
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrido- and benzisothiazolones as inhibitors of histone acetyltransferases (HATs)†
S. D.
Furdas
a,
I.
Hoffmann
a,
D.
Robaa
b,
B.
Herquel
c,
W.
Malinka
d,
P.
Świątek
d,
A.
Akhtar
c,
W.
Sippl
b and
M.
Jung
*a
aInstitute of Pharmaceutical Sciences, Albert-Ludwigs-Universität Freiburg, Albertstraße 25, 79104 Freiburg, Germany. E-mail: manfred.jung@pharmazie.uni-freiburg.de
bInstitute of Pharmacy, Martin-Luther-Universität Halle-Wittenberg, Wolfgang-Langenbeck-Straße 4, 06120 Halle/Saale, Germany
cMax Planck Institute of Immunobiology and Epigenetics, Stübeweg 51, 79108 Freiburg, Germany
dDepartment of Chemistry of Drugs, Wroclaw Medical University, Borowska 211, 50-556 Wrocław, Poland
First published on 11th August 2014
Abstract
Histone acetyltransferases (HATs) are interesting targets for the treatment of cancer and HIV infections but reports on selective inhibitors are very limited. Here we report structure–activity studies of pyrido- and benzisothiazolones in the in vitro inhibition of histone acetyltransferases, namely PCAF, CBP, Gcn5 and p300 using a heterogeneous assay with antibody mediated quantitation of the acetylation of a peptidic substrate. Dependent on the chemical structure distinct subtype selectivity profiles can be obtained. While N-aryl derivatives usually are rather pan-HAT inhibitors, N-alkyl derivatives show mostly a preference for CBP/p300. Selected compounds were also shown to be inhibitors of MOF. The best inhibitors show submicromolar inhibition of CBP. Selected compounds affect growth of HL-60 leukemic cells and LNCaP prostate carcinoma cells with higher potency on the leukemic cells. Target engagement was shown with reduction of histone acetylation in LNCaP cells.
Introduction
Histone acetyltransferases (HATs) catalyse the transfer of an acetyl-group from acetyl coenzyme A to the ε-amino group of a lysine residue within a protein. Their main substrates are histone proteins. Acetylation of histone proteins abolishes the positive charge of lysine side chains in the physiological environment and leads to weaker binding to the negatively charged DNA. Making DNA thus more accessible, histone acetylation is associated with active transcription.1According to their cellular localization, two types of HATs are distinguished: while Type B HATs have a shuttle function between cytoplasm and nucleus, Type A HATs are localized in the nucleus.2,3 Five families of Type A HATs exist. The GNAT-family consists of Gcn5 (KAT2A), PCAF (KAT2B) and ELP3 (KAT9).4 The p300/CBP-family has two members, CBP (KAT3A) and p300 (KAT3B).5 Other families include the MYST family with MOF as an important member, the basal transcription factors and the nuclear receptor cofactors.2
Their influence on transcriptional activity makes HATs epigenetic enzymes. Their counterparts are the histone deacetylases (HDACs), which remove acetyl groups from acetylated lysines. It is of paramount importance for an organism that the equilibrium of histone acetylation and deacetylation is maintained. Both aberrant activity of HATs and HDACs play a role in disease states and thus inhibitors of both enzymes are sought to restore the equilibrium.6,7 While several HDAC inhibitors have already gained FDA approval,8 there is still a wide lack of HAT inhibitors.
Most published HAT inhibitors are natural compounds or derivatives thereof. Their properties are often not druglike and many show inhibitory potential against more than one target. Examples include curcumin, anacardic acid or garcinol.9
Prominent among the few small-molecule HAT inhibitors described is the class of isothiazolones. The first series consisted of simple isothiazolones with N-alkyl and N-aryl substituents. They inhibit PCAF and p300 and suppress growth of HCT116 and HT29 cells. In the course of cellular experiments, the authors noted that the compounds act on HATs in an irreversible manner. According to the postulated mechanism of inhibition, the isothiazolone sulphur forms a disulphide bond to a cysteine residue of the enzyme crucial for acetyltransferase activity.10
The high chemical reactivity of these simple isothiazolones is a drawback to this class of substances, as it makes them likely to act promiscuously. In a study of the effects of substituents in the 4- and 5-position of the isothiazolone ring, an annellated pyridoisothiazolone derivative proved to be the most effective against PCAF with an IC50 value of 6.1 ± 1.9 μM.11 It is still thought that these annellated compounds act as covalent inhibitors but with reduced general bioreactivity. Concurrent studies on a set of pyridoisothiazolones and benzisothiazolones and their PCAF-inhibitory activity was presented by our group shortly afterwards. N-Benzyl compounds were weaker inhibitors of PCAF, but some members of this group affected cancer cell growth, e.g. in neuroblastoma cells more strongly than pyridoisothiazolones.12 Lately we have shown that some N-benzyl derivatives are potent on CBP and p300 while two N-phenyl compounds were pan-inhibitors among the four enzymes tested (PCAF, Gcn5, CBP, p300).13
Here, we present more detailed structure–activity relationships for a selection of pyridoisothiazolones and benzisothiazolones on the human histone acetyltransferases PCAF, Gcn5 and p300 as well as CBP and MOF for selected compounds. We show growth inhibitory effects of several compounds on two human cancer cell lines as well as reduced cellular histone acetylation for one inhibitor.
Results and discussion
Selection of compounds
Starting with the recently identified pyridoisothiazolones12 as a template we screened compounds available in house as well as commercial databases for structurally similar compounds.In vitro tests
Compounds were tested in vitro for inhibitory potential against recombinant human PCAF, Gcn5, p300 and in selected cases CBP and MOF. We used our published HAT assay with a biotinylated histone H3 peptide comprising amino acid 1 to 21 as a substrate. IC50 values or – usually when inhibition did not exceed 70% at a concentration of 50 μM – percent inhibition at a concentration of 50 μM are given in Tables 1 to 4 (see Fig. 1 for general structures). Published HAT inhibitory properties are included for comparison.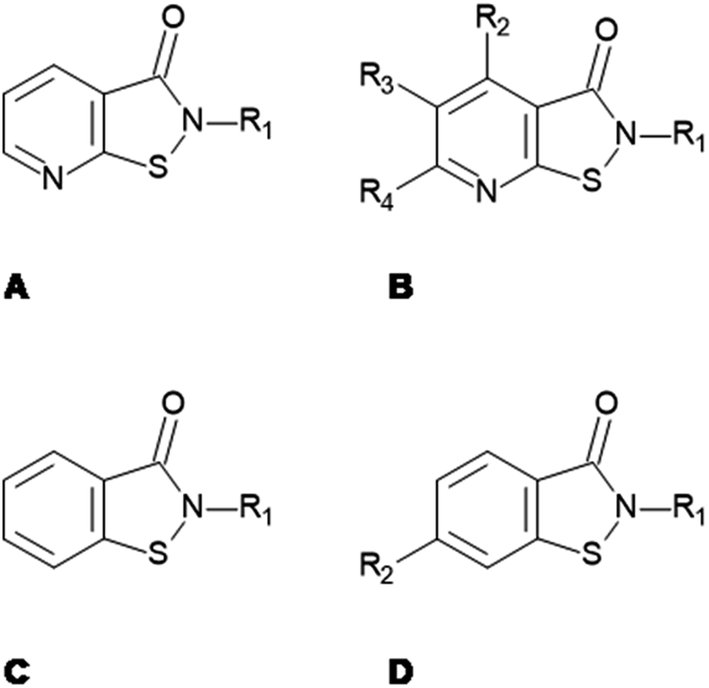 | ||
| Fig. 1 General structure of pyrido- and benzisothiazolones tested for HAT inhibitory activity. | ||
In the compounds with an unsubstituted pyridine ring, N-aryl substituents lead to pan-HAT inhibition in the low micromolar range (Table 1 and Fig. S1†). We see a broad inhibition spectrum also with substituents on the pyridine ring, even a phenyl ring is tolerated there (see e.g. cpd 12, Table 2), or an N-aroyl substituent as in compound 10. N-Benzyl or other alkyl substituents lead to defined subtype selectivity patterns (Tables 1 and 2, Fig. S1 and S2†). Generally, there is a trend to reduced PCAF and Gcn5 inhibition while MOF (only tested for 1–4), CBP and/or p300 inhibition is retained. Compounds with a more flexible linker attached to the isothiazole nitrogen atom show decreased activity on PCAF and GCN5 (e.g.6 and 7) compared to analogs where an aromatic ring is directly connected to the isothiazole ring (e.g.1 and 3). Interestingly, 6 and 7 are highly potent on CBP with pronounced selectivity over p300 despite the high similarity of the two enzymes. Among the benzisothiazolones, N-alkyl derivatives with a carboxylic acid function are inactive (13, 14, see Table 3). Amides are somewhat more potent but also do not reach IC50 values below 10 μM, except for compound 19 on PCAF and 20 for CBP (Table 5). Compounds with small aryl-substituents on benzisothiazolones inhibit all HAT subtypes tested equally well with a pyrimidine being somewhat less effective. Larger moieties lead to a loss of potency against Gcn5 and p300 resulting in a selectivity for PCAF (23, 24, Table 4).
| Name | General structure: A | Inhibition at 50 μM and IC50 [μM] | |||
|---|---|---|---|---|---|
| R1 | PCAF KAT2B | Gcn5 KAT2A | CBP KAT3A | p300 KAT3B | |
| PU139 (1) |
|
93%, 9.74 ± 0.24 | 96%, 8.39 ± 0.22 | 96%, 2.49 ± 0.09 | 98%, 5.35 ± 0.36 |
| PU141 (2) |
|
27%, 130.0 ± 8.49 | 32%, 87.36 ± 7.93 | 96%, 2.85 ± 0.20 | 93%, 5.92 ± 0.17 |
| SF7 (3) |

|
100%, 3.53 ± 0.07 | 96%, 15.47 ± 0.84 | 88%, 5.28 ± 0.23 | 100%, 5.71 ± 0.24 |
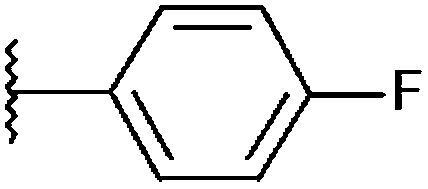
| SF18 (4) |
|
84%, 17.45 ± 1.27 | 30%, 89.91 ± 2.66 | 96%, 1.95 ± 0.35 | 91%, 5.74 ± 0.33 |
| SF19 (5) |
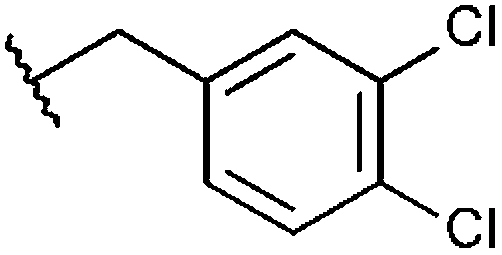
|
100%, 6.94 ± 0.57 | 73%, 23.70 ± 1.05 | 96%, 1.27 ± 0.09 | 91%, 5.32 ± 1.07 |
| Name | General structure: B | Inhibition at 50 μM and IC50 [μM] | ||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | PCAF KAT2B | Gcn5 KAT2A | CBP KAT3A | p300 KAT3B | |
| K783-6798 (6) |
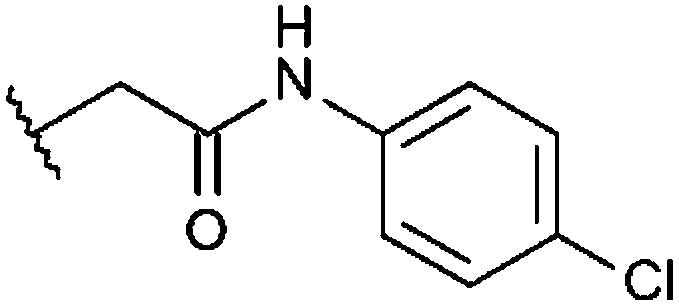
|
–CH3 | H | –CH3 | 38% | 42% | 96%, 0.44 ± 0.32 | 55% |
| P9a (7) |
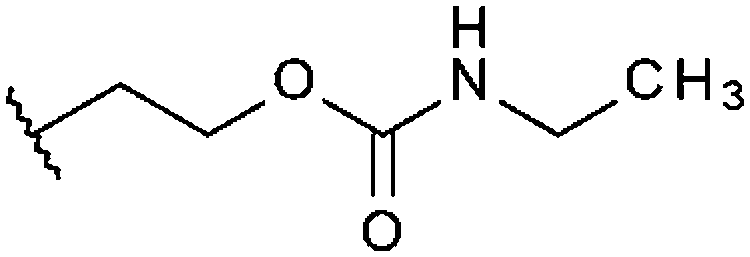
|
–CH3 | H | –CH3 | 14% | 9% | 98%, 1.67 ± 0.46 | 51% |
| P12 (8) |
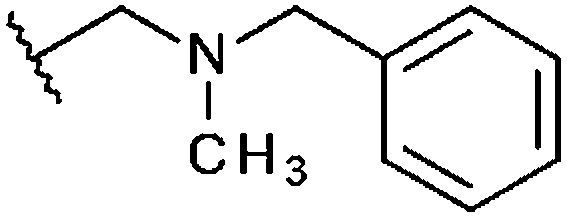
|
–CH3 | H | –CH3 | 14% | 14% | 99%, 5.37 ± 3.33 | 37% |
| HTS12959SC (9) |
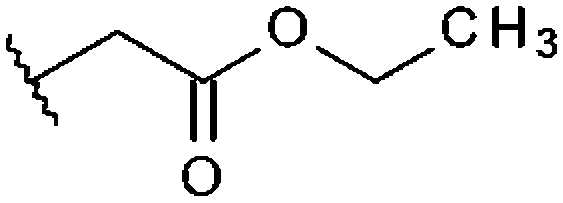
|
–CH3 | H | –CH3 | 97%, 13.6 ± 2.58 | 91%, 8.37 ± 0.63 | n. t. | 31% |
| HTS13039SC (10) |
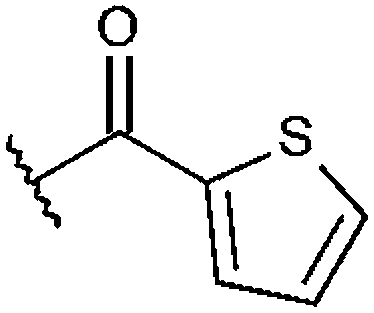
|
–CH3 | H | –CH3 | 97%, 9.30 ± 0.58 | 92%, 6.13 ± 0.57 | n. t. | 97%, 6.74 ± 0.25 |
| NSC694620 (11) |
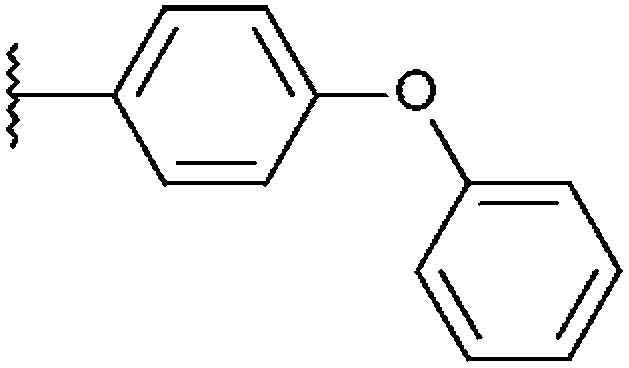
|
H | –NO2 | H | 94%, 5.30 ± 0.13 | 93%, 2.04 ± 0.44 | n. t. | 95%, 2.43 ± 0.17 |
| NSC700864 (12) |
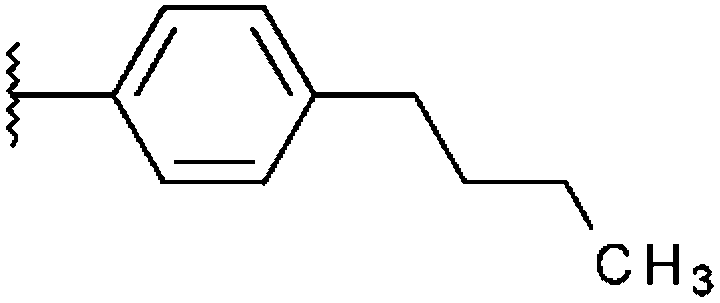
|
H | –Phenyl | H | 54%, 6.64 ± 1.28 | 44%, 8.65 ± 2.76 | n. t. | 68%, 8.28 ± 1.25 |
| Name | General structure: D | Inhibition at 50 μM and IC50 [μM] | |||
|---|---|---|---|---|---|
| R1 | R2 | PCAF KAT2B | Gcn5 KAT2A | p300 KAT3B | |
| STOCK6S-93649 (13) |
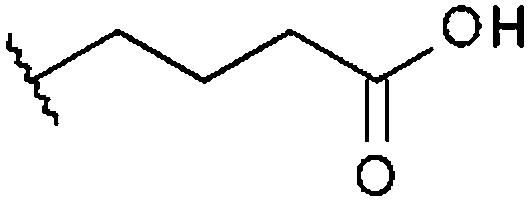
|
H | 36% 67.0 ± 7.40 | 38% 66.7 ± 12.9 | 8.8% |
| STOCK6S-87477 (14) |
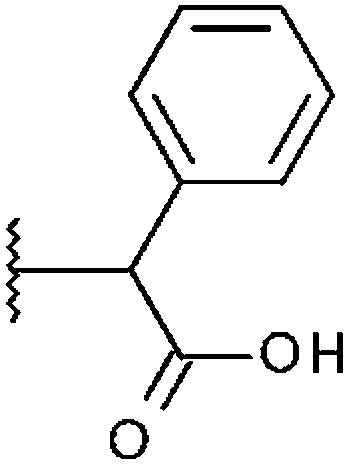
|
H | 11.8% | 28.0% | 16.1% |
| STOCK6S-96311 (15) |
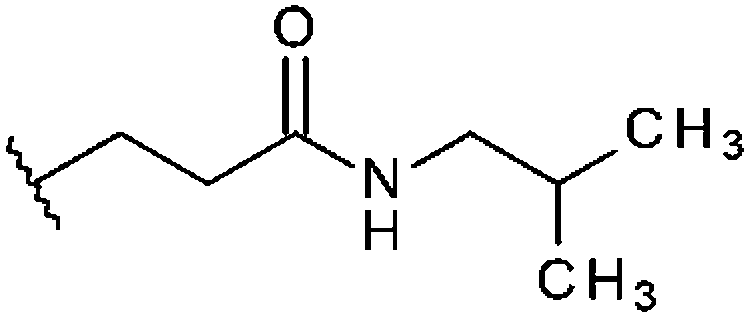
|
H | 59% 41.8 ± 2.48 | 54% 34.9 ± 10.6 | 41.4% |
| STOCK6S-96932 (16) |
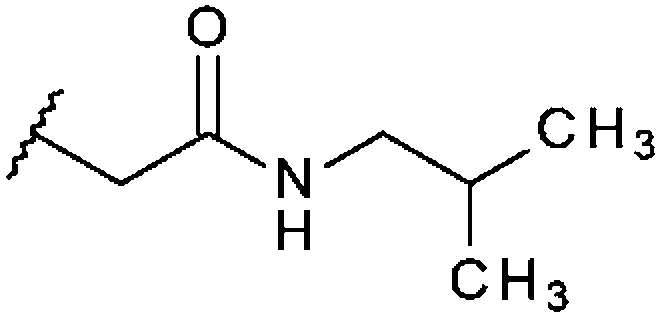
|
H | 95% 10.7 ± 0.52 | 78% 20.8 ± 1.09 | 54.6% |
| STOCK6S-93306 (17) |
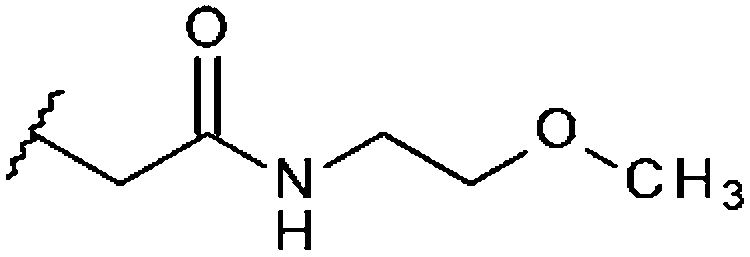
|
H | 92% 11.8 ± 1.40 | 77% 15.5 ± 2.99 | 45.1% |
| 9333079 (18) |

|
H | 91% 15.1 ± 0.75 | 84% 11.2 ± 1.42 | 24% |
| STOCK6S-91460 (19) |
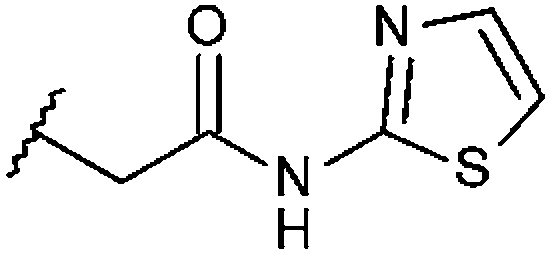
|
H | 97% 9.41 ± 0.18 | 79% 21.7 ± 2.55 | 57.4% |
| A4033-0171899 (20) |
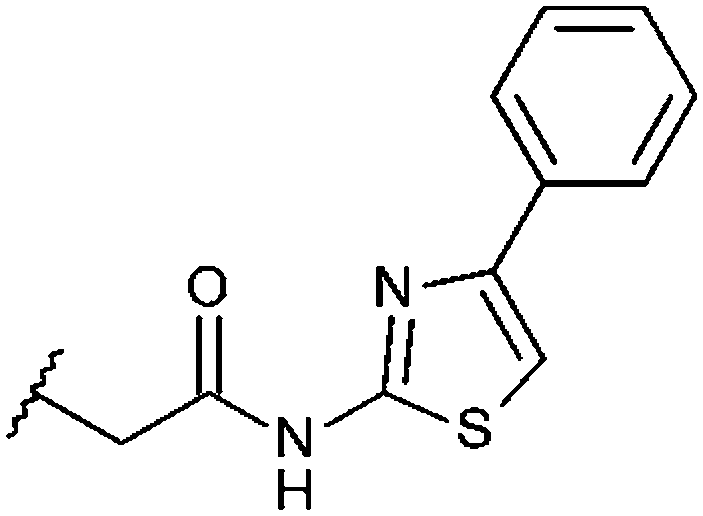
|
F | 21% | 21% | 18% |
| Name | General structure: C | Inhibition at 50 μM and IC50 [μM] | ||
|---|---|---|---|---|
| R1 | PCAF KAT2B | Gcn5 KAT2A | p300 KAT3B | |
| T0504-8862 (21) |
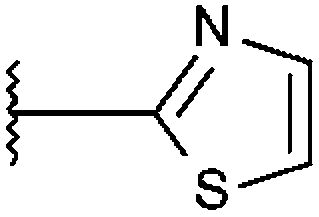
|
94% 5.77 ± 0.39 | 91% 7.87 ± 0.43 | 95% 9.35 ± 0.56 |
| T0504-9122 (22) |
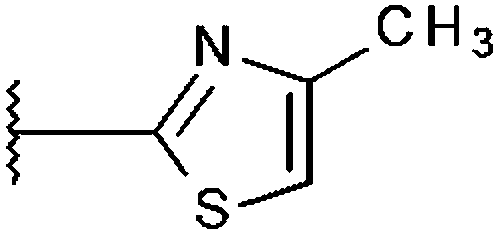
|
92% 6.01 ± 0.24 | 66% 7.11 ± 0.59 | 70% 8.14 ± 1.26 |
| T0504-8865 (23) |
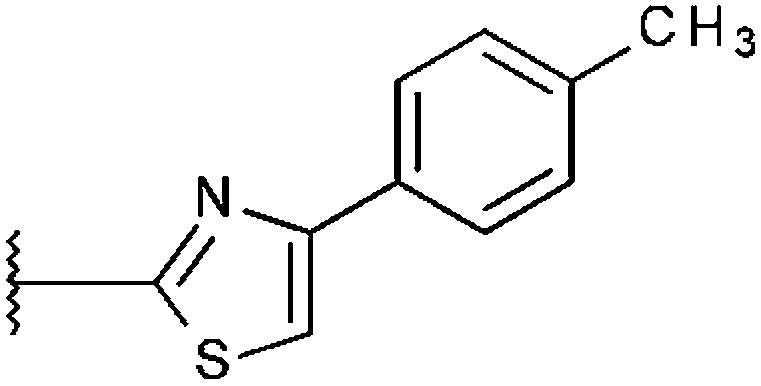
|
70% 5.82 ± 0.62 | 50.3% | 45.8% |
| T0504-9123 (24) |
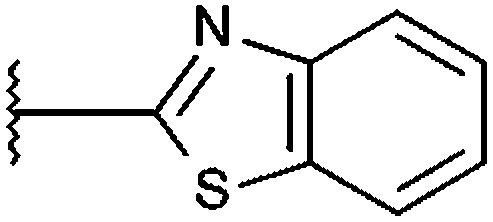
|
73% 4.55 ± 0.34 | 66% 23.3 ± 8.76 | 41.6% |
| T0517-1376 (25) |
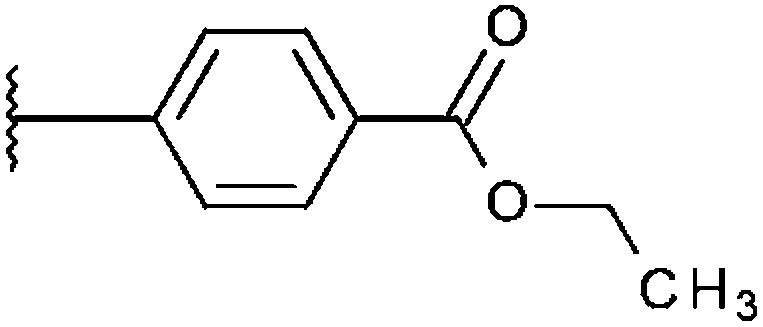
|
95% 6.95 ± 0.27 | 93% 8.53 ± 0.56 | 95% 12.9 ± 1.10 |
| T0504-6395 (26) |
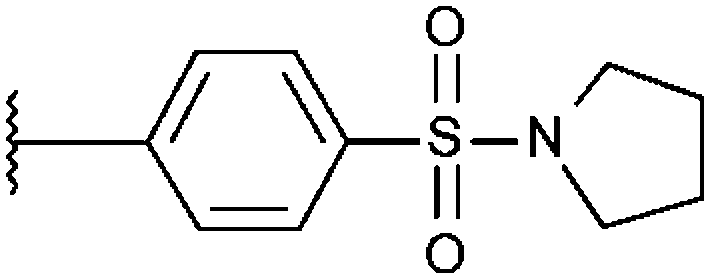
|
90% 6.19 ± 0.43 | 73% 6.27 ± 0.65 | 68% 8.03 ± 0.65 |
| T5227057 (27) |
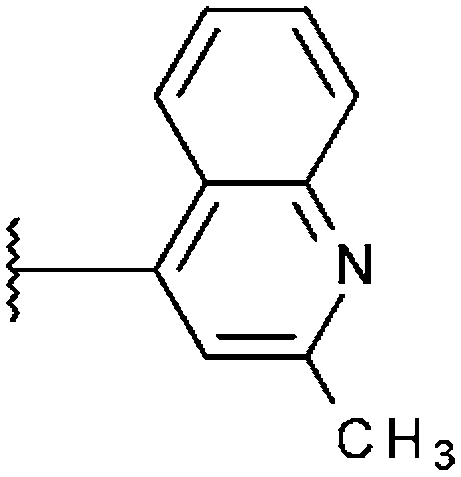
|
2.06% | 2.52% | 20.0% |
| T0515-1684 (28) |
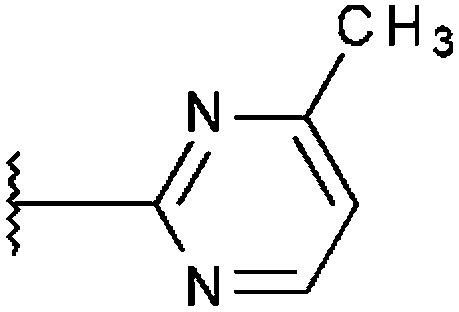
|
90% 13.6 ± 0.35 | 82% 19.9 ± 1.36 | 82% 24.7 ± 1.55 |
Cellular tests
Selected inhibitors were tested for antiproliferative properties on LNCaP prostate cancer and HL-60 leukemic cells using a MTS assay (Table 5). Overall, HL-60 cells are more sensitive to the inhibitors than LNCaP. Selective inhibition of CBP by pyridoisothiazolones hardly affects LNCaP (6, 7) while activity on the leukemic cell line is retained. The combined inhibition of CBP and p300 results in markedly lower GI50-values (e.g.2) than CPB alone. A similar trend has been described by Santer et al. for knockdown experiments in LNCaP.14 Inhibition of PCAF and Gcn5 has only a moderate impact on cell proliferation (9, 18, 23). To show cellular target engagement, acetylation levels of histone H3 were measured after treatment with 1, alone and in combination with the established HDAC inhibitor suberoylanilide hydroxamic acid (SAHA). Basal acetylation levels of histone H3 are very low. Upon treatment with SAHA an increase in acetylation is observed, which is suppressed by addition of 1 (Fig. 2).| Compound | Structure | Inhibition at 50 μM and IC50 [μM] | GI50 [μM] or Inhibition at given concentration | ||||
|---|---|---|---|---|---|---|---|
| PCAF KAT2B | Gcn5 KAT2A | CBP KAT3A | p300 KAT3B | LNCaP | HL-60 | ||
| PU139 (1) |
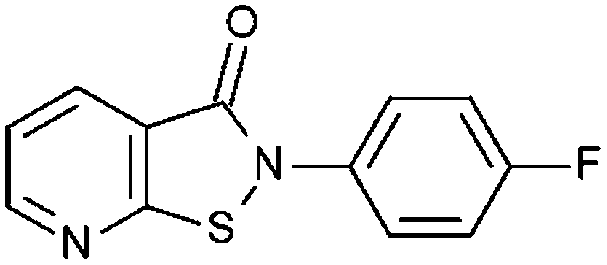
|
93% 9.74 ± 0.24 | 96% 8.39 ± 0.22 | 96% 2.49 ± 0.09 | 98% 5.35 ± 0.36 | 36.2 ± 4.7 | 3.42 ± 1.8 |
| PU141 (2) |
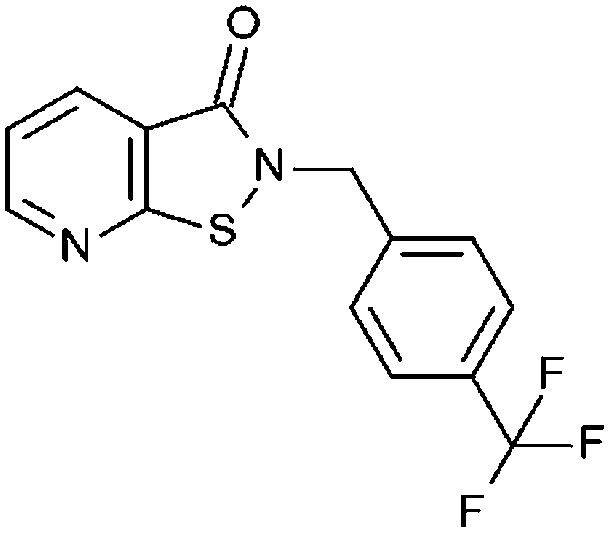
|
27% 130.0 ± 8.49 | 32% 87.36 ± 7.93 | 96% 2.85 ± 0.20 | 93% 5.92 ± 0.17 | 36.9 ± 4.8 | 4.12 ± 1.1 |
| K783-6798 (6) |
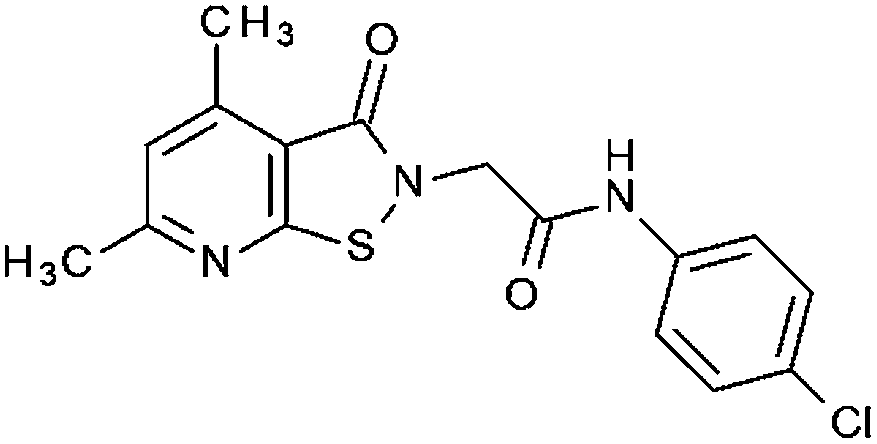
|
38% | 42% | 96% 0.44 ± 0.32 | 55% | 34.5% at 40 μM | 5.60 ± 0.1 |
| 9333079 (18) |
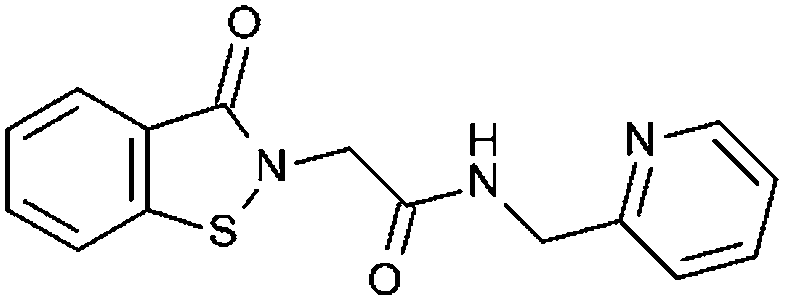
|
91% 15.1 ± 0.75 | 84% 11.2 ± 1.42 | n. t. | 24% | 73.7 ± 6.9 | n. t. |
| T0504-8865 (23) |
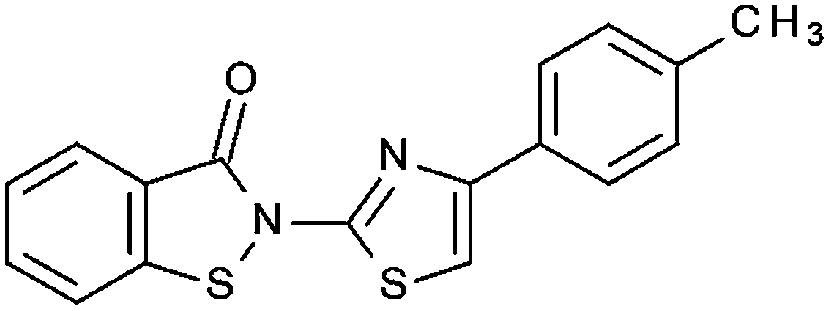
|
70% 5.82 ± 0.62 | 50.3% | n. t. | 45.8% | 67.2 ± 4.7 | n. t. |
| HTS12959SC(9) |
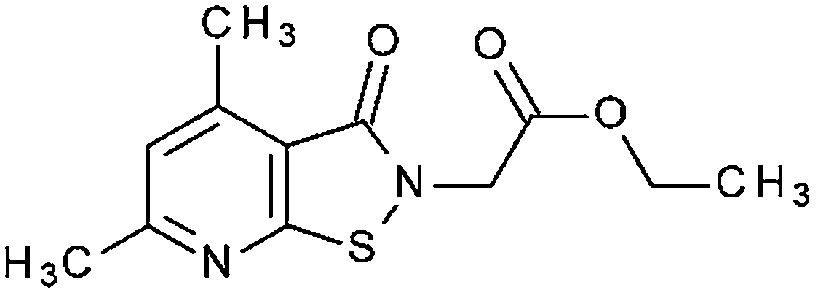
|
97% 13.6 ± 2.58 | 91% 8.37 ± 0.63 | n. t. | 31% | 50.0 ± 3.6 | n. t. |
| A4033-0171899 (20) |
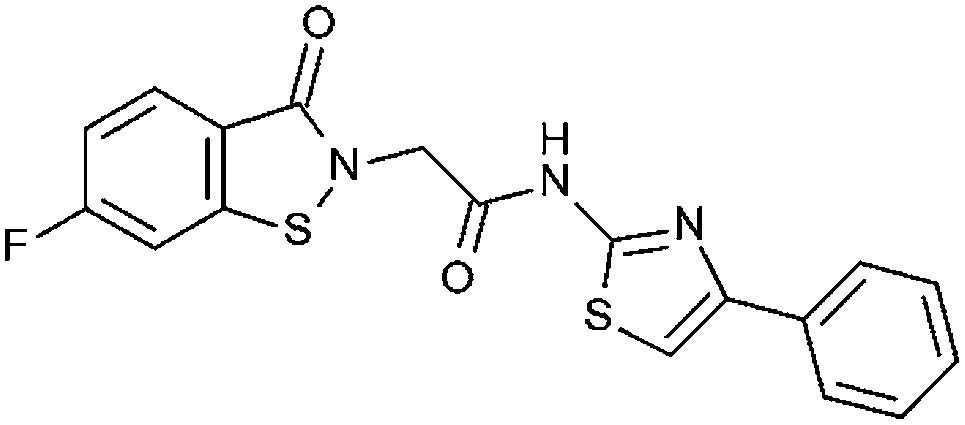
|
21% | 21% | 99% 1.44 ± 1.43 | 18% | No inhibition at 40 μM | 14.5% at 20 μM |
| P9a (7) |
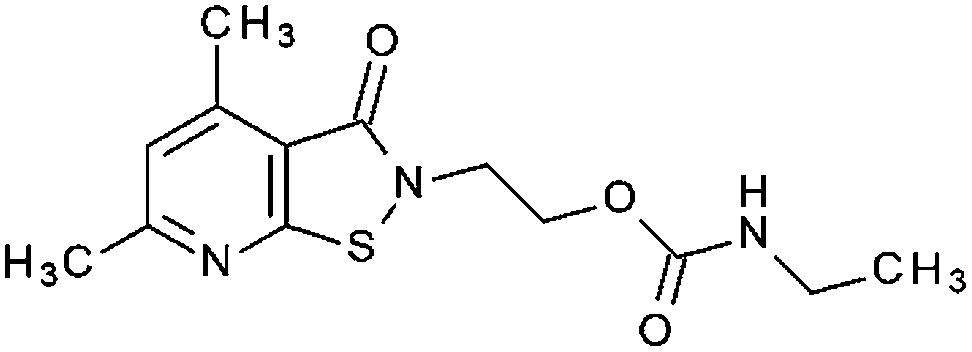
|
14% | 9% | 98% 1.67 ± 0.46 | 51% | n. t. | 6.55 ± 0.2 |
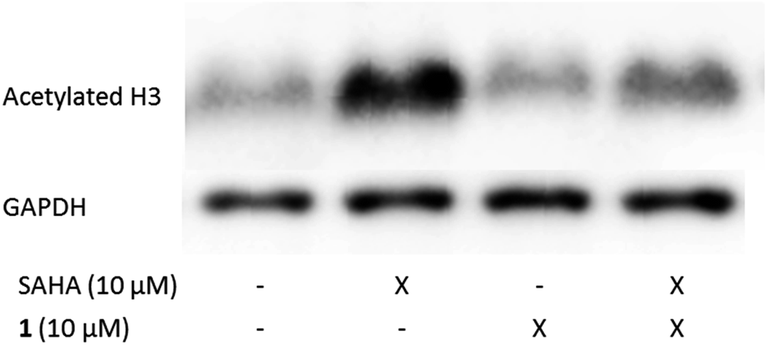 | ||
| Fig. 2 Compound 1 decreases histone H3 acetylation levels in LNCaP cells. Cells were treated with 1 and the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) for 3 hours at the concentrations indicated. Extracts were probed with antibodies against acetylated histone H3 and GAPDH as a loading control. The SAHA induced hyperacetylation is suppressed by compound 1. | ||
Experimental
Inhibitors
Compounds 1 to 5 were synthesized as previously described.126 was purchased from ChemDiverse. 7 and 9 were synthesized as previously described.15,168 was prepared as follows: to a stirred mixture of 10 mmol 2-hydroxymethyl-4,6-dimethylisothiazolo[5,4-b]pyridin-3(2H)-one in 20 mL of ethanol 10 mmol of N-benzyl-N-methylamine was added. Next the mixture was refluxed for 3 hours. After cooling, the ethanol solution was evaporated and the oil residue was cleaned by crystallization with charcoal from hexane to give 8 (70%): mp: 81–83 °C; IR: νmax/cm−1 1660 (CO); δH (300 MHz; CDCl3): 2.39 (3H, s, N–CH3), 2.60 (3H, s, CH3) 2.74 (3H, s, CH3), 3.75 (2H, s, N–CH2–Ar), 4.72 (2H, s, N–CH2), 6.93 (1H, s, Hβ-pyridine), 7.29–7.39 (5H, m, ArH). 10 was purchased from Maybridge. 11 and 12 were a gift from the NCI/DTP Open Chemical Repository. 13 to 17 and 19 were from ibscreen and 18 from Chembridge. 20 was purchased from Chemical Block and 21 to 28 were purchased from Enamine.Histone acetyltransferase assays
Heterogeneous assays based on the DELFIA® technology were performed for in vitro screening of HAT inhibitory activities as reported previously for PCAF (KAT2B) (n = 3).12 The PCAF recombinant catalytic domain used for testing was prepared according to the published protocol for bacterial overexpression of the His-tagged fusion protein.17 Gcn5 (KAT2A) (n = 2) was purchased from BPS Bioscience (catalog # 50070, San Diego, USA), CBP (KAT3A) (n = 2) from Biomol (catalog # SE-452, Hamburg, Germany), p300 (KAT3B) (n = 2) from BPS Bioscience (catalog # 50071, San Diego, USA).12 DMSO was used as solvent control. The substrate for all assays was histone H3 (amino acid 1–21, human) linked to biotin via a c-terminal CGK-linker. The in vitro HAT assay for hMOF was performed as previously described.18Cell culture
LNCaP cells were grown in RPMI-1640 without phenol red supplemented with 20% fetal calf serum and 2 mM L-glutamine. HL-60 were grown in RPMI-1640 supplemented with 10% fetal calf serum, 2 mM L-glutamine and 1% penicillin-streptomycin. All media and media supplements were purchased from PAN-Biotech.Cell proliferation assay
Cells were seeded in 96-well plates at a density of 7000 cells per well for LNCaP and 5000 cells per well for HL-60. Compounds were dissolved in DMSO and added to cells the next day with the final concentration of DMSO being 1% or 0.5% for LNCaP and HL-60 respectively. Pure solvent was used as a control. After an incubation time of 72 hours, the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay was performed according to the manufacturer's instruction (Promega). GI50-values, defined as the compound concentration that reduces the number of metabolically active cells by 50% compared to the DMSO-control, were determined using OriginPro 9.0.0G. GI50-values are given as the average of at least two independent experiments ± standard error of the mean. Standard deviation of individual measurements did not exceed 15 μM.Preparation of cell extracts and western blots
Equal numbers of LNCaP cells were lysed using Biorad loading buffer (tris-HCl, pH 6.8, 62.5 mM, SDS 2% (m/v), glycerol 25% (v/v), bromophenol blue 0.01% (v/v), β-mercaptoethanol 0.05% (v/v)) and boiled for five minutes at 95 °C. Western blots were performed according to standard protocols. As antibodies, anti-acetyl-histone H3 (Merck Millipore, Cat. 06-599), anti-GAPDH (Sigma Aldrich, Cat. G9545) and anti-rabbit HRP (Sigma Aldrich, Cat. A9169) were used.Conclusions
We have shown that by screening of focussed libraries of pyrido- and benzisothiazolones HAT inhibitors with defined subtype specificities can be obtained even without synthetic optimization. These specificities can in turn be correlated to phenotypic activities in a prostate and a leukemic cancer cell line. These results provide a basis for a future optimization of these HAT inhibitors by medicinal chemistry using bioguided synthesis supported by molecular modelling studies. Improved inhibitors will be useful tools to probe distinct HAT pathways and present potential new anticancer drugs.Acknowledgements
M. J. thanks the Deutsche Forschungsgemeinschaft (DFG, Ju-295/9-2 within SPP1463 and CRC992 MEDEP) for funding. A. A. is supported by CRC992 MEDEP and BIOSS. We gratefully acknowledge the NCI/DTP Open Chemical Repository for a gift of compounds NSC694620 and NSC700864 (http://dtp.cancer.gov).Notes and references
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef CAS PubMed.
- S. Y. Roth, J. M. Denu and C. D. Allis, Annu. Rev. Biochem., 2001, 70, 81–120 CrossRef CAS PubMed.
- M. R. Parthun, Oncogene, 2007, 26, 5319–5328 CrossRef CAS PubMed.
- F. Dyda, D. C. Klein and A. B. Hickman, Annu. Rev. Biophys. Biomol. Struct., 2000, 81–103 CrossRef CAS PubMed.
- E. M. Bowers, G. Yan, C. Mukherjee, A. Orry, L. Wang, M. A. Holbert, N. T. Crump, C. A. Hazzalin, G. Liszczak, H. Yuan, C. Larocca, S. A. Saldanha, R. Abagyan, Y. Sun, D. J. Meyers, R. Marmorstein, L. C. Mahadevan, R. M. Alani and P. A. Cole, Chem. Biol., 2010, 17, 471–482 CrossRef CAS PubMed.
- W.-J. Sun, X. Zhou, J.-H. Zheng, M.-D. Lu, J.-Y. Nie, X.-J. Yang and Z.-Q. Zheng, Acta Biochim. Biophys. Sin., 2011, 44, 80–91 CrossRef PubMed.
- Y. Wang, X. Miao, Y. Liu, F. Li, Q. Liu, J. Sun and L. Cai, Oxid. Med. Cell. Longevity, 2014, 2014, 1–11 Search PubMed.
- A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- S. D. Furdas, S. Kannan, W. Sippl and M. Jung, Arch. Pharm. Chem. Life Sci., 2012, 7–21 CrossRef CAS PubMed.
- L. Stimson, M. G. Rowlands, Y. M. Newbatt, N. F. Smith, F. I. Raynaud, P. Rogers, V. Bavetsias, S. Gorsuch, M. Jarman, A. Bannister, T. Kouzarides, E. McDonald, P. Workman and G. W. Aherne, Mol. Cancer Ther., 2005, 4, 1521–1532 CrossRef CAS PubMed.
- S. Gorsuch, V. Bavetsias, M. G. Rowlands, G. W. Aherne, P. Workman, M. Jarman and E. McDonald, Bioorg. Med. Chem., 2009, 17, 467–474 CrossRef CAS PubMed.
- S. D. Furdas, S. Shekfeh, E.-M. Bissinger, J. M. Wagner, S. Schlimme, V. Valkov, M. Hendzel, M. Jung and W. Sippl, Bioorg. Med. Chem., 2011, 19, 3678–3689 CrossRef CAS PubMed.
- V. C. Carneiro, I. C. de Abreu da Silva, E. J. L. Torres, S. Caby, J. Lancelot, M. Vanderstraete, S. D. Furdas, M. Jung, R. J. Pierce and M. R. Fantappié, PLoS Pathog., 2014, 10, e1004116 Search PubMed.
- F. R. Santer, P. P. S. Hoschele, S. J. Oh, H. H. H. Erb, J. Bouchal, I. T. Cavarretta, W. Parson, D. J. Meyers, P. A. Cole and Z. Culig, Mol. Cancer Ther., 2011, 10, 1644–1655 CrossRef CAS PubMed.
- W. Malinka, P. Świątek, M. Śliwińska, B. Szponar, A. Gamian, Z. Karczmarzyk and A. Fruziński, Bioorg. Med. Chem., 2013, 21, 5282–5291 CrossRef CAS PubMed.
- T. Zawisza and W. Malinka, Farmaco, Ed. Sci., 1985, 40, 124–132 CAS.
- S. D. Furdas, S. Shekfeh, S. Kannan, W. Sippl and M. Jung, MedChemComm, 2012, 3, 305 RSC.
- T. Conrad, F. M. G. Cavalli, H. Holz, E. Hallacli, J. Kind, I. Ilik, J. M. Vaquerizas, N. M. Luscombe and A. Akhtar, Dev. Cell, 2012, 22, 610–624 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MOF inhibition data. See DOI: 10.1039/c4md00245h |
| This journal is © The Royal Society of Chemistry 2014 |