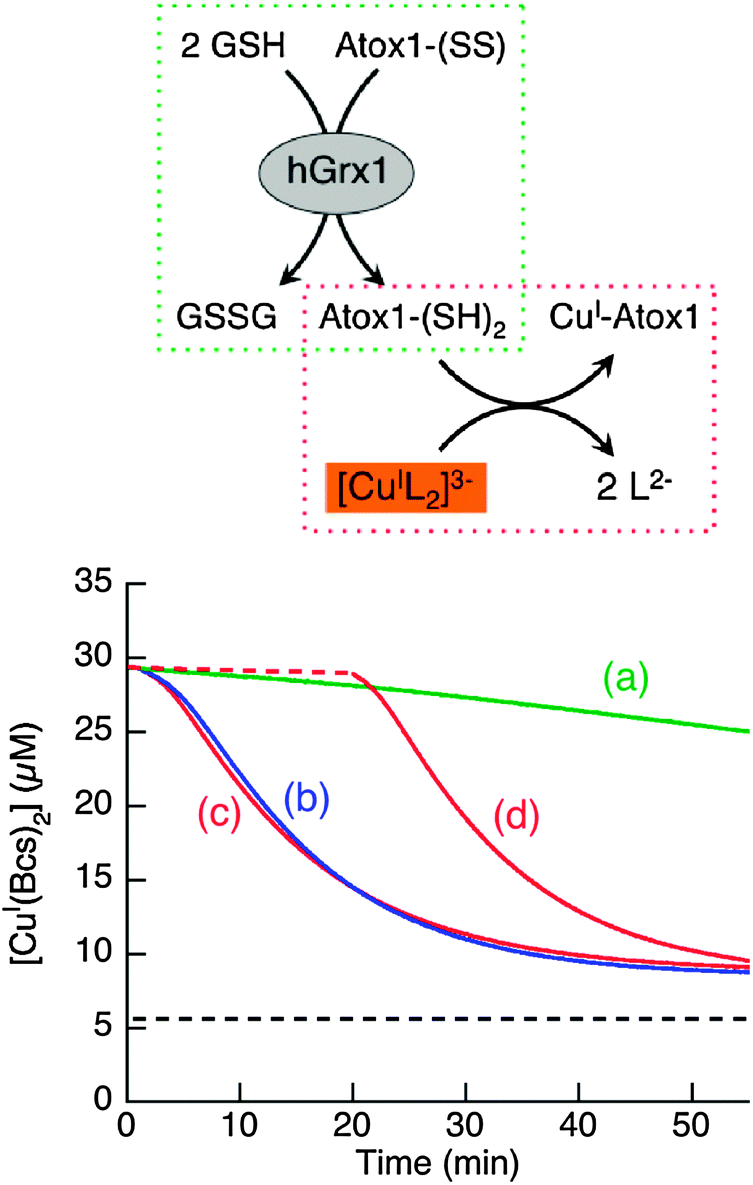 | ||
| Fig. 11 Reduction of Atox1-SS (50 μM) by GSH (800 μM) in KPi (50 mM, pH 7.0) containing the Cu(I) probe [CuI(Bcs)2]3− (composition: [Cu]tot = 37 μM; [Bcs]tot = 100 μM; [NH2OH]tot = 100 μM) with catalyst (each 1.0 μM) to initiate the catalytic reduction: (a) no catalyst (no difference with addition of 1.0 μM hGrx1-C23S); (b) hGrx1-C26S; (c) hGrx1-tm; (d) the same as (c) but the catalyst hGrx1-tm was pre-mixed with Atox1ox and GSH at 105% concentrations in (c) for 20 min (dashed trace) and then the Cu(I) probe [CuI(Bcs)2]3− was added at 20× concentration to the same final solution conditions in (c). The reduction was followed by monitoring Cu(I) transfer from the probe [CuI(Bcs)2]3− to the reduced Atox1 generated in situ as shown in the reaction scheme of inset. The dashed black line shows the [CuI(Bcs)2]3− concentration expected when 50 μM Atox1-SS is fully reduced (see Fig. S6, ESI†). | ||
Reduction of Atox1-(SS) (50 μM) by GSH (800 μM) in metal buffer ([Cu]tot, 37 μM; [Bcs]tot, 100 μM) was slow and unaffected by addition of hGrx1-C23S, an inactive hGrx1 variant (Fig. 11a). The rate of reaction increased upon addition of active catalysts hGrx1-tm or hGrx1-C26S (1.0 μM; Fig. 11b and c). In contrast to the higher catalytic activity of hGrx1-C26S relative to hGrx1-tm for the opposite reaction driven by excess GSSG (Fig. 8; Scheme 2), their activities are comparable under the reducing conditions controlled by excess GSH (800 μM). The initial Cuaq+ concentration of the assay solution was calculated to be p[Cu+] = 15.5, but is decreased rapidly to a final p[Cu+] ∼ 16.7 with Atox1 reduction (Fig. S6, ESI†). Neither form of hGrx1 is able to bind Cu(I) effectively under the conditions (Fig. S6, ESI†) and the inhibited form hGrx1-(SS) appears to be activated efficiently by the high concentration of GSH.
In contrast, in a control reaction that contained Atox1-(SS), GSH and hGrx1-tm but no [CuI(Bcs)2]3−, reduction of Atox1-(SS) was very slow (Fig. 11d, dashed line). However, addition of [CuI(Bcs)2]3− returned the rate of reaction to that observed previously (Fig. 11d (solid line) vs. c). It can be concluded that binding of Cu(I) to the high affinity site Cys–xx–Cys of reduced Atox1 increased the reduction potential for the protein disulfide–thiol couple considerably and that this provided the thermodynamic gradient needed to drive the reduction. However, Atox1-(SS) was not reduced completely under these conditions due to increasing rates of the reverse processes shown in the inset scheme of Fig. 8. The equilibrium concentration of [CuI(Bcs)2]3− predicted for full reduction is indicated by the black dashed line in Fig. 11 (see also Fig. S6, ESI†).
Additional experiments varying the relative concentrations of ligand Bcs2− or GSH further demonstrated that the reduction rate and equilibrium concentrations of Atox1-(SS) are regulated by both GSH concentration and the solution p[Cu+] value (Fig. 12). Apparently, multiple processes and equilibria control the overall reduction process including the differences in reduction potentials between the redox couples Atox1-(SS)/Atox1, hGrx1-(SS)/hGrx1 and GSSG/GSH and competition for Cu(I) between hGrx1, Atox1, probe ligand Bcs2− and GSH.
 | ||
| Fig. 12 Reduction of Atox1-SS (50 μM) by GSH: (a) 200 μM; (b) 400 μM; (c) 800 μM. Reaction was carried out in KPi buffer (50 mM, pH 7.0) containing Cu(I) probe [CuI(Bca)2]3− (composition: [Cu]tot = 40 μM; [Bca]tot = 500 μM; [NH2OH]tot = 100 μM) with catalyst (each 1.0 μM) of hGrx1-tm (blue) and hGrx1-C26S (red). The reduction was followed by monitoring Cu(I) transfer from the probe [CuI(Bcs)2]3− to the reduced Atox1 generated in situ as shown in the reaction scheme of Fig. 11. Note: before the reduction, the total Cu content is bound fully by Bca as visible complex [CuI(Bca)2]3− in (a), but is shared partially by GSH as invisible CuI-GSH complex in (b, c). Thus the reduction rate in (a) is considerably slower than those in (b, c). | ||
In summary, the above experiments demonstrate that: (i) GSH provides a shallow thermodynamic gradient only for reduction of Atox1-(SS). Binding of Cu(I) to the reduced form of Atox1 increases the thermodynamic gradient for reduction. (ii) hGrx1-wt and its variants with the catalytic residue Cys23 in place can readily catalyse reduction of Atox1-(SS) by GSH to CuI-Atox1 but not to apo-Atox1 (dithiol form).
The origins for these observations lie in the differences in reduction potentials of the relevant species and the impact of the binding of Cu(I) on such differences. The proposed catalytic cycle is summarised in Scheme 3. Catalysis is initiated by nucleophilic attack of the Cys23 thiolate of hGrx1 on the Atox1 disulfide bond to yield an intermediate complex featuring a mixed disulfide bond (step i). Reduction of this bond by a monothiol (step ii) and/or dithiol mechanism (step iii) releases fully reduced Atox1 for high affinity Cu(I) binding. The oxidised forms of hGrx1 are regenerated by GSH via mono- or di-thiol mechanisms (steps iv, v) to complete the catalytic cycle.
 | ||
| Scheme 3 Proposed reaction mechanism for GSH reduction of Atox1-(SS) catalysed by hGrx1. | ||
Direct observation of the proposed Atox1-S-S-Grx1 intermediate
Evidence for step (i) of Scheme 3 is provided by ESI-MS analysis of a reaction mixture containing an equimolar ratio of Atox1-(SS) and fully reduced hGrx1-tm (each 10 μM). While more than 95% of the reactants remained intact, the proposed protein complex was detected unambiguously in the gas phase (1–5%; Fig. 13). An accurate molar mass estimated for the complex shown in Scheme 3 excludes the possibility of the single Cys41 in Atox1-(SS) being involved in complex formation (Table S1, ESI†).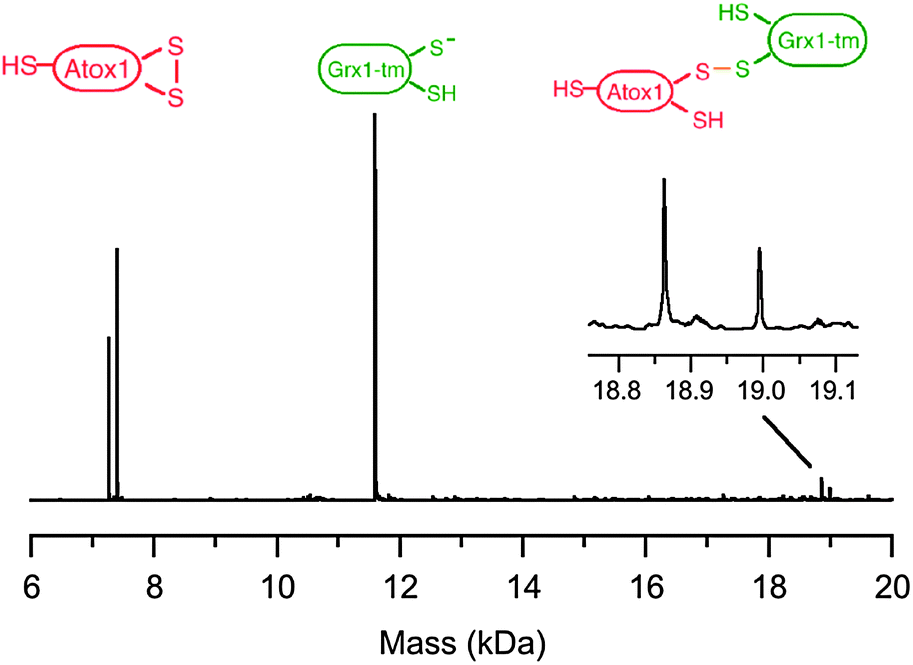 | ||
| Fig. 13 ESI-MS detection of an Atox1-SS-Grx1-tm complex formed from reaction of Atox1-(SS) (10 μM) with reduced hGrx1-tm (10 μM) in KPi buffer (20 mM, pH 7.0). The two ions at 18995.5 and 18964.3 kDa differ in the mass of an N-terminal methionine as the Atox1 reactant was a mixture of two forms. | ||
Notably, step (i) of Scheme 3 is not favoured under equilibrium conditions but is the potential rate-determining step. The underlying thermodynamic reason is that the reduction potential of the couple hGrx1-(SS)/Grx1-(SH)2 is more positive than that of Atox1-(SS)/Atox1-(SH)2 (see Fig. 6) and that the overall catalytic cycle is driven to completion by a combination of the high affinity of the Atox1 protein for Cu(I) (steps (ii, iii)) and regeneration of active hGrx1 enzyme by GSH (steps (iv, v)). However, when Cuaq+ is buffered at relatively high concentrations such as p[Cu+] = 14.8–14.9 in buffer containing lower GSH concentrations (Fig. 12a and b), the catalytic cycle may be inhibited partially by binding of Cu(I) to the catalyst hGrx1. The impact will be different for the different forms of hGrx1: the affinity of hGrx1-C26S for Cu(I) is lower than that of hGrx1-tm (Table 1) and so it is inhibited to a lesser extent (Fig. 12a and b). On the other hand, with GSH at higher concentrations such as in Fig. 12c, access of Cu(I) to either forms of hGrx1 is restricted and furthermore, the reduction is promoted via the monothiol mechanism (step (ii)) and suppressed via the dithiol mechanism (step (iii)). Consequently, inhibition of hGrx1 activity via formation of an internal disulfide bond as observed at high GSSG/GSH ratios in Scheme 2 does not operate in the cases of low GSSG/GSH ratios where both hGrx1-wt and hGrx1-C26S catalyse the reduction cycle via monothiol mechanism with comparable activity (Fig. 12c).
It is widely held that the oxidation and reduction of protein thiols are associated with catalytic glutathionylation and deglutathionylation.41 However, the experimental evidence presented here suggests that this is not always the case. Oxidation of the protein thiols in Atox1 to Atox1-(SS) with hGrx1 as a catalyst involves a two-step process of glutathionylation and deglutathionylation on both the substrate and the catalyst (Scheme 2). The reverse process seems to involve those steps for the catalyst hGrx1 only (Scheme 3). The main reason is that the dominant oxidised form of Atox1 (and likely of all other protein molecules featuring Cys–xx–Cys motifs) features an internal disulfide bond and not a glutathionylated Cys–S–SG group.
Concluding remarks and perspective
This study demonstrates an in vitro correlation between redox sulfur chemistry and copper binding involving hGrx1, Atox1 and GSH (Schemes 2 and 3). hGrx1 is a thiol–disulfide oxido-reductase enzyme in the cytosol and regulates the redox status of protein thiols in biological cells using GSSG/GSH as cofactors.12,42,43 The present work demonstrates that the affinity of hGrx1 for Cu(I) at pH 7.0 is 100 times weaker than that of Atox1 whereas its reduction potential is 70 mV more positive. These differences, plus a very low pKa of Cys23 of hGrx1, allow rationalisation of the reactions proposed in Schemes 2 and 3. The more positive reduction potential of hGrx1 (Fig. 6) is consistent with the enzyme being a robust catalyst for oxidation of apo-Atox1 in Scheme 2 (Fig. 8c) but not for the opposite reaction involving reduction of Atox1-(SS) in Scheme 3 (Fig. 11d, dashed trace). On the other hand, high affinity binding of Cu(I) to Atox1, but not to hGrx1, is expected to invert the order of reduction potentials and to reverse the catalytic preferences of hGrx1 in Schemes 2 and 3 (cf., Fig. 8a versus 11d, solid line trace). It is apparent that the direction of sulfur redox catalysis of Atox1 by hGrx1 may be regulated by both the overall reduction potential and by the availability of Cu(I).The formal oxidant and reductant in Schemes 2 and 3 are GSSG and GSH, respectively. The reduction potential of the couple GSSG/2GSH changes sensitively with both ratio and total concentration. For example, at a fixed total concentration of [GSH] + 2[GSSG] = 5.0 mM at pH 7.0, eqn (3) dictates that the reduction potential changes from −200 mV to −100 mV when the ratio [GSSG]/[GSH] changes from 0.1 to 10. On the other hand, when the ratio is fixed at, say, ∼1% GSSG with total concentration varying between the range 1–10 mM (typical cellular conditions),36 the reduction potential will vary between −210 mV and −240 mV (Fig. 6), as demonstrated recently by in-cell NMR spectroscopy for yeast cells.44 Under these conditions, the dominant forms of both Atox1 and hGrx1 in the cytosol will be their reduced forms. This prediction was confirmed recently.25
However, the potential of the GSSG/2GSH couple changes considerably with the biological status of the cell: ∼−240 mV for proliferation, ∼−200 mV for differentiation and ∼−170 mV for apoptosis.36 A reduction potential of Eo′ = −188 mV for Atox1 suggests that its oxidation level and thus its cellular function will be sensitive to cellular conditions and consequently, in addition to its classic role as a copper chaperone, Atox1 may assume an additional role as a cellular redox regulator. Indeed, its yeast homologue Atx1 was identified originally as an antioxidant (Atx) in yeast cells lacking a functional superoxide dismutase 1 (Sod1).28 Further evidence is accumulating for the antioxidant role of Atox1.45 Intriguingly, this activity relies on supply of copper from Ctrl.28,45 The present study supports the view that Atox1 may work at the intersection between copper homeostasis and cellular redox balance. Such versatility may be extended to hGrx1 as well: it will allow Atox1 to bind Cu(I) even at cell potentials (>−188 mV) that favour its disulfide form (Scheme 3; Fig. 6). This aspect may be relevant to the requirement for copper in Atox1's antioxidant role: a reduced form is required under oxidising conditions.
Reactive oxygen species (ROS) such as HO2 and H2O2 are produced during normal metabolism as by-products of the four-electron reduction of O2 to H2O. In healthy cells, these species are scavenged by the antioxidant enzymes Sod1 and catalase. However, their flux increases in cells under oxidative stress and, if the homeostasis of copper and iron are perturbed, the Haber–Weiss reaction may occur (catalysed by couples CuII/CuI and/or FeIII/FeII from ‘free’ metal ions):
| HO2 + H2O2 → O2 + H2O + HO˙ | (5) |
The ROS species (HO2, H2O2, HO˙) can attack amino acid sidechains (Met, Cys, Trp), nucleic acid bases and lipids. They will also affect the GSSG/GSH ratio of cells via oxidation of GSH and move the cell potential in a positive direction, favouring conversion of Atox1 to its oxidised form Atox1-(SS). However, hGrx1 will always favour its reduced form hGrx1-(SH)2 (Fig. 6).
In ROS-stressed cells and in the presence of available Cuaq+, hGrx1 can catalyse the production of CuI-Atox1 (Fig. 11; Scheme 3). The latter is capable of supplying up to three reducing equivalents (CuII/CuI and Atxo1-(SS)/Atox1-(SH)2) to safely reduce strong oxidants such as HO2, H2O2 and HO˙ to H2O (e.g., Scheme 4). This process is related to the redox sulfur chemistry observed in mitochondrial copper transfer. Metallo-chaperone CuI-Cox17 (with CuI bound in the reduced Cys–xx–Cys site) can transfer both the metal ion and two electrons to the oxidised form of apo-Sco1 (containing a disulfide bond).46
 | ||
| Scheme 4 Proposed mechanism for the antioxidant activity of Atox1. ‘Cu(II)’ refers to a putative transient Cu(II) species that is reductively trapped as Cu(I) rapidly by a cellular reductant such as GSH. This example is for three-electron reduction of HO2 to water. | ||
Copper imported by Ctr1 is delivered to various cellular destinations via a number of copper chaperones in the cytosol. However, many aspects of the processes remain unknown. For example, although Atox1 has been proven to be an important copper delivery vehicle for the Cu-ATPases, this chaperone is not required absolutely for these ATPases to bind copper nor to transfer it across membranes.45 In the case of a photosynthetic bacterium, the copper chaperone Atx1 was not essential for copper delivery but ensured safe delivery without damage to other metallo-protein sites.47 While this observation suggests that ‘free’ Cuaq+ can reach the cellular targets, other molecules may have evolved to deliver copper to the Cu-ATPases as well.
Recent work in human cell lines has revealed that hGrx1 interacts with copper transporters ATP7A/7B whose MBDs feature Cys–xx–Cys motifs as ligands of Cu(I) and that such interactions depend on the availability of both the MBDs and copper ions.20,21 The present study suggests that these interactions are likely to involve multiple events including reductive binding of Cu(I) to P-(SS) sites in the MBDs that is promoted by hGrx1 and GSH (Scheme 3 with Atox1 replaced by MBD) and also possible formation of (transient) Cu(I)-bridged complexes between MBD and hGrx1. Consequently, hGrx1 is a feasible candidate to supplement Atox1 in copper delivery.
The mechanism by which copper chaperones acquire copper from Ctr1 is another puzzle. Recent evidence suggests that GSH may take up that key role.24 Intracellular ‘free’ Cuaq+ is under tight control and is buffered at sub-femtomolar concentrations.18,48,49 Although GSH binds Cu(I) weakly, its cellular concentration is usually at millimolar range. Under such conditions, GSH may buffer free Cuaq+ concentrations in the femtomolar range as well (p[Cu+] = 15.4–16).50 It may form a partnership with Atox1 and other copper chaperones to ensure tight buffering.
In this context, hGrx1 can also buffer free Cuaq+ concentrations in the femtomolar range. Cellular concentrations of hGrx1 have been estimated to be in the μM range in human red blood cells.51 It regulates the redox status of a broad spectrum of protein thiols and is likely to interact directly with many different protein partners. Therefore, while Cys23 is responsible for catalysis, Cys26 may be recruited for Cu(I) binding and hGrx1 has evolved the properties necessary to function in redox regulation, Cu(I) buffering and Cu(I) transport. It is likely to play a key role in the vital copper transport pathway from the plasma membrane to the trans-Golgi network and beyond. The comparative molecular properties of Atox1 and hGrx1 uncovered by the present work are consistent with a sophisticated mechanism of regulation of redox environments and copper homeostasis in cells.
Experimental section
Materials and general procedures
Na2Bca, Na2Bcs, GSH, GSSG were purchased from Sigma-Aldrich. Dtt was purchased from Astral Scientific and Tcep from Fluka. These materials were used as received. Concentrations of Bca and Bcs solutions were standardized routinely as described previously.52 Stock solutions of Dtt and GSH were prepared in deoxygenated Milli-Q water and stored in an anaerobic glove box. Their concentrations based on quantitative dissolution were confirmed and calibrated with the Ellman assay.53 Copper solutions were prepared from CuSO4·5H2O purchased from Sigma-Aldrich.Protein expression and purification
Protein Atox1 was expressed and isolated as described previously.18,30 The plasmid for expression of hGrx1 was constructed by inserting the hGrx1 gene into pET24d expression vector between NcoI and EcoRI restriction sites and was donated kindly by Dr Caryn Outten, the University of South Carolina, USA. Plasmids for expression of its protein variants C23S, C26S, C23,26S and C8,79,83S were generated via the overlapping PCR approach using wild type hGrx1 plasmid as template. Each expression plasmid was transformed into E. coli BL21(DE3) CodonPlus cells for protein expression. The expression cells were grown in 2YT medium supplementary with antibiotics kanamycin (10 mg L−1) and chloramphenicol (34 mg L−1). At an optical density OD600 ≈ 1.0, protein expression was induced with IPTG (0.5 mM) overnight at room temperature. Expressed hGrx1 and its protein variants were purified by gradient elution (0–0.3 M NaCl) from a cation-exchange CM-52 column equilibrated in acetate buffer (20 mM; pH 5.4) containing β-mercaptoethanol (5.0 mM). The final step of purification was elution from a Superdex-75 FPLC gel filtration column in KPi buffer (20 mM; pH 7.0; 150 mM NaCl, 0.5 mM Tcep).The purified hGrx1 proteins were confirmed to be >95% pure by SDS-PAGE analysis and their identities confirmed by ESI-MS (Table S1, ESI†). As reported previously,30 two components (7401.7 and 7270.5 Da) were detected in the Atox1 preparation, corresponding to molecules with and without the first methionine residue, respectively. Their ratio varied from batch to batch. All proteins were isolated in apo-forms with no detectable copper content. Prior to the copper binding studies, apo-proteins were reduced fully by incubation overnight with excess Tcep in a glove box under nitrogen. Excess reductant was removed via a Bio-Gel P-6 DG gel-desalting column (Bio-Rad).
Concentration assays
UV-visible spectra were recorded on a Varian Cary 300 spectrophotometer in dual beam mode with quartz cuvettes of 1.0 cm path length. The concentrations of the apo-protein solutions were estimated by absorbance at 280 nm. The molar absorptivities ε280 derived from protein primary sequences were 2980 M−1 cm−1 for both Atox1 and hGrx1 (and its protein variants). Protein concentrations based on this value match those determined from the thiol assay of the fully-reduced apo-forms using Ellman's reagent DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)).53 All protein samples used for the metal affinity studies were confirmed to be fully reduced by analysis of the free thiol content. Experiments were carried out on samples prepared anaerobically in KPi buffer (40 mM; pH 7.0; NaCl, 100 mM) in the glove-box.Electrospray ionization mass spectrometry
All experiments were conducted on an Agilent time-of-flight mass spectrometer (TOF-MS) (model 6220, Palo Alto, CA) coupled to an Agilent 1200 LC system. All data were acquired and reference mass corrected via a dual-spray electrospray ionisation (ESI) source. Mass spectra were created by averaging the scans across each peak and background subtracted against the first 10 seconds of the TIC. Acquisition was performed using the Agilent Mass Hunter Acquisition software version B.02.01 (B2116.30) and analysis was performed using Mass Hunter version B.05.00 (B5.0.519.0).Mass spectrometer conditions: ionisation mode: electrospray ionisation; drying gas flow: 7 L min−1; nebuliser: 35 psi; drying gas temperature: 325 °C; capillary voltage (Vcap): 4000 V; fragmentor: 250 V; skimmer: 65 V; OCT RFV: 250 V; scan range acquired: 100–3200 m/z; internal reference ions: positive ion mode = m/z = 121.050873 & 922.009798.
Chromatographic separation was performed using an Agilent Zorbax Poroshell SB300-C18 2.1 × 12 mm, 5 μm column (Agilent Technologies, Palo Alto, CA) using an acetonitrile gradient (5% (v/v; 0.1% formic acid) to 75% (v/v; 0.1% formic acid)) over 8 min at 0.25 mL min−1.
Quantification of Cu(I) binding
Two Cu(I) probe ligands, Bca and Bcs were employed for the quantification. They each react with Cu(I) quantitatively to yield well-defined 1:2 complex [CuIL2]3− (L = Bca or Bcs) with characteristic solution spectrum (ε = 7900 M−1 cm−1 at λmax = 562 nm for L = Bca and ε = 13000 M−1 cm−1 at λmax = 483 nm for L = Bcs) and different formation constant (logβ2 = 17.2 and 19.8 for L = Bca and Bcs, respectively).18,31 They were used to define Cu(I) binding stoichiometry under non-competitive condition and Cu(I) binding affinity (expressed as dissociation constant KD) under competitive condition according to eqn (1) and (2).31
The experiments were conducted in an anaerobic glove-box by reaction of apo proteins with [CuIL2]3− (L = Bca, Bcs) in deoxygenated KPi buffer (40 mM; pH 7.0; 100 mM NaCl) as described previously.18,31 Briefly, apo protein was titrated, in various quantities, into a series of [CuIL2]3− solutions of defined molar ratio L:CuI ≥ 2.5 (to ensure the presence of the 1:2 complex [CuIL2]3− with negligible contribution from the 1:1 complex [CuIL]−). All solutions were diluted to a fixed volume to provide a series of solutions with constant total concentrations of Cu(I) and ligand L but varying concentrations of protein P. Transfer of Cu(I) from [CuIL2]3− to protein P was established by the change in solution absorbance. By selection of probe ligands with different Cu(I) affinities and/or by systematic variation of their concentrations, conditions were searched and set that favored either non-competitive or competitive reaction.
The KD values for Atox1 and hGrx1-tm were also determined and compared at different pH within the range 5–7 in buffers Na-Mes (pH 5.0, 5.5, 6.0) and KPi (6.0, 6.5, 7.0) using Cu(I)-Bca as probe and under denatured condition in KPi buffer (pH 7.0) containing urea (7 M) using Cu(I)-Bcs as probe.
Catalytic reduction of model substrate mercaptoethanol disulfide by GSH
The isolated hGrx1 and its protein variants were evaluated for their catalytic functions for several reactions under various conditions. The first is a classic assay based on two coupled enzymatic reactions as shown in Fig. 7a.14,33 The assay was conducted at room temperature in KPi buffer (100 mM, pH 7.5) with following solution compositions: (HOC2H4S)2 (2.5 mM), NADPH (0.25 mM), Grase (1.2 units per mL), GSH (0.5 mM) and hGrx1 at various concentrations (0–25 nM) or different protein variants of hGrx1 at a same concentration of 12.5 nM. The impact of Cu(I) on the reaction was tested by control experiments in the presence and absence of added Cu (3.0 eq. relative to hGrx1). Nonenzymatic background reaction(s) without hGrx1 were subtracted from each assay under the otherwise identical conditions.Catalytic oxidation of protein thiols by GSSG
The second assay was based on catalytic oxidation of Cu(I)-binding protein Atox1 with GSSG as an oxidant (see scheme in Fig. 8). The oxidation was followed by monitoring a loss of the high affinity Cu(I) binding affinity of the target protein Atox1 upon its oxidation. The probes used were chromophoric Cu(I) complexes [CuI(Bca)2]3− and [CuI(Bcs)2]3− which were prepared in situ from reaction of Cu(II) and excess ligand Bca or Bcs in reduced Mops buffer (50 mM, pH 7.0, NH2OH, 1.0 mM). In the assays given in Fig. 8a and b, the probe complex was included in the pre-assay mixture to make up an assay solution which contained typically Atox1 (35 μM), hGrx1 enzyme (2.0 μM), Cu(I) probe [CuIL2]3− (35 μM) with excess free L2− ligand at various concentrations and GSSG (400 μM). The catalysis was started by last addition of the oxidant GSSG and monitored, in real time, by change in solution absorbance for [CuIL2]3− with background control without the enzyme hGrx1 (Fig. 8a and b; Fig. S3a and b, ESI†). In the assays given in Fig. 8c, the probe complex used was the weaker affinity probe [CuI(Bca)2]3− with [Bca]tot/[Cu]tot < 3, but was not added to the catalytic reaction until quantification. Such probe contained relative high concentration free Cuaq+ and was able to saturate the high affinity Cu(I) site in Atox1 rapidly and consequently to quench and quantify the oxidation quantitatively upon its addition. Therefore, these later procedures separated the catalysis and detection into two sequential processes. In practice, the catalysis was started by mixing the fully reduced Atox1 (35 μM) and GSSG (400 μM) in the presence of catalyst hGrx1 (2.0 μM) in KPi (20 mM, pH 7.0) and a small fraction of the assay mixture was withdrawn at various reaction time points and added to an aliquot of probe solution [CuI(Bca)2]3− with Cu:Bca ∼ 1:3. The absorbance at 560 nm for [CuI(Bca)2]3− was recorded for each solution and was found to increase with reaction time due to the Atox1 oxidation (Fig. 8c; Fig. S3c, ESI†). The fraction of the Atox1 oxidation was calculated by dividing the absorbance change observed by the total change expected when the Atox1 was fully oxidised.
An alternative approach of detection is by alkylation and ESI-MS analysis of the protein oxidation products. During the course of the reaction, a small fraction (∼20 μL) of the above assay mixture was removed at various reaction time points and added to an alkylating solution of iodoacetamide in Mops (∼1 μL 100 mM; >50 folds excess). The oxidation levels were quantified by ESI-MS and were found to be consistent with the assay based on Cu(I)-Bca probe (Fig. 8c). This approach also allowed a direct identification of the oxidised protein forms of Atox1 and hGrx1 (Fig. 9 and 10 and Fig. S3, ESI†).
Catalytic reduction of protein disulfide bond by GSH
The third assay was based on reduction of a disulfide bond in Atox1 (i.e., Atox1-SS) by GSH with hGrx1 as catalyst. It was an assay related to the second one but in the reversed direction (cf., schemes in Fig. 8 and 11). Atox1-SS was prepared under anaerobic condition by incubation of Atox1 (10–20 mg mL−1) in KPi (20 mM, pH 7.0) with 2–3 equivalents of K3[Fe(CN)6] for ∼1 h, followed by separation over a desalting column. The catalytic reaction was conducted under anaerobic condition in a mixing cuvette with Atox1-SS (100 μM) being loaded in one half-cell and the rest reaction components (GSH, hGrx1, probe Cu(I)–L complex at specified concentrations) in the other half-cell. After recording the solution absorbance before mixing at 483 nm for L = Bcs or 562 nm for L = Bca, the reaction was started by a rapid mixing and followed by change in the absorbance (note: the initial concentration of each reaction component was halved after the mixing). For control reactions without Cu(I)–L probe, the concentration of each component was increased by 5% and after certain reaction time (∼20 min), a 20-time concentrated Cu(I)–L probe solution was added into the reaction mixture to estimate and follow the reactions before and after the Cu(I) probe addition (Fig. 11d).Determination of reduction potentials for Atox1 and hGrx1
They were determined by equilibration overnight of each protein (∼10 μM) in a series of redox buffers of GSSG/2GSH at mM total concentration under anaerobic condition in Mops buffer (50 mM, pH 7.0). The reduction potential of each buffer was controlled by the ratio of GSSG/2GSH and calculated viaeqn (3) with EGSH′ = −240 mV at pH 7.0.36 The reaction was quenched by alkylation of free thiols with excess iodoacetamide (>10 folds excess). After incubation for >1 h, the oxidation level of each protein was estimated by ESI-MS analysis. Control experiments with extra GSSG being included in the iodoacetamide solution detected essentially the same level of oxidation for either protein, confirming that the alkylation procedure was fast enough to reflect the original redox equilibrium.Acknowledgements
This work was supported by funds from the Australian Research Council under grants DP120100752 and DP130100728. We thank Dr Paul Donnelly (University of Melbourne) for valuable suggestions and discussion.Notes and references
- N. J. Robinson and D. R. Winge, Copper metallochaperones, Annu. Rev. Biochem., 2010, 79, 537–562 CrossRef CAS PubMed.
- S. La Fontaine and J. F. Mercer, Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis, Arch. Biochem. Biophys., 2007, 463, 149–167 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, Structural biology of copper trafficking, Chem. Rev., 2009, 109, 4760–4779 CrossRef CAS PubMed.
- I. Hamza, M. Schaefer, L. W. Klomp and J. D. Gitlin, Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13363–13368 CrossRef CAS.
- D. Achila, L. Banci, I. Bertini, J. Bunce, S. Ciofi-Baffoni and D. L. Huffman, Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5729 CrossRef CAS PubMed.
- L. Banci, I. Bertini, V. Calderone, N. Della-Malva, I. C. Felli, S. Neri, A. Pavelkova and A. Rosato, Copper(I)-mediated protein-protein interactions result from suboptimal interaction surfaces, Biochem. J., 2009, 422, 37–42 CrossRef CAS PubMed.
- P. A. Muller and L. W. Klomp, ATOX1: A novel copper-responsive transcription factor in mammals?, Int. J. Biochem. Cell Biol., 2009, 41, 1233–1236 CrossRef CAS PubMed.
- S. Itoh, K. Ozumi, H. W. Kim, O. Nakagawa, R. D. McKinney, R. J. Folz, I. N. Zelko, M. Ushio-Fukai and T. Fukai, Novel mechanism for regulation of extracellular SOD transcription and activity by copper: role of antioxidant-1, Free Radical Biol. Med., 2009, 46, 95–104 CrossRef CAS PubMed.
- S. M. Vanderwerf, M. J. Cooper, I. V. Stetsenko and S. Lutsenko, Copper specifically regulates intracellular phosphorylation of the Wilson's disease protein, a human copper-transporting ATPase, J. Biol. Chem., 2001, 276, 36289–36294 CrossRef CAS PubMed.
- I. Voskoboinik, R. Fernando, N. Veldhuis, K. M. Hannan, N. Marmy-Conus, R. B. Pearson and J. Camakaris, Protein kinase-dependent phosphorylation of the Menkes copper P-type ATPase, Biochem. Biophys. Res. Commun., 2003, 303, 337–342 CrossRef CAS PubMed.
- N. M. Hasan and S. Lutsenko, Regulation of copper transporters in human cells, Curr. Top. Membr., 2012, 69, 137–161 CAS.
- E. Stroher and A. H. Millar, The biological roles of glutaredoxins, Biochem. J., 2012, 446, 333–348 CrossRef PubMed.
- C. H. Lillig and C. Berndt, Glutaredoxins in thiol/disulfide exchange, Antioxid. Redox Signaling, 2013, 18, 1654–1665 CrossRef CAS PubMed.
- A. Holmgren, Glutathione-dependent synthesis of deoxyribo-nucleotides. Purification and characterization of glutaredoxin from Escherichia coli, J. Biol. Chem., 1979, 254, 3664–3671 CAS.
- C. Johansson, C. H. Lillig and A. Holmgren, Human Mitochondrial Glutaredoxin Reduces S-Glutathionylated Proteins with High Affinity Accepting Electrons from Either Glutathione or Thioredoxin Reductase, J. Biol. Chem., 2004, 279, 7537–7543 CrossRef CAS PubMed.
- C. Sun, M. J. Berardi and J. H. Bushweller, The NMR solution structure of human glutaredoxin in the fully reduced form, J. Mol. Biol., 1998, 280, 687–701 CrossRef CAS PubMed.
- Y. Yang, S. Jao, S. Nanduri, D. W. Starke, J. J. Mieyal and J. Qin, Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity, Biochemistry, 1998, 37, 17145–17156 CrossRef CAS PubMed.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1 and related proteins: detection probes and affinity standards, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef CAS PubMed.
- D. Su, C. Berndt, D. E. Fomenko, A. Holmgren and V. N. Gladyshev, A Conserved cis-Proline Precludes Metal Binding by the Active Site Thiolates in Members of the Thioredoxin Family of Proteins, Biochemistry, 2007, 46, 6903–6910 CrossRef CAS PubMed.
- C. M. Lim, M. A. Cater, J. F. Mercer and S. La Fontaine, Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases, Biochem. Biophys. Res. Commun., 2006, 348, 428–436 CrossRef CAS PubMed.
- W. C. J. Singleton, K. T. McInnes, M. A. Cater, W. R. Winnall, R. McKirdy, Y. Yu, P. E. Taylor, B.-X. Ke, D. R. Richardson, J. F. B. Mercer and S. La Fontaine, Role of Glutaredoxin1 and Glutathione in Regulating the Activity of the Copper-transporting P-type ATPases, ATP7A and ATP7B, J. Biol. Chem., 2010, 285, 27111–27121 CrossRef CAS PubMed.
- M. L. De Benedetto, C. R. Capo, A. Ferri, C. Valle, R. Polimanti, M. T. Carrì and L. Rossi, Glutaredoxin 1 is a major player in copper metabolism in neuroblastoma cells, Biochim. Biophys. Acta, 2014, 1840, 255–261 CrossRef CAS PubMed.
- A. Meister, Glutathione metabolism and its selective modification, J. Biol. Chem., 1988, 263, 17205–17208 CAS.
- E. B. Maryon, S. A. Molloy and J. H. Kaplan, Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1, Am. J. Physiol.: Cell Physiol., 2013, 304, C768–C779 CrossRef CAS PubMed.
- Y. Hatori, S. Clasen, N. M. Hasan, A. N. Barry and S. Lutsenko, Functional Partnership of the Copper Export Machinery and Glutathione Balance in Human Cells, J. Biol. Chem., 2012, 287, 26678–26687 CrossRef CAS PubMed.
- I. Dalle-Donne, R. Rossi, D. Giustarini, R. Colombo and A. Milzani, S-glutathionylation in protein redox regulation, Free Radical Biol. Med., 2007, 43, 883–898 CrossRef CAS PubMed.
- M. D. Shelton and J. J. Mieyal, Regulation by reversible S-glutathionylation: molecular targets implicated in inflammatory diseases, Mol. Cells, 2008, 25, 332–346 CAS.
- S. J. Lin and V. C. Culotta, The ATX1 gene of Saccharomyces cerevisiae encodes a small metal homeostasis factor that protects cells against reactive oxygen toxicity, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3784–3788 CrossRef CAS.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Reassessment of Ellman's reagent, Methods Enzymol., 1983, 91, 49–60 CAS.
- M. Ralle, S. Lutsenko and N. J. Blackburn, X-ray absorption spectroscopy of the copper chaperone HAH1 reveals a linear two-coordinate Cu(I) center capable of adduct formation with exogenous thiols and phosphines, J. Biol. Chem., 2003, 278, 23163–23170 CrossRef CAS PubMed.
- Z. Xiao, L. Gottschlich, R. van der Meulen, S. R. Udagedara and A. G. Wedd, Evaluation of quantitative probes for weaker Cu(I) binding sites completes a set of four capable of detecting Cu(I) affinities from nanomolar to attomolar, Metallomics, 2013, 5, 501–513 RSC.
- S. Allen, A. Badarau and C. Dennison, The influence of protein folding on the copper affinities of trafficking and target sites, Dalton Trans., 2013, 42, 3233–3239 RSC.
- J. J. Mieyal, D. W. Starke, S. A. Gravina and B. A. Hocevar, Thiotransferase in human red blood cells: kinetics and equilibrium, Biochemistry, 1991, 30, 8883–8891 CrossRef CAS PubMed.
- T. Kortemme and T. E. Creighton, Ionisation of Cysteine Residues at the Termini of Model α-Helical Peptides. Relevance to Unusual Thiol pKa Values in Proteins of the Thioredoxin Family, J. Mol. Biol., 1995, 253, 799–812 CrossRef CAS PubMed.
- Control ESI-MS experiments with samples prepared by mixing various molar ratios of pure fully reduced and fully oxidized Atox1 or hGrx1 after pre-alkylation demonstrate that the relative intensities detected in the gas phase match their compositions in the solutions (see Fig. S5, ESI†).
- F. Q. Schafer and G. R. Buettner, Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- J. Sagemark, T. H. Elgan, T. R. Burglin, C. Johansson, A. Holmgren and K. D. Berndt, Redox properties and evolution of human glutaredoxins, Proteins, 2007, 68, 879–892 CrossRef CAS PubMed.
- Z. R. Gan and W. W. Wells, Purification and properties of thioltransferase, J. Biol. Chem., 1986, 261, 996–1001 CAS.
- J. H. Bushweller, F. Aslund, K. Wuthrich and A. Holmgren, Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14–S) and its mixed disulfide with glutathione, Biochemistry, 1992, 31, 9288–9293 CrossRef CAS PubMed.
- S. A. Gravina and J. J. Mieyal, Thioltransferase is a specific glutathionyl mixed-disulfide oxidoreductase, Biochemistry, 1993, 32, 3368–3376 CrossRef CAS PubMed.
- M. M. Gallogly and J. J. Mieyal, Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress, Curr. Opin. Pharmacol., 2007, 7, 381–391 CrossRef CAS PubMed.
- M. M. Gallogly, D. W. Starke and J. J. Mieyal, Mechanistic and kinetic details of catalysis of thiol–disulfide exchange by glutaredoxins and potential mechanisms of regulation, Antioxid. Redox Signaling, 2009, 11, 1059–1081 CrossRef CAS PubMed.
- C. H. Lillig, C. Berndt and A. Holmgren, Glutaredoxin systems, Biochim. Biophys. Acta, 2008, 1780, 1304–1317 CrossRef CAS PubMed.
- S. Y. Rhieu, A. A. Urbas, D. W. Bearden, J. P. Marino, K. A. Lippa and V. Reipa, Probing the Intracellular Glutathione Redox Potential by In-Cell NMR Spectroscopy, Angew. Chem., 2014, 126, 457–460 CrossRef . Note: the numbers on the y-axis of Fig. 3b need to be corrected by adding +88.5 mV (S. Y. Rhieu, private communication).
- Y. Hatori and S. Lutsenko, An expanding range of functions for the copper chaperone/antioxidant protein atox1, Antioxid. Redox Signaling, 2013, 19, 945–957 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Hadjiloi, M. Martinelli and P. Palumaa, Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6803–6808 CrossRef CAS PubMed.
- S. Tottey, C. J. Patterson, L. Banci, I. Bertini, I. C. Felli, A. Pavelkova, S. J. Dainty, R. Pernil, K. J. Waldron, A. W. Foster and N. J. Robinson, Cyanobacterial metallochaperone inhibits deleterious side reactions of copper, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 95–100 CrossRef CAS PubMed.
- T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O'Halloran, Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase, Science, 1999, 284, 805–808 CrossRef CAS PubMed.
- Z. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd, C-terminal domain of the membrane copper transporter Ctr1 from Saccharomyces cerevisiae binds four Cu(I) ions as a cuprous-thiolate polynuclear cluster: sub-femtomolar Cu(I) affinity of three proteins involved in copper trafficking, J. Am. Chem. Soc., 2004, 126, 3081–3090 CrossRef CAS PubMed.
- The Cuaq+ buffer range was calculated from the experimental data presented in Fig. 4 of ref. 18.
- J. J. Mieyal, D. W. Starke, S. A. Gravina, C. Dothey and J. S. Chung, Thioltransferase in human red blood cells: purification and properties, Biochemistry, 1991, 30, 6088–6097 CrossRef CAS PubMed.
- K. Y. Djoko, Z. Xiao, D. L. Huffman and A. G. Wedd, Conserved Mechanism of Copper Binding and Transfer. A Comparison of the Copper-Resistance Proteins PcoC from Escherichia coli and CopC from Pseudomonas syringae, Inorg. Chem., 2007, 46, 4560–4568 CrossRef CAS PubMed.
- G. L. Ellman, Tissue sulfhydryl groups, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experiments for determination of Cu(I) KD for variants hGrx1-C23S and -C26S; Fig. S1–S6; Table S1. See DOI: 10.1039/c4mt00020j |
| This journal is © The Royal Society of Chemistry 2014 |