Molybdenum and tungsten oxygen transferases – structural and functional diversity within a common active site motif
M. Jake
Pushie
a,
Julien J.
Cotelesage
ab and
Graham N.
George
*a
aMolecular and Environmental Science Research Group, Department of Geological Sciences, University of Saskatchewan, Saskatoon, SK S7N 5E2, Canada. E-mail: g.george@usask.ca; Fax: +1 306 966 8593; Tel: +1 306 966 5722
bCanadian Light Source, 101 Perimeter Road, Saskatoon, SK S7N 0X4, Canada
First published on 10th September 2013
Abstract
Molybdenum and tungsten are the only second and third-row transition elements with a known function in living organisms. The molybdenum and tungsten enzymes show common structural features, with the metal being bound by a pyranopterin-dithiolene cofactor called molybdopterin. They catalyze a variety of oxygen transferase reactions coupled with two-electron redox chemistry in which the metal cycles between the +6 and +4 oxidation states usually with water, either product or substrate, providing the oxygen. The functional roles filled by the molybdenum and tungsten enzymes are diverse; for example, they play essential roles in microbial respiration, in the uptake of nitrogen in green plants, and in human health. Together, the enzymes form a superfamily which is among the most prevalent known, being found in all kingdoms of life. This review discusses what is known of the active site structures and the mechanisms, together with some recent insights into the evolution of these important enzyme systems.
 Jake Pushie | Jake Pushie is a physical bioinorganic chemist who received his PhD from the University of Calgary in 2007. His research interests focus on metals in living systems, spanning the anatomic to the atomic. The study of Mo and W enzymes provides a link back to some of the earliest bioinorganic chemistry critical for life on Earth. Dr Pushie is supported by postdoctoral Fellowships from the Canadian Institutes of Health Research (CIHR), the Saskatchewan Health Research Foundation (SHRF) and CIHR-THRUST (training in health research using synchrotron techniques). In 2010 he was awarded SHRF's Top Postdoctoral Researcher in the Biomedical division. |
 Julien J. Cotelesage | Julien Cotelesage is a protein crystallographer by training and received his PhD from the University of Saskatchewan in 2007. He held a postdoctoral fellowship (PDF) at the National Microbiology Laboratory until 2010, after which he took up postdoctoral position joint with the Canadian Light Source and the University of Saskatchewan. Since 2013 he is a Research Associate and a CIHR-THRUST Associate at the Canadian Light Source and the University of Saskatchewan. His research interests exploit a combination of crystallographic methods and X-ray absorption spectroscopy to obtain an in-depth understanding of metalloprotein active sites. |
 Graham N. George | Graham George was educated at King's College London (BSc, 1979) and the University of Sussex (DPhil, 1983). After postdoctoral fellowships at Sussex and Exxon Research & Engineering Co. in New Jersey USA, he continued at Exxon as a Principal Investigator until moving to the Stanford Synchrotron Radiation Laboratory in 1992. In 2003 he became full professor and Canada Research Chair in X-ray Absorption Spectroscopy at the University of Saskatchewan. His research includes a career-long interest in the Mo and W enzymes, development of new methods for understanding metal sites in proteins and applications of synchrotron light to molecular toxicology. |
Introduction
Given the recent growth in the fields of bioinorganic chemistry and metallomics, the literature is surprisingly unclear about what total fraction of all proteins contain a functional metal ion. If one considers both ribozymes1 and enzymes and proteins,2–5 and includes the s-block elements Na, K, Mg and Ca, then close to 100% of these molecules will contain a metal ion of some sort. Narrowing the view to just proteins and excluding the s-block elements, it has been suggested that metal containing proteins constitute between 30%2,3 to 50%4 of the genomic output of most organisms. However, a recent examination of the protein data bank showed that this number may be closer to 22%,5 with almost half of these being zinc-containing proteins. Zn2+ is formally not a transition metal ion because of its filled 3d10 configuration and its lack of d-electron based redox chemistry. Therefore a better estimate of the genomic output incorporating true transition metal ions may be closer to 11%. Setting aside uncertainties concerning the levels of incidence, there is no doubt that nearly all of the most important, industrially relevant and chemically challenging chemistry conducted by living things is catalyzed by transition metal ions as part of enzyme active sites. Molybdenum and tungsten are unique in being the only second and third transition ions that have well defined roles in biology, and tungsten has the further distinction in being the heaviest element with a biological role. Both metals, when contained in their biologically relevant entities (the molybdenum and tungsten enzymes), do the same type of oxo-transferase redox chemistry, forming or breaking bonds between oxygen and carbon where the source or destination of the oxygen is usually water, and the metal cycles between the 6+ and 4+ oxidation states as in eqn (1) in which M represents Mo or W.| M(VI) + H2O + R–H ⇌ M(IV) + R–OH + 2H+ | (1) |
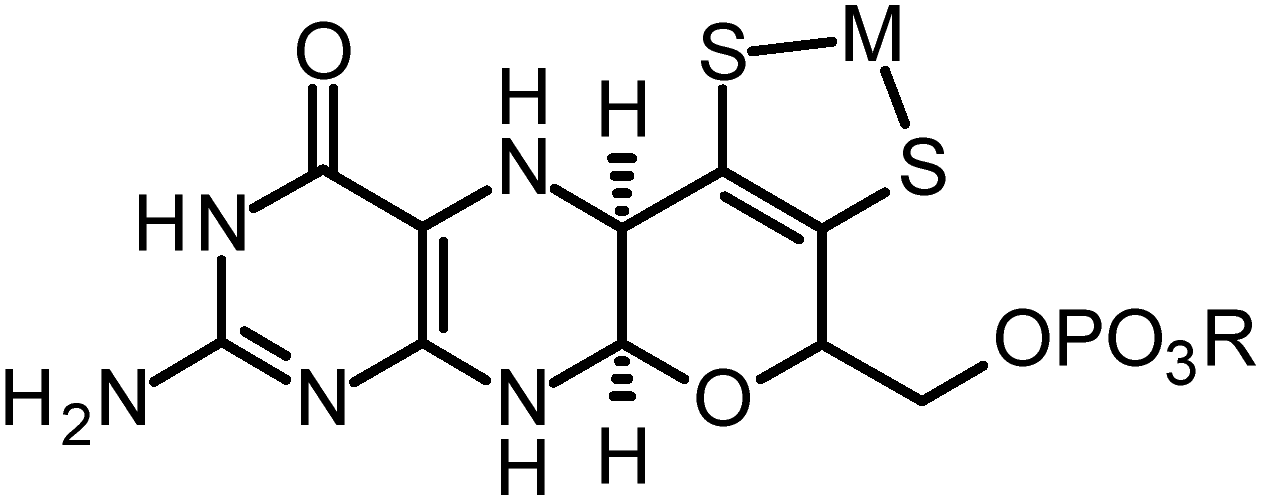 | ||
| Fig. 1 Structure of the molybdopterin cofactor, the metal M is either Mo or W, and the group R can be H (molybdopterin or MPT), guanosine (molybdopterin guanine dinucleotide or MGD), or cystosine (molybdopterin cytidine dinucleotide or MCD). | ||
Despite possessing common structural components, the enzymes show remarkable diversity in the range of chemical reactions that are catalyzed. The functional roles filled by the enzymes are equally diverse; for example, they play essential roles in microbial respiration, in both uptake of nitrogen and in regulation of growth in green plants, in controlling insect eye color, and in human health.11
This review will discuss the Mo and W enzymes, their active site structures as they relate to activity, and how structural and functional diversity are achieved within a common active site motif. In the last part of this review we will discuss the evolution of these enzymes and why they may have been important in early living organisms.
Families of molybdenum and tungsten enzymes
The importance of molybdenum in enzymes has been known since the early 1950's12–15 but the first tungsten enzyme was discovered decades later,16 and the fact that they were related to the Mo enzymes even later still.17 Of the enzymes that have been described to date the vast majority are Mo enzymes, and are ordered within three broad families named for their prototypical members and categorized by the structures of the oxidized Mo(VI) forms.18Fig. 2 summarizes the oxidized active site structures of the different molybdenum enzyme families.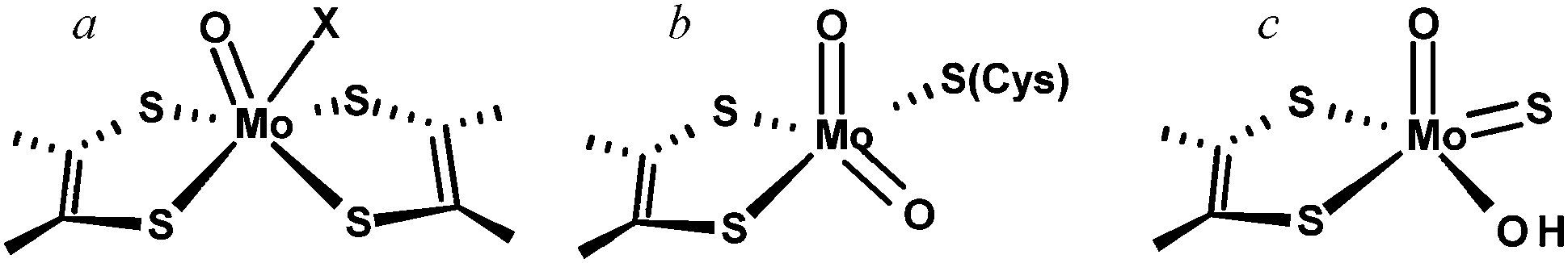 | ||
Fig. 2 Mo & W enzyme families, (a) DMSO reductase family, where individual members of the DMSO reductase family may lack the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and may also have a coordinated amino acid X serine, cysteine, aspartate or selenocysteine. (b) Sulfite oxidase and (c) xanthine oxidase family. O group and may also have a coordinated amino acid X serine, cysteine, aspartate or selenocysteine. (b) Sulfite oxidase and (c) xanthine oxidase family. | ||
The largest and most diverse family of enzymes is known as the dimethylsulfoxide (DMSO) reductase family. Members of this family are found only in the bacteria and the archaea and the family contains both molybdenum enzymes and all tungsten enzymes that have been discovered to date. Two molybdopterins are bound to the metal and many of the enzymes have further metal coordination from an amino acid side chain. The DMSO reductase family has been further divided into three sub-families or types based on sequence homologies and coordination of the metal. Type I enzymes show coordination by either a cysteine (e.g. the periplasmic nitrate reductases, or Nap enzymes)19 or a selenocysteine (e.g. Escherichia coli formate dehydrogenase).20–23 In the Type II enzymes the coordinating residue is an aspartate (e.g. E. coli nitrate reductase),24,25 and in the Type III enzymes a serine is bound (e.g. Rhodobacter sphaeroides DMSO reductase).26,27 Many enzymes of the DMSO reductase family also have a single oxo ligand in their oxidized states (MoO), but some of the more recently characterized members may not.19,28 Sometimes the W enzymes are attributed to a different family, called the AOR family after the prototypical but still incompletely characterized18 W enzyme aldehyde oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. There is, however, considerably more variation between the accepted members of the DMSO reductase family and the tungsten enzymes are probably better placed as members of a larger DMSO reductase enzyme family including the W enzymes.
The other two families are called the sulfite oxidase family and the xanthine oxidase family and both comprise exclusively Mo enzymes with coordination by a single cofactor dithiolene. Eukaryotic and prokaryotic examples of these two families are known. The sulfite oxidase family has a cis-dioxo Mo(VI) structure with a cysteine ligand to Mo, while the xanthine oxidase family members have no amino acid bound to Mo, and an oxo-thio Mo(O)(S).
Oxygen atom transfer chemistry
As we have noted above, and with some notable exceptions, all Mo and W enzymes catalyze two-electron redox reactions in which an oxygen atom is transferred between the metal atom and the substrate molecule. There are only three known exceptions; the first is the W-containing acetylene hydratase from the eubacterial anaerobe Pleobacter acetylenicus which catalyzes a net hydration of acetylene to form acetaldehyde29,30 that is not a redox reaction. A second exception is provided by the Mo containing E.coli formate dehydrogenase H (FDHH), which catalyzes the conversion of formate to carbon dioxide or carbonate. This enzyme has been shown to possess Mo bound by selenocysteineate and to lack exchangeable oxygen ligands at Mo.20–23 We note that there is some controversy concerning the crystal structure of this enzyme,18 with apparent disagreement between conclusions from Mo(V) EPR and the most recent re-interpretation of the crystal structure data of the reduced enzyme.22,31,32 Irrespective of the structure of the reduced active site, stable isotopes have been used to show that this enzyme does not use oxygen atom transfer from Mo to formate.22 Some other FDH enzymes do possess MoO coordination,33 and whether or not these work in a similar manner to E. coli FDHH is at present unknown. The third exception is transhydroxylase from the strictly anaerobic bacterium Pelobacter acidigallici.34,35 Transhydroxylase is a Mo enzyme which catalyzes the conversion of pyrogallol (1,2,3-tri-hydroxybenzene) to phloroglucinol (1,3,5-tri-hydroxybenzene) via a co-substrate (1,2,3,5-tetra-hydroxybenzene) by shuttling the C2–OH from the co-substrate to C5 of the pyrogallol to form phloroglucinol from the co-substrate, and generate co-substrate from the pyrogallol. This enzyme is an exception on two counts – it does not involve redox change at the Mo, and water is not the source of oxygen.34,35 The remainder of this section will focus on the oxygen atom transfer chemistry which almost all Mo and W enzymes utilize to carry out their physiological functions.
Some Mo enzymes, and in particular those of the sulfite oxidase family, have a cis-dioxo Mo(VI) active site structure (Fig. 2). Sulfite oxidases catalyze the oxidation of sulfite to sulfate as in eqn (2):
| SO32− + H2O ⇌ SO42− + 2H+ + 2e− | (2) |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O, and is sometimes shown that way. The consequence of this is the increase in bond-strength of the spectator oxo (i.e. the oxo that is not directly involved in oxygen atom transfer) acts to thermodynamically drive the oxygen atom transfer reaction. For sulfite oxidase family members the dithiolene and cysteineate may serve to appropriately adjust the active site redox and geometry. In particular Izumi et al.37 have suggested that the Ooxo–Mo–S–C torsion angle can modulate the overlap of a sulfur p-orbital with one of the two MoO π* orbitals thereby activating one oxo for transfer to substrate. Sulfur K-edge XAS has been used to show dependency of covalency on such torsion angles in model compounds.38 Peariso et al.39 have suggested that the attack of the sulfite lone pair on the equatorial oxo ligand results in the population of a Mo–O dxy–pπ antibonding orbital which has been hypothesized to labilize the equatorial oxygen atom for transfer to substrate. The proximity and orientation of sulfite relative to the molybdenum in the sulfite oxidase active site is thought to be controlled by basic residues, and in particular three arginines, which orient substrate and/or product in a pocket about 3 Å from the equatorial oxygen that is catalytically transferred to substrate.40 In the crystal structure the observed electron density is consistent with a mixture of sulfite and sulfate in the active site basic pocket, with the three sulfite oxygen atoms oriented away from Mo so that the sulfur lone pair would point in the direction of the metal. Fig. 3 summarizes current views of the sulfite oxidase catalytic mechanism.
O, and is sometimes shown that way. The consequence of this is the increase in bond-strength of the spectator oxo (i.e. the oxo that is not directly involved in oxygen atom transfer) acts to thermodynamically drive the oxygen atom transfer reaction. For sulfite oxidase family members the dithiolene and cysteineate may serve to appropriately adjust the active site redox and geometry. In particular Izumi et al.37 have suggested that the Ooxo–Mo–S–C torsion angle can modulate the overlap of a sulfur p-orbital with one of the two MoO π* orbitals thereby activating one oxo for transfer to substrate. Sulfur K-edge XAS has been used to show dependency of covalency on such torsion angles in model compounds.38 Peariso et al.39 have suggested that the attack of the sulfite lone pair on the equatorial oxo ligand results in the population of a Mo–O dxy–pπ antibonding orbital which has been hypothesized to labilize the equatorial oxygen atom for transfer to substrate. The proximity and orientation of sulfite relative to the molybdenum in the sulfite oxidase active site is thought to be controlled by basic residues, and in particular three arginines, which orient substrate and/or product in a pocket about 3 Å from the equatorial oxygen that is catalytically transferred to substrate.40 In the crystal structure the observed electron density is consistent with a mixture of sulfite and sulfate in the active site basic pocket, with the three sulfite oxygen atoms oriented away from Mo so that the sulfur lone pair would point in the direction of the metal. Fig. 3 summarizes current views of the sulfite oxidase catalytic mechanism.
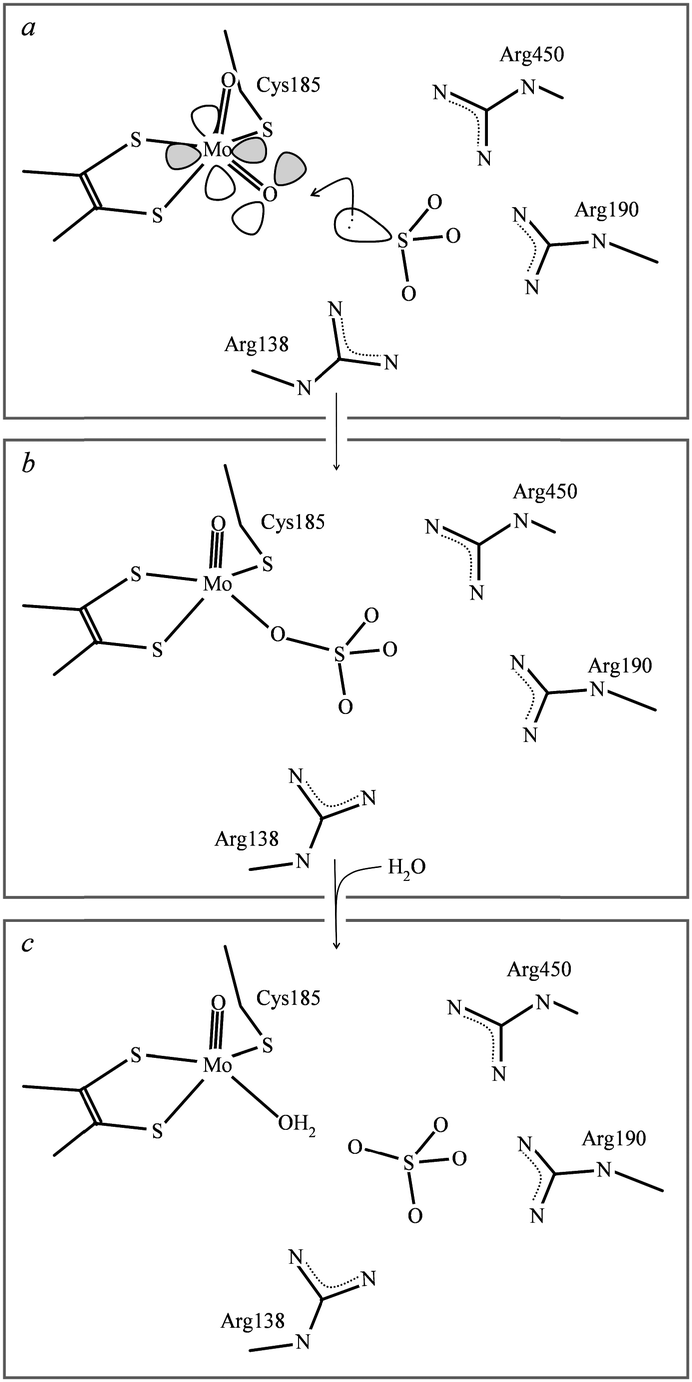 | ||
| Fig. 3 Schematic diagram of sulfite oxidase catalytic mechanism. The enzyme starts in the Mo(VI) oxidation state (a), and is reduced to the Mo(IV) oxidation state (b) and (c). Charges on various groups have been deliberately omitted for clarity. | ||
The plant and fungal nitrate reductases are closely related to the sulfite oxidases and catalyze the conversion of nitrate to nitrite, which is a key part of the nitrogen cycle, eqn (3):
| NO3− + 2H+ + 2e− ⇌ NO2− + H2O | (3) |
In xanthine oxidase, and probably in most other members of this family, the metal-coordinated oxygen that is catalytically transferred to substrate exists in a pH-dependent equilibrium between Mo–OH and MoO, eqn (4):44
| Mo–OH ⇌ MoO + H+ | (4) |
O and Mo–OH, so it is not clear whether it is the Mo–OH or MoO that is actually transferred to the substrate, although most postulated mechanisms suggest that this is Mo–OH. Early EXAFS studies45 only observed the [MoO(OH)S]+ form, and not [MoO2S]0, because the samples were prepared in phosphate buffers, which can change pH on freezing by up to two pH units,46 so that the low-pH protonated form was observed.45
Possibly the most novel members of the xanthine oxidase family are the molybdenum-containing carbon monoxide dehydrogenases. The first of these to be characterized was isolated from the aerobe Oligotropha carboxidovorans.47 The enzyme catalyzes reaction of carbon monoxide with water to carbon dioxide in a reaction that is equivalent to the water gas shift reaction, discovered by Felice Fontana in 1780, eqn (5):
| CO + H2O ⇌ CO2 + 2e− + 2H+ | (5) |
S group forms a bridge with a two-coordinate Cu(I) bound to a nearby cysteine residue to form a novel binuclear Mo–S–Cu–S(Cys) active site. The first crystal structure analyses of two different members of this class of enzyme mistook copper for selenium,48,49 with significant resulting confusion concerning the catalytic mechanism.48 Subsequent work using both crystallography47 and X-ray absorption spectroscopy50 determined the correct structure, with the EXAFS revealing a dioxo [MoO2SCu]+ core structure which was different from the Mo–OH containing core suggested by crystallography. Subsequent resonance Raman studies confirmed the [MoO2SCu]+ core structure.51 This difference is possibly due to pH effects similar to those of xanthine oxidase,44 or to photoreduction. In this protein the substrate CO is thought to bind to Cu positioning the CO adjacent to Mo in an ideal location for oxygen atom transfer from Mo to form CO2.52 It is of interest that enzyme reconstituted with Ag(I) is highly active, supporting a non-redox role for the Cu in the native enzyme.53
Spectator oxo chemistry, which is common to Mo and W species, is not employed by DMSO reductase and related enzymes, as the active forms of these enzymes have mono-oxo M(VI) sites. Kirk and co-workers have pointed out that the presence of axial terminal oxygen, often formulated as MoO, will orient the Mo dxy redox orbital for maximal interaction with in-plane dithiolene sulfur p-orbitals, yielding effective regeneration of the reduced enzyme following oxygen atom transfer. This notion of the oxo acting as a prerequisite for facile electron transfer has been called the oxo gate hypothesis.54,55
The structural studies of the W enzymes that have been reported to date are sparse compared to their Mo counterparts. The data that have been reported seem to indicate a novel cis-dioxo core with approximately four sulfur donors,56 but lacking any elongation in W–S bond-lengths arising trans-direction of WO, which indicates that the geometry must be far from pseudo octahedral.18 At the time of writing the detailed active site structures of the tungsten enzymes still remain to be elucidated.
Irrespective of the mechanistic details, the Mo and W enzymes are both unusual in catalyzing oxygen atom transfer from water at relatively low redox potentials. This capability is likely to have been very important in their evolution, which we will consider in the final section of this review.
Photoreduction and active site structure
X-ray crystallography and X-ray absorption spectroscopy employ ionizing radiation and resulting photo-chemical processes and in particular photoreduction can be a potential problem in both techniques.57 Indeed, with the Mo and W enzymes photoreduced structures may well represent the majority of studies. Thus, even for the best studied Mo enzymes, such as sulfite oxidase, there are almost no crystal structures that are not photoreduced.18,58 With sulfite oxidase the only exceptions are mutants of Cys20759 which have a tri-oxo [MoO3]0 core that is very difficult to reduce.60,61 At the Mo K-edge of 20 keV X-ray absorption spectroscopy is less prone to photoreduction because of the lower inherent X-ray cross-section at high energies, and normally there are no problems with photochemistry in these experiments. In the tungsten enzymes X-ray absorption spectroscopy is usually conducted at the W LIII edge at 10.2 keV at which energy the X-ray absorption cross section is approximately 9 times greater than at the Mo K-edge with a correspondingly increased potential for photochemistry. In either case in XAS the presence of photochemical modification of the active site is evident from changes in the near edge portion of the spectrum. A recent review examining the strengths and weaknesses of both methods concluded that an accurate picture of the active sites can only be obtained from a combined approach.57The role of molybdopterin in the enzymes
The molybdopterin component of the active sites of Mo and W enzymes (Fig. 1) is often partly overlooked in that it is sometimes regarded as only a carrier for the important dithiolene ligands to Mo or W. The cofactor was first discovered as a discrete entity by Johnson, Rajagopalan and co-workers, who studied the oxidized inactive form62 and in a remarkable and elegant series of studies deduced its structure.63 This was initially formulated in the ring-open form (Fig. 4a)63 with the first crystal structure suggesting a closed tricycle form containing a pyran ring (Fig. 4b).64 The history of molybdopterin and its biosynthesis has recently been comprehensively reviewed,65,66 and rather than discussing this here we refer the reader to these excellent reviews,65,66 and restrict our discussion to some of the more ambiguous aspects. The biosynthesis of the cofactor can viewed as taking place in four stages; (i) formation of the pyranopterin tricycle, (ii) addition of the dithiolene group, (iii) adenylation (iv) and molybdenum or tungsten insertion. Details of all four steps have been characterized, and here we will discuss the final step, the transfer and insertion of metal into the cofactor. In bacteria this is conducted by the MogA and MoeE proteins,65,66 while in plants and animals the two-domain Cnx167 and gephyrin68,69 proteins fulfill this function. A role for Cu(I) in this process has been suggested with Cnx1, with a crystal structure copper bound to cofactor dithiolene,70 supposedly providing protection of the reactive dithiolene before insertion of molybdenum.70 This is a somewhat surprising finding because cellular copper is incredibly highly regulated. The reason for this is that the accessibility of the Cu(I)/Cu(II) redox couple means that it can readily indulge in Fenton type chemistry resulting in a very high cellular toxicity. This same chemistry is exploited in metalloenzymes making copper an essential element. The result is that copper is hyper-regulated so that it is nearly always found in association with transporters, chaperones or specific metalloproteins, with essentially no free Cu ions within cells.71 Moreover, adventitious copper is easy to pick up from a number of sources including commercially available metal syringe needles.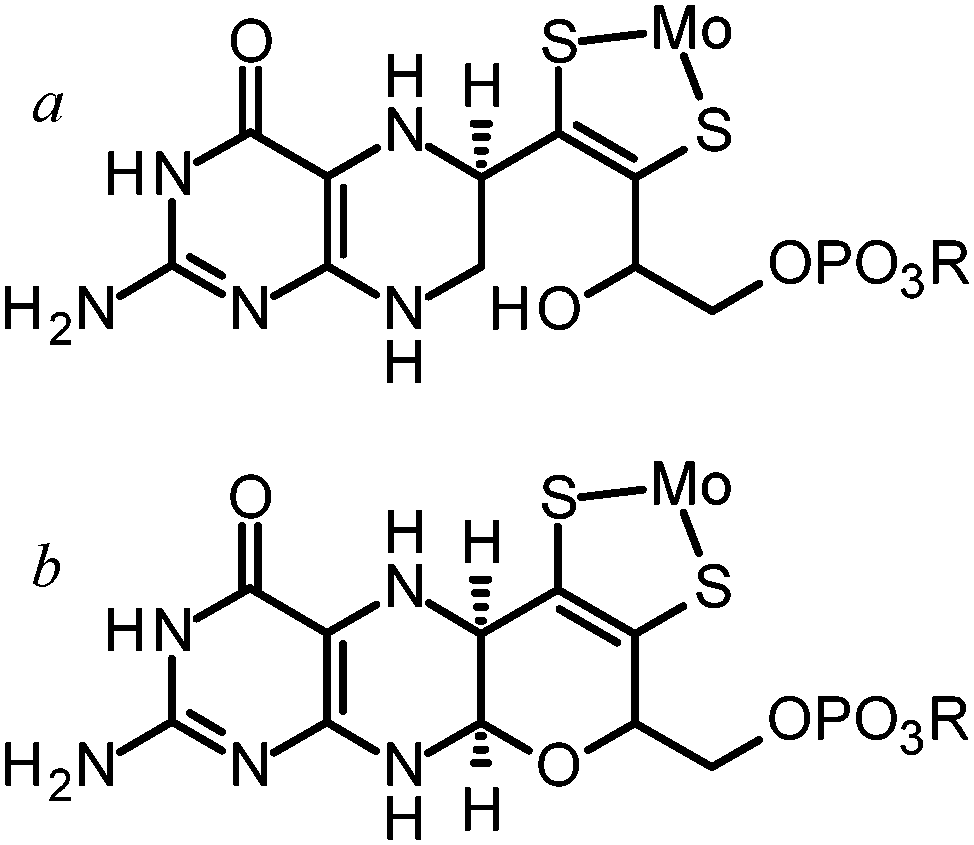 | ||
| Fig. 4 Ring open (a) and ring closed (b) forms of molybdopterin. | ||
As we have discussed above, enzymes belonging to the DMSO reductase family contain two molybdopterins, while members belonging to the xanthine oxidase and sulfite oxidase families contain one molybdopterin. It has been proposed that the molybdopterin may play a purely structural role in organizing the metal binding site for catalysis, and it may have a more direct role in facilitating electron transfer during catalysis, conceptually acting as a “wire” for electrons flowing to or from the metal site. Thus, with some notable exceptions, the molybdopterin often points towards the electron acceptor or donor. We note that these two roles are not necessarily mutually exclusive. Fig. 5 shows the arrangement of redox active centers in the bovine xanthine oxidase,72 in which the single molybdopterin points towards the FeS-I cluster. Electrons are transferred via intramolecular electron transfer from Mo to FeS-I then to FeS-II and then to the flavin moiety (Fig. 5). The molybdopterin system is usually depicted as a syn-4a,10a tetrahydropterin as shown in Fig. 1, but it has a number of potential structural isoforms which differ in redox status and these are summarized in Fig. 6. Recently, Rothery and co-workers73 have examined virtually all of the currently-known mononuclear Mo–W enzymes and have suggested that the three major classes of enzymes each have fine-tuned their respective enzyme binding sites for selection of specific molybdopterin conformations through molecular evolution of the binding pockets. Thus, Rothery et al.73 suggest that the binding pockets exert control over molybdopterin conformations which would facilitate electron transfer, while imposing energetic penalties upon conformations which would hinder catalytic activity through unwanted oxidation of molybdopterin. In particular, they suggest that the conformation of some molybdopterin is more consistent with dihydropterin than tetrahydropterin. We note that the preliminary work in this area does not take into account the influence of the metal ion, nor the local protein environment and H-bonding on the stability of the various molybdopterin structures. Moreover, the limitations of the structural data on which the structural conclusions are based should be considered. In many cases the electron density maps lack the definition to discriminate the different structures. For example the difference map shown in Fig. 7 from Pichia angusta nitrate reductase can accommodate all three different molybdopterins, and the ability of many crystal structures to discriminate between the different pterin redox states is doubtful, and must await higher resolution structures or spectroscopic evidence. While the redox state of the pterin is hard to define in proteins, the recent synthesis of a molybdenum dihydropterin species by Burgmayer and co-workers74 establishes credibility for this species in the enzymes. Moreover, the sensitivity to oxygen of these compounds is similar to that of the intact molybdenum-containing cofactor as reported by Hawkes et al.75
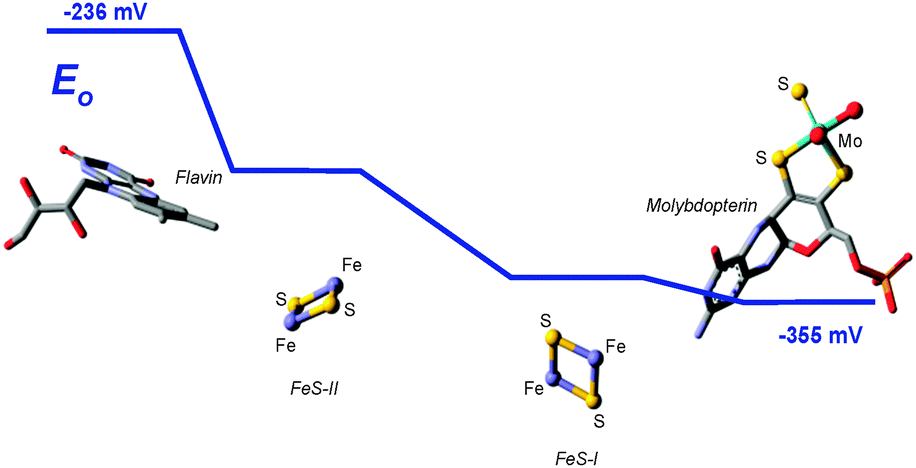 | ||
| Fig. 5 Electron transfer pathway in xanthine oxidase, showing the spatial distribution and orientation of redox active sites within the protein, and the range of their electrochemical mid-point potentials E0. | ||
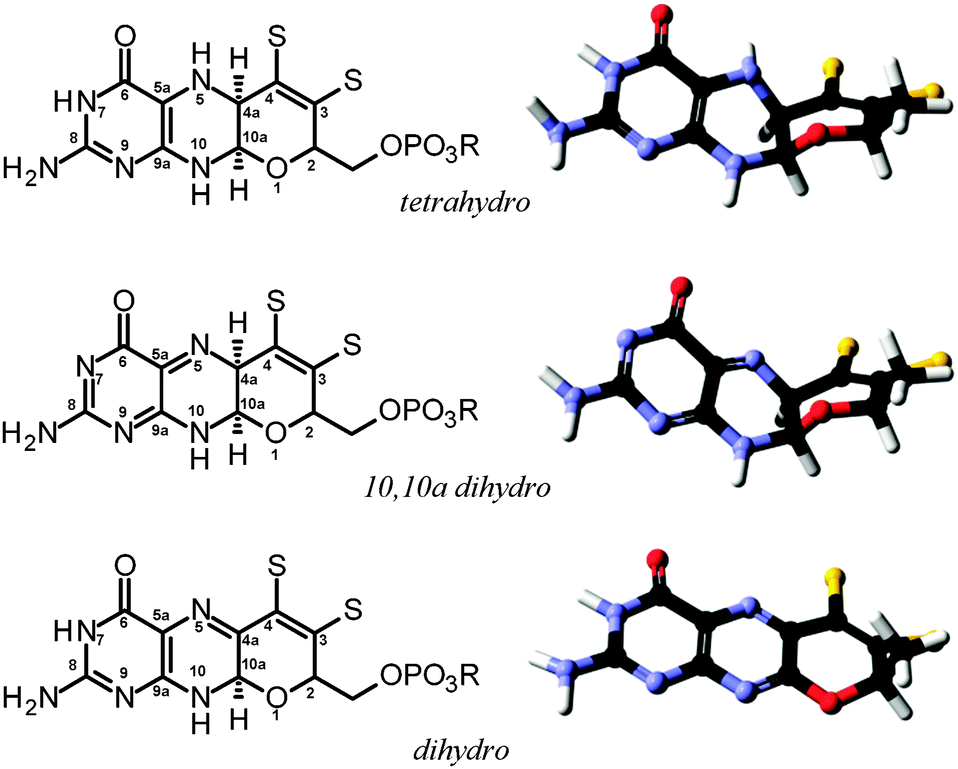 | ||
| Fig. 6 Structural and redox isoforms of molybdopterin, showing both the schematic structure (left) and the geometry optimized energy minimized DFT structure (right). | ||
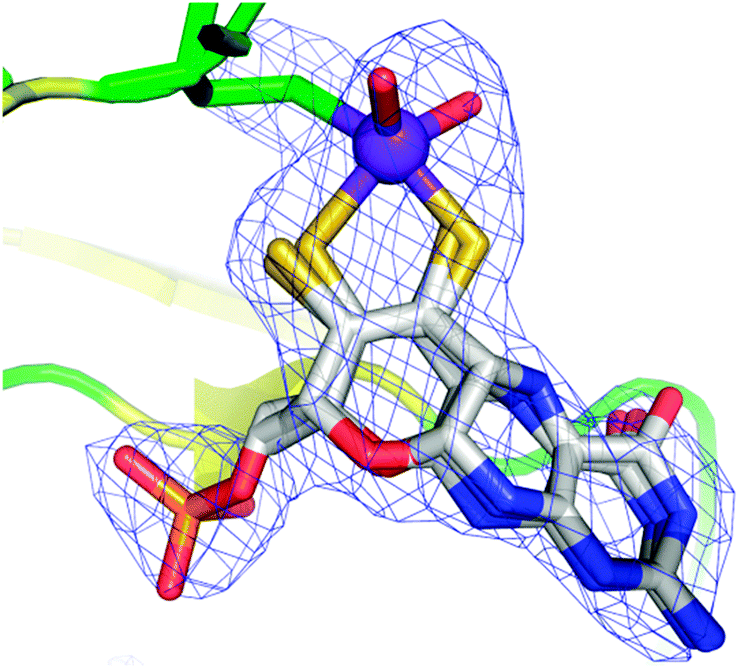 | ||
| Fig. 7 Fit of DFT structures for the different molybdopterin isoforms with electron density. Coordinates and structure factors were taken from Pichia angusta nitrate reductase (2bih), the molydopterin was removed leaving the Mo and phosphate and CCP4 REFMAC used to generate a difference map (contoured at 3σ). The three different molybdopterins can be seen to all adequately fit to the electron density. | ||
As a final consideration in this section we return to the pyran ring-open and ring-closed forms of molybdopterin. The original structures suggested for the cofactor contained no pyran ring as shown in Fig. 4. As we have discussed, the first molybdopterin enzyme structure to be solved was that of Pyrococcus furiosus aldehyde oxidoreductase (AOR) and showed a ring closed form containing a pyran ring.64 Nearly all subsequent structures that have since been determined were also of the ring-closed pyranopterin type. The only exception to date is the respiratory membrane-bound nitrate reductase from E. coli. Two different crystal structures have been reported by two different groups, one showing a ring-open form and the other the expected ring-closed form (Fig. 8).76,77 The electron density unequivocally supports different ring structures (Fig. 8) but insofar as other aspects are concerned, both structures display highly distorted Mo coordinations, with a vacant face at the Mo center and crowded face opposite with apparent bond angles between ∼90° to 70° between an aspartate ligand and one of the cofactor dithiolenes. These may be the result of averaged structures arising from photoreduction of the Mo center during diffraction data collection. Despite these irregularities examination of the electron density maps unmistakably indicates differences in the furan (Fig. 8). How these structural differences might relate to activity is at present unknown, although new evidence from model compound chemistry clearly indicates that ring-open and ring-closed forms can be reversibly formed.74
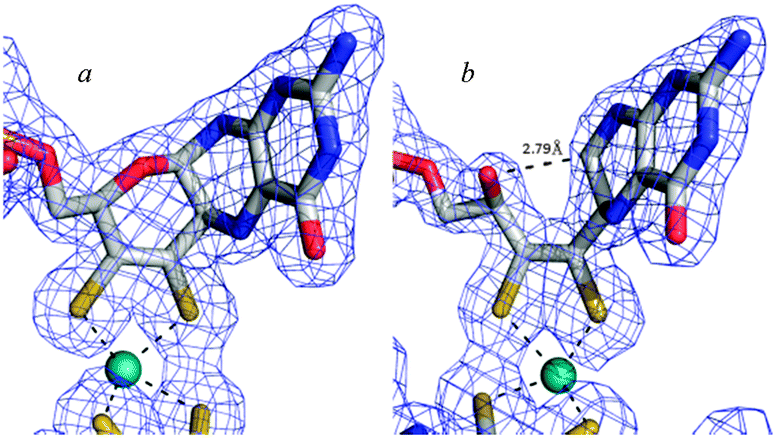 | ||
| Fig. 8 Ring-closed (a) and ring open (b) forms of molybdopterin in two different crystal structures of E. coli nitrate reductase. Coordinates and structure factors were taken from published data (pdb 1Q16 and 1R27). Cofactors were removed, a difference map recalculated (contoured at 3σ) and superimposed with the published coordinates. | ||
The evolution of the Mo and W enzymes
The hyperthermophilic archaea are thought to be some of the most phylogenetically ancient organisms that have been characterized to date.78 The capability to produce Mo–W enzymes is maintained across the archaea. As a general rule the hyperthermophilic archaea do not contain molybdopterin Mo enzymes, and present only W enzymes. We note in passing that Mo-containing proteins may have recently been found in Pyrococcus furiosus although a function for these remains uncertain.9 The dominance of W enzymes in the hyperthermophiles has caused some workers to suggest that the W enzymes were the predecessors of their Mo counterparts.79 Both Mo and W share chemistry that is somewhat non-metallic in character in that both metals form the oxy-dianions molybdate [MoO4]2− and tungstate [WO4]2− both of which are stable at lower redox potentials.The abundance of Mo and W in the earth's crust are 1.1 and 1.9 ppm, respectively,80 and the ocean crust has 0.9 ppm Mo and 0.09 ppm W.81 The levels of Mo and W in ambient seawater must reflect some combination of weathering of these sources together hydrothermal input,82 and with influence from the redox status of the systems concerned. Archaean seawater is likely to have been dominated by the latter two sources as it is thought that there was much continental crust but high average elevation of the emerged continental crust above sea level.
The group 6 metal based oxy-dianions [MO4]2− are tetrahedral species bearing a marked similarity to sulfate [SO4]2−, which poses problems for cells in specific uptake of molybdate or tungstate amidst a relative abundance of sulfate. Molybdate and tungstate are 19% and 20% larger than sulfate, respectively. This difference is sufficient for the metal-specific transporters to discriminate the anions. However, due to their chemical similarity many of these transporters have the ability to transport more than one chemical species. To date, all of the systems for transport of this family of ions are members of the adenosine triphosphate binding cassette (ABC) transporter family. ABC transporters are ubiquitous throughout all kingdoms of life, suggesting that they evolved very early. The molybdate transporter ModABC, for example, uses a rigid anion binding pocket to discriminate between molybdate and sulfate, in which looser-fitting sulfate is discriminated against because the miss-match with cavity size prevents stabilizing interactions and because of a greater desolvation penalty.83 Conversely, sulfate transporters can effectively exclude molybdate or tungstate because their anion binding pockets are too small.83 Modern oceans contain approximately 28 mM sulfate, 100 nM molybdate and only 60 pM tungstate.84 We note that many of the tungsten-dependent hyperthermophilic archaebacteria grow in the vicinity of marine hydrothermal vents, which expel water containing relatively high tungsten levels.82 Thus, in modern organisms, molybdate and tungstate transporters exploit the subtle structural differences between the oxy-dianions to selectively bind and take up the metal-based anion into the cell, even in the presence of what might seem to be an overwhelming excess of sulfate.
In E. coli the expression of the ModABC transport system is regulated by the ModE repressor protein, which contains an N-terminal DNA binding domain and a molybdate binding C-terminal domain. This mechanism of regulation appears to occur in only a small fraction of bacteria and archaea (below 30% and 20%, respectively).85 An analogous repressor has been identified in Campylobacter jejuni which represses ModABC in response to both molybdenum and tungsten, and TupABC in response to tungsten.86 The lack of universality of ModE-type regulation of ModABC suggests alternate regulatory pathways may exist in many organisms. Until recently little was known about the analogous transport systems and their regulation in eukaryotes. MOT1 is a Mo transporter and member of the sulfate transporter superfamily, and first characterized in Arabodopsis thaliana87 and Chlamydomonas reinhardtii.88 An analogous molybdate transporter in humans, MOT2, has also recently been identified.89
While such transporters clearly might have developed their current levels of specificity once a dependency on molybdenum or tungsten had been established, it is most unlikely that such specificity would have evolved in the first place in the presence of competition from high levels of sulfate in the environment. It is apparent, therefore, that the molybdenum and/or tungsten enzymes must have been well established in the period before sulfate became important in the ocean's chemistry. Life on earth is believed to have begun approximately 3.7 × 109 years ago (or 3.7 Ga), but the period in which aerobic life evolved is the subject of controversy. The conflicting views are that aerobic life evolved towards the end of the first billion years, with the first noticeable large-scale environmental effects at around 2.4 Ga,90,91 or substantially earlier at around 3.4 Ga and thus much closer to life's origins.92–94 Irrespective of the timing the oceans in pre-oxic earth are thought to have been very low in sulfate.95 Probably the best estimates for the evolution of molybdenum and tungsten enzymes come from work using a protein folding approach to understand evolution at a molecular level.96 Recent work using this method indicated that, like the iron–sulfur proteins,97 the molybdenum and tungsten enzymes are incredibly ancient,98,99 and likely existed in the so-called LUCA (last universal common ancestor) sometimes called the cenancestor, the progenitor of all life on earth.98 If Mo enzymes or more likely their W counterparts were important in early life, what were they used for? Very probably they carried out much the same kind of biochemistry as they carry out in modern organisms. In the reducing environment of the primordial world oxygen atom transfer was likely somewhat more challenging than in our oxic modern world, with a considerably more limited set of biochemical approaches available. Williams and Fraústo da Silva100 have suggested that a primary use of the Mo–W enzymes in early microorganisms may have been to reversibly handle the aldehyde oxido reductase chemistry, eqn (6):
| R–CHO + H2O ⇌ RCOOH + 2e− + 2H+ | (6) |
Evolved selection vs. environmental availability
The notion that W enzymes are more ancient than the Mo enzymes is also consistent with planetary redox history, with an overall more reducing environment on the early earth.90–100 Under sulfidic reducing conditions where highly insoluble MoS2 will precipitate tungsten compounds remain soluble.101 As previously pointed out, it is perhaps significant that the major mineral source for Mo is molybdenite (MoS2) while those for W are scheelite (CaWO4) and wolframite ([Fe, Mn]WO4).100 Thus Mo levels in the early ocean may have been low, with W levels relatively high, supporting increased environmental availability for the latter. This same chemistry is responsible for the low Mo and relatively high W levels in marine hydrothermal vent waters,82 from which most hyperthermophilic archaea known today have been isolated.We now turn to the differences between the chemical capabilities expected for the molybdenum and tungsten. Relativistic effects are significant in the chemistry of molybdenum and tungsten and these are much more important for tungsten. Relativistic contraction of the core orbitals enhances bond dissociation energies, while destabilization of the metal centered d-orbitals lowers associated redox potentials. Thus, comparison of the electrochemistry of iso-structural Mo and W compounds shows that W compounds have lower redox potentials than their Mo counterparts. Moreover, W complexes also have somewhat enhanced bond strengths (and therefore higher bond vibrational frequencies) compared to analogous Mo complexes. W complexes are also more sensitive to O2 than equivalent Mo complexes. Taken together, these trends make W better suited to catalyzing lower-potential redox reactions under anaerobic conditions, and at higher operating temperatures.102 In agreement with this notion, tungsten enzymes catalyze some of the lowest redox potential reactions known in biology.103 The combination of oxygen sensitivity, enhanced thermal stability and activity profiles that tend to lower redox potentials together make a compelling chemical argument for the importance of W, as opposed to Mo, in the primordial environment in which life first evolved.
Conclusions
The molybdenum and tungsten enzymes represent a large superfamily of substantially related enzymes, almost all of which catalyse oxygen transferase reactions, coupled with two electron redox reactions involving the metal. A detailed understanding of the active site structures of the molybdenum and tungsten enzymes is essential to an in-depth understanding of the catalytic mechanism. The enzymes operate under a broad envelope of redox potentials, including highly reducing conditions. The enzymes are thought to have been important in early life because of their ability to catalyse oxygen atom transfer reactions. Life and its supporting cellular machinery evolved under conditions that are very different from those at present, and modern organisms have inherited a genetic heritage of elaboration and retooling of more ancient machinery, which in some cases, was fine tuned for alternate purposes and selection of different chemical species.Acknowledgements
Work carried out at the University of Saskatchewan was supported by the Natural Sciences and Engineering Research Council of Canada, with other support from a Canada Research Chair (G.N.G.), the Canadian Institutes of Health Research (CIHR) and the Saskatchewan Health Research Foundation (SHRF). M.J.P. is a Fellow in the CIHR Training grant in Health Research Using Synchrotron Techniques (CIHR-THRUST) and J.J.H.C. is a CIHR-THRUST research associate. M.J.P. is a CIHR Postdoctoral Fellow and is also supported by SHRF. We thank our esteemed colleague Professor Robert Kerrich of the University of Saskatchewan for stimulating discussions on the conditions upon the primordial earth. We mark with sadness his untimely death from Cancer on April 17th 2013, during the preparation of this manuscript.Notes and references
- M. J. Fedor, Curr. Opin. Struct. Biol., 2002, 12, 289–295 CrossRef CAS PubMed.
- C. Swart, P. Fisicaro, H. Goenaga-Infante and S. Zakel, Metallomics, 2012, 4, 1137–1140 RSC.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 6, 25–35 CrossRef PubMed.
- A. J. Thomson and H. B. Gray, Curr. Opin. Chem. Biol., 1998, 2, 155–158 CrossRef CAS PubMed.
- J. J. H. Cotelesage, M. J. Pushie, P. Grochulski, I. J. Pickering and G. N. George, J. Inorg. Biochem., 2012, 115, 127–137 CrossRef CAS PubMed.
- D. C. Rees, A. Tezcan, C. A. Haynes, M. Y. Walton, S. Andrade, O. Einsle and J. B. Howard, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 971–984 CrossRef CAS PubMed.
- G. N. George, I. J. Pickering, E. Y. Yu, R. C. Prince, S. A. Bursakov, O. Y. Gavel, I. Moura and J. J. G. Moura, J. Am. Chem. Soc., 2000, 132, 8321–8322 CrossRef.
- M. G. Rivas, M. S. Carepo, C. S. Mota, M. Korbas, M.-C. Durand, A. T. Lopes, C. D. Brondino, A. S. Pereira, G. N. George, A. Dolla, J. J. G. Moura and I. Moura, Biochemistry, 2009, 48, 873–882 CrossRef CAS PubMed.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole II, F. E. Jenney Jr, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779–782 CrossRef CAS PubMed.
- F. J. Hine, A. J. Taylor and C. D. Garner, Coord. Chem. Rev., 2010, 254, 1570–1579 CrossRef CAS.
- J. L. Johnson and M. Duran, Molybdenum Cofactor Deficiency and Isolated Sulfite Oxidase Deficiency, in The Metabolic and Molecular Bases of Inherited Disease, ed. C. R. Scriver, et al., McGraw-Hill, New York, 8th edn, 2001, pp. 3163–3177 Search PubMed.
- E. C. De Renzo, E. Kaleita, P. Heytler, J. J. Oleson, B. L. Hutchings and J. H. Williams, J. Am. Chem. Soc., 1953, 75, 753 CrossRef CAS.
- D. E. Green and H. Beinert, Biochim. Biophys. Acta, 1953, 11, 599–600 CrossRef CAS.
- D. J. Nicholas and A. Nason, J. Biol. Chem., 1954, 207, 353–360 CAS.
- P. G. Avis, F. Bergel, R. C. Bray and K. V. Shooter, Nature, 1954, 173, 1230–1231 CrossRef CAS PubMed.
- L. G. Ljungdahl, Trends Biochem. Sci., 1976, 1, 63–65 CAS.
- G. N. George, R. C. Prince, S. Mukund and M. W. W. Adams, J. Am. Chem. Soc., 1992, 114, 3521–3523 CrossRef CAS.
- M. J. Pushie and G. N. George, Coord. Chem. Rev., 2011, 255, 1055–1084 CrossRef CAS.
- C. Coelho, P. J. Gonzalez, J. Trincao, A. L. Carvalho, S. Najmudin, T. Hettman, S. Dieckman, J. J. G. Moura, I. Moura and M. J. Romão, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2007, 63, 516–519 CrossRef CAS PubMed.
- F. Zinoni, A. Birkmann, T. C. Stadtman and A. Bock, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4650–4654 CrossRef CAS.
- J. C. Boyington, V. N. Gladyshev, S. V. Khangulov, T. C. Stadtman and P. D. Sun, Science, 1997, 275, 1305–1308 CrossRef CAS PubMed.
- S. V. Khangulov, V. N. Gladyshev, G. C. Dismukes and T. C. Stadtman, Biochemistry, 1998, 37, 3518–3528 CrossRef CAS PubMed.
- G. N. George, C. M. Colangelo, J. Dong, R. A. Scott, S. V. Khangulov, V. N. Gladyshev and T. C. Stadtman, J. Am. Chem. Soc., 1998, 120, 1267–1273 CrossRef CAS.
- M. G. Bertero, R. A. Rothery, M. Palak, C. Hou, D. Lim, F. Blasco, J. H. Weiner and N. C. J. Strynadka, Nat. Struct. Biol., 2003, 10, 681–687 CrossRef CAS PubMed.
- M. Jormakka, D. Richardson, B. Byrne and S. Iwata, Structure, 2004, 12, 95–104 CrossRef CAS PubMed.
- H. K. Li, C. Temple, K. V. Rajagopalan and H. Schindelin, J. Am. Chem. Soc., 2000, 122, 7673–7680 CrossRef CAS.
- G. N. George, J. Hilton, C. Temple, R. C. Prince and K. V. Rajagopalan, J. Am. Chem. Soc., 1999, 121, 1256–1266 CrossRef CAS.
- S. Najmudin, P. J. Gonzalez, J. Trincão, C. Coelho, A. Mukhopadhyay, C. C. Romão, I. Moura, J. J. G. Moura, C. D. Brondino and M. J. Romão, JBIC, J. Biol. Inorg. Chem., 2008, 13, 737–753 CrossRef CAS PubMed.
- B. M. Rosner and B. Schink, J. Bacteriol., 1995, 177, 5767–5772 CAS.
- G. B. Seiffert, G. M. Ullmann, A. Messerschmidt, B. Schink, P. M. H. Kroneck and O. Einsle, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3073–3077 CrossRef CAS PubMed.
- H. Raaijmakers, S. Macieira, J. M. Dias, S. Teixeira, S. Bursakov, R. Huber, J. J. Moura, I. Moura and M. J. Romão, Structure, 2002, 10, 1261–1273 CrossRef CAS PubMed.
- H. Raaijmakers and M. J. Romão, JBIC, J. Biol. Inorg. Chem., 2006, 11, 849–854 CrossRef CAS PubMed.
- G. N. George, C. Costa, J. J. G. Moura and I. Moura, J. Am. Chem. Soc., 1999, 121, 2625–2626 CrossRef CAS.
- A. Messerschmidt, H. Niessen, D. Abt, O. Einsle, B. Schink and P. M. H. Kroneck, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11571–11576 CrossRef CAS PubMed.
- M. Boll, B. Schink, A. Messerschmidt and P. M. H. Kroneck, Biol. Chem., 2005, 386, 999–1006 CAS.
- A. K. Rappé and W. A. Goddard, J. Am. Chem. Soc., 1982, 104, 3287–3294 CrossRef.
- Y. Izumi, T. Glaser, K. Rose, J. McMaster, P. Basu, J. H. Enemark, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1999, 121, 10035–10046 CrossRef CAS.
- K. Peariso, M. E. Helton, E. N. Duesler, S. E. Shadle and M. L. Kirk, Inorg. Chem., 2007, 46, 1259–1267 CrossRef CAS PubMed.
- K. Peariso, R. L. McNaughton and M. L. Kirk, J. Am. Chem. Soc., 2002, 124, 9006–9007 CrossRef CAS PubMed.
- C. Kisker, H. Schindelin, A. Pacheco, W. A. Wehbi, R. M. Garrett, K. V. Rajagopalan, J. H. Enemark and D. C. Rees, Cell, 1997, 91, 973–983 CrossRef CAS PubMed.
- K. Fischer, G. Barbier, H.-J. Hecht, R. R. Mendel, W. H. Campbell and G. Schwarz, Plant Cell, 2005, 17, 1167–1179 CrossRef CAS PubMed.
- G. N. George, J. A. Mertens and W. A. Campbell, J. Am. Chem. Soc., 1999, 121, 9730–9731 CrossRef CAS.
- J. A. Qiu, H. L. Wilson and K. V. Rajagopalan, Biochemistry, 2012, 51, 1134–1147 CrossRef CAS PubMed.
- C. J. Doonan, A. Stockert, R. Hille and G. N. George, J. Am. Chem. Soc., 2005, 127, 4518–4522 CrossRef CAS PubMed.
- S. P. Cramer, R. Wahl and K. V. Rajagopalan, J. Am. Chem. Soc., 1981, 103, 7721–7727 CrossRef CAS.
- D. L. Williams-Smith, R. C. Bray, M. J. Barber, A. D. Tsopanakis and S. P. Vincent, Biochem. J., 1977, 167, 593–600 CrossRef CAS PubMed.
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15971–15976 CrossRef CAS PubMed.
- H. Dobbek, L. Gremer, O. Meyer and R. Huber, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8884–8889 CrossRef CAS.
- P. Hanzelmann, H. Dobbek, L. Gremer, R. Huber and O. Meyer, J. Mol. Biol., 2000, 301, 1221–1235 CrossRef CAS PubMed.
- M. Gnida, R. Ferner, L. Gremer, O. Meyer and W. Meyer-Klaucke, Biochemistry, 2003, 42, 222–230 CrossRef CAS PubMed.
- B. Zhang, C. F. Hemann and R. Hille, J. Biol. Chem., 2010, 285, 12571–12578 CrossRef CAS PubMed.
- M. Hofmann, J. K. Kassube and T. Graf, JBIC, J. Biol. Inorg. Chem., 2005, 10, 490–495 CrossRef CAS PubMed.
- J. Wilcoxen, S. Snider and R. Hille, J. Am. Chem. Soc., 2011, 133, 12934–12936 CrossRef CAS PubMed.
- F. E. Inscore, R. McNaughton, B. L. Westcott, M. E. Helton, R. Jones, I. K. Dhawan, J. H. Enemark and M. L. Kirk, Inorg. Chem., 1999, 38, 1401–1410 CrossRef CAS.
- R. L. McNaughton, M. E. Helton, N. Rubie and M. L. Kirk, Inorg. Chem., 2000, 39, 4386–4387 CrossRef CAS.
- G. N. George, R. C. Prince, S. Mukund and M. W. W. Adams, J. Am. Chem. Soc., 1992, 114, 3521–3523 CrossRef CAS.
- G. N. George, I. J. Pickering, M. J. Pushie, K. Nienaber, M. J. Hackett, I. Ascone, B. Hedman, K. O. Hodgson, J. B. Aitken, A. Levina, C. Glover and P. A. Lay, J. Synchrotron Radiat., 2012, 19, 875–886 CrossRef CAS PubMed.
- G. N. George, I. J. Pickering and C. Kisker, Inorg. Chem., 1999, 38, 2539–2540 CrossRef CAS.
- J. A. Qiu, H. L. Wilson, M. J. Pushie, C. Kisker, G. N. George and K. V. Rajagopalan, Biochemistry, 2010, 49, 3989–4000 CrossRef CAS PubMed.
- G. N. George, R. M. Garrett, R. C. Prince and K. V. Rajagopalan, J. Am. Chem. Soc., 1996, 118, 8588–8592 CrossRef CAS.
- G. N. George, R. M. Garrett, R. C. Prince and K. V. Rajagopalan, Inorg. Chem., 2004, 43, 8456–8460 CrossRef CAS PubMed.
- J. L. Johnson, B. E. Hainline and K. V. Rajagopalan, J. Biol. Chem., 1980, 255, 1783–1786 CAS.
- J. L. Johnson and K. V. Rajagopalan, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 6856–6860 CrossRef CAS.
- M. K. Chan, S. Mukund, A. Kletzin, M. W. W. Adams and D. C. Rees, Science, 1995, 267, 1463–1469 CrossRef CAS PubMed.
- S. Leimkühler, M. M. Wuebbens and K. V. Rajagopalan, Coord. Chem. Rev., 2011, 255, 1129–1144 CrossRef PubMed.
- R. R. Mendel and G. Schwarz, Coord. Chem. Rev., 2011, 255, 1145–1158 CrossRef CAS.
- G. Schwarz, J. Schulze, F. Bittner, T. Eilers, J. Kuper, G. Bollmann, A. Nerlich, H. Brinkmann and R. R. Mendel, Plant Cell, 2000, 12, 2455–2472 CrossRef CAS.
- B. Stallmeyer, G. Schwarz, J. Schulze, A. Nerlich, J. Reiss, J. Kirsch and R. R. Mendel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1333–1338 CrossRef CAS.
- A. A. Belaidi and G. Schwarz, Biochem. J., 2013, 450, 149–157 CrossRef CAS PubMed.
- J. Kuper, A. Llamas, H.-J. Hecht, R. R. Mendel and G. Schwarz, Nature, 2004, 430, 803–806 CrossRef CAS PubMed.
- S. C. Leary and D. R. Winge, EMBO Rep., 2007, 8, 224–227 CrossRef CAS PubMed.
- C. Enroth, B. T. Eger, K. Okamoto, T. Nishino, T. Nishino and E. F. Pai, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10723–10728 CrossRef CAS.
- R. A. Rothery, B. Stein, M. Solomonson, M. L. Kirk and J. H. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14773–14778 CrossRef CAS PubMed.
- B. R. Williams, Y. Fu, G. P. A. Yap and S. J. N. Burgmayer, J. Am. Chem. Soc., 2012, 134, 19584–19587 CrossRef CAS PubMed.
- T. R. Hawkes and R. C. Bray, Biochem. J., 1984, 219, 481–493 CrossRef CAS PubMed.
- M. G. Bertero, R. A. Rothery, M. Palak, C. Hou, D. Lim, F. Blasco, J. H. Weiner and N. C. J. Strynadka, Nat. Struct. Biol., 2003, 10, 681–687 CrossRef CAS PubMed.
- M. Jormakka, D. Richardson, B. Byrne and S. Iwata, Structure, 2004, 12, 95–104 CrossRef CAS PubMed.
- K. O. Stetter, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 1837–1843 CrossRef CAS PubMed.
- A. Kletzin and M. W. W. Adams, FEMS Microbiol. Rev., 1996, 18, 5–63 CrossRef CAS PubMed.
- R. L. Rudnick and S. Gao, The Composition of the Continental Crust, in The Crust (ed. R. L. Rudnick) Vol. 3, Treatise on Geochemistry, ed. H. D. Holland and K. K. Turekian, Elsevier-Pergamon, Oxford, 2003, pp. 1–64 Search PubMed.
- R. Arevalo and W. F. McDonough, Chem. Geol., 2010, 271, 70–85 CrossRef CAS.
- K. Kishida, Y. Sohrin, K. Okamura and J. Ishibashi, Earth Planet. Sci. Lett., 2004, 222, 819–827 CrossRef CAS.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2004, 126, 10296–10305 CrossRef CAS PubMed.
- Y. Sohrin, K. Isshiki, T. Kuwamoto and E. Nakayama, Mar. Chem., 1987, 22, 95–103 CrossRef CAS.
- Y. Zhang and V. N. Gladyshev, J. Mol. Biol., 2008, 379, 881–899 CrossRef CAS PubMed.
- M. E. Taveirne, M. L. Sikes and J. W. Olson, Mol. Microbiol., 2009, 74, 758–771 CrossRef CAS PubMed.
- H. Tomatsu, J. Takano, H. Takahashi, A. Watanabe-Takahashi, N. Shibagaki and T. Fujiwara, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18807–18812 CrossRef CAS PubMed.
- M. Tejada-Jiménez, A. Llamas, E. Sanz-Luque, A. Galvan and E. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20126–20130 CrossRef PubMed.
- M. Tejada-Jiménez, A. Galván and E. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6420–6425 CrossRef PubMed.
- A. L. Sessions, D. M. Doughty, P. V. Welander, R. E. Summons and D. K. Newman, Curr. Biol., 2009, 19, R567 CrossRef CAS PubMed.
- R. Buick, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 2731–2743 CrossRef CAS PubMed.
- M. Hoashi, D. C. Bevacqua, T. Otake, Y. Watanabe, A. H. Hickman, S. Utsunomiya and H. Ohmoto, Nat. Geosci., 2009, 2, 301–306 CrossRef CAS.
- R. Kerrich and N. Said, Chem. Geol., 2011, 280, 232–241 CrossRef CAS.
- R. Kerrich, N. Said, C. Manikyamba and D. Wyman, Gondwana Res., 2013, 23, 506–525 CrossRef CAS.
- J. W. Jamieson, B. A. Wing, J. Farquhar and M. D. Hannington, Nat. Geosci., 2013, 6, 61–64 CAS.
- M. Wang, Y.-Y. Jiang, K. M. Kim, G. Qu, H.-F. Ji, J. E. Mittenthal, H.-Y. Zhang and G. Caetano-Anollés, Mol. Biol. Evol., 2011, 28, 567–582 CrossRef CAS PubMed.
- E. J. Milner-White and M. J. Russell, Origins Life Evol. Biospheres, 2005, 35, 19–27 CrossRef CAS.
- B. Schoepp-Cothenet, R. van Lis, P. Philippot, A. Magalon, M. J. Russell and W. Nitschke, Sci. Rep., 2012, 2, 263 Search PubMed.
- E. Lebrun, M. Brugna, F. Baymann, D. Muller, D. Liévremot, M.-C. Lett and W. Nitschke, Mol. Biol. Evol., 2003, 20, 686–693 CrossRef CAS PubMed.
- R. J. P. Williams and J. J. R. Fraústo da Silva, Biochem. Biophys. Res. Commun., 2002, 292, 293–299 CrossRef CAS PubMed.
- S. Licht, J. Electrochem. Soc., 1988, 135, 2971–2975 CrossRef CAS.
- J. Wiegel and M. W. W. Adams, Protein Sci., 2008, 8, 1564–1565 Search PubMed.
- J. W. Kung, S. Baumann, M. von Bergen, M. Mueller, P.-H. Hagedoorn, W. R. Hagen and M. Boll, J. Am. Chem. Soc., 2010, 132, 9850–9856 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |