
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of α-brominated phosphonates and their application as phosphate bioisosteres
A. Michael
Downey†
and
Christopher W.
Cairo
*
Alberta Glycomics Centre, Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: ccairo@ualberta.ca; Fax: +1-780-492-8231; Tel: +1-780-492-0377
First published on 5th September 2014
Abstract
Substrate phosphorylation is a key modulator of signal transduction. Abnormal phosphorylation in vivo is implicated in many diseases including cancer, diabetes, and Alzheimer's disease. Inhibitors of phosphate-recognizing proteins have potential as medicinal agents as well as tools to study phosphorylation pathways. A well-known and common inhibition strategy is to synthetically replace the labile phosphate moiety with a non-hydrolyzable phosphonate. Fully saturated, α-fluoro and α,α-difluorophosphonates are often effective phosphate bioisosteres and have been well studied. More recently α-brominated phosphonates have begun to emerge as inhibitors of phosphate recognizing enzymes, some of which operate by irreversible mechanisms. Herein we discuss the synthetic approaches to aliphatic and benzylic α-bromophosphonates and their biological activities.
1. Introduction: phosphonates and α-halophosphonates as phosphate bioisosteres
Substrate phosphorylation is a pivotal modulator of cellular function through its regulation of signal transduction pathways. It is estimated that one-third of all cellular proteins are modified through phosphorylation, making this the second most prevalent post-translational modification after glycosylation. Phosphorylation of target molecules is carried out by kinases, acting in opposition to phosphatases, which dephosphorylate these same targets.1 A large number of kinases and phosphatases are found in humans which generate or modify phospho-serine, threonine, tyrosine, or histidine residues.1,2 Additionally, phosphorylation pathways on lipid- and carbohydrate-based substrates play fundamental biochemical roles in metabolic processing and protein–carbohydrate or lipid recognition.3,4Due to the ubiquitous nature of substrate phosphorylation in cell signaling networks, these pathways are important to physiological processes including cellular differentiation, migration, metabolism, and apoptosis.2,5,6 Aberrant phosphorylation pathways can have devastating physiological consequences and may play a role in pathologies including cancer,6 diabetes,7 and Alzheimer's disease.8 As a result of these wide ranging effects, general strategies for the design of improved inhibitors and probes of this class of enzymes, or the receptors that recognize them, is of continued interest to medicinal chemists. Novel functional groups that can be exploited for inhibitor design may be valuable components of research tools or therapeutic strategies.
It has been known for many years that a general strategy for producing competitive inhibitors of phosphate recognizing proteins is to create substrate mimics that replace the hydrolyzable C–O–P bond of a phosphate with a non-hydrolyzable C–C–P bond to yield a phosphonate moiety. Phosphonates are not only resistant to enzymatic cleavage, hydrolysis of the functional group is also dramatically decreased allowing for synthetic manipulation of the molecule after installation of the phosphonate.9 However, there are obvious shortcomings to this strategy. Firstly, replacement of an electronegative oxygen atom with a non-polar methylene group may reduce affinity for the phosphate-recognizing-protein active site. Solubility of the compounds in water may also be decreased. The phosphonate group may also exhibit changes in charge state, for example if the pKa of the phosphonic acid is increased above physiological pH;10 this can make salt bridge or hydrogen bond contacts less favorable in an enzyme active site.9,11
An early approach to address this issue was the installation of fluorine atoms on the phosphonate α-carbon, thus making the phosphonate a more effective electronic isostere of the phosphate group.12,13 α-Fluorophosphonates also reduce the pKa of the phosphonic acid protons below physiological pH (pKa2 ∼ 5.6), restoring the group to the same charge state as a native phosphate (pKa2 ∼ 6.4) at physiological pH.11,12
Medicinal chemists have more recently exchanged fluorine for bromine to determine the activity of α-bromophosphonates as inhibitors. Typical C–X bond lengths within α-halophosphonate compounds are 1.41 Å (C–F),14 1.79 Å (C–Cl),15 and 1.93 Å (C–Br).16 Thus, α-bromophosphonates may have different steric requirements than their fluoro-derived counterparts. Additionally, while the bromine atom does not offer the same electronegativity as fluorine, it may act as a good leaving group offering the potential to create covalent labels of enzyme active sites that contain potent nucleophiles.17,18 This unique combination of features makes α-bromophosphonates a useful tool for the medicinal chemist. Herein we will discuss the synthesis and examples of applications which have exploited α-bromophosphonate derivatives in biological studies.
2. Design, synthesis and evaluation of α-bromophosphonates as bioactive compounds
α-Bromophosphonates were first described in the literature over 90 years ago. The earliest example of an α-bromophosphonate was isolated as an α,β-dibromo phosphonic acid intermediate (1, Fig. 1) which was decomposed in the presence of base to provide a β-bromo-α,β-unsaturated phosphonic acid.19 However, as the use of phosphonates expanded in medicinal chemistry, additional reports of the synthesis of α-bromophosphonates as synthetic targets began to emerge. Early examples of α-bromophosphonates appeared in a 1953 study of pKa values of α-substituted phosphonic acids (2). The pKa values of compound 2 were found to be 1.14 and 6.52, respectively,20 indicating that the α-bromophosphonates provide a pKa lowering effect over CH2-phosphonates (pKa2 = 7.7–9.0)10 which more closely matches the acidity of the phosphonate to that of naturally occurring phosphates. This also indicates that the α-bromophosphonate group is 10-fold more basic than the α-fluorophosphonate moiety.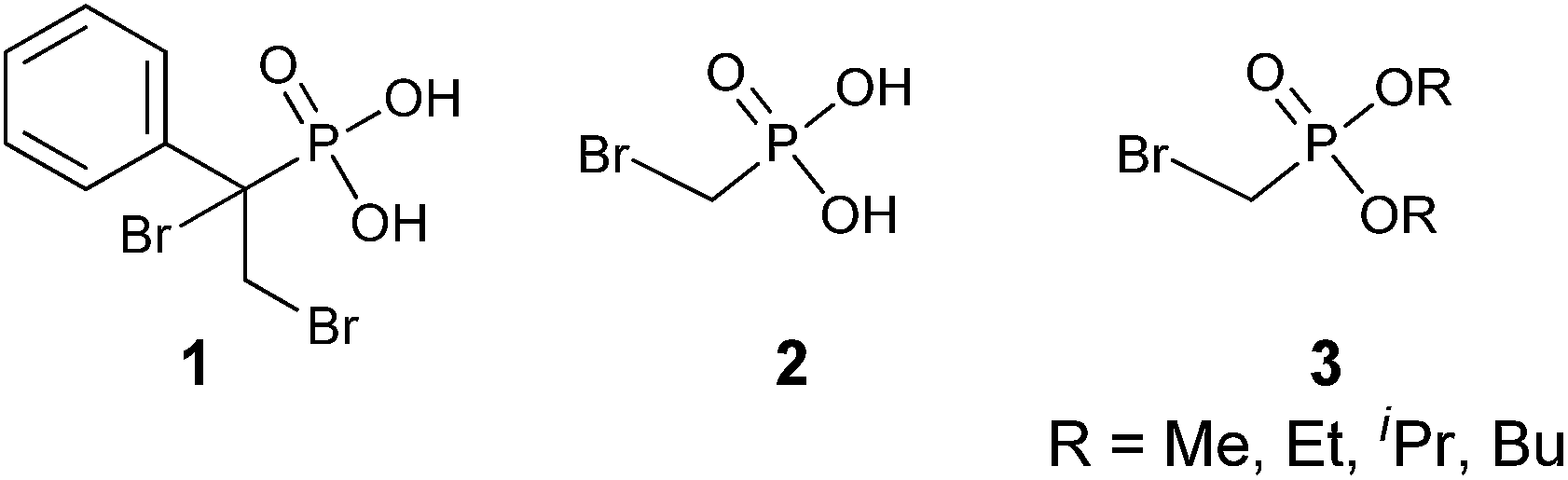 | ||
| Fig. 1 Early examples of α-bromophosphonates. | ||
The first biological studies of alkyl α-bromophosphonates (3) were conducted with α-bromophosphonates tested as agricultural chemicals. These compounds were found to be active in killing worms, insects, and bacteria.21
2.1. Benzylic α-bromophosphonates
Synthetic access to benzylic α-bromophosphonates (BBP) has been well studied, and these are generally easier to access than their aliphatic counterparts (Scheme 1). This functional group has been exploited as a bioisostere of phosphotyrosine residues in medicinal chemistry. We highlight examples of BBP in the literature, and discuss general strategies for their synthesis below.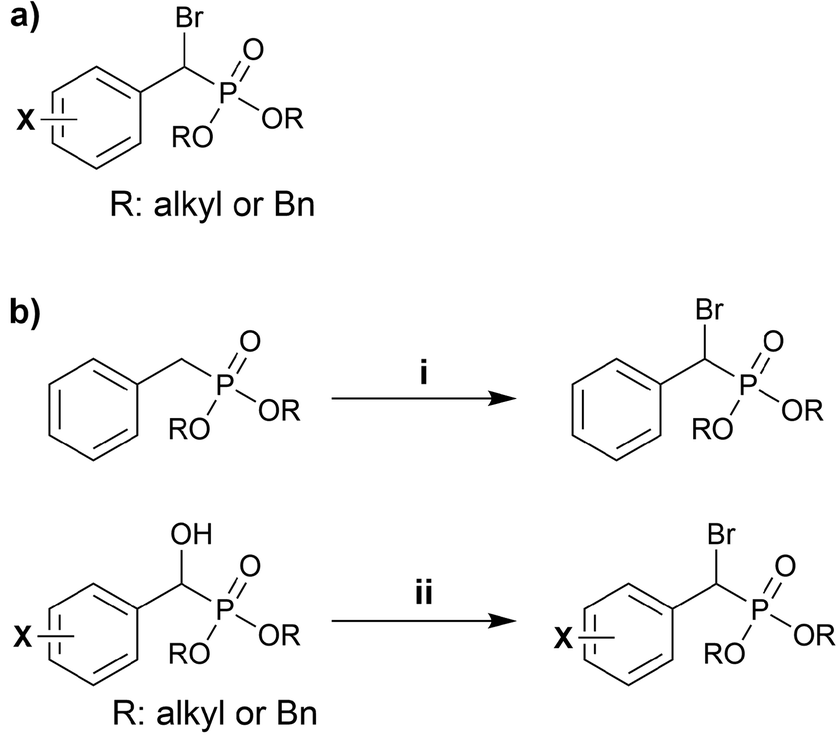 | ||
| Scheme 1 (a) A generic benzylic α-bromophosphonate (BBP). (b) Synthetic strategies for obtaining BBPs. | ||
A summary of reported conditions used through pathway (i) (Scheme 1b) is given in Table 1. The fully saturated phosphonate precursor is typically converted to the α-bromophosphonate through either radical bromination using N-bromosuccinimide (entries 1–4, Table 1) or through deprotonation of the α-proton to form a phosphorus-stabilized carbanion that is subsequently quenched with an electrophilic bromine source (entry 5, Table 1). Benzyl diethylphosphonate was subjected to three equivalents of lithium hexamethyldisilazide (LiHMDS) at −78 °C and the phosphorus-stabilized anion was quenched with trimethylsilyl chloride (TMSCl) to temporarily install a TMS group at the α position in situ (entry 5). Under these conditions, a second deprotonation occurred simultaneously which was subsequently quenched with an electrophilic bromine source, 1,2-dibromotetrachloroethane (TCDBE). The reaction was then warmed to 0 °C and lithium ethoxide was added to cleave the TMS group, furnishing the BBP adduct in good yield.29 Importantly, for all entries in Table 1, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers at the α position is expected in the absence of any directing groups.
:1 mixture of diastereomers at the α position is expected in the absence of any directing groups.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | R | Conditions | Yielda | Reference |
| a The R group shown is for the highest yielding analog. b 1,2-Dibromotetrachloroethane. | |||||
| 1 |
|
Me | (PhCO)2, NBS, 76 °C | 41% | Collins et al., 1984 (ref. 24) |
| 2 |
|
Et | NBS | 55% | Gross et al., 1990 (ref. 25) |
| Me | 71% | Andriyashin et al., 2012 (ref. 26) | |||
| 3 |
|
iPr | (PhCO)2, NBS, 76 °C | 82% | Chakraborty et al., 1991 (ref. 27) |
| 4 |
|
Et | NBS, hλ | N/A | Pieper et al., 2000 (ref. 28) |
| 5 |
|
Et | (1) LiHMDS, TMSCl, −78 °C | 87% | Iorga et al., 1999 (ref. 29) |
| (2) TCDBE,b −78 °C | |||||
| (3) LiOEt, 0 °C | |||||

The phosphorous-stabilized carbanion (Scheme 2) has been examined by several groups. The pKa of the alkyl phosphonate α proton is relatively high (∼29)22 and deprotonation requires the use of a strong and non-nucleophilic base to produce the anion.12 Cantat et al. confirmed that anionic C–P bonds are shorter than C–P bonds of the corresponding saturated compounds using crystallography. Computational analysis suggested stabilization of the anion through hyperconjugation resulting from p(C) → σ*(P–O) donation. It was concluded that contact with the metal cation is shared between the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–C atoms.23
P–C atoms.23
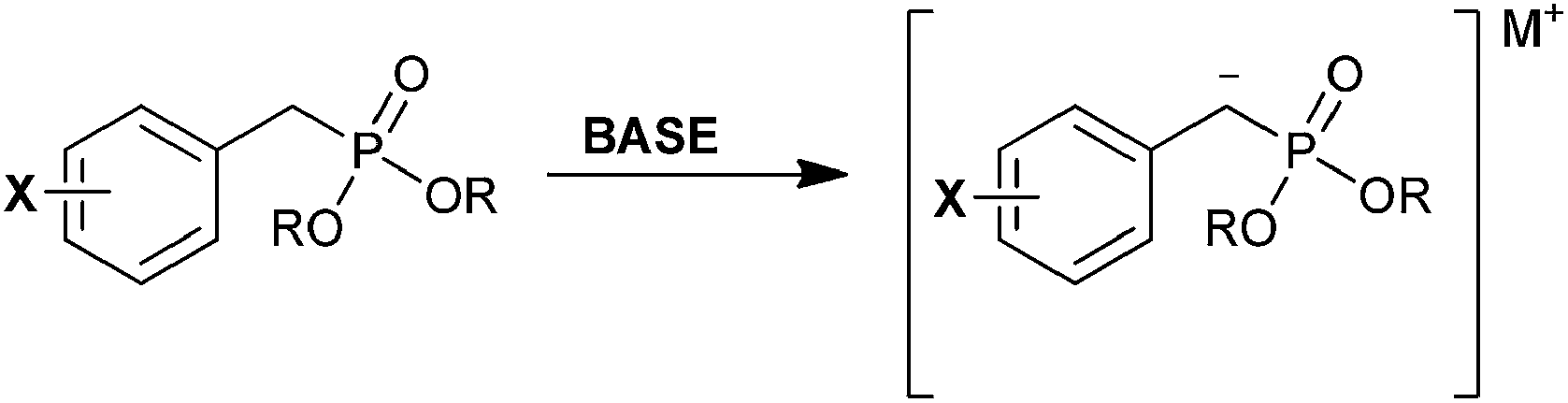 | ||
| Scheme 2 Deprotonation of a benzylic phosphonate to form a phosphorus-stabilized carbanion. | ||
The more common synthetic approach to form BBP is via an α-hydroxyphosphonate intermediate (pathway (ii), Scheme 1b). This strategy typically involves milder conditions than the strong basic conditions required for formation of the phosphorus-stabilizied carbanion. Shown in Scheme 3 is a general approach to the synthesis of the parent α-hydroxy adduct via Pudovik reaction.30 In this transformation, a benzyaldehyde derivative is treated with a deprotonated dialkyl- or diarylphosphite to furnish a diastereomeric mixture of the α-hydroxy analog.
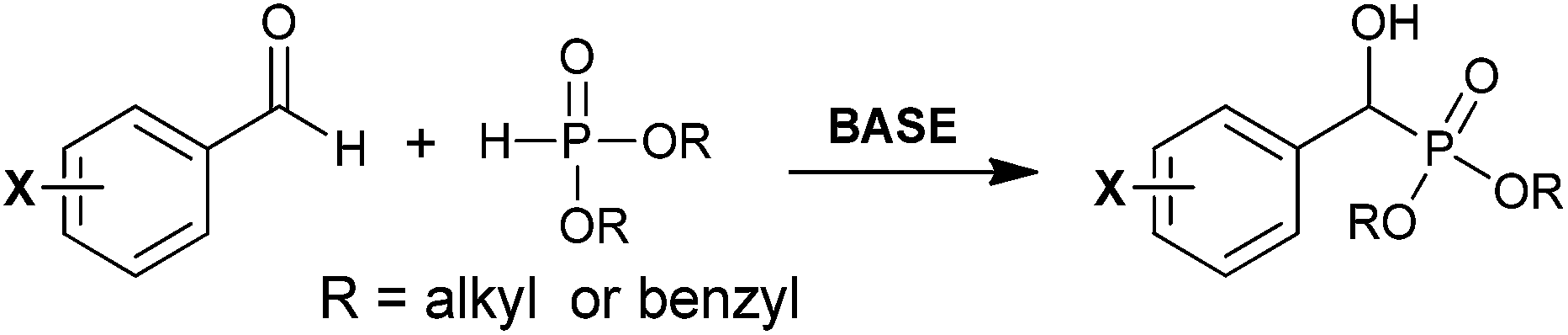 | ||
| Scheme 3 General form of the Pudovik reaction to generate a benzylic α-hydroxyphosphonate. | ||
After obtaining the α-hydroxy precursor, the subsequent bromination reaction can be performed using a variety of conditions. Several methods used to achieve this transformation are summarized in Table 2. Entries 1–8 are examples that achieved benzylic α-bromination with an electrophilic bromide source, with yields ranging from 42% to 100%. Entries 9–13 summarize conditions employed to obtain α-brominated phosphonates which were ultimately screened for activity against phosphate recognizing proteins. In all instances, the syntheses required no more than three chemical steps to obtain the target compound. Several analogs tested for activity from the references included in Table 2 are illustrated in Fig. 2. We discuss the biological activity of these species below.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | R | Conditions | Yielda | Reference |
| a The R group shown is for the highest yielding analog. b N,N-Carbonyldiimidazole. c 4-Aminophenyldiphenylphosphinite (APDPP). | |||||
| 1 |
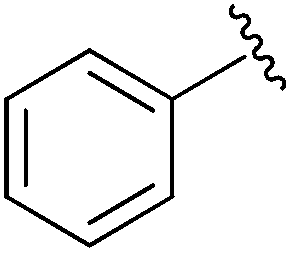
|
Et | CBr4, PPh3, 80 °C | 42% | Gajda, 1990 (ref. 31) |
| 2 |
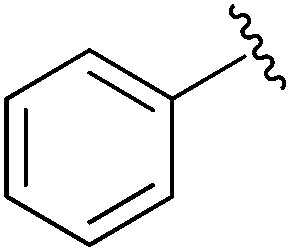
|
Et | PPh3/Br2, pyr | 42% | Gajda, 1990 (ref. 31) |
| 3 |
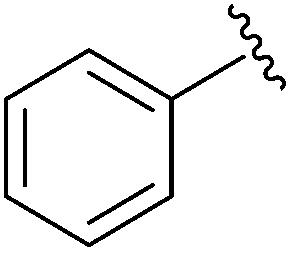
|
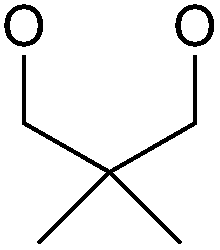
|
SOBr2 or PBr3 | 79% | Kumaraswamy et al., 1997 (ref. 32) |
| 4 |
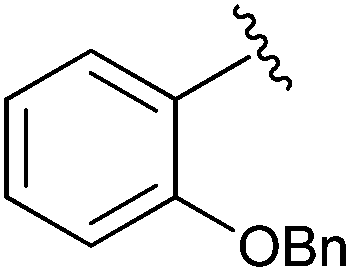
|
Et | SOBr2, pyr | 88% | Gross et al., 1993 (ref. 33) |
| 5 |
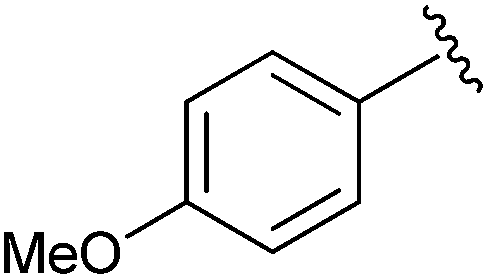
|
Et | CDI,b allyl bromide | 100% | Green et al., 1996 (ref. 34) |
| 6 |
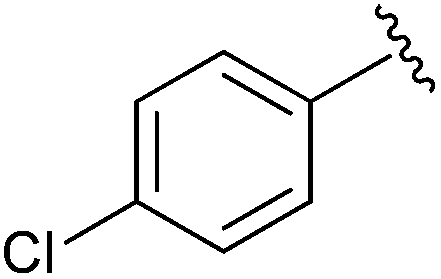
|
Et | n-BuNBr, PPh3, DDQ | 98% | Firouzabadi et al., 2004 (ref. 35) |
| 7 |
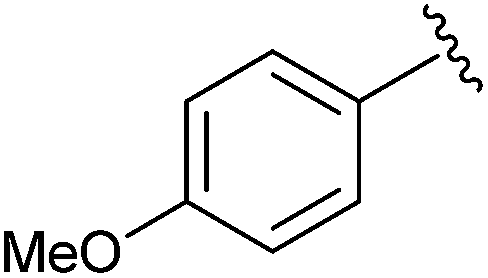
|
Et | APDPP,c Br2 | 100% | Iranpoor et al., 2006 (ref. 36) |
| 8 |
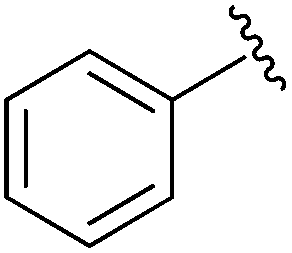
|
Et | SOBr2 | 80% | Balczewski et al., 2006 (ref. 37) |
| 9 |
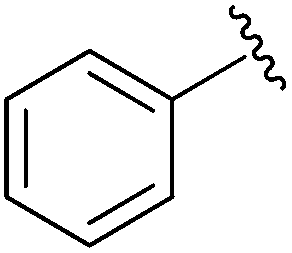
|
Et | NBS, pyr | N/A | Taylor et al., 1996 (ref. 38) |
| 10 |
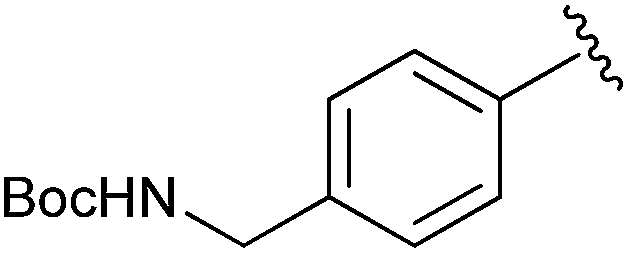
|
Et | PPh3/Br2, pyr | 51% | Kumar et al., 2004 (ref. 39) |
| 11 |
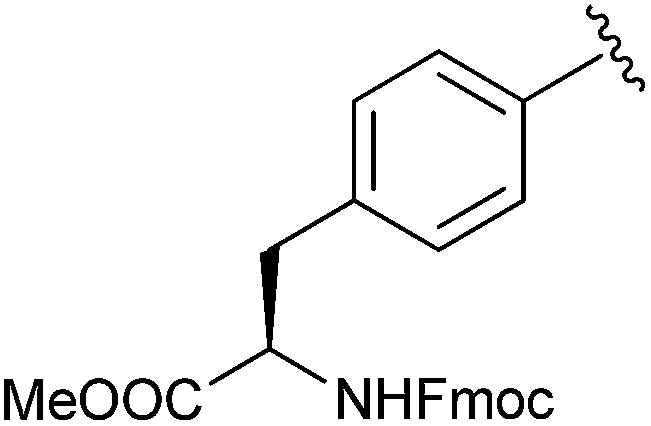
|
Me | SOBr2, pyr | 88% | Tulsi et al., 2010 (ref. 40) |
| 12 |
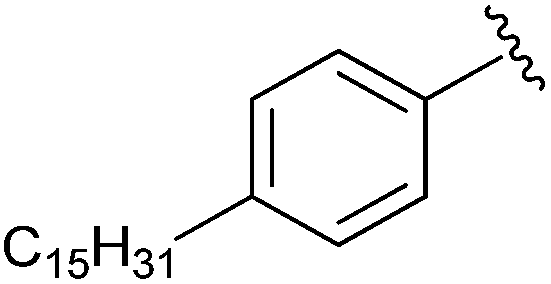
|
Me | PBr3 | N/A | Gupte et al., 2011 (ref. 41) |
| 13 |
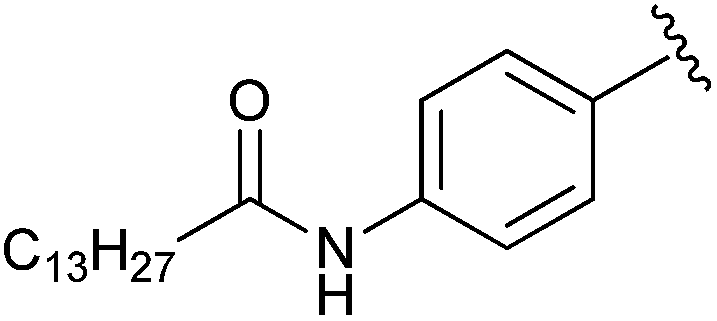
|
Et | CBr4, PPh3 | 81% | Jiang et al., 2011 (ref. 42) |

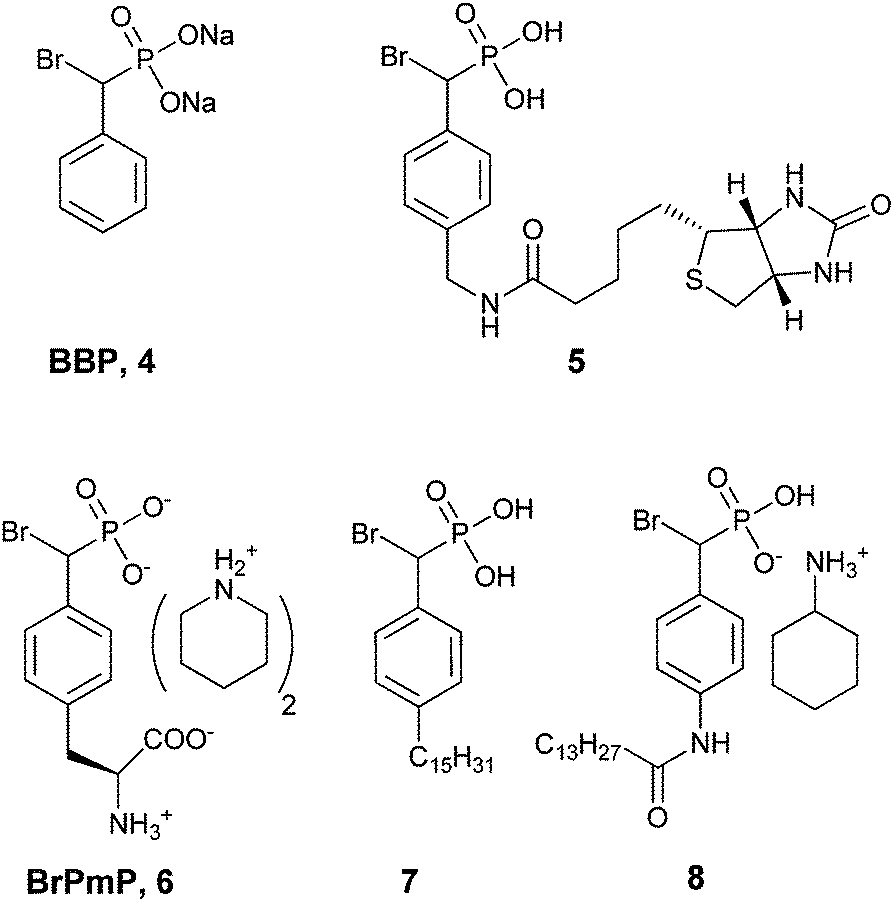 | ||
| Fig. 2 Biologically active BBPs.43 | ||
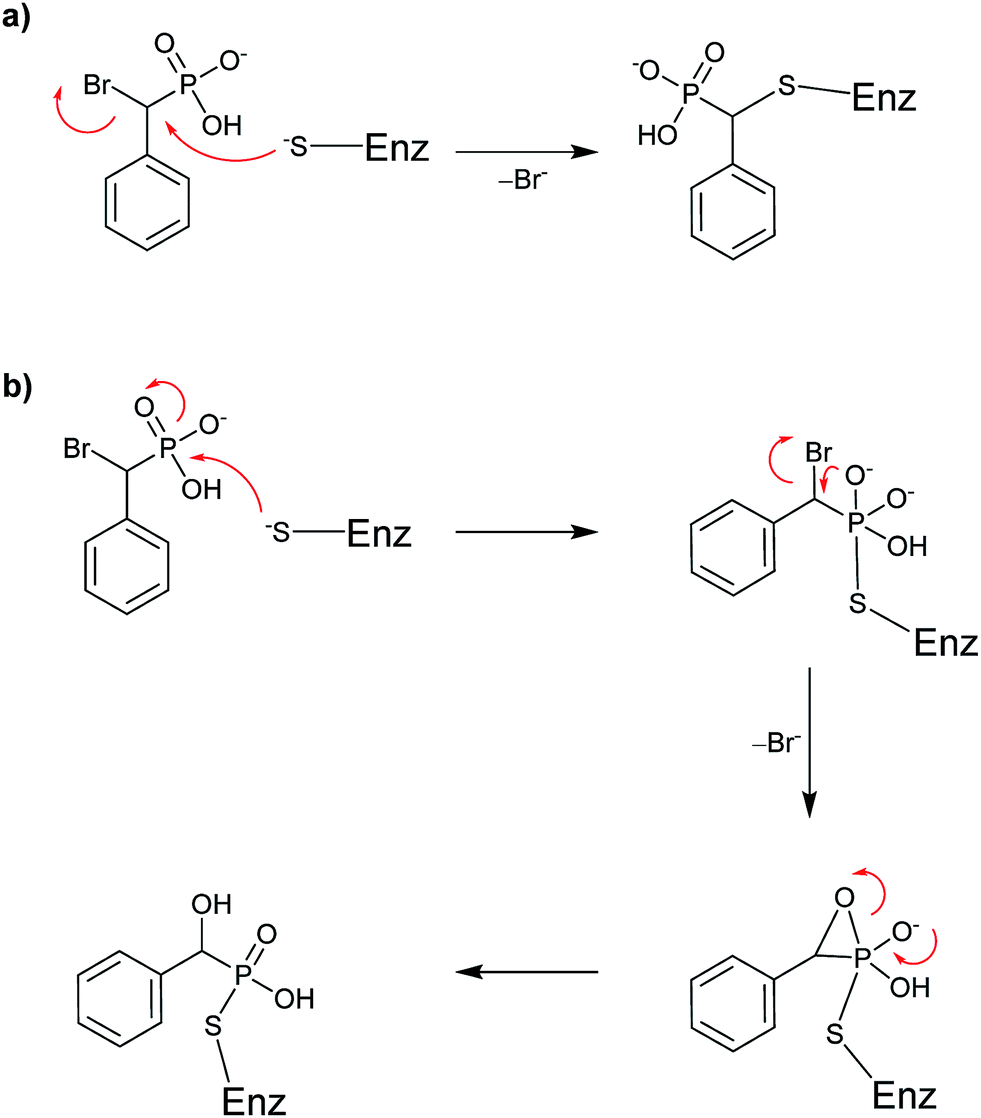 | ||
| Scheme 4 Proposed mechanisms for covalent inhibition of a PTP by BBP: (a) SN2 displacement of bromide or (b) intramolecular formation of a three-membered oxaphosphetane-like intermediate. | ||
Taylor et al. concluded that BBP 4 was an active site-directed covalent inhibitor of Yop51 at low mM concentration.38 The absence of activity for probe 4 with alkaline phosphatase confirmed its specificity for PTPs. Zhang and co-workers expanded on this finding by constructing BBP analog 5,39 which was proposed as an activity-based proteomic probe (ABPP) to covalently tag phosphatase enzymes.44,45 Analog 5 built on the original BBP design by including an affinity tag, biotin, linked through the para-position of the aromatic ring. This modification enabled streptavidin to be used for detection of bioconjugates formed in the reaction of 5 with its targets. Testing of analog 5 as an irreversible inhibitor of YopH, another PTP from Yersinia, showed that its activity was comparable to 4 (Table 3). Compound 5 followed a similar inactivation time course to 4; however, it displayed 5-fold improved specificity for the enzyme.39
| Analog | K I (μM) | k i (min−1) |
|---|---|---|
| 4 | 4100 | 0.11 |
| 5 | 740 | 0.17 |
| 9 & 10 | 690 | 0.52 |
Importantly, when probe 5 was introduced into a mixture PTPs (YopH, PTP1b, HePTP, SHP2, FAP-1, PTPα, DEP-1, VHR, Cdc14 and PRL-3) at 1 mM concentration, covalent adducts were formed with all enzymes. Specificity of the reagent toward PTPs was confirmed by comparison to other phosphatase enzymes (alkaline, potato, prostatic acid, DSP and λ phosphatases) in vitro as well and among the milieu of cellular proteins found in the cell lysate of E. coli.39 These studies were the first to demonstrate that α-bromobenzylphosphonates could be used successfully for selective, irreversible inhibition of PTPs as an enzyme class. More recently α-BBP analog 5 was used by Boivin et al. to identify PTPs and monitor platelet-derived growth factor (PDGF) receptor signaling in an angiomyolipoma cancer cell model.46 It is worth noting that these studies also raise the concern that, while extremely active, the α-BBP functional group alone does not provide specificity among different PTPs.39
Fluorescent tags (9 and 10, Fig. 3) have been incorporated into α-BBP derivatives to detect PTPs in breast, lung, liver, ovarian and cervical cancer cell lines. The cell lines could be specifically labeled using these fluorescent probes at a concentration of 1 mM. These rhodamine-based regioisomers of α-BBP were isolated as a mixture and were not separated (Fig. 3). These analogs offered a slight improvement in KI over analog 5 (Table 3), but provided 1000-fold more sensitivity for detection of labeled enzyme.47
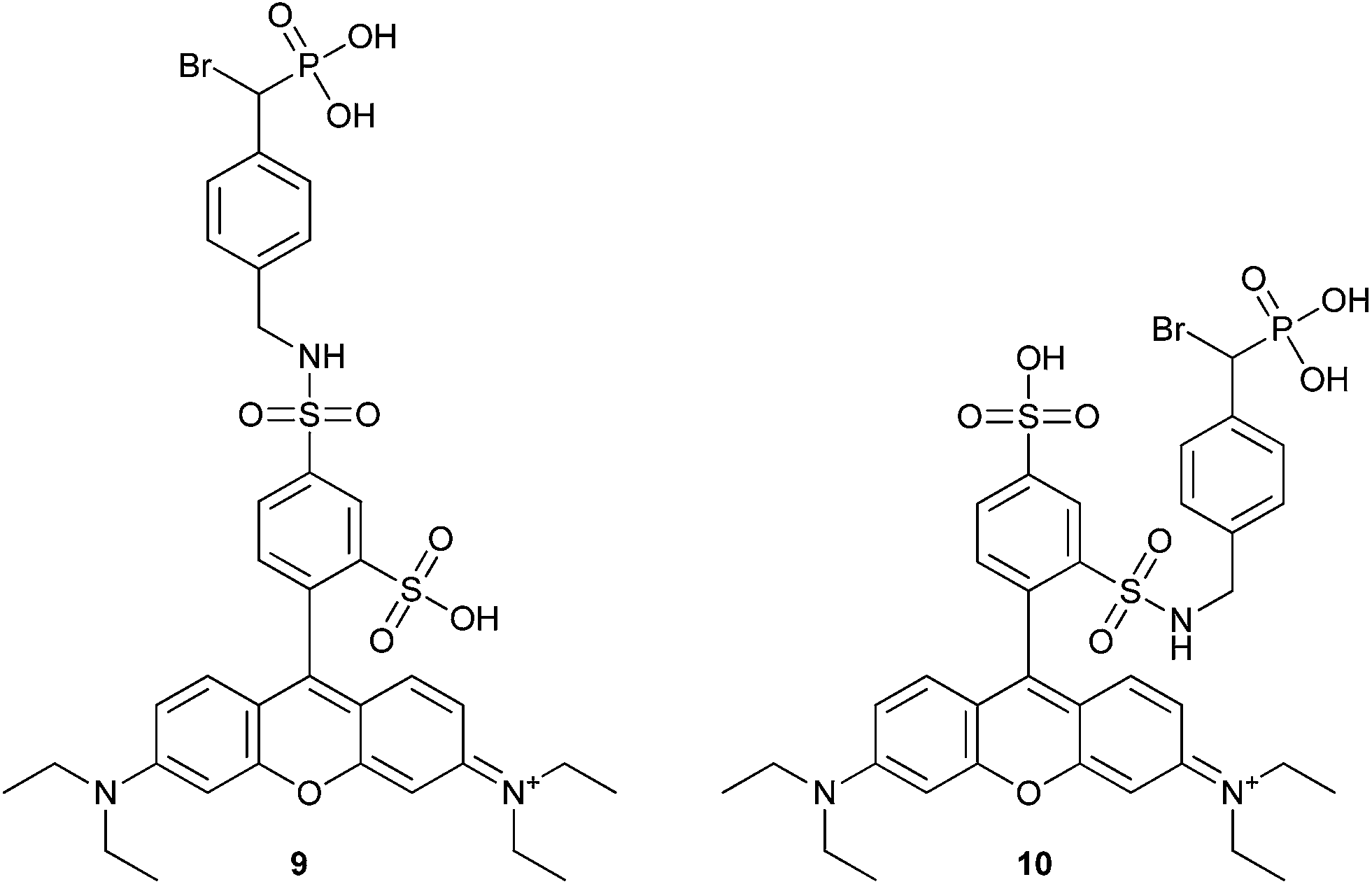 | ||
| Fig. 3 The structure of fluorescent rhodamine-based α-bromophosphonate regioisomers 9 and 10.47 | ||
More recently Tulsi et al. developed an improved strategy for the design of BBP-based inhibitors to address the problem of selectivity among PTP enzymes. They approached this issue by developing a method to incorporate α-BBP-based analogs into peptides, offering the possibility of increased specificity for individual PTP enzymes and modular synthesis of new inhibitors. The analog was synthesized as a protected phosphotyrosine analog incorporating the α-BBP functional group, L-bromophosphonomethylphenylalanine (BrPmp, 6). BrPmp was the first attempt to incorporate BBP into an amino acid derivative. BrPmp was synthesized from L-tyrosine in 14 steps and 22% overall yield. It was then incorporated into the tripeptide sequence, Asp-BrPmp-Leu (11, Fig. 4) using solid phase peptide synthesis. This sequence was selected as an example of a short sequence that contained an acidic residue N-terminal to the phosphotyrosine site, and a hydrophobic residue at the C-terminal side—both of which are features commonly found in phosphopeptide substrates of the human PTP enzyme, CD45. While BBP 4 was only a weak inhibitor, BrPmp inhibited CD45 both as an individual amino acid and as part of tripeptide analog 11.40
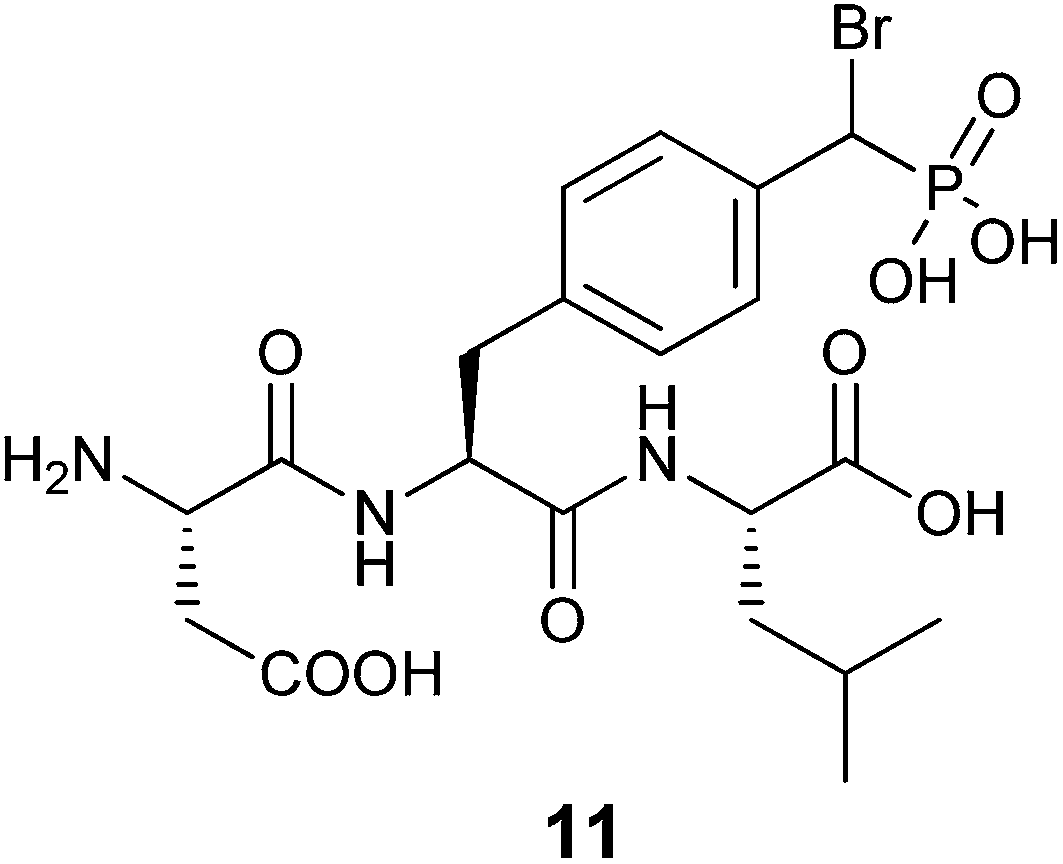 | ||
| Fig. 4 Synthetic tripeptide incorporating BrPmp shown to have specificity for immune PTP, CD45.40 | ||
Tulsi et al. confirmed that 6 was indeed a covalent inhibitor of CD45 using a Kitz–Wilson analysis with a similar rate of inactivation to that of tripeptide 11. Importantly, they found that BrPmp in the context of the tripeptide had 4-fold increased specificity for the enzyme over BrPmp alone (Table 4).40 These results strongly suggest that an appropriate BrPmp peptide sequence could be used to target an individual PTP enzyme with high potency. The increased specificity of BrPmp in a peptide sequence provides support for this strategy as a means to design new inhibitors or probes that target individual PTPs.
| Analog | K I (μM) | k i (min−1) |
|---|---|---|
| 6 | 40 ± 8 | 0.041 ± 0.001 |
| 11 | 16 ± 4 | 0.048 ± 0.003 |
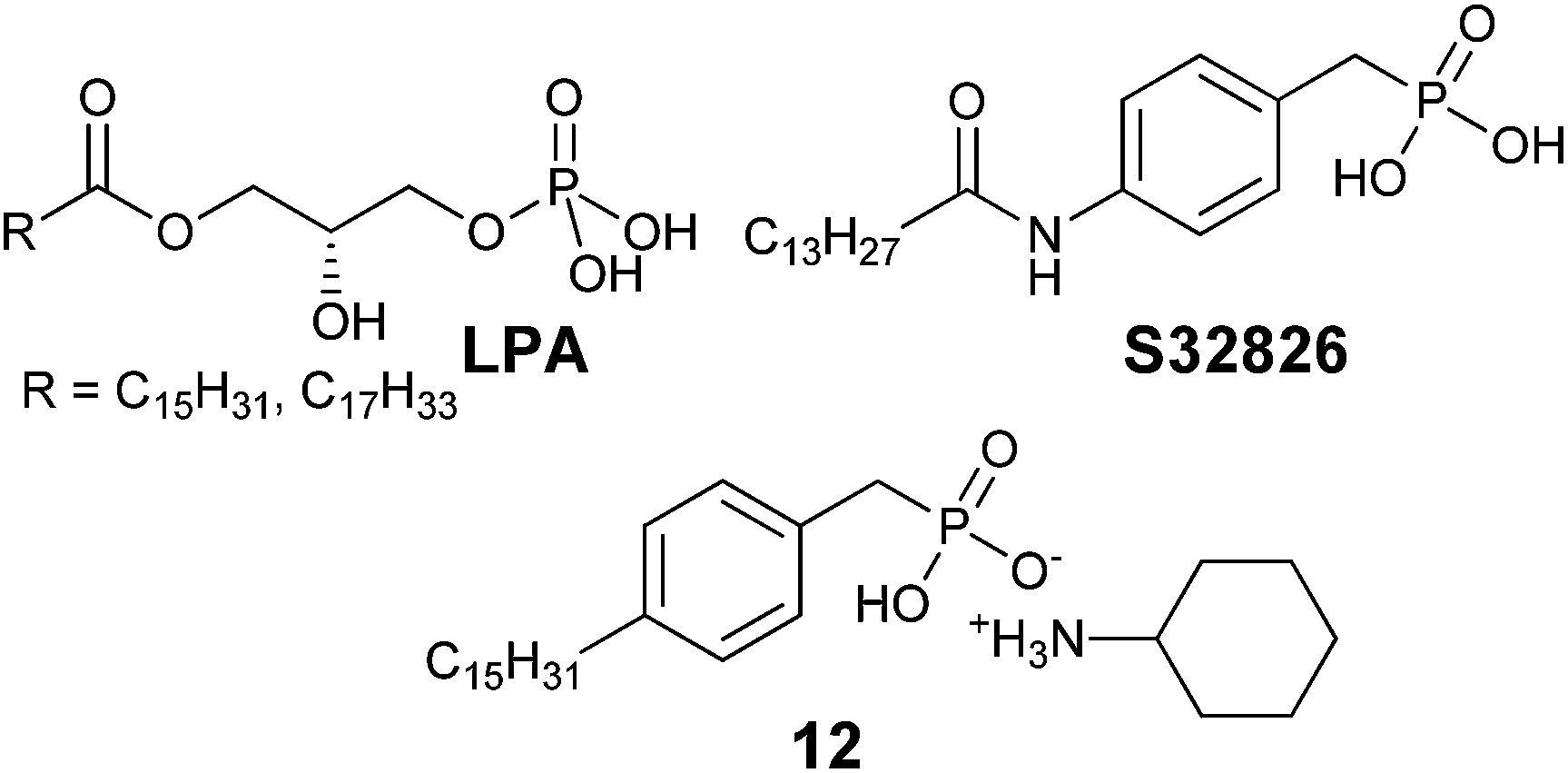 | ||
| Fig. 5 LPA and related synthetic analogs. | ||
In 2008, ATX inhibitor S32826 (Fig. 5) was identified as the first nanomolar inhibitor of ATX (KI = 9.0 nM)42 by high-throughput screening; however, it showed no inhibitory activity in vitro or in vivo.41 Gupte et al.41 postulated that the reason for this lack of activity was due to cleavage of the amide bond by other enzymes. The authors synthesized a library of S32826-based analogs, including α-bromophosphocholine 7, which offered the advantage of a non-hydrolyzable aliphatic chain para to the alkyl chain. Compound 7 demonstrated a mixed mode of inhibition for ATX (IC50 = 10.1 μM) with a KI value of 6.10 μM and K′I value of 2.97 μM.51 Compound 7 was a much better inhibitor of ATX than the α-chloro derivative and was similar to the α-fluoro analog of the same scaffold. Interestingly, the analogous fully saturated phosphonate 12 (Fig. 5) was a much more potent inhibitor of ATX (IC50 = 0.17 μm, KI = 0.27 μm, and K′I = 0.28 μm) and was therefore tested more rigorously. Analog 12 showed significantly (p < 0.05) decreased LPA-dependent MM1 heptocarcinoma cell invasion of human umbilical vein endothelial cells (HUVEC), at a concentration of 3 μM. The authors observed that compound 12 exhibited anti-metastatic properties in B16-F10 melanoma cells, with a reduction in metastasis (p < 0.05) at concentrations of 0.5 mg kg−1 day−1 for 21 days.41
The Prestwich group tested an α-BBP derivative of S32826, compound 8. They argued that the lack of in vitro and in vivo activity was due to the low solubility of S32826. As a result, they opted to preserve the amide functionality and reduce the aliphatic chain length found in S32826. One resulting compound, α-bromophosphonate 8, was found to inhibit ATX with nearly a 1000-fold improvement in potency (KI = 8.1 nM) over compound 7. This activity was also a 20-fold improvement over compound 12 and offered slight improvement over S32826.42 These data suggest that the amide bond is crucial to potency toward ATX. We speculate that the limited activity of 12 and S32826 in vitro or in vivo could also be due to the reduced stability of these compounds, as hypothesized by Gupte et al.41
Although there are an appreciable number of early synthetic examples that incorporate the α-BBP functionality, interest in their application to inhibit phosphate recognizing proteins was not explored until the mid-nineties. We believe that the examples discussed above highlight the untapped potential of benzylic α-bromophosphonates as irreversible inhibitors and probes of enzyme activity.
2.2. Aliphatic α-bromophosphonates
There are far fewer synthetic examples of aliphatic α-bromophosphonates relative to α-benzylbromophosphonates; however, several biologically active analogs have been identified and tested. We summarize the synthesis and biological properties of these below.An early example of an aliphatic α-bromophosphonate synthesis was reported by Teulade and Savignac, where they synthesized a series of five aliphatic α-bromophosphonates. The synthesis of propyl α-bromophosphonate derivative 16 is shown in Scheme 5. Compound 14 was generated via silylation of a phosphorus-stabilized carbanion intermediate and subsequent alkylation in one pot using two equivalents of n-BuLi, TMSCl, and ethyl iodide. Subsequent treatment of analog 14 with n-BuLi in the presence of 1,2-dibromoethane resulted in halogen–lithium exchange of the chlorine atom (15). In a second one-pot procedure, the α-bromo-α-silylphosphonate was converted to the final α-bromo product by removal of the TMS group with sodium ethoxide in ethanol to furnish 16 in 78% yield as a diastereomeric mixture.52
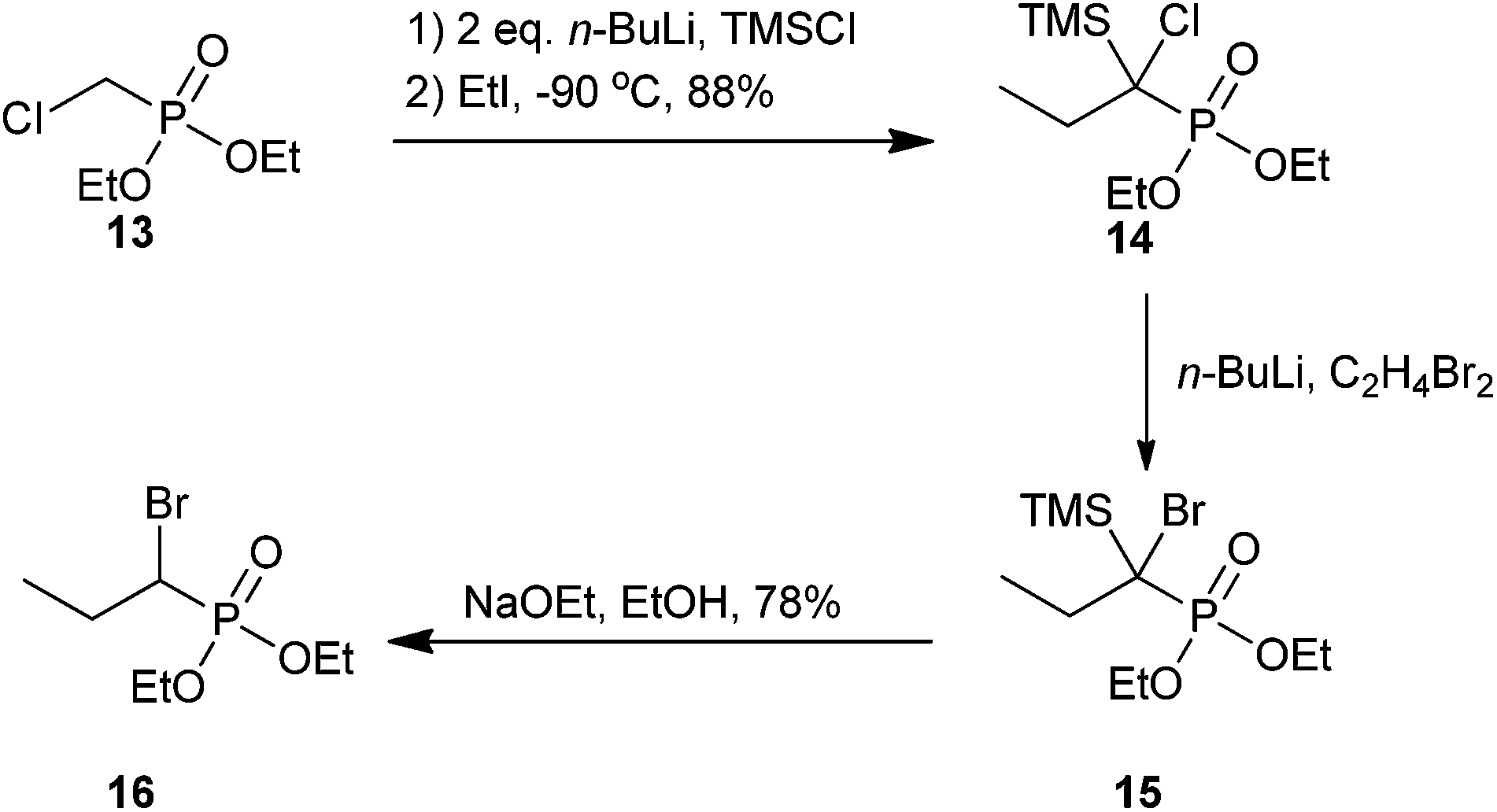 | ||
| Scheme 5 The synthesis of an aliphatic α-bromophosphonate derivative.52 | ||
Savignac and co-workers later reported an improvement to this aliphatic bromination strategy that avoided the need for α-chlorophosphonate 13 (Scheme 6).53 Instead, they utilized the Michaelis–Arbuzov reaction by treatment of triethylphosphite with an alkyl halide under reflux to obtain the starting alkyl phosphonate.54 A number of alkyl phosphonates were synthesized using this method; however, Scheme 6 only depicts the synthesis of a single representative ethyl adduct, 20. Treatment of compound 17 with 2 equivalents of lithium diisopropyl amide (LDA) to generate a phosphorus-stabilized carbanion, followed by dropwise addition of TMSCl, produced intermediate 18 which was confirmed through 31P NMR studies. Lithium–bromine exchange was performed using tetrachlorodibromoethane. Finally, intermediate 19 was desilylated using lithium ethoxide in ethanol to furnish α-bromophosphonate 20 as a diastereomeric mixture in nearly quantitative yield from 17.53 In 2007, the Fu group also synthesized 20 using this procedure, but reported a more modest yield of 70%.55
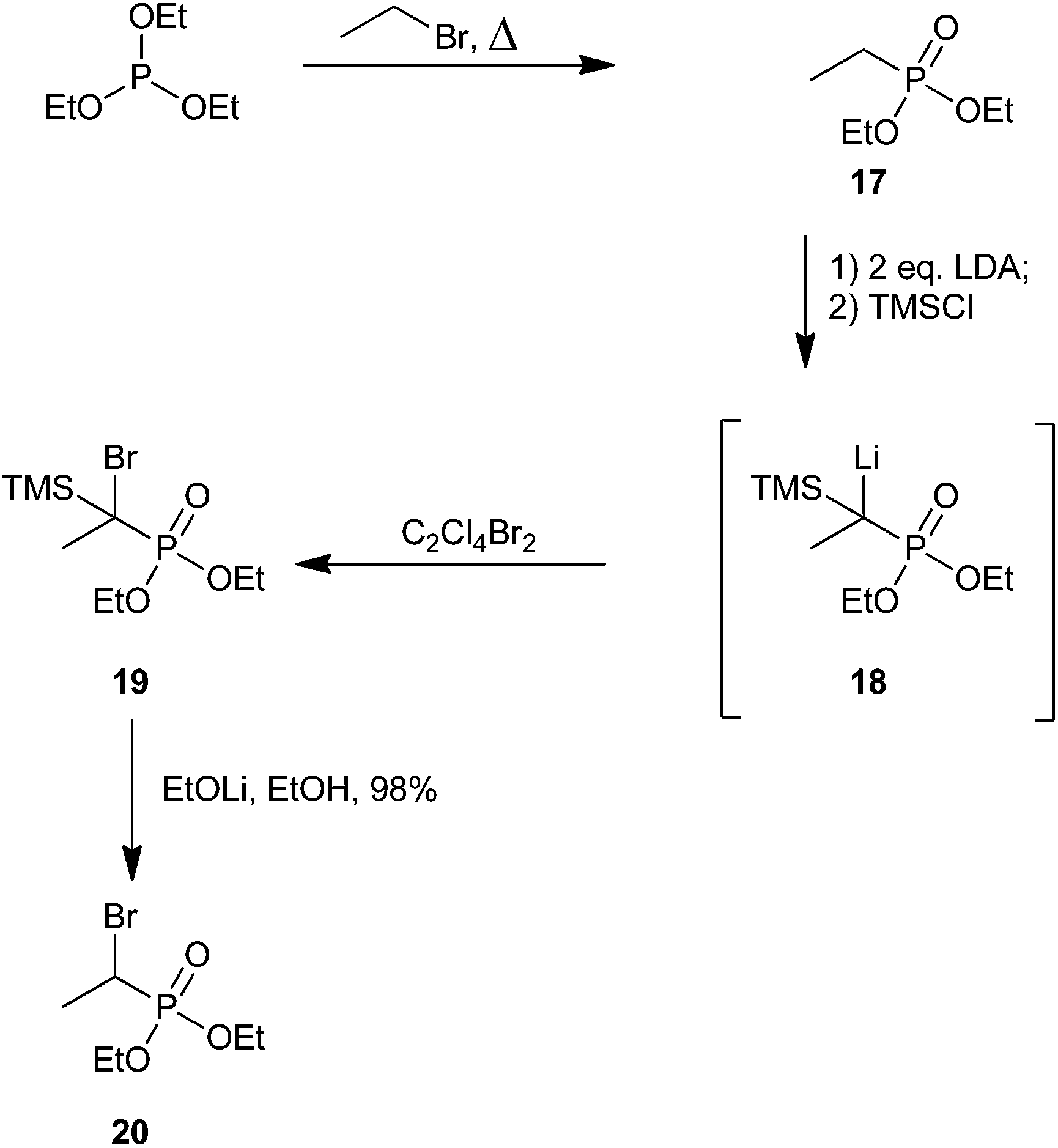 | ||
| Scheme 6 An improved synthetic strategy to alkyl α-bromophosphonates.53 | ||
To the best of our knowledge, the only other use of this methodology has been reported by Wnuk et al. Lithium–bromine exchange was used to convert saturated phosphonate 21 into α-bromo analog 22 as a precursor to radical cyclization (Scheme 7).56
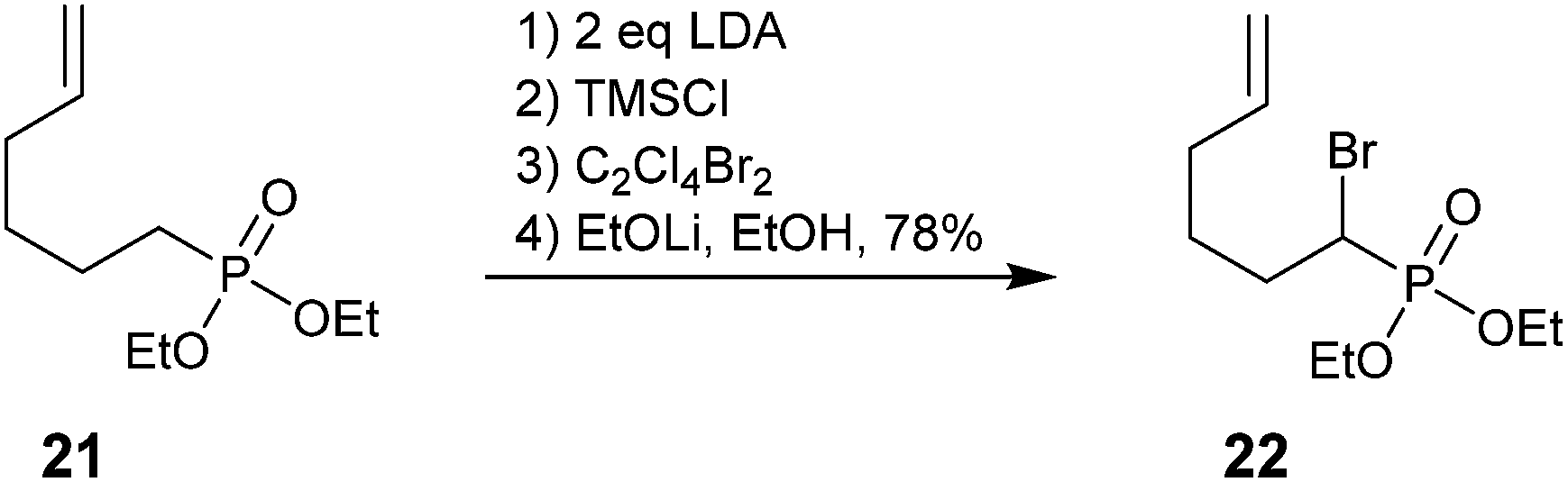 | ||
| Scheme 7 An α-bromophosphonate as a precursor for radical cyclization.56 | ||
Although methodology for obtaining aliphatic α-bromophosphonates via an α-hydroxy intermediate has not been explored as thoroughly as for the α-BBP moiety, it has been used with moderate to good yields. This conversion generally utilizes far milder conditions and provides the opportunity to obtain enantiopure products, which has not yet been achieved via lithium–bromine exchange.
Fu and co-workers conducted a methodological study on this transformation by bromination of a library of α-hydroxyphosphonates using mild conditions. Scheme 8 shows an example using an alkyl phosphonate. α-Hydroxypropylphosphonate 23 was brominated to obtain 24 using either PPh3/Br2 and pyridine or PPh3, DDQ and Bu4NBr.57 Mechanistic studies appear to rule out a radical pathway.35 Analog 24 was used as an intermediate for subsequent fluorination via a phosphorus-stabilized carbanion using N-fluorobenzenesulfonimide (NFSI). Although the authors discussed the potential importance of these α-bromo-α-fluorophosphonate analogs as inhibitors of PTPs, they did not report biological testing.57
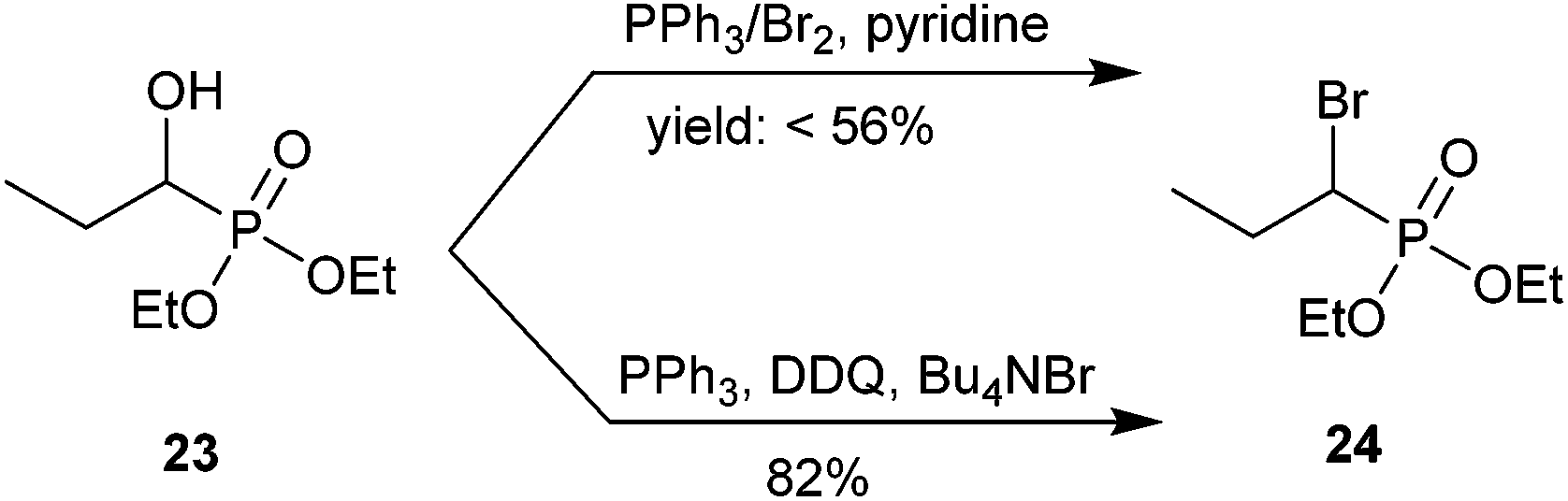 | ||
| Scheme 8 Electrophilic bromination of aliphatic α-bromophosphonates.57 | ||
We focus now on examples of aliphatic α-bromophosphonates that have been assayed as inhibitors of phosphate recognizing proteins.
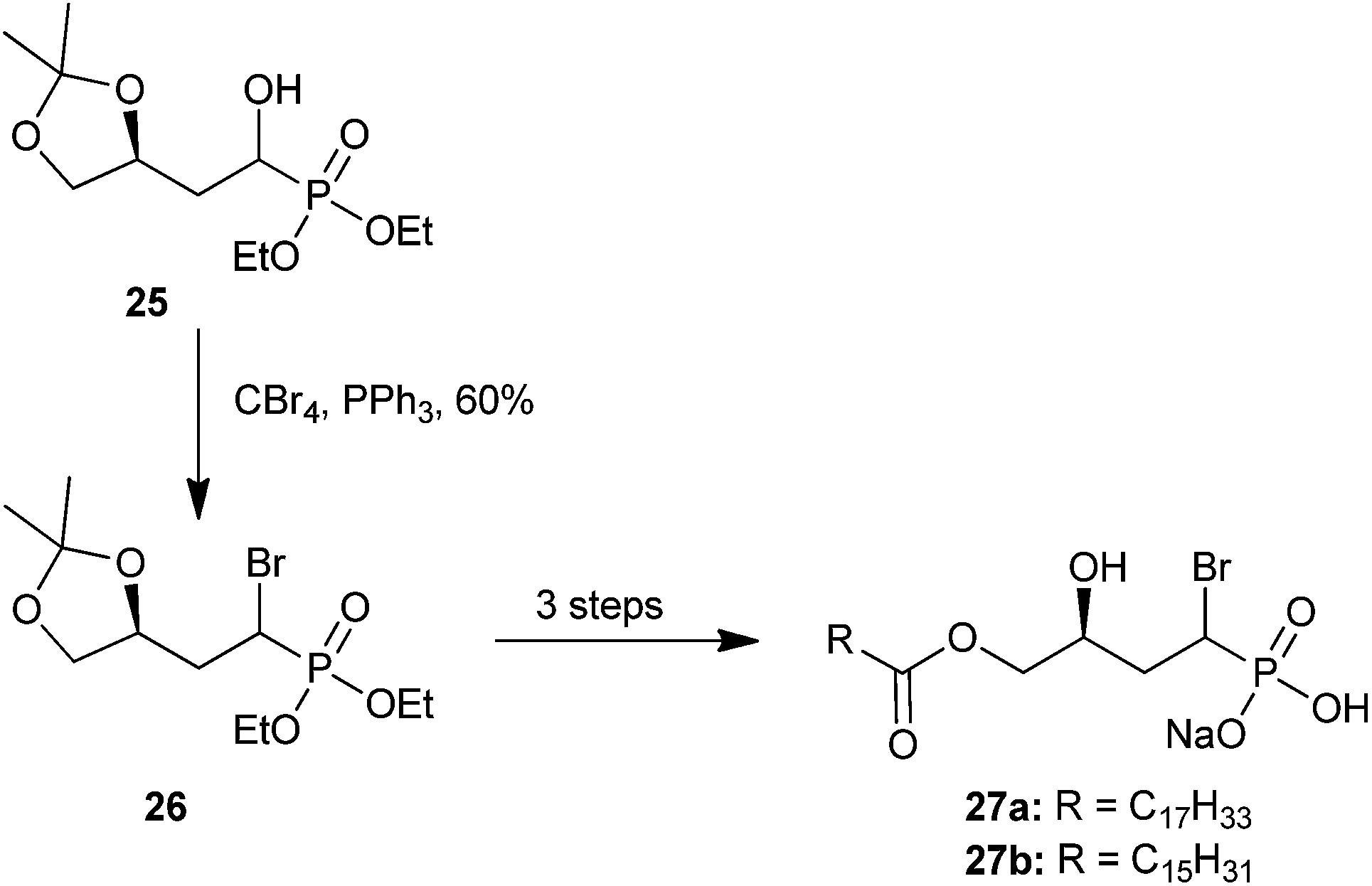 | ||
| Scheme 9 The synthesis of biologically active α-bromo analogs 27a and 27bvia α-hydroxy intermediate 25.50 | ||
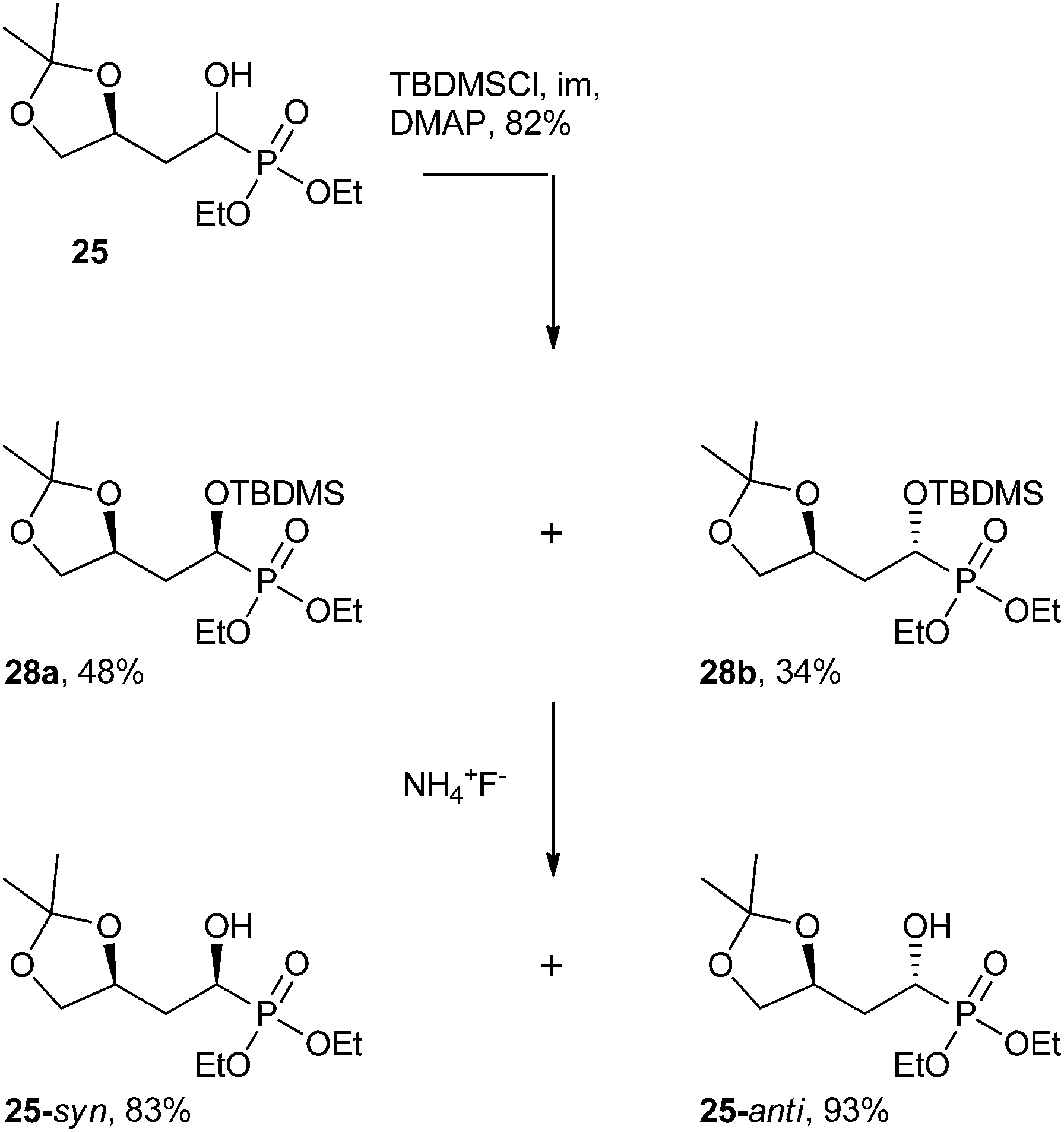 | ||
| Scheme 10 The separation of α-hydroxy derivative 25 into its two diastereomers.50 | ||
By treating analog 25 with tert-butyldimethylsilyl chloride (TBDMSCl) under basic conditions, the resulting bulky siloxane product could be separated using flash chromatography. The TBDMS group was then cleaved using ammonium fluoride (NH4F) to furnish the isolated α-hydroxy diastereomers (25-syn, 25-anti, Scheme 10).50
In a follow up study, Prestwich and co-workers tested the activity of diastereomeric α-bromo derivatives 27b-syn and 27b-anti in LPA–ATX signal transduction (Fig. 6). They termed the collective syn and anti isomers of analog 27b as Br–LPA. In initial tests of the syn and anti-isomers as separate compounds against LPA1–4 receptors, both were found to be inhibitors of the GPCRs on their own, as had been predicted using molecular modeling. When screened against ATX, the anti-isomer (IC50 = 22 nM) was determined to be a better inhibitor than the syn-isomer (IC50 = 165 nM); however, both isomers inhibited ATX in a dose-dependent manner. Furthermore, compounds 27b-syn, 27b-anti, and Br–LPA knocked out 98% of ATX activity at a concentration of 10 μM, but were only weakly active against platelet-activating LPA5.5
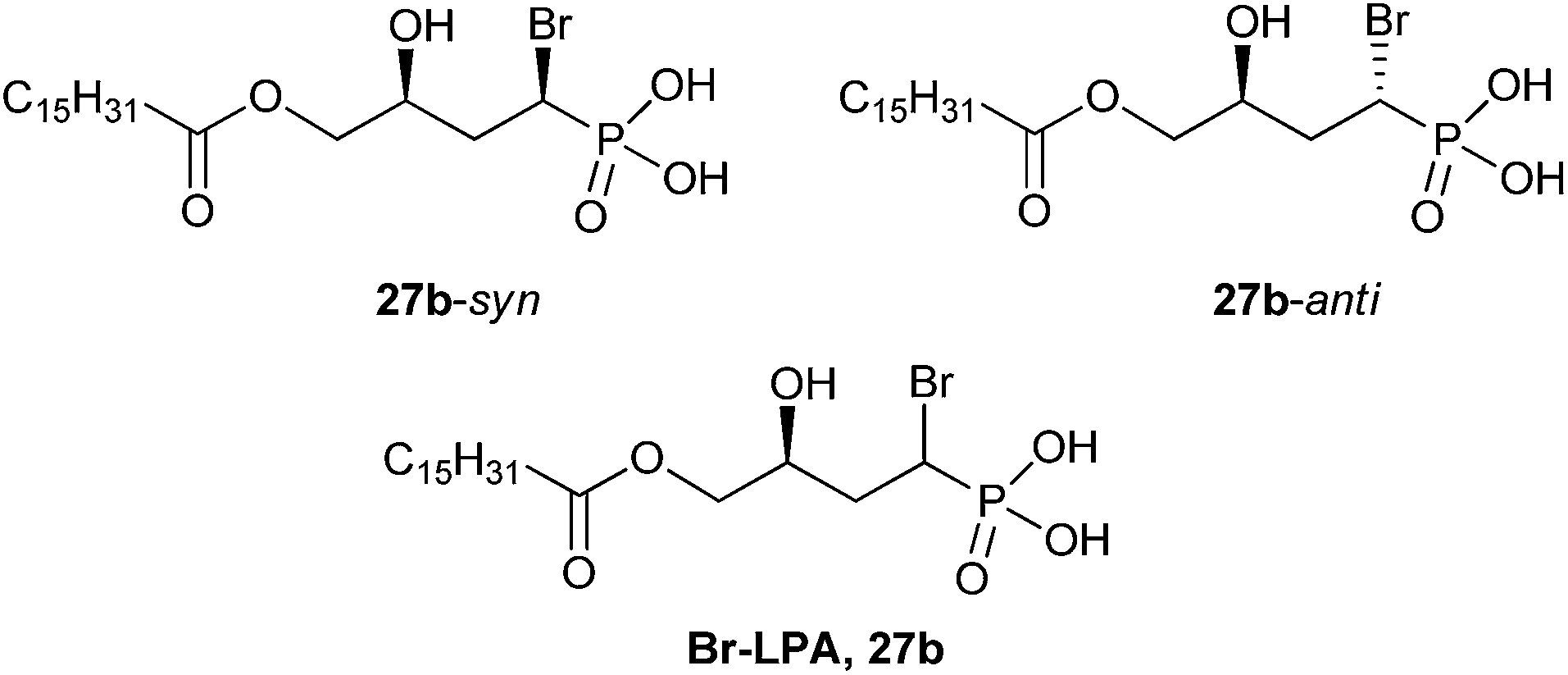 | ||
| Fig. 6 The structures of two LPA-based α-bromophosphonate diastereomers, 27b-syn and 27b-anti. A 1:1 mixture of 27b-syn and 27b-anti was termed Br–LPA (27b).5 | ||
Further exploration of Br–LPA in breast and lung cancers in vitro and in vivo have suggested these compounds may have therapeutic potential.58 Compounds 27b-syn and 27b-anti were tested as inhibitors of LPA-dependent cancer cell migration. Both analogs reduced migration of MDA-MB-231 breast cancer cells at a concentration of 40 μM, using a scratch wound assay. Both isomers also reduced MDA-MB-231 breast cancer cell invasion by ∼50%; interestingly, the potency of the two diastereomers were not significantly different.5 Follow-up studies have since found that Br–LPA can reduce tumor size in breast5 and lung59 tumor models in mice, and the compound had limited toxicity.
2.3. α-Bromophosphonates on a carbohydrate scaffold
Phosphonate-based mimics of carbohydrates are also well known in the literature.9 The first example of phosphonate synthesis in carbohydrates appeared over 50 years ago by Griffin and Burger when they synthesized glucose-6-phosphate analog, 29 (Fig. 7).61 Since that time, a range of carbohydrate phosphonate analogs have been targeted with general access available at all positions of pyranosides and furanosides (e.g.30).9,62,63 Work to date towards glycosyl α-halophosphonate analogs has focused on the introduction of α-fluoro and α,α-difluorophosphonates at various positions of O- and C-glycosides (e.g.31).12,64 Important advances in the synthesis of both α-fluorinated and α,α-difluorinated glucosyl-6-phosphonates have been described by the Berkowitz group (Scheme 11). By treating α-hydroxyphosphonate 32 with diethylaminosulfur trifluoride (DAST) α-fluorophosphonate 33 was obtained in moderate yield, which could be resolved as a diastereomeric mixture by column chromatography.11 To obtain the related α,α-difluorophosphonate, Berkowitz and co-workers displaced triflate 34 using a phosphorus-stabilized carbanion, [diethylphosphono(difluoromethyl)]lithium, to provide α,α-difluorophosphonate 35 in good yield.11,65–67 These examples illustrate the accessibility of α-fluorophosphonates; however, until recently there was no corresponding precedent to the α-brominated phosphonates on a carbohydrate scaffold.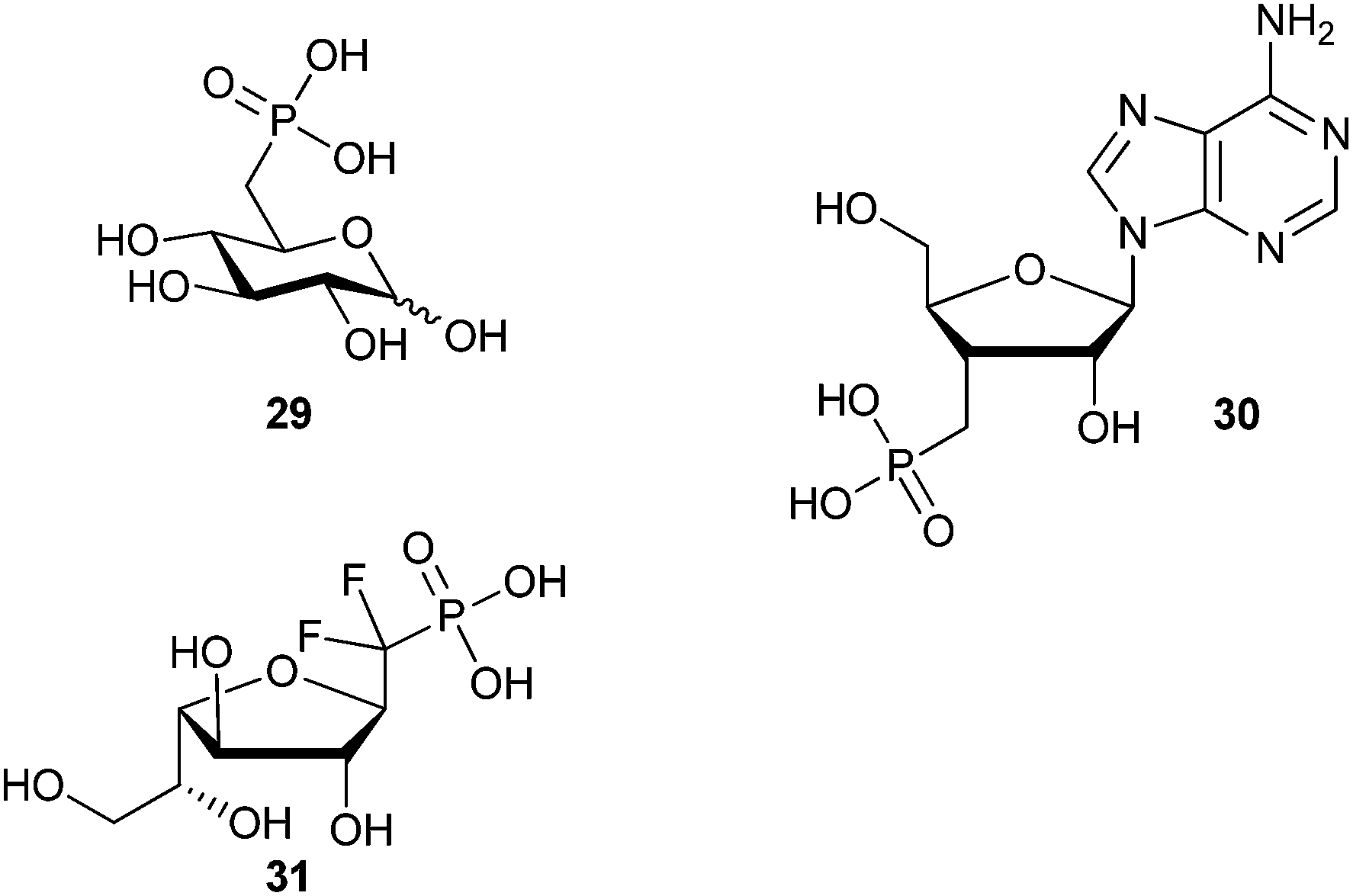 | ||
| Fig. 7 Examples of known carbohydrate-based phosphonates. | ||
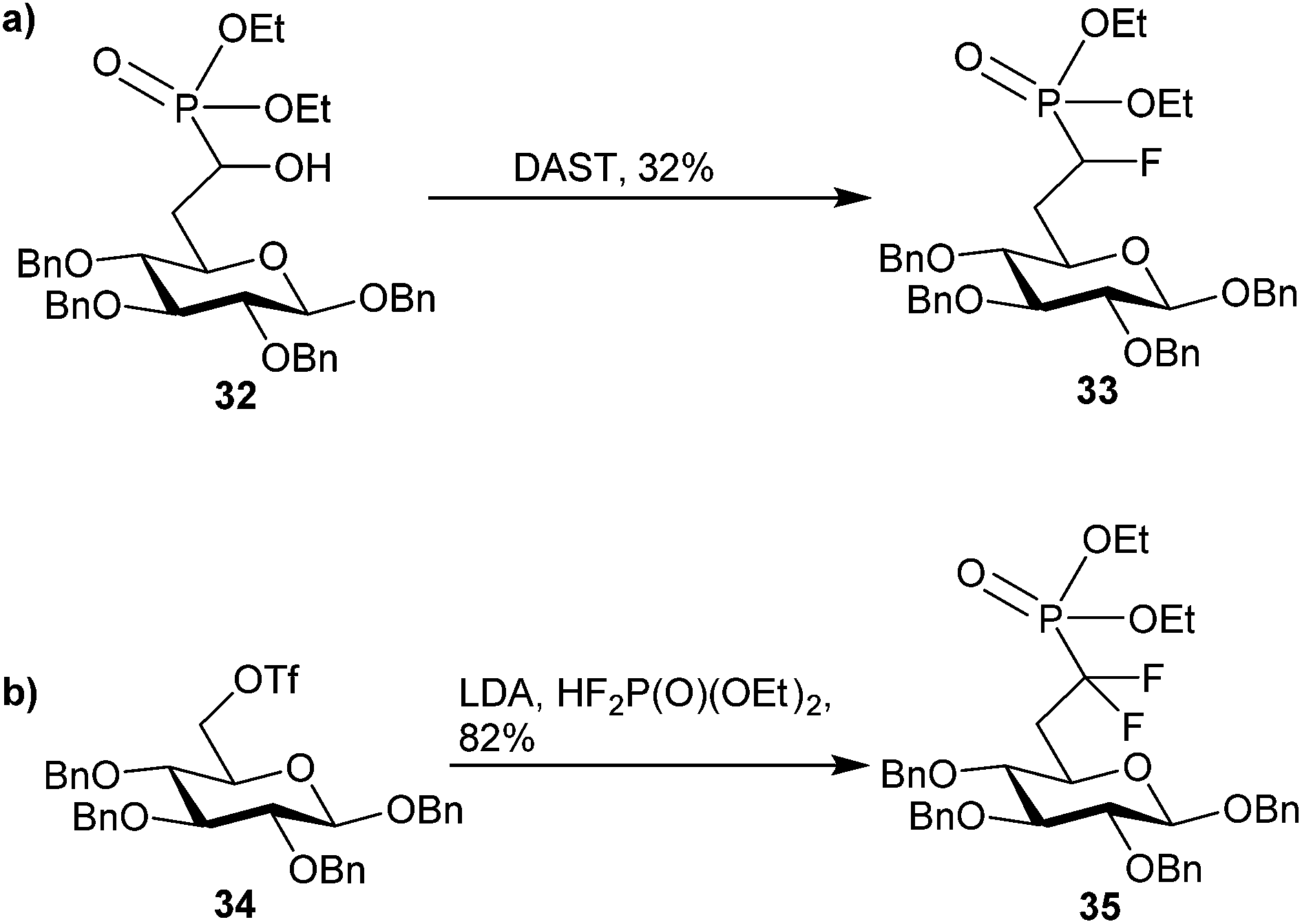 | ||
| Scheme 11 Known methods to accessing (a) α-fluoro and (b) α,α-difluorophosphonates on glucose scaffolds.11,65–67 | ||
We recently approached this problem by developing synthetic methods to access glucose-6-phosphate (G6P) derived α-bromophosphonates (36, Fig. 8) as inhibitors of glucose-6-phosphatase (G6Pase).68 Specific inhibitors of G6Pase are of interest to both medicinal and biochemists69 as G6Pase activity is upregulated in type II diabetes and no crystal structures of human G6Pase are available.7,70 G6Pase is known to contain a nucleophilic histidine and G6Pase analog 37 was hypothesized to be a potent or even irreversible inhibitor of the enzyme.68
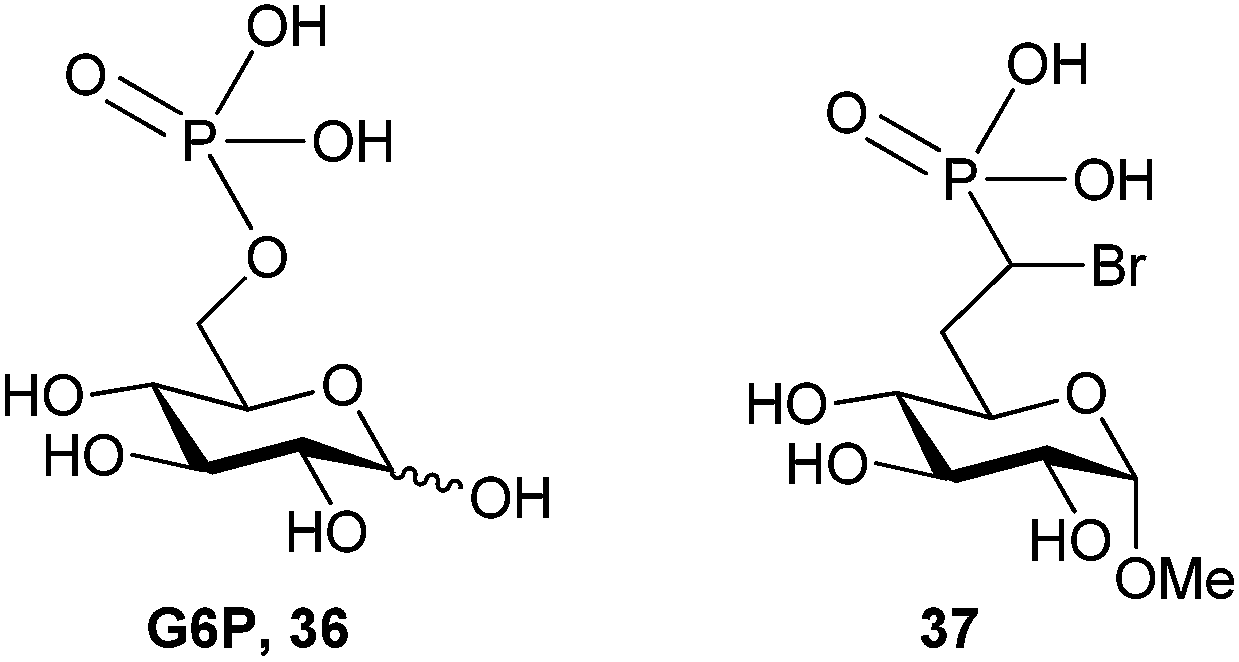 | ||
| Fig. 8 Structures of G6P (36) and targeted G6P analog 37.68 | ||
Initial access to compound 37 was attempted via reduction of an α-bromo-α,β-unsaturated phosphonate intermediate (38, Scheme 12) using standard hydrogenation conditions. This failed due to concomitant hydrogenolysis of the C–Br bond;68 however, the α-bromo-α,β-unsaturated phosphonate scaffold has been shown to be biologically active in other systems71 and was unprecedented in carbohydrates.
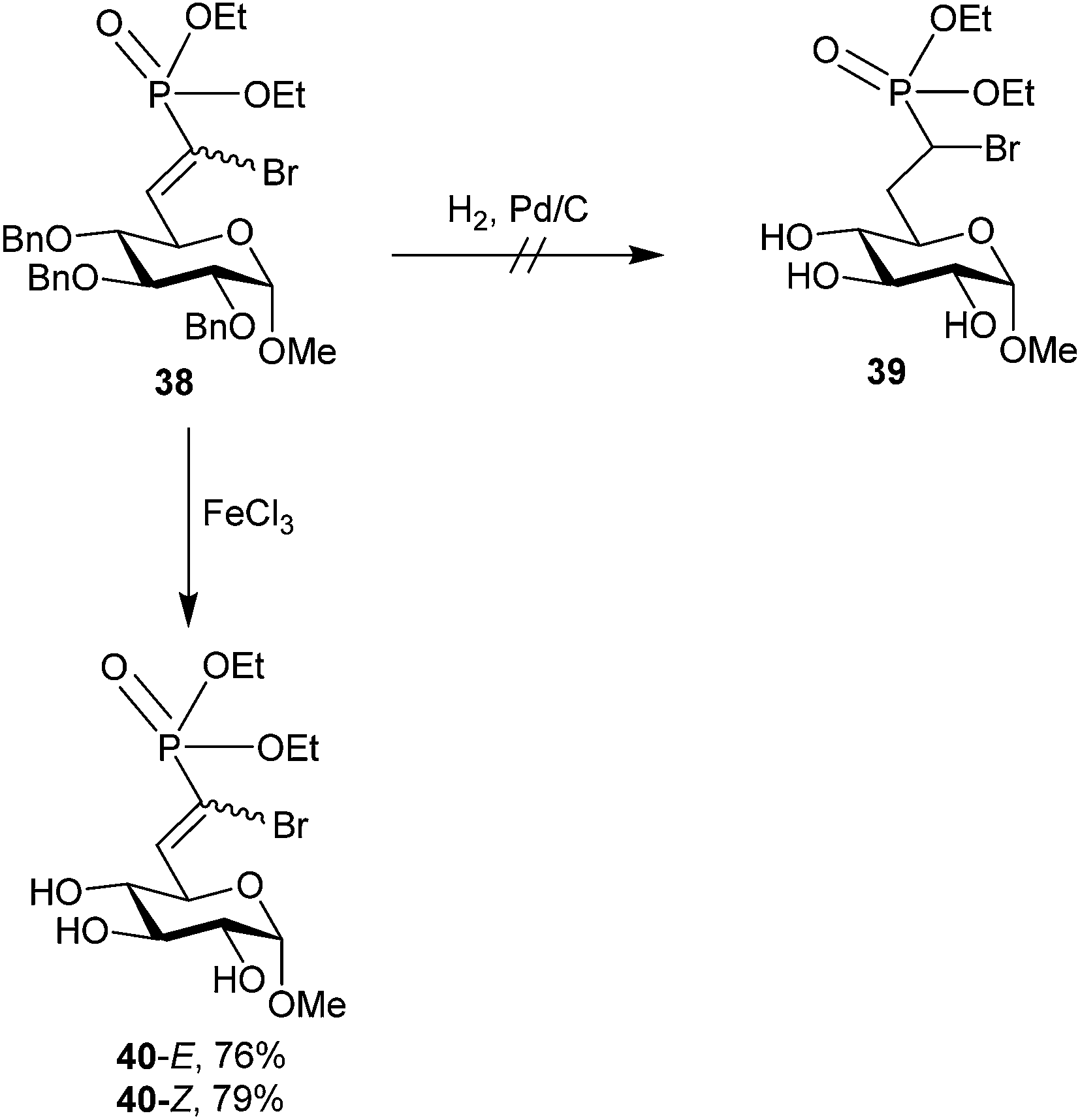 | ||
| Scheme 12 Attempted access to α-bromo G6P analog 37via standard olefinic reduction. Biologically active E and Z isomers of α-bromo-α,β-unsaturated phosphonate 40 could be isolated via debenzylation using FeCl3.68 | ||
Access to compound 37 was ultimately gained through a phosphorus-stabilized carbanion generated by n-BuLi that was quenched with Br2 at −98 °C (Scheme 13). Deprotection of the 2-, 3-, and 4-OH groups and the phosphonate ester furnished target compound 37 in an additional two steps (not shown). Interestingly, the unique α,α-dibromophosphonate analog 43 could be isolated by subsequent treatment of the α-bromo analog 42 with LDA and quenching with Br2 (Scheme 13). Similar methodology to provide an α,α-dibromophosphonate is known, but not on a carbohydrate scaffold.72
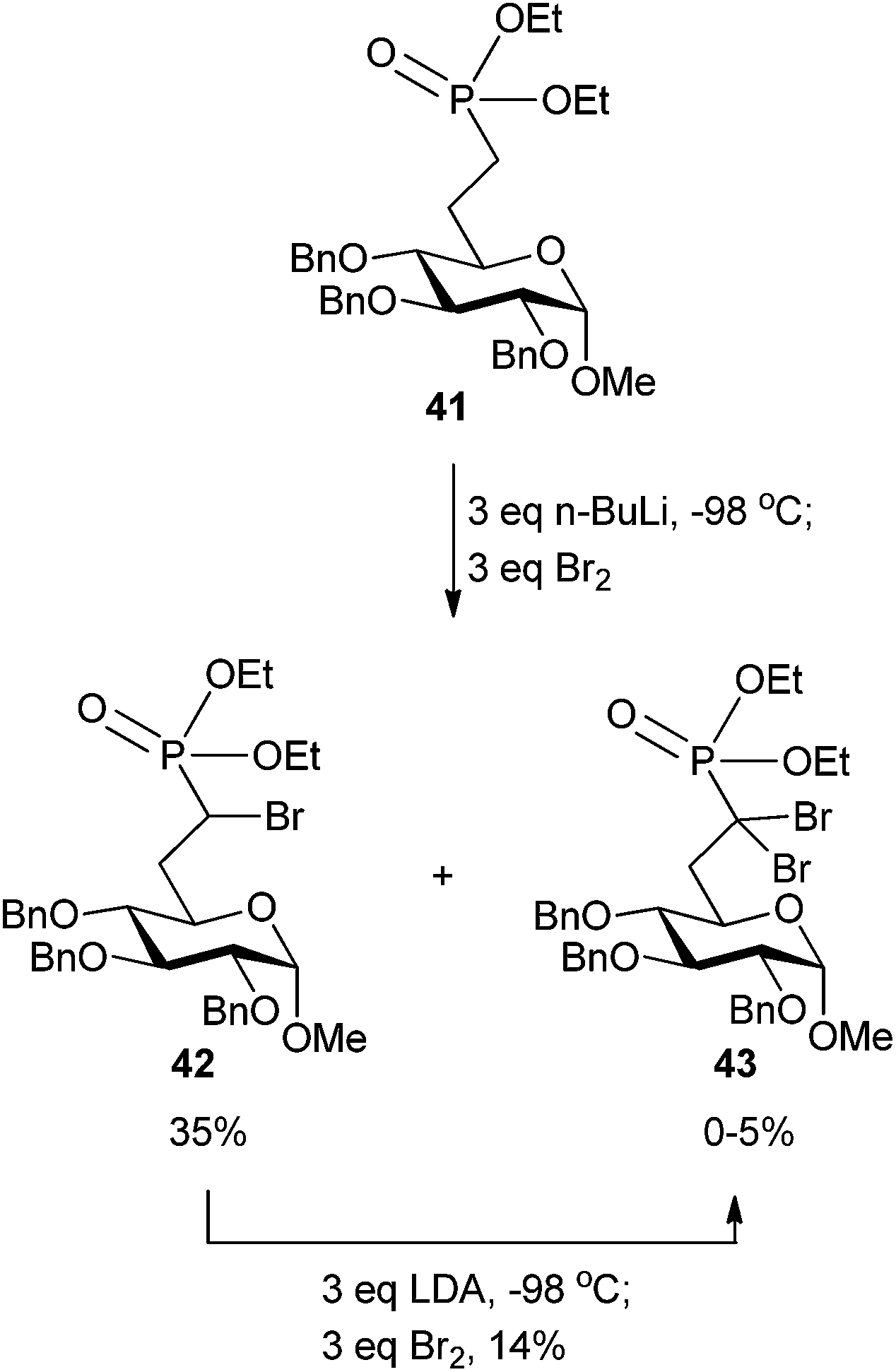 | ||
| Scheme 13 Partial synthesis of α-brominated phosphonate 37 with access to the α,α-dibromophosphonate 43.68 | ||
Screening of target compound 37 displayed greater potency for G6Pase than the native G6P substrate in Wister rat liver microsomes; however, a 60 h incubation was required to achieve inhibition. The phosphonate ester 44 (Fig. 9) also displayed potency that exceeded the Km of G6P, but with an incubation time of only 1 h. The α,α-dibromo analog 45 also exhibited similar potency under the same conditions, indicating that α,α-dibromophosphonates are worthy of exploration as inhibitors of phosphate recognizing enzymes. Of note, the stereoisomers of α-bromo-α,β-unsaturated phosphonate 40 also had similar potency against G6Pase.
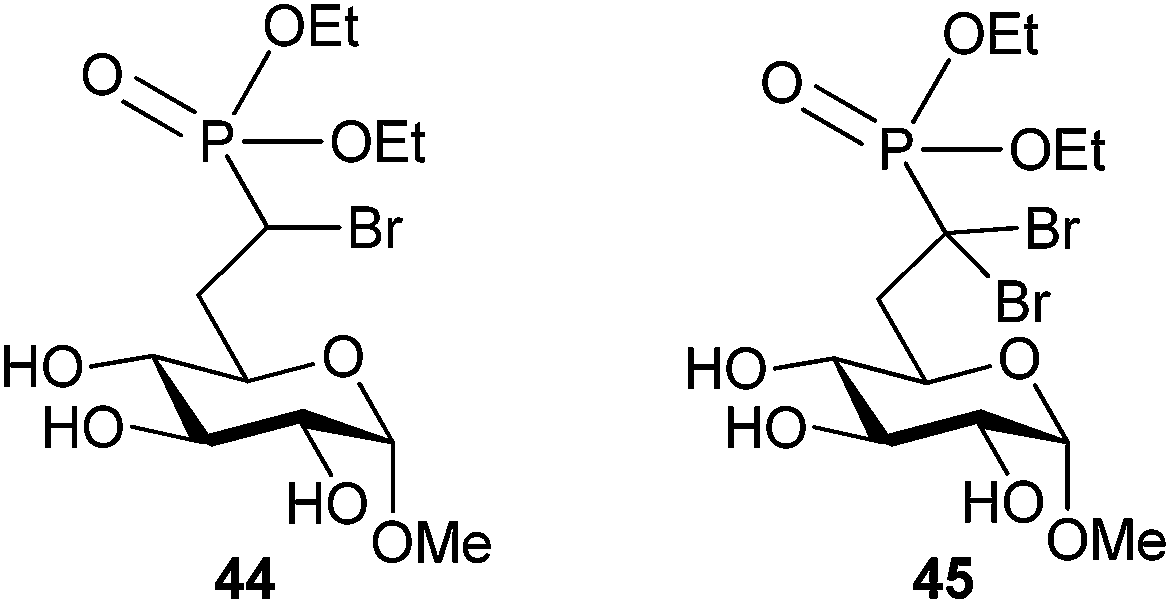 | ||
| Fig. 9 Phosphonate ester-based inhibitors of G6Pase.68 | ||
To explore the activity of compound 44, 31P NMR was used to confirm that the phosphonate ester could be hydrolyzed to compound 37 when incubated for 12 hours in rat liver microsomes. This finding suggested that inhibition may be a result of phosphonic acid analog 37 after cleavage by a native esterase. There was no evidence of irreversible inhibition of G6Pase by any of the α-bromophosphonates screened, and future work will be required to confirm the mode of inhibition.68
2.4. α-Bromo-α,β-unsaturated phosphonates
The α-bromo-α,β-unsaturated phosphonate functional group of compound 40 generated active inhibitors of G6Pase.68 This novel functional group has been tested for biological activity in only one other instance. Fosmidomycin (46, Fig. 10) has shown promise in clinical trials for malaria by targeting the mevalonate-independent isoprenoid biosynthesis pathway in this bacterium.71,73,74 Fosmidomycin inactivates the essential 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) enzyme in this pathway (IC50 = 35 nM in recombinant E. coli DXR). Devreux et al. synthesized a small library of α-arylphosphonates (not shown) using either Stille or Suzuki coupling via fully protected α-bromophosphonate 47. The results of assays with the α-functionalized phosphonates against recombinant E. coli DXR found that only α-bromophosphonate 47 had a sub-micromolar IC50 (450 nM).71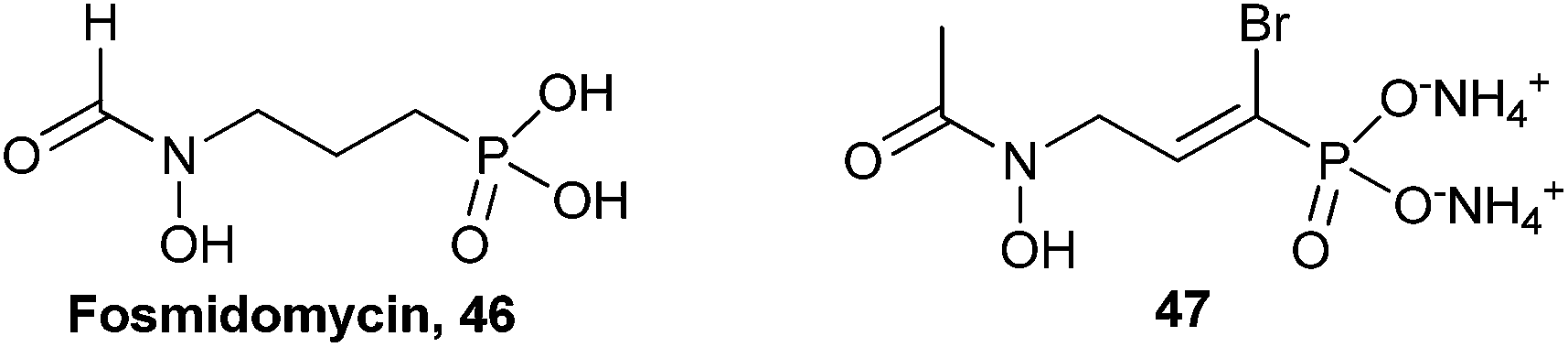 | ||
| Fig. 10 Structures of fosmidomycin (46) and α-bromo-α,β-unsaturated phosphonate 47 that was screened against recombinant E. coli DXR.71 | ||
2.5. α-Bromophosphonocarboxylates and α-bromobisphosphonates
Phosphonocarboxylates and bisphosphonates have a host of medicinally important effects ranging from the prevention of bone resorption to cancer chemotherapy as bioisosteres of pyrophosphate.75,76 Bisphosphonates are well known to have affinity for bone.76 The reactivity of both bisphosphonate and phosphonocarboxylate analogs is dramatically increased over their monophosphonate counterparts. Additionally, both mono- and dibrominated bisphosphonates have been reported. We briefly highlight synthetic access to α-bromophosphonocarboxylates and α-bromobisphosphonates and offer a discussion of their applications in medicinal chemistry.Synthetic efforts by McKenna and co-workers have demonstrated di- or mono-bromination of triethyl phosphonoacetate (48), in one or two steps, respectively (Scheme 14). Treatment of 48 with NaOBr provided triethyl dibromophosphonoacetate (49) in good yield, and the product could be reduced to the mono-brominated analog 50 in the presence of 0.96 equiv. of SnCl2 if desired.77 Several researchers have utilized this methodology to obtain α-bromo and α,α-dibromophosphonoacetates on similar scaffolds with moderate to excellent yields.78–86 Examples of access to brominated bisphosphonates using similar conditions are available.87–89
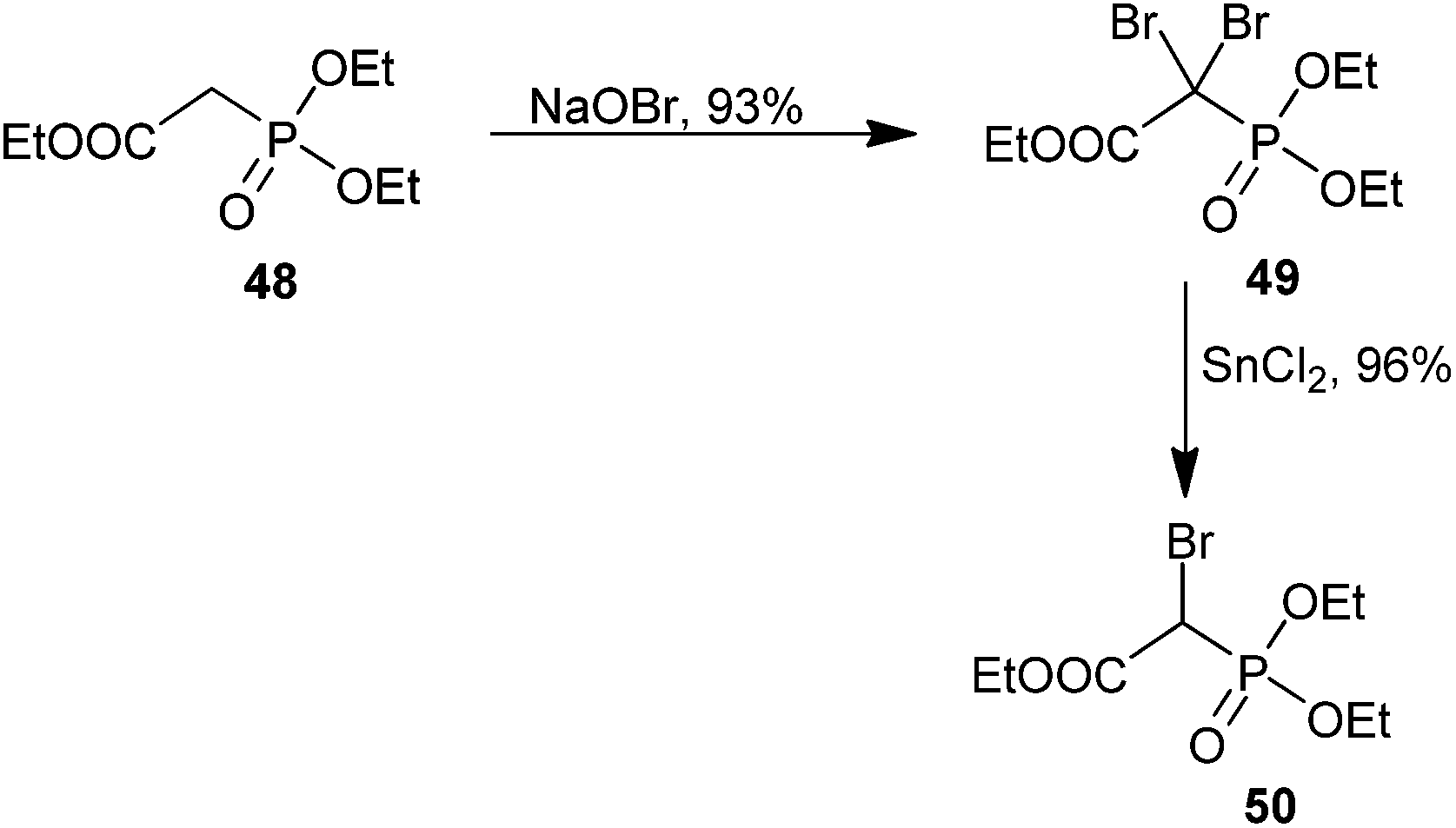 | ||
| Scheme 14 Access to α-bromo and α,α-dibromophosphonoacetates. | ||
We note that the pKa of the α-proton in phosphonocarboxylates and bisphosphonates is significantly lowered, allowing for the formation of either a bisphosphonate-stabilized carbanion or phosphorus–carboxylate-stabilized carbanion to occur under milder conditions than in monophosphonates. Quenching the anion with an electrophilic bromine source subsequently provides the expected α-brominated phosphonocarboxylate and bisphosphonate analog. Suitable bases include n-BuLi,90 LDA,91 LiHMDS,92 and NaH.68,81,93–95
The McKenna group was able to develop α-halo derivatives of α-hydroxy compounds 53a and 53b (Scheme 15), which are known anti-osteoporotic agents that act through blockage of prenylation. These compounds are also known to bind bone minerals.75,76,96 Protected benzylic bisphosphonates and phosphonocarboxylates were brominated via a stabilized carbanion generated by treatment with NaH and quenched with NBS. This strategy provided analogs 52a and 52b in good yields (Scheme 15).75,96 Unfortunately, testing of the biologically active, free acid form of α-bromo analogs 52a and 52b indicated they were generally less active than the previously studied α-OH analogs 53a and 53b.75,96
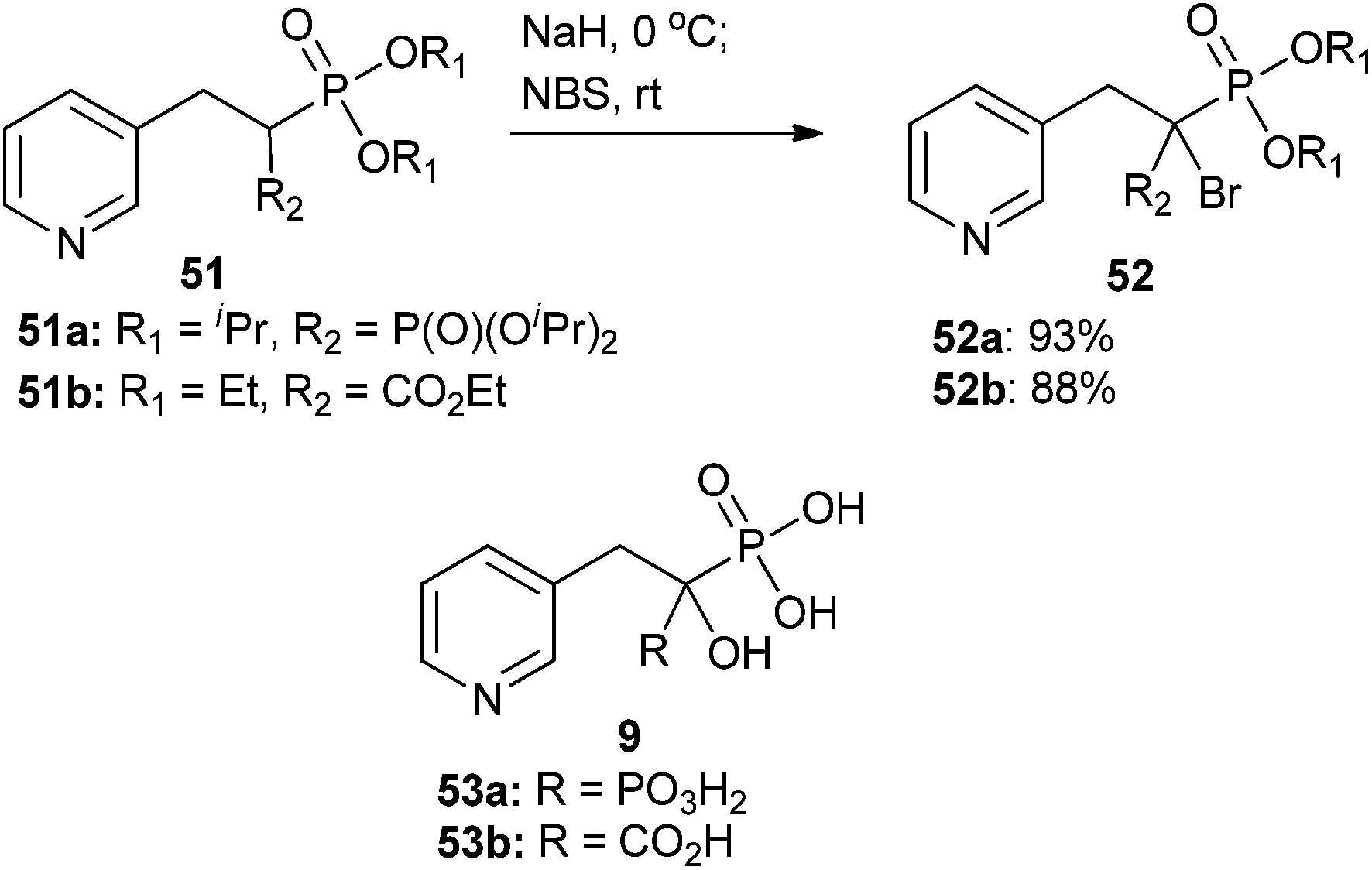 | ||
| Scheme 15 α-Bromination of benzyl bisphosphonates and phosphonocarboxylates via a bisphosphonate-stabilized (51a to 52a) or phosphorus–carboxylate-stabilized (51b to 52b) carbanion.75,96 | ||
2.6. Mono- and dibrominated polyphosphonates
Both nucleoside and non-nucleoside polyphosphonate analogs have been proposed for use in medicinal applications. We highlight the syntheses that incorporate dibromo-polyphosphonate moieties into this class of molecules (Scheme 16). General access to these seemingly intricate brominated polyphosphonate analogs begins from bisphosphonate (54) and a carbodiimide (CDI)-activated phosphonate (5597,98 or 5799). Yields are generally moderate, and both the adenine and guanine analogs of carbocyclic dinucleoside 57 were synthesized (e.g.58). In all instances the NBu4+ phosphonate salts were isolated.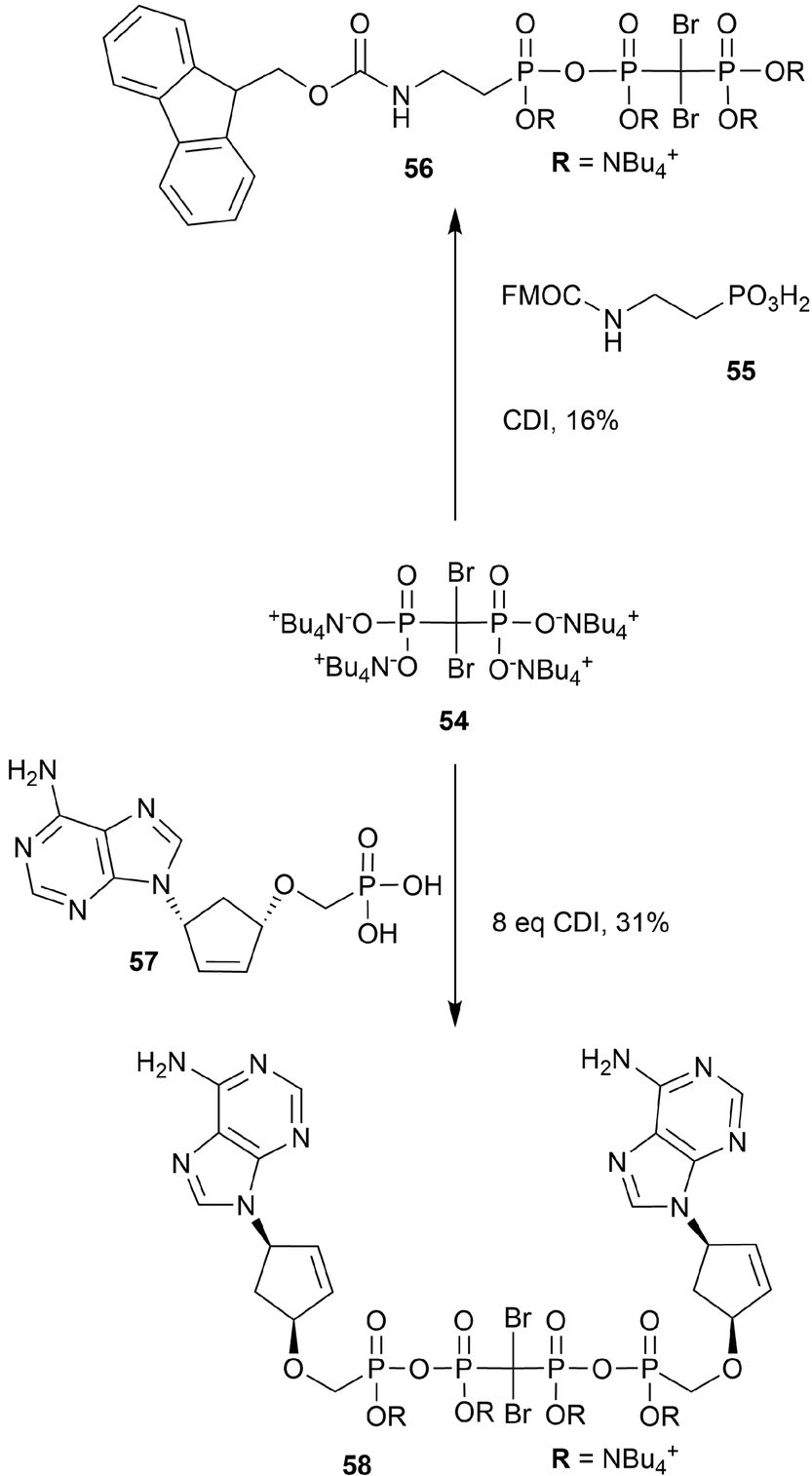 | ||
| Scheme 16 Synthesis of dibromo-polyphosphonate analogs 56 and 58.97–99 | ||
Non-nucleoside triphosphate analogs are inhibitors of terminal deoxynucleotidyl transferase (TDT), which has upregulated expression in leukemia.98,100 Arzumanov et al. screened dibromo species 56 against TDT but it was found to have lower affinity for TDT than the corresponding P–CF2–P or P–O–P triphosphonate analog.98
Carbocyclic dinucleoside polyphosphonates have shown activity against HIV reverse transcriptase-catalyzed DNA synthesis and hence demonstrate antiviral activity.97,99 Polyphosphonate 58 displayed weaker anti-HIV activity than the equivalent CF2 and P–O–P tetraphosphonate analogs.99
It is important to note here that synthetic access to biologically interesting cyclic and acyclic α-fluorophosphonate and α,α-difluorophosphonate nucleoside(-tide) analogs has been well studied and reviewed (e.g.59,10160,10261,10362,104 and 63,105Fig. 11).12 Analog 63 was tested as an inhibitor of purine nucleoside phosphorylase (PNP) which catalyzes the phosphorolysis of (deoxy)guanosine and (deoxy)inosine into guanidine and hypoxanthine. This class of inhibitors for PNP are of interest as potential immunosuppressive agents.12 The corresponding fully-saturated phosphonate of compound 63 was found to inhibit human erythrocyte PNP with a KI of 174 nM, while 63 had substantially increased activity.105 These examples demonstrate the potential of α-halogenated phosphonates as immunosuppressive agents, and the corresponding α-bromo phosphonate compounds remain to be explored.
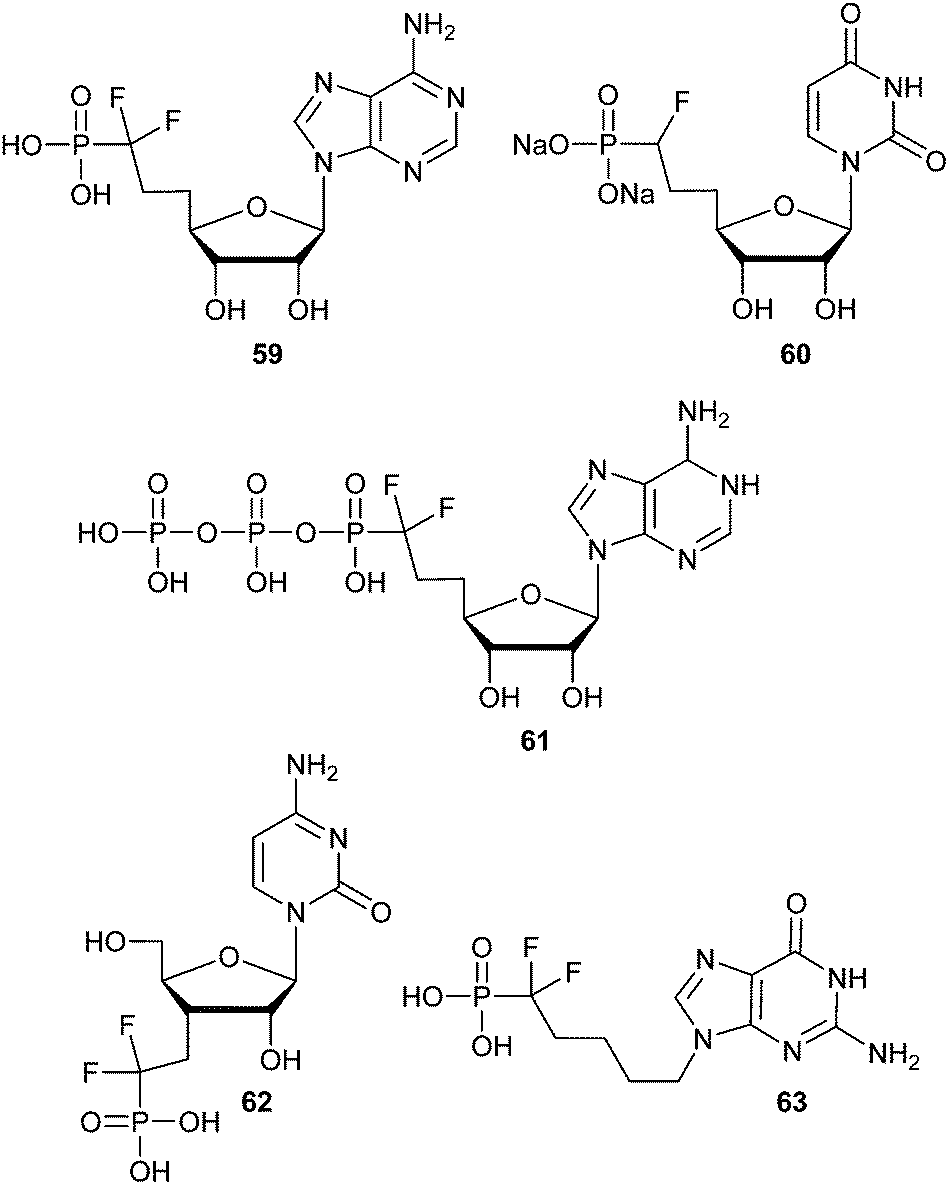 | ||
| Fig. 11 Examples of α-fluorinated phosphonate-based nucleosides(tides). | ||
More recently it has been demonstrated that β,γ-methylene-dGTP analogs can act as substrate mimics of dNTPs for DNA polymerase β.106–109 Monobromo dGTP analog 66 was synthesized in one step by coupling N,N′-dicyclohexylcarbodiimide (DCC)-activated dGMP with bisphosphonic acid (65) in 40% yield (Scheme 17). This synthetic strategy mirrors that of the nucleosides presented above (Scheme 16). The dibromo dGMP analog has also been reported, which was accessed via phosphonic acid 54 in 40% yield (not shown).107 To demonstrate the ability of β,γ-methylene-dGTP analogs to function as substrate mimics of DNA polymerase β, the authors obtained crystal structures of numerous halogenated β,γ-methylene-dGTP–DNA polymerase β ternary complexes, including analog 66.109 They used this data to study the fidelity of DNA polymerase β by looking at specific alterations in the active site due to binding of foreign nucleoside analogs.107,109 Recently, these authors expanded the library of β,γ-methylene-dNTP to include halogenated thymidine and cytidine analogs. They showed that binding of analog 66 and its dibrominated counterpart, as well as other halogenated β,γ-methylene dNTPs have tremendous effects on the transition state energies of DNA polymerase β-mediated catalysis.110 These results certainly show promise for a broadened utility of β,γ-bromomethylene dNTP analogs to function as substrate mimics for other polymerases and other enzyme systems.
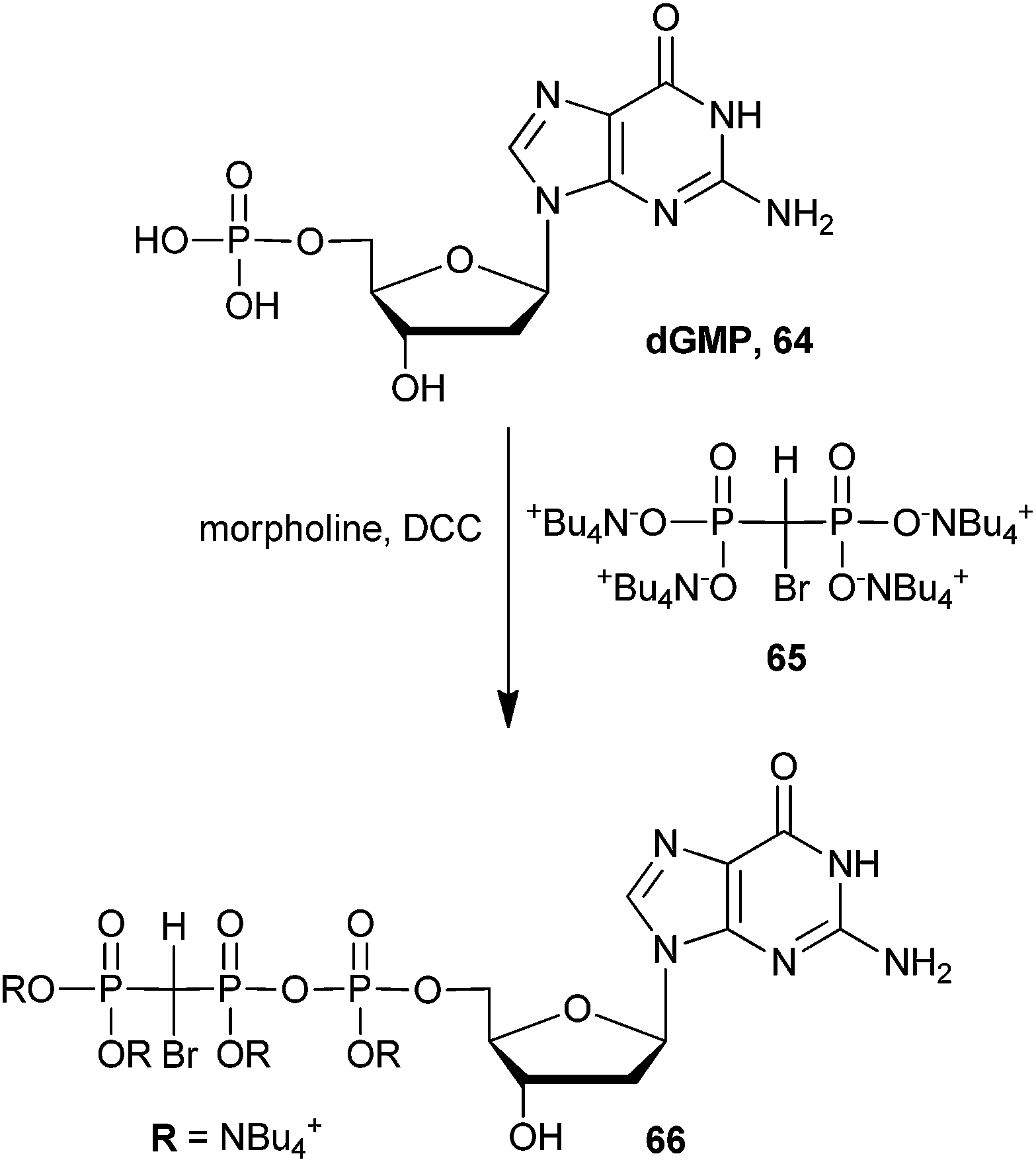 | ||
| Scheme 17 Synthesis bromo-GTP analog 66 as a substrate for DNA polymerase β.107 | ||
3. Conclusions and outlook
There are a range of available methods to synthesize α-bromophosphonates, and these allow versatile access to this functional group from alkyl, α-hydroxy, or benzyl substituted phosphonate starting materials. The reports summarized here provide a survey of the known biological activities observed for α-bromophosphonates and α,α-dibromophosphonates. This functional group has become an important bioisostere of the phosphate moiety, which can be used in the development of inhibitors and probes of phosphate-recognizing proteins. The ability of α-bromophosphonates to act as specific covalent labels in PTP systems has enormous potential in the identification of new enzymes, and in the development of new specific inhibitors. The scope of the inhibitor activity of α-bromophosphonates has recently been broadened to include GPCRs, ion channels, and glucose-6-phosphatase. The application of these compounds in mechanistic studies of enzymes, e.g. as dNTP mimics with polymerases, remains an area primed for expansion. The α-brominated phosphonates are an important functional group in the toolbox of medicinal chemists and chemical biologists.Acknowledgements
CWC would like to acknowledge support from the Natural Sciences and Engineering Research Council of Canada. AMD would like to thank the University of Alberta for a Queen Elizabeth II Graduate Scholarship.Notes and references
- D. M. Virshup and S. Shenolikar, Mol. Cell, 2009, 33, 537–545 CrossRef CAS PubMed.
- P. G. Besant and P. V. Attwood, Biochem. Soc. Trans., 2012, 40, 290–293 CrossRef CAS PubMed.
- R. C. Nordlie, J. D. Foster and A. J. Lange, Annu. Rev. Nutr., 1999, 19, 379–406 CrossRef CAS PubMed.
- T. Maehama, F. Okahara and Y. Kanaho, Biochem. Soc. Trans., 2004, 32, 343–347 CrossRef CAS PubMed.
- H. L. Zhang, X. Y. Xu, J. Gajewiak, R. Tsukahara, Y. Fujiwara, J. X. Liu, J. I. Fells, D. Perygin, A. L. Parrill, G. Tigyi and G. D. Prestwich, Cancer Res., 2009, 69, 5441–5449 CrossRef CAS PubMed.
- G. Chan, D. Kalaitzidis and B. G. Neel, Cancer Metastasis Rev., 2008, 27, 179–192 CrossRef CAS PubMed.
- J. Y. Kim-Muller and D. Accili, Science, 2011, 331, 1529–1531 CrossRef CAS PubMed.
- L. Martin, X. Latypova and F. Terro, Neurochem. Int., 2011, 58, 458–471 CrossRef CAS PubMed.
- R. Engel, Chem. Rev., 1977, 77, 349–367 CrossRef CAS.
- P. C. Crofts and G. M. Kosolapoff, J. Am. Chem. Soc., 1953, 75, 3379–3383 CrossRef CAS.
- D. B. Berkowitz, M. Bose, T. J. Pfannenstiel and T. Doukov, J. Org. Chem., 2000, 65, 4498–4508 CrossRef CAS PubMed.
- V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868–3935 CrossRef CAS PubMed.
- G. M. Blackburn, Chem. Ind., 1981, 134–138 CAS.
- M. Ruiz, V. Ojea, J. M. Quintela and J. J. Guillin, Chem. Commun., 2002, 1600–1601 RSC.
- M. B. Gazizov, R. A. Khairullin, A. I. Alekhina, I. A. Litvinov, D. B. Krivolapov, S. K. Latypov, A. A. Balandina, R. Z. Musin and O. G. Sinyashin, Mendeleev Commun., 2008, 18, 262–264 CrossRef CAS.
- C. Schnaars and T. Hansen, Org. Lett., 2012, 14, 2794–2797 CrossRef CAS PubMed.
- A. P. Bento and F. M. Bickelhaupt, J. Org. Chem., 2008, 73, 7290–7299 CrossRef CAS PubMed.
- C. J. M. Stirling, Acc. Chem. Res., 1979, 12, 198–203 CrossRef CAS.
- J. B. Conant and B. B. Coyne, J. Am. Chem. Soc., 1922, 44, 2530–2536 CrossRef CAS.
- P. C. Crofts and G. M. Kosolapoff, J. Am. Chem. Soc., 1953, 75, 5738–5740 CrossRef CAS.
- T. Sagawa, S. Hirano, H. Takahasi, K. Hosoda, K. Togashi, M. Takeuchi, Y. Iwane and Y. Murakami, Japan Pat., JP 48001133 B, 1973 Search PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- T. Cantat, L. Ricard, P. Le Floch and N. Mézailles, Organometallics, 2006, 25, 4965–4976 CrossRef CAS.
- D. J. Collins, P. F. Drygala and J. M. Swan, Aust. J. Chem., 1984, 37, 1009–1021 CrossRef CAS.
- H. Gross and S. Ozegowski, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 1–5 CrossRef CAS.
- V. V. Andriyashin, Y. V. Bakhtiyarova, R. A. Cherkasov, V. I. Galkin and I. V. Galkina, Zh. Org. Khim., 2012, 48, 1603–1604 Search PubMed.
- S. K. Chakraborty and R. Engel, Synth. Commun., 1991, 21, 1039–1046 CrossRef CAS.
- T. Pieper and B. K. Keppler, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 165, 77–82 CrossRef CAS.
- F. Eymery, B. Iorga and P. Savignac, Tetrahedron, 1999, 55, 13109–13150 CrossRef CAS.
- A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81–96 CrossRef CAS.
- T. Gajda, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 53, 327–331 CrossRef CAS.
- S. Kumaraswamy, R. S. Selvi and K. C. K. Swamy, Synthesis, 1997, 207–212 CrossRef CAS.
- H. Gross, B. Costisella, S. Ozegowski, I. Keitelé and K. Forner, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 84, 121–128 CrossRef CAS.
- D. Green, S. Elgendy, G. Patel, J. A. Baban, E. Skordalakes, W. Husman, V. V. Kakkar and J. Deadman, Tetrahedron, 1996, 52, 10215–10224 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and S. Sobhani, Synthesis, 2004, 290–294 CrossRef CAS.
- N. Iranpoor, H. Firouzabadi and M. Gholinejad, Can. J. Chem., 2006, 84, 1006–1012 CrossRef.
- P. Balczewski, A. Szadowiak and T. Bialas, Heteroat. Chem., 2006, 17, 22–35 CrossRef CAS.
- W. P. Taylor, Z. Y. Zhang and T. S. Widlanski, Bioorg. Med. Chem., 1996, 4, 1515–1520 CrossRef CAS PubMed.
- S. Kumar, B. Zhou, F. B. Liang, W. Q. Wang, Z. H. Huang and Z. Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7943–7948 CrossRef CAS PubMed.
- N. S. Tulsi, A. M. Downey and C. W. Cairo, Bioorg. Med. Chem., 2010, 18, 8679–8686 CrossRef CAS PubMed.
- R. Gupte, R. Patil, J. X. Liu, Y. H. Wang, S. C. Lee, Y. Fujiwara, J. Fells, A. L. Bolen, K. Emmons-Thompson, C. R. Yates, A. Siddam, N. Panupinthu, T. C. T. Pham, D. L. Baker, A. L. Parrill, G. B. Mills, G. Tigyi and D. D. Miller, ChemMedChem, 2011, 6, 922–935 CrossRef CAS PubMed.
- G. W. Jiang, D. Madan and G. D. Prestwich, Bioorg. Med. Chem. Lett., 2011, 21, 5098–5101 CrossRef CAS PubMed.
- All chemically synthesized phosphonic acids are presented in the text as salts as they were originally isolated, or as a doubly protonated species if a counterion was not specified.
- A. Saghatelian and B. F. Cravatt, Nat. Chem. Biol., 2005, 1, 130–142 CrossRef CAS PubMed.
- A. M. Sadaghiani, S. H. L. Verhelst and M. Bogyo, Curr. Opin. Chem. Biol., 2007, 11, 20–28 CrossRef CAS PubMed.
- B. Boivin, S. Zhang, J. L. Arbiser, Z. Y. Zhang and N. K. Tonks, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9959–9964 CrossRef CAS PubMed.
- S. Kumar, B. Zhou, F. Liang, H. Yang, W. Q. Wang and Z. Y. Zhang, J. Proteome Res., 2006, 5, 1898–1905 CrossRef CAS PubMed.
- M. Umezu-Goto, Y. Kishi, A. Taira, K. Hama, N. Dohmae, K. Takio, T. Yamori, G. B. Mills, K. Inoue, J. Aoki and H. Arai, J. Cell Biol., 2002, 158, 227–233 CrossRef CAS PubMed.
- G. B. Mills and W. H. Moolenaar, Nat. Rev. Cancer, 2003, 3, 582–591 CrossRef CAS PubMed.
- G. W. Jiang, Y. Xu, Y. Fujiwara, T. Tsukahara, R. Tsukahara, J. Gajewiak, G. Tigyi and G. D. Prestwich, ChemMedChem, 2007, 2, 679–690 CrossRef CAS PubMed.
- K I is the affinity of the compound for the enzyme and K′I is the affinity for the compound for the enzyme–substrate complex.
- M. P. Teulade and P. Savignac, J. Organomet. Chem., 1988, 338, 295–303 CrossRef CAS.
- B. Iorga, F. Eymery and P. Savignac, Synthesis, 2000, 576–580 CrossRef CAS.
- A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS.
- N. A. Strotman, S. Sommer and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 3556–3558 CrossRef CAS PubMed.
- S. F. Wnuk, L. A. Bergolla and P. I. Garcia, J. Org. Chem., 2002, 67, 3065–3071 CrossRef CAS PubMed.
- J.-P. Fu, Y.-H. He, J. Zhong, Y. Yang, X. Deng and Z. Guan, J. Fluorine Chem., 2011, 132, 636–640 CrossRef CAS.
- X. Y. Xu, G. H. Yang, H. L. Zhang and G. D. Prestwich, Prostaglandins Other Lipid Mediators, 2009, 89, 140–146 CrossRef CAS PubMed.
- X. Y. Xu and G. D. Prestwich, Cancer, 2010, 116, 1739–1750 CrossRef CAS PubMed.
- A. Nieto-Posadas, G. Picazo-Juárez, I. Llorente, A. Jara-Oseguera, S. Morales-Lázaro, D. Escalante-Alcalde, L. D. Islas and T. Rosenbaum, Nat. Chem. Biol., 2012, 8, 78–85 CrossRef CAS PubMed.
- B. Smith-Griffin and A. Burger, J. Am. Chem. Soc., 1956, 78, 2336–2338 CrossRef.
- H. P. Albrecht, G. H. Jones and J. G. Moffatt, J. Am. Chem. Soc., 1970, 92, 5511–5513 CrossRef CAS PubMed.
- A. Vasella, G. Baudin and L. Panza, Heteroat. Chem., 1991, 2, 151–161 CrossRef CAS.
- J. Kovensky, M. McNeil and P. Sinay, J. Org. Chem., 1999, 64, 6202–6205 CrossRef CAS.
- D. B. Berkowitz, M. Eggen, Q. Shen and D. G. Sloss, J. Org. Chem., 1993, 58, 6174–6176 CrossRef CAS.
- D. B. Berkowitz and D. G. Sloss, J. Org. Chem., 1995, 60, 7047–7050 CrossRef CAS.
- D. B. Berkowitz, D. Bhuniya and G. Peris, Tetrahedron Lett., 1999, 40, 1869–1872 CrossRef CAS.
- A. M. Downey and C. W. Cairo, Carbohydr. Res., 2013, 381, 123–132 CrossRef CAS PubMed.
- L. K. Charkoudian, B. P. Farrell and C. Khosla, MedChemComm, 2012, 3, 926–931 RSC.
- A. Ghosh, J. J. Shieh, C. J. Pan, M. S. Sun and J. Y. Chou, J. Biol. Chem., 2002, 277, 32837–32842 CrossRef CAS PubMed.
- V. Devreux, J. Wiesner, H. Jomaa, J. Van der Eycken and S. Van Calenbergh, Bioorg. Med. Chem. Lett., 2007, 17, 4920–4923 CrossRef CAS PubMed.
- A. M. Polozov and S. E. Cremer, J. Organomet. Chem., 2002, 646, 153–160 CrossRef CAS.
- J. Zeidler, J. Schwender, C. Müller, J. Wiesner, C. Weidemeyer, E. Beck and H. Jomaa, Z. Naturforsch., C: J. Biosci., 1998, 53, 980–986 CAS.
- T. Kuzuyama, T. Shimizu, S. Takahashi and H. Seto, Tetrahedron Lett., 1998, 39, 7913–7916 CrossRef CAS.
- M. S. Marma, Z. D. Xia, C. Stewart, F. Coxon, J. E. Dunford, R. Baron, B. A. Kashemirovli, F. H. Ebetino, J. T. Triffitt, R. G. G. Russell and C. E. McKenna, J. Med. Chem., 2007, 50, 5967–5975 CrossRef CAS PubMed.
- F. H. Ebetino, A.-M. L. Hogan, S. Sun, M. K. Tsoumpra, X. Duan, J. T. Triffitt, A. A. Kwaasi, J. E. Dunford, B. L. Barnett, U. Oppermann, M. W. Lundy, A. Boyde, B. A. Kashemirov, C. E. McKenna and R. G. G. Russell, Bone, 2011, 49, 20–33 CrossRef CAS PubMed.
- C. E. Mckenna and L. A. Khawli, J. Org. Chem., 1986, 51, 5467–5471 CrossRef.
- A. Nakata, K. Kobayashi and H. Kogen, Chem. Pharm. Bull., 2013, 61, 108–110 CrossRef CAS PubMed.
- S. Zhao, Y.-H. He, D. Wu and Z. Guan, J. Fluorine Chem., 2010, 131, 597–605 CrossRef CAS.
- B. M. Baron, R. J. Cregge, R. A. Farr, D. Friedrich, R. S. Gross, B. L. Harrison, D. A. Janowick, D. Matthews, T. C. McCloskey, S. Meikrantz, P. L. Nyce, R. Vaz and W. A. Metz, J. Med. Chem., 2005, 48, 995–1018 CrossRef CAS PubMed.
- T. Olpp and R. Brückner, Synthesis, 2004, 2135–2152 CAS.
- K. Tago and H. Kogen, Tetrahedron, 2000, 56, 8825–8831 CrossRef CAS.
- K. Tago and H. Kogen, Org. Lett., 2000, 2, 1975–1978 CrossRef CAS PubMed.
- C. D. Vanderwal, D. A. Vosburg, S. Weiler and E. J. Sorensen, J. Am. Chem. Soc., 2003, 125, 5393–5407 CrossRef CAS PubMed.
- C. D. Vanderwal, D. A. Vosburg and E. J. Sorensen, Org. Lett., 2001, 3, 4307–4310 CrossRef CAS PubMed.
- F.-L. Qing and X. Zhang, Tetrahedron Lett., 2001, 42, 5929–5931 CrossRef CAS.
- J. Vepsäläinen, H. Nupponen, E. Pohjala, M. Ahlgren and P. Vainiotalo, J. Chem. Soc., Perkin Trans. 2, 1992, 835–842 RSC.
- D. W. Hutchinson and G. Semple, J. Organomet. Chem., 1985, 291, 145–151 CrossRef CAS.
- O. T. Quimby, J. D. Curry, D. A. Nicholson, J. B. Prentice and C. H. Roy, J. Organomet. Chem., 1968, 13, 199–207 CrossRef CAS.
- C. Lai, C. Xi and Y. Feng, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 449–455 CrossRef CAS.
- T. Ageno, T. Okauchi, T. Minami and M. Ishida, Org. Biomol. Chem., 2005, 3, 924–931 CAS.
- B. Iorga and P. Savignac, J. Organomet. Chem., 2001, 624, 203–207 CrossRef CAS.
- V. P. Gubskaya, F. G. Sibgatullina, V. V. Yanilkin, V. I. Morozov, A. V. Toropchina, V. V. Zverev, N. M. Azancheev and I. A. Nuretdinov, Russ. Chem. Bull., 2005, 54, 1424–1429 Search PubMed.
- P. Balczewski and M. Mikolajczyk, Org. Lett., 2000, 2, 1153–1155 CrossRef CAS PubMed.
- P. Balczewski, Tetrahedron, 1997, 53, 2199–2212 CrossRef CAS.
- K. M. Błażewska, F. Ni, R. Haiges, B. A. Kashemirov, F. P. Coxon, C. A. Stewart, R. Baron, M. J. Rogers, M. C. Seabra, F. H. Ebetino and C. E. Mckenna, Eur. J. Med. Chem., 2011, 46, 4820–4826 CrossRef PubMed.
- E. A. Shirokova, A. L. Khandazhinskaya, Y. S. Skoblov, L. Y. Goryunova, R. S. Beabealashvilli and A. A. Krayevsky, Nucleosides Nucleotides, 2001, 20, 1033–1036 CAS.
- A. A. Arzumanov, L. S. Victorova and M. V. Jasko, Nucleosides Nucleotides, 2000, 19, 1787–1793 CAS.
- A. L. Khandazhinskaya, E. A. Shirokova, Y. S. Skoblov, L. S. Victorova, L. Y. Goryunova, R. S. Beabealashvilli, T. R. Pronyaeva, N. V. Fedyuk, V. V. Zolin, A. G. Pokrovsky and M. K. Kukhanova, J. Med. Chem., 2002, 45, 1284–1291 CrossRef CAS PubMed.
- R. P. McCaffrey, T. A. Harrison, R. Parkman and D. Baltimore, N. Engl. J. Med., 1975, 292, 775–780 CrossRef CAS PubMed.
- J. Matulic-Adamic and N. Usman, Tetrahedron Lett., 1994, 35, 3227–3230 CrossRef CAS.
- S. F. Wnuk and M. J. Robbins, J. Am. Chem. Soc., 1996, 118, 2519–2520 CrossRef CAS.
- J. Matulic-Adamic, P. Haeberli and N. Usman, J. Org. Chem., 1995, 60, 2563–2569 CrossRef CAS.
- C. Lopin, A. Gautier, G. Gouhier and S. R. Piettre, J. Am. Chem. Soc., 2002, 124, 14668–14675 CrossRef CAS PubMed.
- C. E. Nakamura, S.-H. Chu, J. D. Stoeckler and R. E. Parks, Nucleosides Nucleotides, 1989, 8, 1039–1040 Search PubMed.
- C. A. Sucato, T. G. Upton, B. A. Kashemirov, P. Batra, V. Martínek, Y. Xiang, W. A. Beard, L. C. Pedersen, S. H. Wilson, C. E. Mckenna, J. Florián, A. Warshel and M. F. Goodman, Biochemistry, 2007, 46, 461–471 CrossRef CAS PubMed.
- C. A. Sucato, T. G. Upton, B. A. Kashemirov, J. Osuna, K. Oertell, W. A. Beard, S. H. Wilson, J. Florián, A. Warshel, C. E. Mckenna and M. F. Goodman, Biochemistry, 2008, 47, 870–879 CrossRef CAS PubMed.
- C. E. Mckenna, B. A. Kashemirov, T. G. Upton, P. Batra, M. F. Goodman, L. C. Pedersen, W. A. Beard and S. H. Wilson, J. Am. Chem. Soc., 2007, 129, 15412–15413 CrossRef CAS PubMed.
- V. K. Batra, L. C. Pedersen, W. A. Beard, S. H. Wilson, B. A. Kashemirov, T. G. Upton, M. F. Goodman and C. E. Mckenna, J. Am. Chem. Soc., 2010, 132, 7617–7625 CrossRef CAS PubMed.
- K. Oertell, B. T. Chamberlain, Y. Wu, E. Ferri, B. A. Kashemirov, W. A. Beard, S. H. Wilson, C. E. Mckenna and M. F. Goodman, Biochemistry, 2014, 53, 1842–1848 CrossRef CAS PubMed.
Footnote |
| † Present address: Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Gilead Sciences & IOCB Research Center, Flemingovo nám. 2, 16610 Prague 6 (Czech Republic) |
| This journal is © The Royal Society of Chemistry 2014 |