Characterization of UO22+ binding to osteopontin, a highly phosphorylated protein: insights into potential mechanisms of uranyl accumulation in bones†
Lei
Qi
ab,
Christian
Basset
a,
Olivier
Averseng
a,
Eric
Quéméneur
ac,
Agnès
Hagège
ad and
Claude
Vidaud
*a
aCEA/DSV/iBEB/SBTN, Laboratoire d'Etude des Protéines Cibles, BP 17171, 30207 Bagnols sur Cèze Cédex, France. E-mail: claude.vidaud@cea.fr; Tel: +33(0) 4 66 79 67 62
bState Key Laboratory of Agro-biotechnology and College of Biological Sciences, China Agricultural University, Beijing, China
cCEA/DSV/DIR, 18, route du Panorama, 95265 Fontenay aux Roses, France
dCNRS UMR 7265, CEA/DSV/iBEB, 13108 St. Paul Les Durance, France
First published on 19th November 2013
Abstract
Bones are one of the few organs in which uranyl (UO22+) accumulates. This large dioxo-cation displays affinity for carboxylates, phenolates and phosphorylated functional groups in proteins. The noncollagenous protein osteopontin (OPN) plays an important role in bone homeostasis. It is mainly found in the extracellular matrix of mineralized tissues but also in body fluids such as milk, blood and urine. Furthermore, OPN is an intrinsically disordered protein, which, like other proteins of the SIBLING family, contains a polyaspartic acid sequence and numerous patterns of alternating acidic and phosphorylated residues. All these properties led to the hypothesis that this protein could be prone to UO22+ binding. In this work, a simple purification procedure enabling highly purified bovine (bOPN) and human OPN (hOPN) to be obtained was developed. Various biophysical approaches were set up to study the impact of phosphorylations on the affinity of OPN for UO22+ as well as the formation of stable complexes originating from structural changes induced by the binding of this metal cation. The results obtained suggest a new mechanism of the interaction of UO22+ with bone metabolism and a new role for OPN as a metal transporter.
Introduction
Natural uranium is a low alpha emitter and its chemical, rather than its radiological, toxicity is a subject of concern. In aqueous media it is mainly found as hexavalent uranyl ions UO22+. Once in the body, it reaches the blood and accumulates preferentially in the kidneys (∼12%) and the bones (∼22%) where it remains for years, leading to inhibition of bone formation.1–3 The mechanisms leading to this accumulation are still unknown. In bones, UO22+ exhibits a prolonged half-life of several years and it has been shown that the bone remodeling process enables only a low release into the blood and elimination from the body.4 No decorporation agent has been effective to date, and knowledge of these mechanisms would help in the development of new therapeutic approaches.It has been previously shown that UO22+ can affect expression of the SPP1 gene, which codes for osteopontin (OPN).5 This major noncollagenous protein is present in the extracellular matrix of mineralized tissues and is involved in bone homeostasis, with a high affinity for both calcium and hydroxyapatite.6 But it is also found in different concentrations in various body fluids including blood, milk and urine, and performs multiple functions such as wound healing, immune responses and tumorigenesis.7–9 OPN is a SIBLING protein (Small Integrin–Binding LIgand, N-linked Glycoprotein) which displays an integrin-binding motif (G-D-R-G). This highly conserved glycoprotein presents a polyaspartate region (D-D-L-D-D-D-D-D for bovine OPN, D-M-D-D-E-D-D-D-D for human OPN) and high rates of phosphorylations,10,11 arranged in clusters of three to five, which constitute potential binding sites for minerals and calcium salts. OPN is an Intrinsically Disordered Protein (IDP), and the phosphorylation clusters could be responsible for induction of structures and scaffolds that mediate OPN binding to hydroxyapatite in mineralized tissues.11
Therefore, the relative plasticity of OPN, as well as its special composition in acidic clusters and numerous phosphorylations, may be particularly well suited for the binding of the large UO22+ cation.
UO22+ is an actinyl ion, which adopts a bi-pyramidal geometry with 3 to 5 ligands in an equatorial plane perpendicular to its oxo axis. As a hard Lewis metal it reacts mainly with carboxylate, phenolate, phosphate and phosphonate groups. Several studies have also highlighted that phosphorylations are greatly involved in some UO22+–protein interactions.12–14
OPNs are also known for their calcium binding ability and this cation was often presented as a substitute for UO22+, although this analogy is based only on similar behaviors and biodistributions at the cellular level. Previous studies dedicated to calcium binding properties determined that OPN can bind 8 Ca2+, probably in the aspartic region, and that dephosphorylated OPN seems to conserve its Ca2+-binding ability.15 Other studies found that up to 50 Ca2+ per OPN molecule can be bound to the protein.16 In both cases, there was no isolation of the complex formed. However, there are no studies dedicated to specific interactions between metals and OPNs, while the question of phosphorylation-dependent metal binding was addressed for other protein fragments.17 We recently demonstrated that both phosphoryl and carboxyl functional groups of a selected peptide (pS-D-E-pS-D-E, code name H8V) from OPN were involved in the binding of UO22+.18
With the aim of deciphering the UO22+ mineralization process, this study first addressed the question of whether human (hOPN) and bovine (bOPN) osteopontins have affinity for the uranyl cation in physiological conditions by surface plasmon resonance (SPR) investigation. The potential role of phosphorylations and acidic regions in this binding was also considered. An assessment of the stoichiometry of the complexes formed was performed by ICP-MS after their isolation by size exclusion chromatography. Then the structural modifications that OPNs undergo when they are in contact with UO22+ were investigated by circular dichroism (CD). Our results are consistent with a strong and specific UO2–OPN binding leading to stable complexes with the native phosphorylated proteins.
Experimental
All the chemicals were purchased from SIGMA-Aldrich (Lyon, France). All aqueous solutions were prepared with pure water (18.2 MΩ cm resistivity; Milli-Q station; Millipore). The solution of natural uranyl acetate (0.1 M) was from the CEA. Phosvitin was from SIGMA, controls of hOPN were from Abcam (France, #ab81549) and bOPN from R&D System (France). The LF-166 anti-hOPN antibody was kindly provided by Larry. W. Fisher19 from NIH (Bethesda, Maryland) for immuno-detection.20Under our Material Transfer Agreement, Lacprodan®10 was provided from Arla Foods Ingredients a.m.b.a., (Viby, Denmark). H8V and poly-D or A peptides were purchased from PolyPeptide-NEOMPS, Strasbourg, France.
Purification of OPNs
The chromatographic processes were performed using an ÄKTA Purifier 10 System (GE Healthcare).The OPNs were obtained from milk, where they remain highly phosphorylated.10,21
Bovine OPN (bOPN)
bOPN was purified from Lacprodan®10. According to the manufacturer's information, the sample contained 83% of bovine milk proteins, with 25% of these entire OPN (EbOPN) and 75% a shorter fragment (FbOPN). The full length protein and its fragment were separated by SEC (TSK-GEL G3000SW semi-preparative column, 21.5 mm × 30 cm, Tosoh Bioscience) equilibrated with 50 mM Tris-HCl, 150 mM NaCl, pH 7.4 buffer at 66 cm h−1 linear velocity. The purified proteins were stored in aliquots at −20 °C.Human OPN (hOPN)
Human milk was obtained from three healthy volunteer mothers.The purification process was optimized from previously published results22 and led to highly purified OPNs in three single steps (acidic precipitation, ion exchange (IEX) and size exclusion chromatography (SEC)).
The milk was first centrifuged (4000g, 10 min at 4 °C, Multifuge 3SR, HERAEUS). The skimmed milk (100 mL) was brought to room temperature, its pH adjusted to 4.6 (1 M HCl) and again centrifuged (3000g, 20 min at 4 °C) to eliminate casein. The supernatant was dialysed (Spectra/Por tubing MWCO 6-8000, Spectrum Laboratories) against a 100 mM sodium acetate pH 5.0 buffer.
DEAE-Sephacel (30 mL, GE Healthcare), pre-equilibrated with sodium acetate buffer, was brought into contact with the whey overnight at 4 °C under stirring. The mixture was then packed into an XK26/20 column (GE Healthcare). The bound proteins were eluted by a five-step gradient from 0.1 to 0.5 M NaCl in sodium acetate buffer.
The fraction containing hOPN was further purified by SEC (TSK-GEL G3000SW column, 21.5 mm × 30 cm, Tosoh Bioscience) equilibrated with 50 mM Tris-HCl buffer, 150 mM NaCl, pH 7.4, at 66 cm h−1 linear velocity. The purified OPN was stored in aliquots at −20 °C.
Concentration determination and control of the purified OPNs
UV-Vis protein spectra were recorded with a Cary 50 Probe UV-visible Spectrometer (Varian, France). The specific absorption coefficient (Lg−1 cm−1) at 280 nm was fixed at 0.68 for hOPN (22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 820 M−1 cm−1, P10451, ProtParam ExPASy). The specific absorption coefficient at 280 nm was calculated at 0.21 for EbOPN and 0.42 for FbOPN (calculation details in the Results and discussion).
820 M−1 cm−1, P10451, ProtParam ExPASy). The specific absorption coefficient at 280 nm was calculated at 0.21 for EbOPN and 0.42 for FbOPN (calculation details in the Results and discussion).
All the purified proteins were controlled by SDS-PAGE and identified by Western blots (NuPAGE, Bis-Tris Gel 4–12%, Novex®, Life Technologies) with commercial controls of hOPN and bOPN, and with the LF-166 anti-hOPN antibody.
Mass spectra analysis
Protein sequences were identified by peptide analysis after trypsin digestion and mass spectrometry (MS) analysis (LTQ-Orbitrap XL hybrid mass spectrometer, ThermoFisher Scientific, coupled to an UltiMate 3000 LC system, Dionex). Briefly, peak lists were generated using MASCOT DAEMON software (Matrix Science, 2.3.2). From SwissProt_2012_0, the search parameters were as follows: 2 maximum mis-cleavages, 5 ppm as mass tolerances (parent ion) and 0.5 Da for MS/MS, fixed modification for carbamidomethylated Cys, variable modification for oxidized Met, and phosphorylated Ser, Thr, and Tyr residues. The p value was set at 0.01, and the proteins validated with at least 2 different peptides detected.De-phosphorylation of OPNs
Protein phosphorylations were removed by Lambda Protein Phosphatase (Lambda PP, Biolabs, P0753L and 400000 U mL−1).23,24 Dephosphorylation assays were first performed on small amounts of EbOPN prior to application on larger protein amounts. The purified proteins and the enzyme were mixed in the buffer (50 mM HEPES, 0.1% Brij 35, 20 mM DTT and 10 mM MnCl2, pH 7.5), and incubated at 30 °C for 30 min. An enzyme activity of 250 U mL−1 was found to be optimum to remove the phosphorylations (data not shown). The phosphatases and by-products were then separated from dephosphorylated proteins by SEC (TSK-GEL G3000SW column, 21.5 mm × 30 cm, Tosoh Bioscience).
ICP-MS analysis
Phosphorus, sulfur and uranium contents of the protein samples were measured by ICP-MS (7700, Agilent Technologies) after mineralization at 70 °C for 8 h using HNO3/H2O2 2/1 and subsequent dilution by HNO3 5%. Uranium and phosphorus were detected at m/z = 238 and 31 respectively, and protein was quantified through sulfur measurements at m/z = 34. Each measurement was at least the average value of 3 replicates.Determination of apparent affinities by surface plasmon resonance (SPR) studies
Determination of apparent KD of OPNs for UO22+ was performed by SPR (T100 biosensor system, Biacore, GE Healthcare Biosciences), following the method described elsewhere.25 All the experiments were carried out in duplicate and with at least two experimental replicates.Circular dichroism (CD) studies
Incidence of UO22+ additions on the OPN structures was studied by CD (CD-JASCO J-810, Jasco, UK, equipped with a 0.1 cm path length cell). OPN samples (from 1.5 to 2.5 μM depending on the experiments) were prepared in a 5 mM Tris-HCl buffer, 15 mM NaCl, pH 7.4, which was subtracted as a blank. A uranyl solution (1 mM) was prepared in 10 mM NaHCO3 solution. This solution was cumulatively added to the sample from 1 to 50 UO22+: OPN ratios. CD spectra were recorded over the 190–250 nm range at 1 nm intervals, at 20 °C. The spectra are the average of 5 cumulative scans of 3 experimental replicates. No precipitate was observed whatever the UO22+ ratios used in the assays.Isolation of UO2–OPN complexes by size exclusion chromatography (SEC)
The isolation of UO2–OPN complexes was performed according to previously published work.26 Briefly, OPNs were placed in contact overnight at 4 °C and under gentle stirring with a UO22+ solution. Various excesses (and at least an excess of 50) of UO22+versus OPNs, supplemented with a 10NaHCO3:1UO22+ ratio were used. The UO2–OPN complexes were separated from unbound UO22+ by injecting the samples (200 μL) onto a G25 column (10 mm × 30 cm, GE Healthcare) equilibrated in a 50 mM Tris-HCl, 150 mM NaCl, pH 7.4 buffer. Gel cleaning was performed between each run by 0.2 M NaHCO3 solution. The stoichiometry of the purified complexes was measured by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS).
Results and discussion
Obtaining highly purified bovine and human OPNs and dephosphorylated OPNs
Bovine OPN was purified from a Lacprodan®10 sample. Quality control documents indicated the presence of the full length protein (EbOPN) and a shorter N-terminal fragment (FbOPN). The proteins were separated into 4 peaks by SEC (Fig. 1, left). This single step enabled the separation of the entire protein from its fragment, but also the elimination of other residual milk proteins. The purified EbOPN and the expected FbOPN fragment were identified in peaks 2 and 3 respectively with ∼50 kDa and ∼35 kDa as apparent MW in SDS PAGE controls (Fig. 1, right). Protein concentration needed to be precisely measured. A 1 mg mL−1 Lacprodan®10 solution in MilliQ H2O led to an estimated specific absorption coefficient (Lg−1 cm−1) at 280 nm around 0.37. A precise OPN concentration measurement remains difficult by classical methods, as stated for other unstructured proteins.27,28 Absorbance at 280 nm recorded during the SEC indicated that peak 2 and peak 3 represented respectively 15% and 85% of the two main peak areas. This EbOPN/FbOPN proportion was stable all through the different purification runs performed for this study. Therefore, based on both the manufacturer's indications and these observations, the specific absorption coefficients at 280 nm were calculated at 0.21 and 0.42 respectively for EbOPN and FbOPN. For EbOPN, the value was slightly lower than the theoretical one (0.29) calculated from ProtParam (ExPASy). From the amino acid sequence, the EbOPN theoretical MW is 29.3 kDa. But the real MW is difficult to determine for OPNs, which are both highly post-translationally modified and intrinsically disordered. A wide disparity is observed in published results, with MW ranking from 36.3 kDa up to 60 kDa.29,30 A 50 kDa was attributed to the bOPN, in agreement with our SDS PAGE results and corresponding to a 40% protein glycosylation. EbOPN presents one methionine residue in its sequence. To confirm the determination of EbOPN concentration, the sulfur content of EbOPN was then controlled by ICP-MS, and was found to be ∼1 for 1 equivalent of EbOPN calculated with a 50 kDa MW and a specific absorption coefficient of 0.21.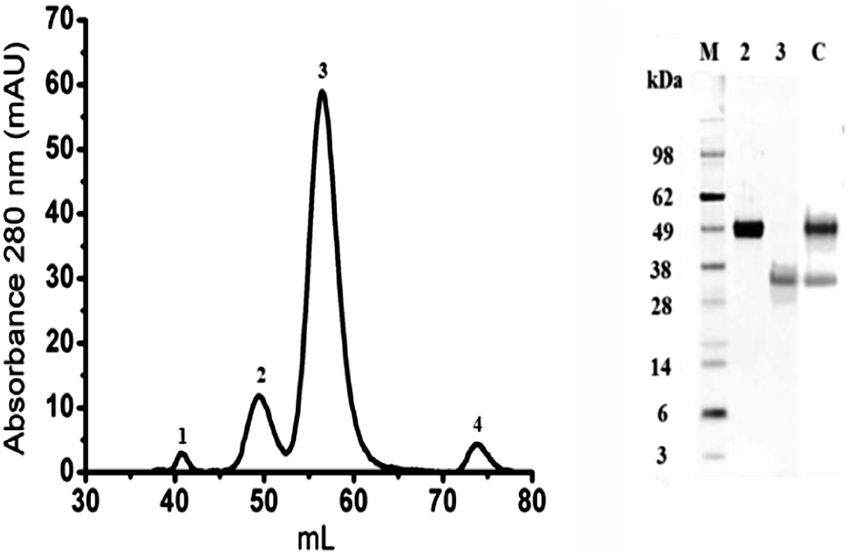 | ||
| Fig. 1 EbOPN and FbOPN purification from Lacprodan®10 by SEC. Left: the Lacprodan®10 proteins were separated by SEC on a TSK-GEL G3000 SW semi-preparative column with 50 mM Tris, 150 mM NaCl pH 7.4 as the mobile phase. The elution profile was recorded by monitoring the absorbance variations at 280 nm against the elution volume. Right: silver-stained SDS-PAGE controls of eluted fractions corresponding to EbOPN and FbOPN. Lane M: molecular markers (SeeBlue Plus2 Pre-stained Standards, Invitrogen); lane 2: purified EbOPN (peak 2); lane 3 purified FbOPN (peak 3); lane C: control (bovine OPN, R&D System). | ||
Protein purity was checked by mass spectra analysis on an LTQ-Orbitrap XL system from electrophoresis spots corresponding to the purified proteins. They confirmed that bOPN was the only protein present in the selected fractions. The very good sequence coverage and the absence of any other detected proteins in the spots indicated a very high purity level (Table 1).
| hOPN analysis on human OPN sequence | |||||
|---|---|---|---|---|---|
|
|
|||||
| Protein | Primary acc. numb. | Score | Mass (Da) | Coverage | #Peptides |
| OSTP_HUMAN | P10451 | 392 | 35401 |
32.8 | 8 |
| Peptide sequence | |||||
| 57 | YPDAVATWLNPDPSQK | ||||
| 46 | QLYNKYPDAVATWLNPDPSQK | ||||
| 71 | QNLLAPQNAVSSEETNDFKQETLPSK | ||||
| 48 | AIPVAQDLNAPSDWDSR | ||||
| 37 | GDSVVYGLR | ||||
| 45 | RPDIQYPDATDEDITSHMESEELNGAYK | ||||
| 52 | FRISHELDSASSEVN | ||||
| 44 | FRRPDIQYPDATDEDITSHMESEELNGAYK | ||||

| EbOPN analysis on bovine OPN sequence | |||||
|---|---|---|---|---|---|
|
|
|||||
| Protein | Primary acc. numb. | Score | Mass (Da) | Coverage | #Peptides |
| OSTP_BOVIN | P31096 | 295 | 30885 |
25 | 5 |
| Peptide sequence | |||||
| 100 | QTFLAPQNSVSSEETDDNK | ||||
| 50 | QTFLAPQNSVSSEETDDNKQNTLPSK | ||||
| 37 | SNVQSPDATEEDFTSHIESEEMHDAPK | ||||
| 48 | RSNVQSPDATEEDFTSHIESEEMHDAPK | ||||
| 61 | IRISHELDSASSEVN | ||||

| FbOPN analysis on bovine OPN sequence | |||||
|---|---|---|---|---|---|
|
|
|||||
| Protein | Primary acc. numb. | Score | Mass (Da) | Coverage | #Peptides |
| OSTP_BOVIN | P31096 | 273 | 30885 |
23.7 | 5 |
| Peptide sequence | |||||
| 47 | YPDAVATWLKPDPSQK | ||||
| 80 | QTFLAPQNSVSSEETDDNK | ||||
| 50 | QTFLAPQNSVSSEETDDNKQNTLPSK | ||||
| 59 | GDSVAYGLK | ||||
| 37 | IRISHELDSASSEVN | ||||

The case of FbOPN was quite different, because not only N-terminal peptides but also a C-terminal peptide, with low occurrence, were identified by mass spectra analysis. This result was consistent with low remaining sulfur content (∼0.15) estimated by ICP-MS. Plasmin cleavages can occur naturally in milk,29,31 and cleavage sites are present in the C-terminal part of the protein where methionine is found. One of these sites (S-R-S-K-K-E-R-R for bOPN) is located after the integrin-binding domain RGD. Therefore, the C-terminal fragment could be undetectable by SDS-PAGE, but shown by mass spectrum analysis. Again, a slight contamination by EbOPN could lead to the same result, and in both cases, this might be consistent with very low sulfur content in the FbOPN fraction. An average molecular weight was set at 35 kDa, corresponding to the apparent MW observed on the spots of PAGE-SDS controls.
hOPN was purified in our laboratory. The purification process was adapted from previous studies22 after its optimization on bovine milk. It led to highly purified OPNs in three single steps (acidic precipitation, ion exchange (IEX) and size exclusion chromatography (SEC)). From IEX chromatography, the fraction eluted with 0.3 M NaCl contained mainly full length hOPN, leading to an estimated purity of ∼90% by SDS-PAGE (Fig. 2A). Further purification of this fraction by SEC led to separation of the proteins into one main broad peak and two small peaks (Fig. 2B). Full length hOPN was detected in 4 sub-fractions of the main broad peak and SDS-PAGE controls demonstrated that the MW differed in these groups, decreasing from 55 kDa to 43 kDa. A theoretical 49 kDa was chosen, corresponding to the average apparent molecular weights on the SDS-PAGE, in agreement with a protein glycosylation estimated at around 33%.32 By taking 0.68 as the theoretical specific absorption coefficient at 280 nm (ProtParam, ExPaSy), 2 mg of highly purified full length hOPN were obtained from human milk in 3 simple steps.
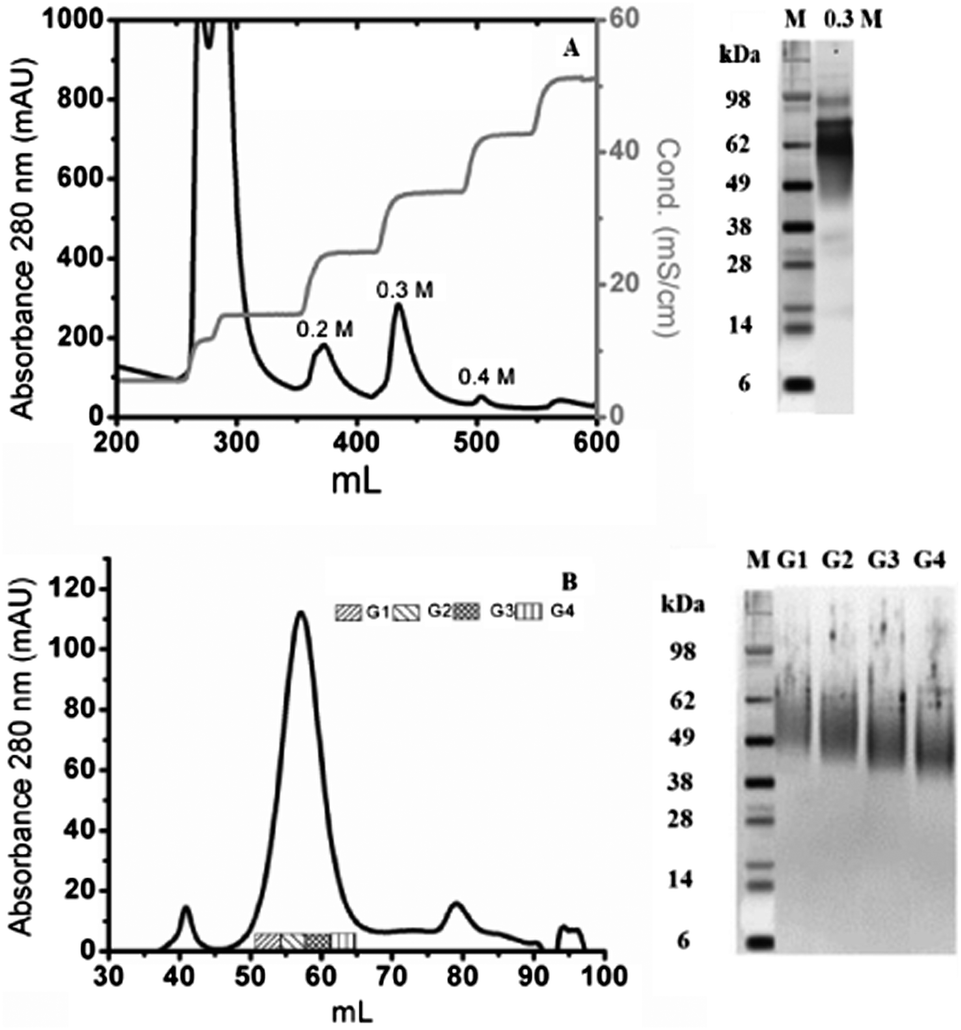 | ||
| Fig. 2 hOPN purification by IEX and SEC. (A) Acidic whey was injected on an IEX column (DEAE-Sephacel) and proteins were eluted by a step-gradient of NaCl. The elution profile was recorded by monitoring the absorbance variations at 280 nm (black line) and the conductivity (grey line) against the elution volume. (B) The proteins of the 0.3 M fraction were fractionated by SEC on a TSK-GEL G3000 SW semi-preparative column with 50 mM Tris, 150 mM NaCl pH 7.4 as the mobile phase. The silver-stained PAGE-SDS controls of the eluted proteins are indicated on the right of each chromatogram with M: molecular markers (SeeBlue Plus2 Pre-stained Standards, Invitrogen). | ||
Protein purity was also checked by mass spectrum analysis from electrophoresis spots corresponding to the purified proteins of the 4 sub-fractions. Again, hOPN was the only protein present in these sub-fractions. The very good sequence coverage and the absence of any other detected proteins in the spots indicated a very high purity level (Table 1).
The question of the involvement of phosphorylations on UO22+ binding was raised during the study. EbOPN and FbOPN available in larger amounts were used in these experiments, since the OPN sequence is highly conserved. After preliminary assays, OPNs were dephosphorylated (S1, ESI†), and the phosphorus contents of the OPNs were determined by ICP-MS (Table 2). A ratio of 26 ± 2 phosphorylations/molecule was found for hOPN, lower than the average level of 32 phosphorylation/molecule expected;11 while bOPN phosphorylation content (28 ± 2) was in agreement with published results.10 Dephosphorylated EbOPN and FbOPN presented around 15% of residual phosphorus content (ICP-MS) after the enzymatic reaction.
| hOPN | EbOPN | FbOPN | dP-EbOPN | dP-FbOPN | |
|---|---|---|---|---|---|
| P/OPN | 26 ± 2 | 28 ± 2 | 20 ± 2 | 4.5 ± 1 | 5.5 ± 1 |
| K D (nM) | 3.6 ± 1.3 | 3.0 ± 1.2 | 30 ± 2 | 185 ± 10 | 380 ± 70 |
| K D, 24h (nM) | 5.8 ± 1.3 | 3.2 ± 1.2 | 49 ± 2 | 315 ± 15 | 580 ± 90 |
OPN affinities for UO22+: impact of phosphorylation
The determination of the apparent affinity constant (KD) of proteins for uranyl from their corresponding dose-response curves has been described previously.25 Briefly the KD are deduced from an accurate quantification of free UO22+ at equilibrium by SPR analysis. KD values were determined for the OPNs, as illustrated for hOPN (Fig. 3, left). They were respectively 3.6 ± 1.3 nM, 3.2 ± 1.2 nM and 30 ± 2 nM for hOPN, bOPN and FbOPN (Table 2). These values remained stable and within the same range after 24 h, except for FbOPN for which a decreased KD was observed. This observation confirmed that EbOPN and hOPN interact strongly with UO22+in vitro, the FbOPN having ∼10 times less affinity than the full length proteins. This reduced affinity for uranyl ions might indicate a role of the C-terminal part of the OPN. Phosvitin, for which uranyl binding properties have been evidenced,13 also displayed a KD within the nanomolar range in our test, while albumin and transferrin were in the μM range33 (Fig. 3, right). Because the ionic strength is high in this test (50 mM Tris, 150 mM NaCl), these strong interactions cannot be explained only by simple ionic bonds. | ||
| Fig. 3 OPN affinity for uranyl: determination and comparison with other proteins. Left: example of apparent affinity determination of hOPN for UO22+ by SPR; signal variations (expressed in resonance units-RU) are plotted against log of hOPN concentration. Biological replicates at J0 (grey squares) and J+1 (dark triangles). The corresponding fitted curves are indicated on the graph. The inflection points correspond to the EC50 (apparent KD) and are identical for both experiments. Right: ranking of apparent KD of EbOPN, FbOPN, hOPN, dP-EbOPN and dP-FbOPN for UO22+, in comparison with albumin and transferrin. | ||
Even though the bOPNs were not fully dephosphorylated, their affinities were strongly altered, in particular for the dP-EbOPN, which presented a KD ∼60 times lower than that of EbOPN. A deterioration of the affinity was also observed after 24 h on board, with the dP-EbOPN displaying a KD ∼100 times lower. An affinity decrease was also observed for dP-FbOPN, but to a lesser extent (∼10–15 times lower).
UO22+ and OPN conformational modifications: CD analysis
Like other IDPs, OPNs are depleted in aromatic residues. This lack of a definite-ordered 3D structure gives the flexibility and plasticity of these proteins, which undergo structural transitions when they interact with their partners. CD is a method of choice to study structural modifications of proteins, and the question of UO22+ binding was addressed by analyzing potential structural changes induced by the cation.The impact of UO22+ additions on the OPN structure was monitored at pH 7.4. Due to experimental constraints, the ionic strength of the buffer was low in these experiments (5 mM Tris-HCl buffer, 15 mM NaCl). A typical spectrum obtained from UO2 titrations of EbOPN is given in Fig. 4A. The CD signatures are consistent with a randomly coiled polypeptide chain, characteristic of OPN conformations and flexible structures as previously published27 with a main large negative band centered at ∼200 nm and a slightly negative value at ∼210–230 nm. The intensity of the ∼200 nm band was higher for the FbOPN than for full length human or bovine OPN. Additions of UO22+ reduced the intensity of the minimum at 200 nm by 15% for hOPN, and by 22% for both EbOPN and FbOPN. It was accompanied by a slight red shift (2 nm for EbOPN and hOPN). Furthermore, the negative ellipticity in the 210–230 nm region was increased, reflecting a metal-induced secondary structure formation for the full length proteins,34,35 and probably an increased stability of helical regions of the proteins. For all the OPNs, an isodichroic point (IP) at ∼207 ± 1 nm was observed corresponding to two-state dichroic models and indicative of structural rearrangements. Typical CD variations at 200 and 222 nm and expressed in S/S0 (S and S0 in the presence and absence of UO22+ respectively) were plotted versus UO22+ additions. The signal variations at 200 and 222 nm displayed biphasic profiles, reaching a plateau at ∼35 equivalent UO22+ additions for hOPN, ∼30 for EbOPN and ∼20 for FbOPN (Fig. 4B–D).
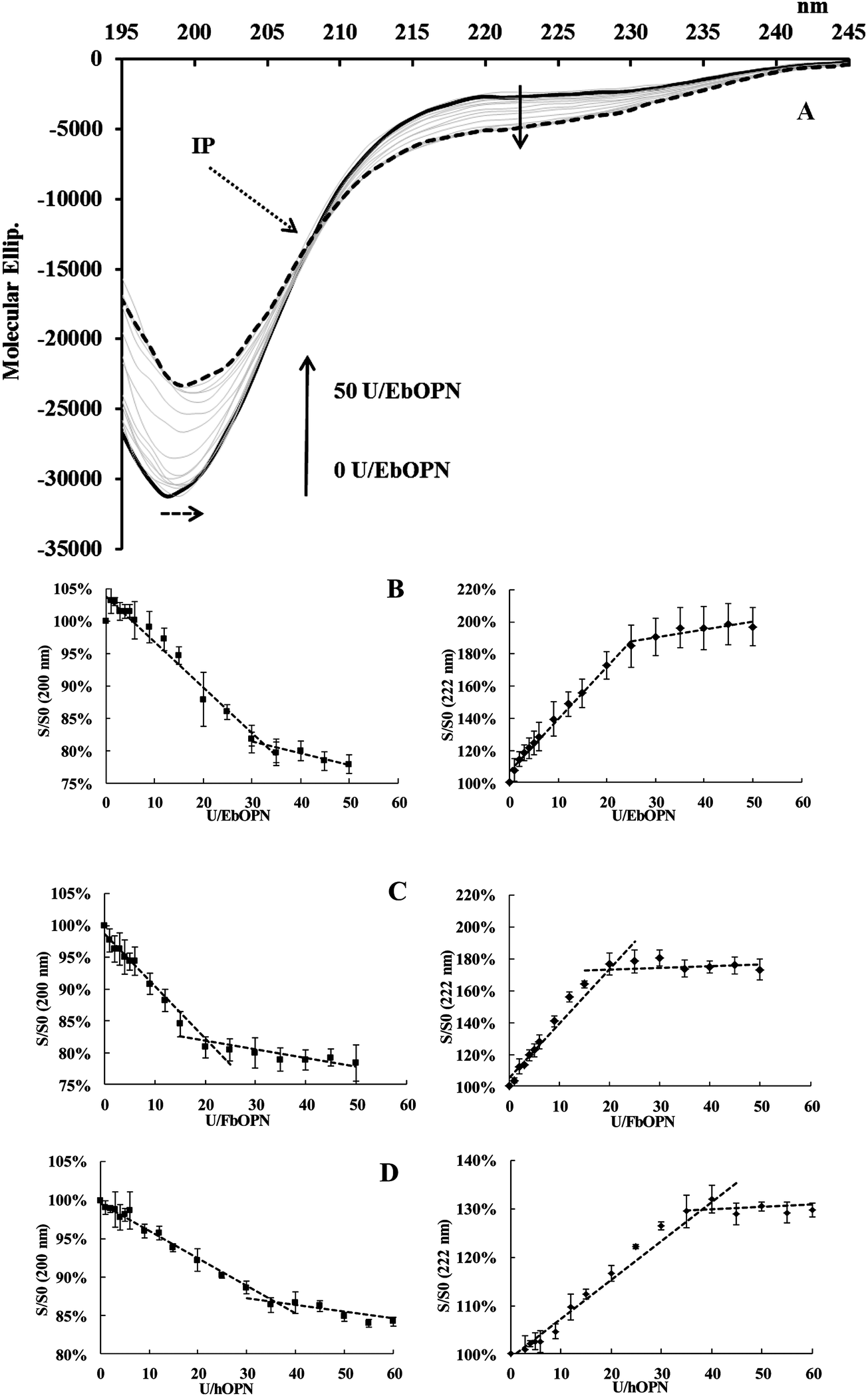 | ||
| Fig. 4 Evolution of CD spectra of OPN versus uranyl additions. (A) Molecular ellipticity variations vs. wavelength for bOPN upon uranyl additions. “U” represents UO22+. Continuous thick line: native EbOPN; dashed black line: 50U/EbOPN; continuous grey lines correspond to intermediate uranyl additions; the dotted black arrow indicates the isodichroic point (IP), and the dashed black arrow a λ shift at the valley. (B–D) Plots of CD signal variations at 222 nm and at 200 nm versus UO22+ additions for EbOPN, FbOPN and hOPN (S and S0 are the signals with and without UO22+; “U” represents UO22+). | ||
The CD spectra of UO22+ titration with dP-EbOPN and dP-FbOPN were different (Fig. 5A). Additions of UO22+ also reduced the intensity of the band at 200 nm (around 15–20%), and the S/S0 variations displayed a biphasic profile, with intercepts at ∼18 and ∼15 equivalent UO22+ additions for dP-EbOPN and dP-FbOPN respectively (Fig. 5B and C). But this was not accompanied by an isodichroic point around 207 nm. Even though formations of UO2–dP-OPN complexes cannot be totally excluded, UO22+ additions induced no real dP-OPN structuration at 222 nm, as suggested by the CD spectra evolutions.
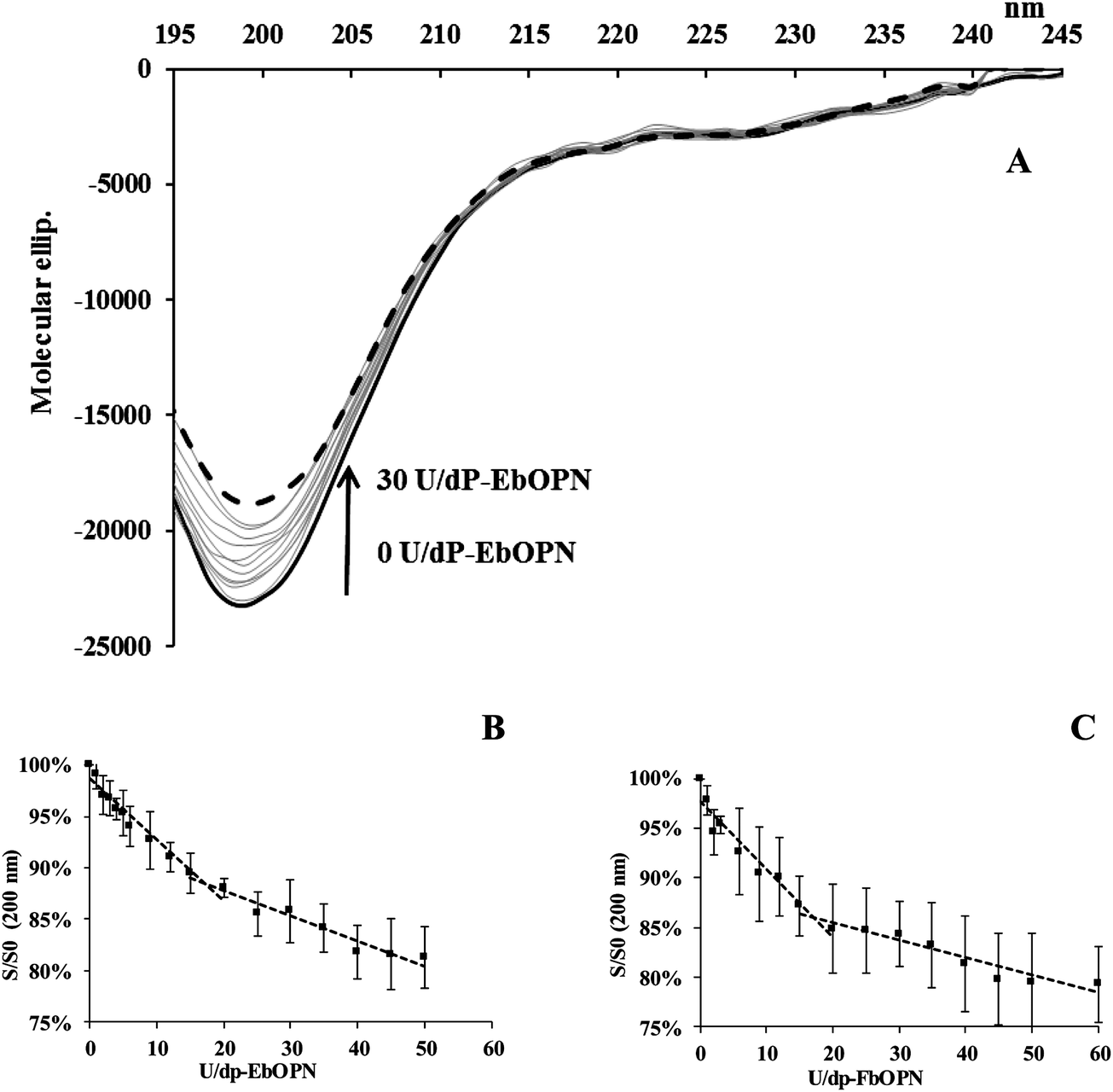 | ||
| Fig. 5 Evolution of CD spectra of dP-EbOPN and -FbOPN versus uranyl additions. (A) Molecular ellipticity variations vs. wavelength for bOPN upon uranyl additions. “U” represents UO22+. Continuous thick line: native dP-EbOPN; dashed black line: 30U/dP-EbOPN; continuous grey lines correspond to intermediate uranyl additions; (B, C) plots of CD signal variations at 200 nm versus UO22+ additions for dP-EbOPN and dP-FbOPN (S and S0 are the signals with and without UO22+; “U” represents UO22+). | ||
Thus, phosphorylated or not, the stabilizations of OPN CD spectra were only observed after additions of relatively high amounts of UO22+, these amounts being smaller for the dephosphorylated than for the phosphorylated OPNs. This observation raised the questions of specificity and binding strength. Chen and colleagues16 have observed that calcium binding sites in OPN were inversely proportional to the salt concentrations, with 50 calcium atoms bound per OPN molecule for 60 mM KCl and only 19 calcium atoms bound per OPN molecule for 150 mM NaCl. In both cases, OPN displayed a low affinity for calcium close to the mM range. These authors concluded “low affinity and high calcium capacity of OPN, consistent with a predominant electrostatic mode of metal binding”. It was also speculated that “the presence of phosphate modulates the lack of structure to some extent on the basis that the neighbouring electrostatic group repulsions might stiffen the protein backbone somewhat and increase the polypeptide chain persistence length”.36
In our CD experiments, and therefore under low ionic strength (5 mM Tris, 15 mM NaCl, pH 7.4), the UO22+ addition and binding could promote a partial folding of OPN due to neutralization of charge–charge repulsion. For native hOPN, EbOPN and FbOPN, the repulsive forces generated by the numerous phosphorylations are higher than in the case of dephosphorylated proteins, thus necessitating larger metal amounts to stabilize their conformation. Following UO22+ additions, different conformational behaviours were observed for native and dephosphorylated OPNs, as indicated by CD spectra variations at 200 and 222 nm. Numerous binding sites, displaying strong and weak affinities, and involving the phosphorylations or not, could explain these results. High apparent affinities of native OPNs for UO22+ were determined by SPR experiments, with a strongly reduced affinity for dephosphorylated OPNs. The ionic strength was higher in SPR experiments (50 mM Tris, 150 mM NaCl, pH 7.4) than in CD experiments, and these conditions could lead to the selection of binding sites displaying high affinities only. Whatever the case, either through OPN conformational changes monitored by CD or through quantification of unbound UO22+ to determine OPN affinities for the cation in SPR experiments, the results are given at equilibrium in both situations. Therefore to settle the question of “a high affinity or a high capacity of OPNs for uranyl”, the stability of the UO2–protein complexes formed was investigated.
Phosphorylated but not dephosphorylated OPNs form stable UO2–OPN complexes
In SPR experiments, the apparent KD was determined at equilibrium. SEC is a mild method which can be used to separate UO2–OPN complexes from the unbound part of UO22+ without causing major changes in the equilibrium. Therefore, the UO2–OPN complexes were isolated by SEC to differentiate unspecific weak bonds from more specific ones. Their uranium content was determined by ICP-MS. First of all, most of the UO22+ was eliminated by SEC, as observed on the chromatogram elution profiles (data not shown), indicative of a dissociation of weak bonds occurring in the separation process. The UV-vis spectra of the eluted UO2–OPN complexes were carefully observed. UO22+ solutions absorb within the 260–280 nm range, with 260nmA/280nmA = 1.64 ± 0.02, whereas proteins exhibit 260nmA/280nmA below 1. The 260nmA/280nmA were checked for the phosphorylated and un-phosphorylated proteins after SEC. The average results of at least 4 different runs are presented (Table 3). These ratios were multiplied by 1.5–1.6 for UO2–EbOPN, UO2–hOPN and UO2–FbOPN, but remained quite similar (1.05–1.1) for the dephosphorylated OPNs, suggesting that UO2–OPN stable complexes were formed only with phosphorylated proteins.| OPN | UO2–OPN complexes | U/complex | ||
|---|---|---|---|---|
| 260nm A/280nmA | 260nm A/280nmA | |||
| EbOPN | 0.79 ± 0.03 | UO2–EbOPN | 1.29 ± 0.02 | 6 ± 1 |
| FbOPN | 0.74 ± 0.02 | UO2–FbOPN | 1.13 ± 0.05 | 5 ± 1 |
| hOPN | 0.63 ± 0.02 | UO2–hOPN | 0.97 ± 0.03 | 9 ± 1 |
| UO2–dP-EbOPN | 0.83 ± 0.05 | (<1) | ||
| UO2–dP-FbOPN | 0.80 ± 0.02 | (<1) |
The remaining uranium contents of the purified complexes were then determined by ICP-MS (Table 3). The complexes from native OPNs conserved high UO2–protein ratios, with 9 ± 1 UO2 for hOPN, 6 ± 1 UO2 for EbOPN, and 5 ± 1 UO2 for the FbOPN. No aggregation occurred, since no complex presented any absorbance in the 400–600 nm region. The UO2–protein ratios were very low (around 0.15) for the dephosphorylated OPNs, consistent with the UV-vis spectra analysis.
These results confirmed the major role of the phosphorylations in establishing strong bonds with UO22+, inducing stable structuration of the bovine and human OPNs with respectively ∼6 and 9 UO2 equivalents per EbOPN and hOPN molecule. The OPN C-terminal part, which also presents some phosphorylated serine residues, could enhance the stabilisation of the complex, since 5 UO2 equivalents were bound per FbOPN compared to 6 for the EbOPN. Sorensen et al. indicated that “the phosphorylations are clustered in groups of approximately three, spanned by unphosphorylated regions”.10 Strikingly, the UO2vs. phosphorus ratios were found to be around 4.5 for EbOPN and FbOPN, and 3 for hOPN.
Compared to previous studies indicating that OPN can bind 8 Ca2+, probably in the aspartic region, and that dephosphorylated OPN seemed to conserve its Ca2+-binding ability,15 this study demonstrated that phosphorylations play an essential role in UO22+ binding since the dephosphorylated OPNs had by far less affinity for UO22+ and did not lead to stable UO2–OPN complexes. In the presence of UO22+ cations, the polyaspartic region and/or the acidic residues still present in the dephosphorylated bOPNs could not lead to their structuration and the complexes formed were not stable enough to be isolated after SEC. To confirm this hypothesis, the affinities of varied peptides were determined by SPR. The H8V peptide (H-pS-D-E-pS-D-E-V) displayed a 4.7 ± 0.5 μM apparent affinity within the same range as the binding constant determined by ITC with logK = 5.20.18 But no affinity could be measured either for D-D-D-D-D-D, or for E-E-E-E-E-E or E-D-E-D-E-D peptides. The EXAFS study of the UO2–H8V complex indicated that 1 oxygen atom from the phosphorylated serine, two from the carboxylate group of the neighbouring aspartate and two water molecules were present in the first coordination sphere. A very similar coordination environment was also observed with the bOPN contacted with 1 UO22+ equivalent per molecule. Of the 36 potentially phosphorylable serine residues in hOPN, 29 are located in pS-X-E/pS sequences10,36 identical to those of H8V, suggesting that these basic patterns are probably essential in UO22+ binding, inducing affinities from the micro to the nanomolar range depending on their location and number.
Conclusions
Transcriptomic analysis has highlighted the involvement of OPN in the UO22+ toxicity mechanisms.5 This major non-collagenous protein involved in the organo-mineral homeostasis of the bone attracted attention because it presents a specific composition in acidic clusters associated with numerous phosphorylations, and also a relative plasticity as an intrinsically disordered protein. All these properties seemed favourable to the binding of the particularly large UO22+ cation. In this study, it has been demonstrated that native phosphorylated OPNs bind UO22+ with a nanomolar affinity, and that this binding induces conformational changes enabling the formation of stable complexes with 6 and 9 equivalents of uranium per mol of protein for bovine and human OPN, respectively. Compared to previous studies indicating that OPN dephosphorylation does not impede its Ca2+-binding ability,15 we demonstrated that these groups play an essential role in the affinity of OPN for UO22+ and the establishment of stable complexes. Serine residues are usually not fully phosphorylated, except for milk OPN. Thus, the uranium content found in this study has probably reached the upper limit of uranyl binding capacity of OPNs. It could be speculated that the location of the phosphorylated residues has an influence on the affinity for the metal ion and on the stability of the complex.This leads to two considerations for the future. First, the impact of the metal ion on the function of IDP has been documented for those involved in neurodegenerative diseases.35,41 There is some preliminary evidence that IDP might be involved in the control of mineralization.39,40 To our knowledge, this is the first report on the involvement of a SIBLING protein. This further exemplifies the impact of phosphorylations on IDP function.37,38 Second, the feature of multiple phosphorylations alternating with acidic clusters in OPN is shared with other noncollagenous SIBLING proteins involved in mineralization processes. This is particularly true for bone sialoproteins, which present very similar sequences and a poly-A motif instead of the poly-D motif, but also for dentin matrix protein DMP1, dentin sialophosphoprotein DSPP, and matrix extracellular phosphoglycoprotein MEPE.36,42 All these proteins may be potential uranyl targets. Therefore it could be interesting to evaluate their affinity and analyze whether they are correlated to similar/different sequences, phosphorylation degrees, etc., with OPN. This would provide more precise information on the uranyl-binding sites. The more accurate the data on uranyl binding sites, the better the design of new chelating agents to prevent uranyl accumulation can be. Further studies are in progress.
Acknowledgements
Lei Qi was sponsored by the China Scholarship Council. We thank the CEA Toxicologie Nucleaire Program for funding. We acknowledge Jean Charles Gaillard's aid for mass spectrometry analysis of proteins and Sophie Plantevin for ICP-MS controls. We thank the Arla Foods Group (Viby J, Denmark) who provided the Lacprodan®10 sample.Notes and references
- T. C. Pellmar, A. F. Fuciarelli, J. W. Ejnik, M. Hamilton, J. Hogan, S. Strocko, C. Emond, H. M. Mottaz and M. R. Landauer, Distribution of uranium in rats implanted with depleted uranium pellets, Toxicol. Sci., 1999, 49, 29–39 CrossRef CAS PubMed.
- O. Pible, P. Guilbaud, J. L. Pellequer, C. Vidaud and E. Quemeneur, Structural insights into protein-uranyl interaction: towards an in silico detection method, Biochimie, 2006, 88, 1631–1638 CrossRef CAS PubMed.
- P. W. Durbin, Actinides in Animals and Man, in The Chemistry of the Actinide and Transactinide Elements, 2006, vol. 3, 3339–3440 Search PubMed.
- C. Vidaud, D. Bourgeois and D. Meyer, Bone as Target Organ for Metals: The Case of f-Elements, Chem. Res. Toxicol., 2012, 25, 1161–1175 CrossRef CAS PubMed.
- O. Prat, F. Berenguer, G. Steinmetz, S. Ruat, N. Sage and E. Quemeneur, Alterations in gene expression in cultured human cells after acute exposure to uranium salt: involvement of a mineralization regulator, Toxicol. In Vitro, 2012, 24, 160–168 CrossRef PubMed.
- P. V. Azzopardi, J. O'Young, G. Lajoie, M. Karttunen, H. A. Goldberg and G. K. Hunter, Roles of electrostatics and conformation in protein-crystal interactions, PLoS One, 2010, 5, e9330 Search PubMed.
- D. T. Denhardt and X. Guo, Osteopontin: a protein with diverse functions, FASEB J., 1993, 7, 1475–1482 CAS.
- T. Standal, M. Borset and A. Sundan, Role of osteopontin in adhesion, migration, cell survival and bone remodeling, Exp. Oncol., 2004, 26, 179–184 CAS.
- M. D. McKee, C. E. Pedraza and M. T. Kaartinen, Osteopontin and Wound Healing in Bone, Cells Tissues Organs, 2011, 194, 313–319 CrossRef CAS PubMed.
- E. S. Sorensen, P. Hojrup and T. E. Petersen, Posttranslational Modifications of Bovine Osteopontin – Identification of 28 Phosphorylation and 3 O-Glycosylation Sites, Protein Sci., 1995, 4, 2040–2049 CrossRef CAS PubMed.
- B. Christensen, M. S. Nielsen, K. F. Haselmann, T. E. Petersen and E. S. Sorensen, Post-translationally modified residues of native human osteopontin are located in clusters: identification of 36 phosphorylation and five O-glycosylation sites and their biological implications, Biochem. J., 2005, 390, 285–292 CrossRef CAS PubMed.
- A. Dedieu, F. Berenguer, C. Basset, O. Prat, E. Quemeneur, O. Pible and C. Vidaud, Identification of uranyl binding proteins from human kidney-2 cell extracts by immobilized uranyl affinity chromatography and mass spectrometry, J. Chromatogr., A, 2009, 1216, 5365–5376 CrossRef CAS PubMed.
- B. Li, J. Raff, A. Barkleit, G. Bernhard and H. Foerstendorf, Complexation of U(VI) with highly phosphorylated protein, phosvitin A vibrational spectroscopic approach, J. Inorg. Biochem., 2010, 104, 718–725 CrossRef CAS PubMed.
- R. Pardoux, S. Sauge-Merle, D. Lemaire, P. Delangle, L. Guilloreau, J.-M. Adriano and C. Berthomieu, Modulating uranium binding affinity in engineered calmodulin EF-hand peptides: effect of phosphorylation, PLoS One, 2012, 7, e41922 CAS.
- K. Singh, D. Deonarine, V. Shanmugam, D. R. Senger, A. B. Mukherjee, P. L. Chang, C. W. Prince and B. B. Mukherjee, Calcium-Binding Properties of Osteopontin Derived from Nonosteogenic Sources, J. Biochem., 1993, 114, 702–707 CAS.
- Y. Chen, B. S. Bal and J. P. Gorski, Calcium and collagen binding properties of osteopontin, bone sialoprotein, and bone acidic glycoprotein-75 from bone, J. Biol. Chem., 1992, 267, 24871–24878 CAS.
- L. L. Liu and K. J. Franz, Phosphorylation-dependent metal binding by alpha-synuclein peptide fragments, J. Biol. Inorg. Chem., 2007, 12, 234–247 CrossRef CAS PubMed.
- S. Safi, G. Creff, A. Jeanson, L. Qi, C. Basset, J. Roques, P. L. Solari, E. Simoni, C. Vidaud and C. Den Auwer, Osteopontin: A Uranium Phosphorylated Binding-Site Characterization, Chemistry, 2013, 19, 11261–11269 CrossRef CAS PubMed.
- L. W. Fisher, J. T. Stubbs and M. F. Young, Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins, Acta Orthop. Scand., Suppl., 1995, 66, 61–65 CrossRef.
- M. F. Young, J. M. Kerr, J. D. Termine, U. M. Wewer, M. G. Wang, O. W. McBride and L. W. Fisher, cDNA cloning, mRNA distribution and heterogeneity, chromosomal location, and RFLP analysis of human osteopontin (OPN), Genomics, 1990, 7, 491–502 CrossRef CAS PubMed.
- L. Schack, A. Lange, J. Kelsen, J. Agnholt, B. Christensen, T. E. Petersen and E. S. Sorensen, Considerable variation in the concentration of osteopontin in human milk, bovine milk, and infant formulas, J. Dairy Sci., 2009, 92, 5378–5385 CrossRef CAS PubMed.
- N. Azuma, A. Maeta, K. Fukuchi and C. Kanno, A rapid method for purifying osteopontin from bovine milk and interaction between osteopontin and other milk proteins, Int. Dairy J., 2006, 16, 370–378 CrossRef CAS.
- S. Zhuo, J. C. Clemens, R. L. Stone and J. E. Dixon, Mutational analysis of a Ser/Thr phosphatase, Identification of residues important in phosphoesterase substrate binding and catalysis, J. Biol. Chem., 1994, 269, 26234–26238 CAS.
- P. T. Cohen and P. Cohen, Discovery of a protein phosphatase activity encoded in the genome of bacteriophage lambda. Probable identity with open reading frame 221, Biochem. J., 1989, 260, 931–934 CrossRef CAS PubMed.
- O. Averseng, A. Hagege, F. Taran and C. Vidaud, Surface plasmon resonance for rapid screening of uranyl affine proteins, Anal. Chem., 2010, 82, 9797–9802 CrossRef CAS PubMed.
- C. Vidaud, A. Dedieu, C. Basset, S. Plantevin, I. Dany, O. Pible and E. Quemeneur, Screening of human serum proteins for uranium binding, Chem. Res. Toxicol., 2005, 18, 946–953 CrossRef CAS PubMed.
- L. W. Fisher, D. A. Torchia, B. Fohr, M. F. Young and N. S. Fedarko, Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin, Biochem. Biophys. Res. Commun., 2001, 280, 460–465 CrossRef CAS PubMed.
- E. Szollosi, E. Hazy, C. Szasz and P. Tompa, Large systematic errors compromise quantitation of intrinsically unstructured proteins, Anal. Biochem., 2007, 360, 321–323 CrossRef CAS PubMed.
- N. Bissonnette, P. L. Dudemaine, C. Thibault and G. Robitaille, Proteomic analysis and immunodetection of the bovine milk osteopontin isoforms, J. Dairy Sci., 2012, 95, 567–579 CrossRef CAS PubMed.
- A. P. Yamniuk, H. Burling and H. J. Vogel, Thermodynamic characterization of the interactions between the immunoregulatory proteins osteopontin and lactoferrin, Mol. Immunol., 2009, 46, 2395–2402 CrossRef CAS PubMed.
- B. Christensen, L. Schack, E. Klaning and E. S. Sorensen, Osteopontin is cleaved at multiple sites close to its integrin-binding motifs in milk and is a novel substrate for plasmin and cathepsin D, J. Biol. Chem., 2010, 285, 7929–7937 CrossRef CAS PubMed.
- J. Sodek, B. Ganss and M. D. McKee, Osteopontin, Crit. Rev. Oral Biol. Med., 2000, 11, 279–303 CAS.
- C. Basset, O. Averseng, P. J. Ferron, N. Richaud, A. Hagege, O. Pible and C. Vidaud, Revision of the biodistribution of uranyl in serum: is fetuin-a the major protein target?, Chem. Res. Toxicol., 2013, 26, 645–653 CrossRef CAS PubMed.
- J. P. Gorski, E. Kremer, J. Ruiz-Perez, G. E. Wise and A. Artigues, Conformational analyses on soluble and surface bound osteopontin, Ann. N. Y. Acad. Sci., 1995, 760, 12–23 CrossRef CAS PubMed.
- L. Breydo and V. N. Uversky, Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases, Metallomics, 2011, 3, 1163–1180 RSC.
- A. George and A. Veis, Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed.
- P. Tompa, Intrinsically unstructured proteins, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS PubMed.
- V. N. Uversky and A. K. Dunker, Understanding protein non-folding, Biochim. Biophys. Acta, 2010, 1804, 1231–1264 CrossRef CAS PubMed.
- A. Gericke, C. Qin, L. Spevak, Y. Fujimoto, W. T. Butler, E. S. Sorensen and A. L. Boskey, Importance of phosphorylation for osteopontin regulation of biomineralization, Calcif. Tissue Int., 2005, 77, 45–54 CrossRef CAS PubMed.
- G. K. Hunter, J. O'Young, B. Grohe, M. Karttunen and H. A. Goldberg, The flexible polyelectrolyte hypothesis of protein-biomineral interaction, Langmuir, 2010, 26, 18639–18646 CrossRef CAS PubMed.
- A. Santner and V. N. Uversky, Metalloproteomics and metal toxicology of alpha-synuclein, Metallomics, 2010, 2, 378–392 RSC.
- C. Qin, O. Baba and W. T. Butler, Post-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis, Crit. Rev. Oral Biol. Med., 2004, 15, 126–136 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Silver stained PAGE-SDS controls of EbOPN and FbOPN dephosphorylations. See DOI: 10.1039/c3mt00269a |
| This journal is © The Royal Society of Chemistry 2014 |