In-cell NMR: an emerging approach for monitoring metal-related events in living cells
Hongyan
Li
* and
Hongzhe
Sun
*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: hylichem@hku.hk; hsun@hku.hk; Fax: +86 852 2857 1586; Fax: +86 852 2859 8974
First published on 11th October 2013
Abstract
In-cell NMR, an isotope-assisted multi-dimensional NMR technique, has been proven to be successful in the investigation of protein dynamics, folding, conformational changes induced by binding events, posttranslational modification in the complex native environments, as well as in vivo drug screening, even de novo 3D protein structure determination in living cells. This technique was initially applied to bacterial cells, and subsequently has been extended to various other cells including eukaryotic cells. In this review, we briefly summarize the methodology and application of in-cell NMR with a focus on its application in metallomics and metalloproteomics. This emerging technique is anticipated to be an excellent tool for studying metal-associated events in complex native environments of living cells.
 Hongyan Li | Hongyan Li is a senior research associate at the University of Hong Kong. She received her PhD in inorganic biochemistry from the University of Edinburgh in 2000. She then worked as a postdoc at The Hong Kong Polytechnic University. Her current research interests lie in biological NMR, structural biology and metalloproteins. |
 Hongzhe Sun | Hongzhe Sun is a professor of chemistry at the University of Hong Kong. His current research interests focus on metalloproteins metallodrugs, as well as metallomics/metalloproteomics. He has published over 130 papers and edited a book entitled “Biological chemistry of arsenic, antimony and bismuth”. He is the recipient of Crocher Senior Research Fellow. |
1. Introduction
NMR spectroscopy has been proven to be extremely useful for studying protein dynamics and structures at atomic resolution under physiologically relevant conditions.1 However, in vitro NMR, in most cases carried out in buffers optimized for the purpose of observation of NMR parameters, may sometimes not fully reflect the situation for a protein in vivo. The native crowded and complex environments in which proteins reside inside cells often affect their biological processes such as protein–protein interactions, protein folding, dynamics, and aggregation behavior as well as protein stability.2 For example, an in vitro intrinsically disordered protein FIg M gains structure inside living E. coli cells;2b while, in contrast, macromolecular crowding keeps α-synuclein disordered in E. coli cells.3 Therefore observation of proteins or other macromolecules in living cells is indispensable for explicit understanding of the biological function of proteins and subsequently their involvement in other cellular events.The first observation of proteins by NMR in living cells was achieved over a decade ago by two research groups independently. The N-terminal domain of the bacterial mercury-detoxification protein (NmerA) was observed in E. coli cytoplasm by two-dimensional [1H, 15N] HSQC spectroscopy,4 and in the meantime, the mobility of osmoregulated periplasmic glycans of Ralstonia solanacearum was also investigated in the bacterial periplasm by high-resolution [1H, 13C] HSQC NMR under magic angle spinning conditions.5 Different from conventional in vivo NMR (usually one dimensional NMR), by which small molecules in living organisms e.g. metabolites or metal ions could be observed from suspensions of bacteria and other cells to entire perfused organs,6 in-cell NMR spectroscopy takes advantage of the multi-dimensional NMR technique together with the aid of isotope-labelling of targeted proteins, allowing researchers to characterize conformations and dynamics as well as other biological events of proteins and other macromolecules in their natural environments at the atomic level.7 Owing to the fact that 3D structures of proteins do not vary significantly with their environments, in-cell NMR experiments are not designed for direct structure determination of proteins in the cellular environments despite such an attempt has been made successfully for certain proteins.8 Instead, in-cell NMR takes advantage of the chemical shifts being sensitive towards changes in the environments to obtain information of macromolecules in their native surroundings. Since its inception, in-cell NMR has been used extensively to study protein dynamics,7c,9 folding,2b,d binding events,4a,7b,10in vivo protein maturation,11 intrinsically disordered proteins,2e,12 as well as protein post-translational modifications in living cells.13 Moreover, this technique has also been proven to be an important tool for in vivo drug screening by the so-called STINT-NMR. This approach involves sequential expression of two or more proteins within single bacteria cells and subsequently monitors their interactions with potential drugs.10a,14 Very recently, in-cell NMR has also been implicated as a lipidomic tool to investigate structural changes within the mobile lipid pool.15
Recently, in-cell NMR has been used to study metalloproteins,11 metallodrug binding,10c as well as metal binding inside living cells.4b,7b,11b Metalloproteins represent a large share of the proteome, account for nearly one-third of all human proteins with the intrinsic metal ions playing critical roles in protein functions.16 Given the importance of metal ions as well as metalloproteins in life processes, metallomics and metalloproteomics, an emerging field in parallel to genomics and proteomics has received increasing attention.17 However, understanding the function of metalloproteins often comes from individual characterization of the protein structures as well as of metal centres. In spite of numerous efforts being done on metalloproteins and metal binding events, most studies were performed in vitro by various biochemical and biophysical techniques.18 Attempts have also been made to match metals to proteins in cells and monitor metals in a single pathogen.19 Previously NMR has been used extensively in the study of structures, dynamics and metal-binding properties of metalloproteins.20 Recently, it has been clearly demonstrated that metal selectivity of metalloproteins in vivo is different from that in vitro7b and this may hold true for metalloprotein folding.21 Therefore, to understand in vivo biological functions of these proteins, subsequently their involvement in other important biological or disease processes, investigation of metalloproteins as well as metal binding properties of proteins inside living cells should provide invaluable information. In the present review, we will briefly summarize the methodology of in-cell NMR as detailed information can be found elsewhere.22 We will mainly focus on its application for monitoring metalloproteins and metal-binding properties in living cells.
2. Methodology
In-cell NMR spectroscopy deals with biological macromolecules such as proteins and thus requires the NMR signals of target molecules of interest to be distinguishable from others of cellular components. Similar to those in vitro experiments, the prerequisite of applying NMR techniques to proteins inside living cells is that the protein of interest must be over-expressed to a certain level and also adequately labelled with NMR active isotopes (e.g.13C, 15N),7a,22a and the sample preparation procedure as summarized in Fig. 1 for in-cell NMR is different from that for in vitro experiments, depending on the type of cell used e.g. prokaryotic or eukaryotic cells. In eukaryotic cells, direct overexpression of the protein of interest though is possible. However, in most cases, the protein of interest is first overexpressed in bacterial cells and purified, subsequently introduced into eukaryotic cells via different approaches (Fig. 1B). In order to obtain reasonable NMR signals, the protein of interest must tumble freely inside the cell which means its rotational correlation time must be within a certain range, allowing narrow resonance lines to be observed. In spite of higher intracellular viscosity (ca. 8-fold) in different cells compared with that of water, this appeared not to be a limiting factor for observation of proteins inside living cells, especially with the use of high-field NMR spectrometers equipped with a cryogenic probe, combined with advance NMR techniques such as TROSY-HSQC, SOFAST-HMQC NMR spectroscopy. Instead, transient interaction with cytoplasmic components affects the mobility of the proteins thus their NMR detectability.10b Finally, different from in vitro experiments, in-cell NMR requires the cells to be alive during the course of experiments to ensure biological significance of the studies. Various approaches such as encapsulation and immobilization of culture cells at high density, addition of cell protectants such as glycerol or sucrose or alginate have been attempted to extend cell viability.22b Alternatively, flow cells or bioreactors that supply cells with nutrients and gases have also been used for in-cell NMR experiments.23 | ||
| Fig. 1 Summary of in-cell NMR sample preparation procedures. (A) Prokaryotic cells. Similar to conventional sample preparation for in vitro NMR, the protein of interest is prepared by growing the cells in unlabeled medium to a certain optical density, followed by induction in isotopically labelled medium and the slurry is used for in-cell NMR detection directly. (B) Eukaryotic cells. Top panel: the protein of interest is overexpressed in bacterial cells and purified, followed by introduction into the eukaryotic cells via either microinjection (e.g. Xenopus laevis oocytes), or cell penetrating peptides (CCP) (e.g. twin arginine peptide) covalently linked to the proteins to allow entry of the protein which subsequently was released inside the cells, or through cell-permeabilizing toxins to form a pore which enables the protein to be diffused into the cells through a concentration gradient. Bottom panel: the protein of interest is directly overexpressed in eukaryotic cells and in-cell NMR was carried out in these cells similarly as in prokaryotic cells. | ||
2.1 In-cell NMR sample preparation in prokaryotic cells
Similar to in vitro NMR experiments, observation of NMR signals of a specific protein in living bacterial cells such as E. coli cells relies on labelling of the protein of interest with NMR-active isotopes as well as overexpression of the protein to a certain level. The same rational of sample over-expression and isotopic labelling in bacterial cells is applicable for prokaryotic in-cell NMR spectroscopy. 15N, 13C and 19F have been used for such labelling with 19F giving virtually zero background due to low abundance of fluorine compounds in cells.24 However, 19F labelling requires introducing an unnatural amino acid which may interfere with the biological function of the protein. Full 13C labelling usually gives rise to high background noises owing to higher abundance of carbon than nitrogen in small molecules in cellular components, which makes such labelling less useful than 15N labelling for in-cell NMR studies.22d,25 Nevertheless, selective labelling with [13C]-methyl groups on methionine (–εCH3) and alanine was proven to provide excellent sensitivity with low background levels and can be used to detect proteins as well as their interactions with other molecules in cells.Majority of in-cell NMR experiments were carried out in bacterial cells with the aid of 15N labelling of the protein of interest.4b,7b,8,26 The well-established techniques used for overexpression and labelling proteins in bacteria have been demonstrated to be adaptable for sample preparation for in-cell NMR experiments.4b,7a In the case of E. coli (Fig. 1A), the cells harbouring the expression plasmid are firstly grown in unlabelled LB medium to attain certain optical density and then transferred to isotope-enriched culture medium (with 15NH4Cl as the sole nitrogen source) prior to induction by IPTG and the cell slurry (ca. 30%) can be transferred directly into NMR tubes for spectral acquisition.4b,22d In comparison with 13C labelling, 15N labelling usually gives rise to less intense background noises. The quality of the in-cell NMR signals of 15N-labelled target proteins highly depends on the behaviour of individual proteins such as expression levels and oligomerization states. 15N-labelling of certain types of amino acids can also be used to resolve peak overlapping both for in vitro and in-cell NMR.4b
2.2 In-cell NMR sample preparation in eukaryotic cells
Over-expression and isotopic labelling in the systems other than E. coli have been shown to be difficult owing to the lack of strong promoters capable of producing appropriate amounts of isotopically labelled proteins for in-cell NMR detection, thus the protein of interest often needs to be produced in a similar approach to in vitro experiments and the purified isotopically labelled protein is subsequently introduced into living eukaryotic cells using various approaches as shown in Fig. 1B. In eukaryotic cells, in-cell NMR experiments were performed in most cases by injection of isotopically labelled highly concentrated proteins into Xenopus laevis oocytes or eggs,21,27 which has been successfully used for observation of the protein of interest. Such an approach enabled in situ monitoring of protein phosphorylation by in-cell NMR.13 For cultured mammalian cells, cell-penetrating peptides (CPP) and pore-forming cell-permeable toxin have been used to deliver proteins successfully.28 Recently, attempts have been made to overexpress and observe proteins in eukaryotic cells directly by in-cell NMR (Fig. 1B). In-cell NMR observation of yeast ubiquitin overexpressed in Pichia pastoris has allowed metabolic changes to be monitored in yeast vesicles.29 Structural analysis of the Streptococcus protein GB1 domain overexpressed in insect cells using the sf9 cell–baculovirus system has also been demonstrated to be feasible.30 Moreover, in-cell NMR has been successfully used to monitor the maturation process of superoxide dismutase 1 (SOD1) in human embryonic kidney cells (HEK293T).11a The protein was expressed directly in HEK293T cells through transient expression to attain high concentrations (vide infra).3. Application of in-cell NMR in metallomics/metalloproteomics
Despite that in-cell NMR was introduced about a decade ago, there are very limited reports on its application to metalloproteins as well as to monitoring metal binding properties of metalloproteins in living cells. This is largely due to the complexity of such studies since the structures, folding, aggregation behaviour, stability and even biological functions of metalloproteins are often highly dependent on the features of metal ions bound, i.e. the type of metal ion, the oxidation states, spin states and corresponding coordination geometries,16 which might be distinct inside living cells from those in vitro experiments.7b,11b Thus to fully understand the properties and biological function of metalloproteins, it is necessary to study these proteins in their native complex environments. Up to now, in-cell NMR has been attempted to investigate the binding mode (motif) of metallodrugs to metallochaperones, metal selectivity of metalloproteins as well as metal induced metalloprotein folding as illustrated in Fig. 2.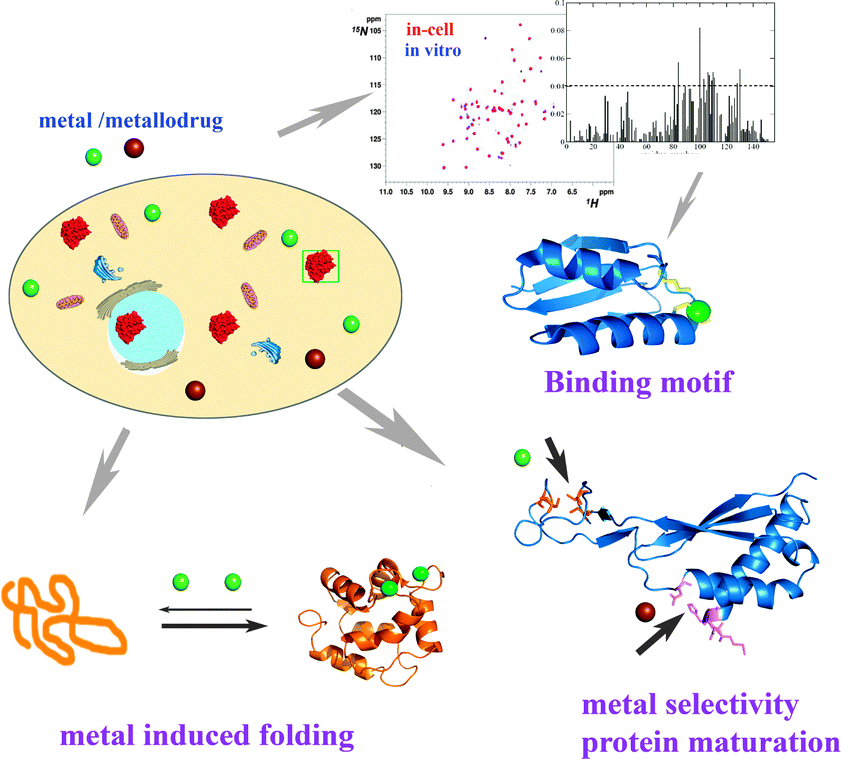 | ||
| Fig. 2 Schematic overview of application of in-cell NMR for the study of metal-relevant biochemical events. In-cell NMR is able to detect the interaction of metallodrugs or metal ions in living cells such as the metal binding motif and kinetics of the binding. Metal selectivity as well as metal-induced protein folding and conformational changes can also be examined by this technique. | ||
3.1 Probing metallodrug–protein interaction
Cisplatin (cis-Pt(NH3)2Cl2) appears to be one of the most successful metallodrugs in clinics for the treatment of various cancers. The mechanism of action of the drug has been attributed to the formation of stable adducts with DNA, thus interfering with replication and transcription processes.31 Cellular uptake and activation of the drug were unexpectedly found to be associated with copper(I) transporters 1 (Ctr1).32 Besides Ctr1, many other copper proteins have also been demonstrated to play important roles in sequestration and efflux of the drug from cells.33 However, up to now, most of the studies have been carried out in vitro, and there appear to be very limited studies in the complex cellular environments though such studies are essential for understanding the binding mode or motif as well as binding kinetics of the drugs under real cellular conditions. Owing to the fact that metallodrugs might be toxic to the cells at high concentrations, it is much complicated to probe the interaction of metallodrugs with the potential protein targets by in-cell NMR. The concentration of metallodrugs must be optimized to a level that is not toxic to the cells, but simultaneously allows observable chemical shift changes to be attained upon their interactions with the targets inside living cells.Atox1 is a cytosolic copper chaperone mediating Cu+ delivery and binds cisplatin at its Cu-binding site.34 Recently, in-cell NMR has been used to monitor the interaction of cisplatin with this protein, as well as to probe intracellular drug delivery.10c The protein was first overexpressed (with IPTG induction) and 15N-labelled in E. coli BL21-Gold (DE3) cells with any paramagnetic metal ions were removed to prevent broadening of NMR signals. The SOFAST-HMQC spectroscopy was applied which allows the observation of Atox1 signals with a high signal-to-noise ratio within a few seconds. By comparison with its in vitro NMR spectrum, Atox1 exhibits reduced apo-state signals in live cells with most of the signals being superimposable on those of the in vitro maps.10c
To study intracellular interaction of the protein with cisplatin, Atox1-expressing E. coli cells were grown in LB medium, then harvested and re-suspended in 15N-minimal medium. Upon induction by IPTG in the absence or presence of 10 μM cisplatin, which is the upper-limit concentration for maintaining good cell viability for in-cell NMR experiments, cells were washed and re-suspended in fresh 15N-minimal medium to obtain a concentrated in-cell NMR sample.10c Comparison of the peak intensities of the protein and the chemical shifts with and without cisplatin (Fig. 3A) showed that the CxxC metal-binding motif is amongst the most affected signals, indicating that these residues are involved in the drug binding with the peaks of Cys12, Cys15 and Ala16 shifting in a direction similar to those in vitro experiments, Fig. 3B. This appears to be the first direct evidence to “visualize” that cisplatin crosses the cell membrane and interacts with the CxxC metal-binding motif of Atox1. Moreover, the study also demonstrated that the reaction of Atox1 with cisplatin in cells is much faster than that in vitro, possibly due to intracellular macromolecular crowding which increases the effective protein concentration.10c This study clearly demonstrated the feasibility of in-cell NMR in monitoring metallodrug–protein interactions in the native complex environments.
 | ||
| Fig. 3 Illustration of Atox1 binding to cisplatin in E. coli cells by in-cell NMR spectroscopy adapted with permission from ref. 10c. Copyright (2011) American Chemical Society. (A) Overlaid [1H, 15N] SOFAST-HMQC spectra of 15N-labelled Atox1 overexpressed in E. coli cells after 4 h of IPTG induction in the absence (red) and presence (blue) of 10 μM cisplatin. The cross-peaks for residues Cys12, Cys15 and Ala16 are indicated by arrows. (B) Comparison of chemical shift changes of Cys15, Ala16 and Cys12 induced by binding of 1 molar equivalent of cisplatin in vitro (Left) and 10 μM cisplatin in E. coli cells (Right). | ||
3.2 Tracking metal binding properties
Metal ions play essential roles in biological systems. Not only do they participate in many biological processes serving as cofactors of metalloenzymes to regulate their functions, but also they have been implicated in many disease processes if metalloproteins are misfolded or metal ions are inserted inappropriately.35 Thus each metal ion has to be regulated tightly in the complex biological systems. In spite of tremendous advances being made in understanding the mechanism and biological roles of metal ions, nearly all the studies were carried out in vitro. Only recently, the metal binding properties of metalloproteins and their folding in live cells were studied by in-cell NMR spectroscopy.7b,11b,21 Such studies might provide an insight into how metal ions regulate protein and enzyme functions under their native environments.In-cell NMR has been used to probe conformation or dynamic changes induced by the binding of metal ions in living cells. Comparison of the [1H, 15N] HSQC spectrum of the N-terminal domain of the bacterial mercury-detoxification protein NmerA in live cells with that in vitro showed that the metal binding loop experienced different chemical shift changes in living cells. More specifically, Cys11 shifted by 0.19 and 0.53 ppm in the 1H- and 15N-dimensions respectively. Similar phenomena were also observed for Ala15, which is also located in the metal binding loop,4a,7a indicative of a different magnetic environment or dynamics behaviour of the protein in living cells. The in-cell NMR spectrum of selectively labelled lysine residues of human calmodulin gave rise to more signals than those in vitro, possibly due to conformational differences resulting from differences in the occupation of numbers of binding sites of the protein in vivo and in vitro. In vitro experiments showed that the protein binding sites were fully occupied by calcium (Ca2+), while calmodulin mainly existed as an apo-form in living E. coli cells,4b suggesting that the intracellular concentration of calcium is not high enough to turn the protein into the metal-bound form. Similarly, when 15N-labelled calmodulin was injected into Xenopus oocytes, the [1H, 15N] HSQC spectrum exhibited the Ca2+-free form; while upon co-injection of the protein with excess of Ca2+, the intensities of the cross-peaks in the in-cell NMR spectrum decreased dramatically; in contrast, the pattern of the cross-peaks showed similarity to that of the Ca2+-bound form.21
In-cell NMR has also been applied for tracking metal selectivity in living cells. For example, CheY, a small protein involved in chemotaxis in bacteria, binds several physiologically available metal ions such as Mg2+ and Ca2+ with different functions in vitro. The 2D [1H, 15N] HSQC spectrum of CheY in living E. coli cells showed that the protein is well folded as compared with that in vitro, and more importantly, comparison of the in-cell NMR spectrum of cells re-suspended in Ca2+ and Mg2+ free buffer with those of the in vitro protein with different metal ions bound demonstrated that the protein preferentially binds Mg2+ over Ca2+ in the E. coli cytoplasm as judged from the similar chemical shift changes.7b The additional small changes observed between the spectra of in-cell and in vitro might indicate further interactions of the protein with other components.7b Apart from CheY, in-cell NMR has also been used to examine the folding state of human copper, zinc superoxide dismutase 1 (hSOD1) accompanied by metal binding in E. coli cells.11b The in-cell [1H, 15N]-SOFAST-HMQC spectrum of hSOD1 exhibits a reduced metal-free state with partially folded structure. Interestingly, when the protein was overexpressed in the medium with supplement of excess of zinc, the in-cell NMR spectrum of hSOD1 showed significant difference from that of the apo-protein, but is similar to that of E, Zn-hSOD1 with one zinc bound to the zinc binding site, not to that of Zn, Zn-hSOD1 with two zinc ions bound to both metal binding sites. This clearly demonstrated that the cytoplasm hSOD1 binds zinc only in its specific site and not the copper site even in the presence of excess of zinc, different from in vitro experiments in which hSOD1 is able to bind two equivalents of zinc per subunit under physiological conditions. Such phenomena may indicate the specificity of metal binding for proteins and subsequently the unique biological function of metalloproteins in the native cellular context.
3.3 Monitoring metalloprotein maturation
The proper function of metalloproteins depends heavily on the insertion of correct metal ions into the functional catalytic sites of metalloenzymes at the right time. The maturation of metalloproteins and/or metalloenzymes is a very complicated process, requiring a battery of accessory metallochaperones to participate such as the cases in the maturation of urease and hydrogenase. Monitoring such a process in the physiologically cellular environment will significantly advance our knowledge on the biological role of metalloproteins as well as their potential involvement in certain diseases.Recently, NMR has been used to monitor the post-translational maturation process of human superoxide dismutase 1 (SOD1) directly in live human cells (human embryonic kidney HEK293T cells).11a As demonstrated previously, SOD1 plays a fundamental role in cellular defence against oxidative stress, and to reach its maturation form, SOD1 requires incorporation of one Zn2+ ion and one Cu+ ion per subunit and the formation of an intra-molecular disulfide bridge between Cy357 and Cys146. Such a process largely depends on the CCS protein, a copper chaperone for SOD1 (CCS).36 2D [1H, 15N]-SOFAST-HMQC spectra of SOD1 expressed in living HEK293 cells at a level of physiologically relevant concentration were obtained with or without supplements of zinc or copper in the medium. The protein was found mainly in the cytoplasm and exhibits two forms i.e. the monomeric metal free species (E, E-SOD1) and the dimeric species with one Zn2+ ion bound to each subunit (E, Zn-SOD1) when the cells were grown without supplements of zinc or copper ions as shown in Fig. 4A. Subsequent analysis indicated that the E, Zn-SOD1 form results from residual zinc in culture medium. Upon addition of zinc to the culture medium, the signals for apo-SOD1 decrease in their intensities and those for E, Zn-SOD1 increase in their intensities (Fig. 4B), indicating that zinc ions are efficiently taken up by cells in culture and bind specifically at the zinc site of dimeric SOD1 stoichiometrically. Such site-specific selectivity of Zn2+ binding was also observed in living E. coli cells.11b In prokaryotic cells, addition of Cu2+ to cell culture led to the formation of Cu(I), Zn-SOD1 upon reduction of Cu2+ and intra-subunit disulfide bond (Cys57–Cys146) in ca. 50% of the protein. However, in eukaryotic cells, such a process is more complicated owing to a number of specific chaperones involved in copper uptake and delivery. Supplementation of Cu2+ in human cell culture caused Cu+ to be incorporated into only a quarter of the protein at its specific site with less portion of the disulfide bond formed and the remaining SOD1 exists in the form of E, Zn-SOD1 (Fig. 4E).11a The incorporation of Cu+ into SOD1 to a lower extent might be attributed to the lower amounts of endogenously expressed CSS protein given that copper incorporation into SOD1 in eukaryotes is dependent on the CSS protein.36 Simultaneous overexpression and isotopic enrichment of both SOD1 and CSS were attempted to verify this hypothesis. Unexpectedly, coexpression of SOD1 and CSS in zinc supplemented medium resulted in mainly dimeric zinc-containing SOD1 species with no difference in the SOD1 metal content compared with those for the cells expressing basal levels of CCS protein. Surprisingly, the proportion of the protein with intra-subunit disulfide bonds was elevated to ca. 50% (Fig. 4C), consistent with previous observation that CCS-mediated SOD1 disulfide bond formation does not require incorporation of copper into SOD1, different from the mechanism observed in vitro.37 Supplementation of Cu2+ to the cells coexpressing SOD1 and CCS resulted in a higher ratio of Cu+, Zn-SOD1 to E, Zn-SOD1 (Fig. 4E) and complete formation of the SOD1 intra-subunit disulfide bond (Fig. 4D), suggesting that the CCS promotes the incorporation of copper into SOD1. Upon lowering concentrations of both SOD1 and CCS to the levels of physiological relevance (45 and 15 μM respectively), the compete formation of oxidized Cu, Zn-SOD1 was detected, demonstrating that the abundance of the proteins may affect detection of the intermediate maturation steps, but not the maturation process. This appears to be the first observation using NMR spectroscopy to visualize the protein maturation process in atomic detail in living human cells. Such an approach might open up a new horizon in the investigation of other protein targets for structural, dynamics, interaction studies at the molecular level in living cells.
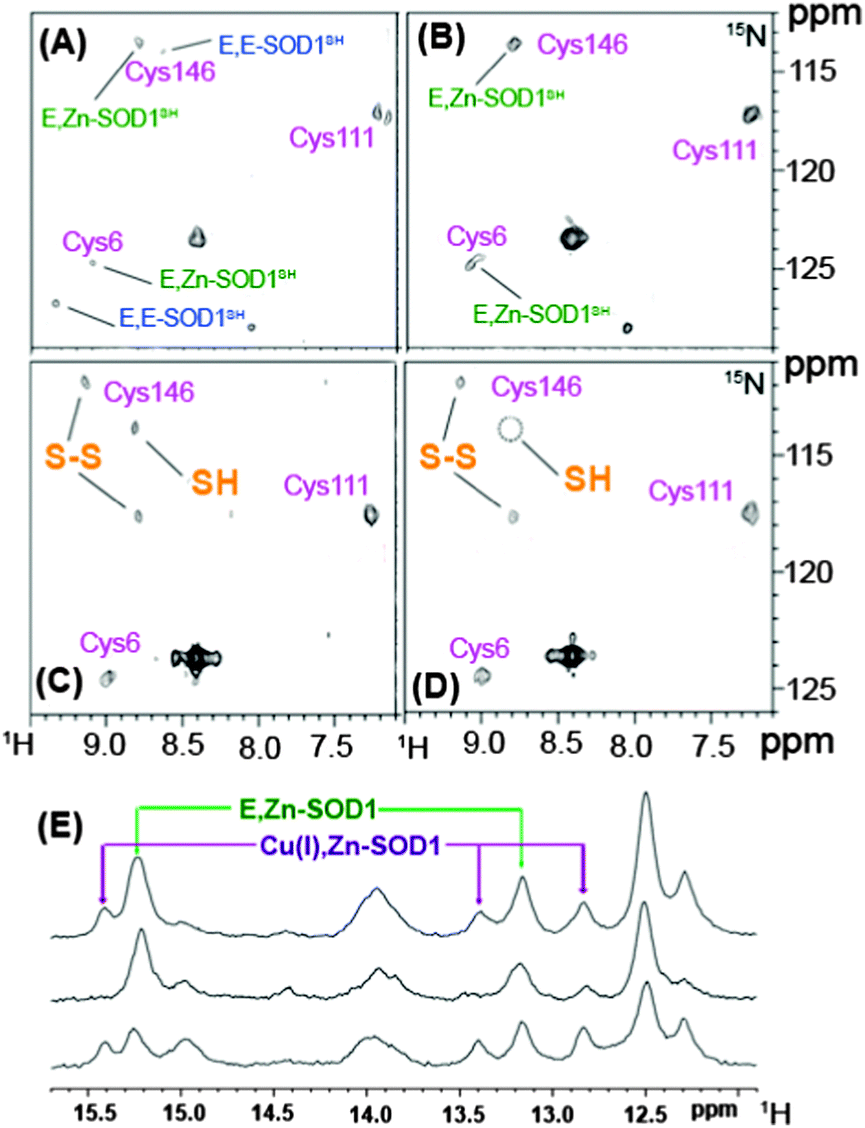 | ||
| Fig. 4 Illustration of SOD1 maturation in human HEK293T cells by in-cell NMR spectroscopy adapted with permission from Macmillan Publishers Ltd on behalf of Nature Publishing Group: [Nat. Chem. Biol.] (ref. 11a), copyright (2013). SOD1 was overexpressed in human HKE293T cells with cysteine residues labelled with 15N and [1H, 15N] SOFAST HMQC spectra were recorded without supplementing Zn2+ or Cu2+ in culture medium (A) and with addition of Zn2+ (B); with coexpressing CCS in Zn2+ supplemented medium without Cu2+ (C) and with Cu2+ (D). Assigned cysteine residues are labelled in magenta; E, E-SOD1SH and E, Zn-SOD1SH represent reduced apo-SOD1 and reduced SOD1 containing one zinc per subunit respectively. (E) Histidine regions of 1H NMR spectra of human cells expressing unlabelled SOD1 in Zn2+-supplemented medium with (top) and without (middle) addition of Cu2+; with CCS coexpressed in Zn2+ supplemented medium with incubation of Cu2+ (bottom) enhancing the protein maturation. | ||
4. Conclusions and perspectives
In-cell NMR spectroscopy appears to be one of the few techniques that can provide a detailed and comprehensive molecular view of biologically important cellular events. In spite of extensive studies being made on the investigation of structure, dynamic and interactions of certain biological macromolecules in the native environments, the technique is still in its infancy and each protein has to be examined individually for its feasibility to be detected by in-cell NMR. Both the expression level and interaction-network of the target protein may restrict the application of this technique. Moreover, this technique is commonly applied in prokaryotic cells and it encounters huge challenges when extended to eukaryotic cells, where expression levels of the protein are often lower and intracellular interactions are more complicated. Such a problem needs to be resolved before in-cell NMR can be routinely used.Up to now, there are only a few studies in which the in-cell NMR technique was used to investigate metalloproteins and metal-related events e.g. metal-dependent folding and metal selectivity of the proteins. This is possibly owing to the complexity of such studies. Nevertheless, such studies have been initiated and proven to be extremely useful for providing invaluable information on in vivo metal selectivity of metalloproteins, metalloprotein folding and the maturation process.17c,d,f,38 With increasing awareness of significance and maturity of the in-cell NMR technique as well as the improved knowledge of the properties of metal ions or metalloproteins, we anticipate that this technique will find growing applications in metallomics and metalloproteomics and will enormously benefit the research in this field. To further promote such research, interdisciplinary collaboration among bioinorganic chemists, protein chemists, biophysicists and molecular biologists is necessary to integrate advanced technologies, which allows characterization of proteins/metalloproteins within their cellular milieu and subsequently unveiling their biological functions.
Acknowledgements
This research was supported by the Research Grants Council of Hong Kong (705310P, 704612P and 703913P), NSFC/RGC joint research scheme (N_HKU75209) and the University of Hong Kong (for the emerging Strategic Research Themes on Integrative Biology).Notes and references
- K. Wüthrich, Science, 1989, 243, 45–50 Search PubMed.
- (a) G. Rivas, J. A. Fernandez and A. P. Minton, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3150–3155 CrossRef CAS PubMed; (b) M. M. Dedmon, C. N. Patel, G. B. Young and G. J. Pielak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12681–12684 CrossRef CAS PubMed; (c) D. M. Hatters, A. P. Minton and G. J. Howlett, J. Biol. Chem., 2002, 277, 7824–7830 CrossRef CAS PubMed; (d) A. P. Schlesinger, Y. Q. Wang, X. Tadeo, O. Millet and G. Pielak, J. Am. Chem. Soc., 2011, 133, 8082–8085 CrossRef CAS PubMed; (e) C. Li, L. M. Charlton, A. Lakkavaram, C. Seagle, G. Wang, G. B. Young, J. M. Macdonald and G. J. Pielak, J. Am. Chem. Soc., 2008, 130, 6310–6311 CrossRef CAS PubMed.
- B. C. McNulty, G. B. Young and G. J. Pielak, J. Mol. Biol., 2006, 355, 893–897 CrossRef CAS PubMed.
- (a) Z. Serber, A. T. Keatinge-Clay, R. Ledwidge, A. E. Kelly, S. M. Miller and V. Dötsch, J. Am. Chem. Soc., 2001, 123, 2446–2447 CrossRef CAS; (b) Z. Serber, R. Ledwidge, S. M. Miller and V. Dötsch, J. Am. Chem. Soc., 2001, 123, 8895–8901 CrossRef CAS PubMed.
- J. M. Wieruszeski, A. Bohin, J. P. Bohin and G. Lippens, J. Magn. Reson., 2001, 151, 118–123 CrossRef CAS PubMed.
- B. S. Szwergold, Annu. Rev. Physiol., 1992, 54, 775–798 CrossRef CAS PubMed.
- (a) Z. Serber and V. Dötsch, Biochemistry, 2001, 40, 14317–14323 CrossRef CAS PubMed; (b) J. A. Hubbard, L. K. MacLachlan, G. W. King, J. J. Jones and A. P. Fosberry, Mol. Microbiol., 2003, 49, 1191–1200 CrossRef CAS; (c) J. E. Bryant, J. T. Lecomte, A. L. Lee, G. B. Young and G. J. Pielak, Biochemistry, 2005, 44, 9275–9279 CrossRef CAS PubMed; (d) P. Selenko and G. Wagner, J. Struct. Biol., 2007, 158, 244–253 CrossRef CAS PubMed.
- T. Ikeya, A. Sasaki, D. Sakakibara, Y. Shigemitsu, J. Hamatsu, T. Hanashima, M. Mishima, M. Yoshimasu, N. Hayashi, T. Mikawa, D. Nietlispach, M. Walchli, B. O. Smith, M. Shirakawa, P. Guntert and Y. Ito, Nat. Protocols, 2010, 5, 1051–1060 CAS.
- (a) Y. Takaoka, Y. Kioi, A. Morito, J. Otani, K. Arita, E. Ashihara, M. Ariyoshi, H. Tochio, M. Shirakawa and I. Hamachi, Chem. Commun., 2013, 49, 2801–2803 RSC; (b) C. Li and M. Liu, FEBS Lett., 2013, 587, 1008–1011 CrossRef CAS PubMed.
- (a) D. S. Burz, K. Dutta, D. Cowburn and A. Shekhtman, Nat. Protocols, 2006, 1, 146–152 CrossRef CAS PubMed; (b) Q. Wang, A. Zhuravleva and L. M. Gierasch, Biochemistry, 2011, 50, 9225–9236 CrossRef CAS PubMed; (c) F. Arnesano, L. Banci, I. Bertini, I. C. Felli, M. Losacco and G. Natile, J. Am. Chem. Soc., 2011, 133, 18361–18369 CrossRef CAS PubMed.
- (a) L. Banci, L. Barbieri, I. Bertini, E. Luchinat, E. Secci, Y. Zhao and A. R. Aricescu, Nat. Chem. Biol., 2013, 9, 297–299 CrossRef CAS PubMed; (b) L. Banci, L. Barbieri, I. Bertini, F. Cantini and E. Luchinat, PLoS One, 2011, 6, e23561 CAS.
- R. Hansel, S. Foldynova-Trantirkova, F. Lohr, J. Buck, E. Bongartz, E. Bamberg, H. Schwalbe, V. Dötsch and L. Trantirek, J. Am. Chem. Soc., 2009, 131, 15761–15768 CrossRef PubMed.
- P. Selenko, D. P. Frueh, S. J. Elsaesser, W. Haas, S. P. Gygi and G. Wagner, Nat. Struct. Mol. Biol., 2008, 15, 321–329 CAS.
- D. S. Burz, K. Dutta, D. Cowburn and A. Shekhtman, Nat. Methods, 2006, 3, 91–93 CrossRef CAS PubMed.
- P. Ramm Sander, M. Peer, M. Grandl, U. Bogdahn, G. Schmitz and H. R. Kalbitzer, PLoS One, 2013, 8, e56360 Search PubMed.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 7, 25–35 CrossRef CAS PubMed.
- (a) R. Ge, X. Sun, Q. Gu, R. M. Watt, J. A. Tanner, B. C. Wong, H. H. Xia, J. D. Huang, Q. Y. He and H. Sun, JBIC, J. Biol. Inorg. Chem., 2007, 12, 831–842 CrossRef CAS PubMed; (b) X. S. Sun, C. N. Tsang and H. Sun, Metallomics, 2009, 1, 25–31 RSC; (c) S. Freilich, R. Zarecki, O. Eilam, E. S. Segal, C. S. Henry, M. Kupiec, U. Gophna, R. Sharan and E. Ruppin, Nat. Commun., 2011, 2, 589 CrossRef PubMed; (d) W. Shi and M. R. Chance, Curr. Opin. Chem. Biol., 2011, 15, 144–148 CrossRef PubMed; (e) S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119–1138 RSC; (f) H. Sun and Z. F. Chai, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2010, 106, 20–38 RSC.
- (a) W. Xia, H. Li, K. H. Sze and H. Sun, J. Am. Chem. Soc., 2009, 131, 10031–10040 CrossRef CAS PubMed; (b) S. Cun and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4943–4948 CrossRef CAS PubMed.
- (a) L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y. T. Lai, G. Jiang and H. Sun, Angew. Chem., Int. Ed., 2013, 52, 4916–4920 CrossRef CAS PubMed; (b) C. N. Tsang, K. S. Ho, H. Sun and W. T. Chan, J. Am. Chem. Soc., 2011, 133, 7355–7357 CrossRef CAS PubMed.
- (a) H. Li and H. Sun, Top. Curr. Chem., 2012, 326, 69–98 CrossRef CAS; (b) H. Wang, Q. Zhang, B. Cai, H. Li, K. H. Sze, Z. X. Huang, H. M. Wu and H. Sun, FEBS Lett., 2006, 580, 795–800 CrossRef CAS PubMed; (c) W. Xia, H. Li, X. Yang, K. B. Wong and H. Sun, J. Biol. Chem., 2012, 287, 6753–6763 CrossRef CAS PubMed.
- T. Sakai, H. Tochio, T. Tenno, Y. Ito, T. Kokubo, H. Hiroaki and M. Shirakawa, J. Biomol. NMR, 2006, 36, 179–188 CrossRef CAS PubMed.
- (a) S. Reckel, F. Lohr and V. Dötsch, ChemBioChem, 2005, 6, 1601–1606 CrossRef CAS PubMed; (b) A. Y. Maldonado, D. S. Burz and A. Shekhtman, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 197–212 CrossRef CAS PubMed; (c) Y. Ito and P. Selenko, Curr. Opin. Struct. Biol., 2010, 20, 640–648 CrossRef CAS PubMed; (d) Z. Serber, L. Corsini, F. Durst and V. Dötsch, Methods Enzymol., 2005, 394, 17–41 CrossRef CAS.
- N. G. Sharaf, C. O. Barnes, L. M. Charlton, G. B. Young and G. J. Pielak, J. Magn. Reson., 2010, 202, 140–146 CrossRef CAS PubMed.
- (a) C. Li, G. F. Wang, Y. Wang, R. Creager-Allen, E. A. Lutz, H. Scronce, K. M. Slade, R. A. Ruf, R. A. Mehl and G. J. Pielak, J. Am. Chem. Soc., 2010, 132, 321–327 CrossRef CAS PubMed; (b) J. T. Hammill, S. Miyake-Stoner, J. L. Hazen, J. C. Jackson and R. A. Mehl, Nat. Protocols, 2007, 2, 2601–2607 CrossRef CAS PubMed.
- Z. Serber, W. Straub, L. Corsini, A. M. Nomura, N. Shimba, C. S. Craik, P. O. de Montellano and V. Dötsch, J. Am. Chem. Soc., 2004, 126, 7119–7125 CrossRef CAS PubMed.
- V. Dötsch, Top. Curr. Chem., 2008, 273, 203–214 CrossRef.
- (a) P. Selenko, Z. Serber, B. Gadea, J. Ruderman and G. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11904–11909 CrossRef CAS PubMed; (b) Z. Serber, P. Selenko, R. Hänsel, S. Reckel, F. Löhr, J. E. Ferrell Jr, G. Wagner and V. Dötsch, Nat. Protocols, 2006, 1, 2701–2709 CrossRef CAS PubMed.
- (a) K. Inomata, A. Ohno, H. Tochio, S. Isogai, T. Tenno, I. Nakase, T. Takeuchi, S. Futaki, Y. Ito, H. Hiroaki and M. Shirakawa, Nature, 2009, 458, 106–109 CrossRef CAS PubMed; (b) S. Ogino, S. Kubo, R. Umemoto, S. Huang, N. Nishida and I. Shimada, J. Am. Chem. Soc., 2009, 131, 10834–10835 CrossRef CAS PubMed.
- K. Bertrand, S. Reverdatto, D. S. Burz, R. Zitomer and A. Shekhtman, J. Am. Chem. Soc., 2012, 134, 12798–12806 CrossRef CAS PubMed.
- J. Hamatsu, D. O'Donovan, T. Tanaka, T. Shirai, Y. Hourai, T. Mikawa, T. Ikeya, M. Mishima, W. Boucher, B. O. Smith, E. D. Laue, M. Shirakawa and Y. Ito, J. Am. Chem. Soc., 2013, 135, 1688–1691 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- (a) X. H. Wang, X. B. Du, H. Y. Li, D. S. B. Chan and H. Sun, Angew. Chem., Int. Ed., 2011, 50, 2706–2711 CrossRef CAS PubMed; (b) X. H. Wang, H. Y. Li, X. B. Du, J. Harris, Z. J. Guo and H. Sun, Chem. Sci., 2012, 3, 3206–3215 RSC; (c) S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef CAS PubMed.
- M. Komatsu, T. Sumizawa, M. Mutoh, Z. S. Chen, K. Terada, T. Furukawa, X. L. Yang, H. Gao, N. Miura, T. Sugiyama and S. Akiyama, Cancer Res., 2000, 60, 1312–1316 CAS.
- M. E. Palm, C. F. Weise, C. Lundin, G. Wingsle, Y. Nygren, E. Bjorn, P. Naredi, M. Wolf-Watz and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6951–6956 CrossRef CAS PubMed.
- N. P. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- V. C. Culotta, M. Yang and T. V. O'Halloran, Biochim. Biophys. Acta, 2006, 1763, 747–758 CrossRef CAS PubMed.
- L. Banci, I. Bertini, F. Cantini, T. Kozyreva, C. Massagni, P. Palumaa, J. T. Rubino and K. Zovo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13555–13560 CrossRef CAS PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |