
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prekinamycin and an isosteric-isoelectronic analogue exhibit comparable cytotoxicity towards K562 human leukemia cells†
Glenn L.
Abbott
a,
Xing
Wu
b,
Zhufeng
Zhao
c,
Lei
Guo
c,
Vladimir B.
Birman
c,
Brian B.
Hasinoff
b and
Gary I.
Dmitrienko
*a
aDepartment of Chemistry, University of Waterloo, 200 University Ave. West, Waterloo, Ontario N2L 3G1, Canada. E-mail: dmitrien@uwaterloo.ca; Fax: +1-519-746-0435; Tel: +1-519-888-4567 ext. 84642
bFaculty of Pharmacy, Apotex Centre, University of Manitoba, 750 McDermot Ave., Winnipeg, Manitoba R3E 0T5, Canada. E-mail: B_Hasinoff@Umanitoba.ca; Fax: +1-204-474-7617; Tel: +1-204-474-8325
cDepartment of Chemistry, Washington University, 750 Campus Box 1134, One Brookings Drive, St Louis, Missouri 63130, USA. E-mail: birman@wustl.edu
First published on 25th June 2014
Abstract
The synthesis of N-cyanobenzo[b]carbazoloquinone 4, an isosteric-isoelectronic analogue of prekinamycin, is described. Cytotoxicity studies with K562 human leukemia cells reveal that the cyanamide analogue has a bioactivity profile similar to that of prekinamycin. These results indicate that the diazo functionality may not be an absolute requirement for bioactivity.
Introduction
The kinamycins 1 (Fig. 1) are bacterial metabolites isolated from the soil bacterium Streptomyces murayamaensis sp. nov. Hata et al. Ohtani by Omura and co-workers.1–5 The Omura group proposed that kinamycins A, B, C and D were derivatives of N-cyanobenzo[b]tetrahydrocarbazole 2a–d and reported that they possessed antitumour and antibacterial activity. Based on their synthetic efforts in this area, the Echevarren group cast some doubt on the validity of the structural assignment to the kinamycins.6 Later, our laboratory7 and that of Gould8 established the kinamycins as diazobenzo[b]tetrahydrofluorenes 1, through synthetic, spectroscopic and single crystal X-ray crystallographic studies.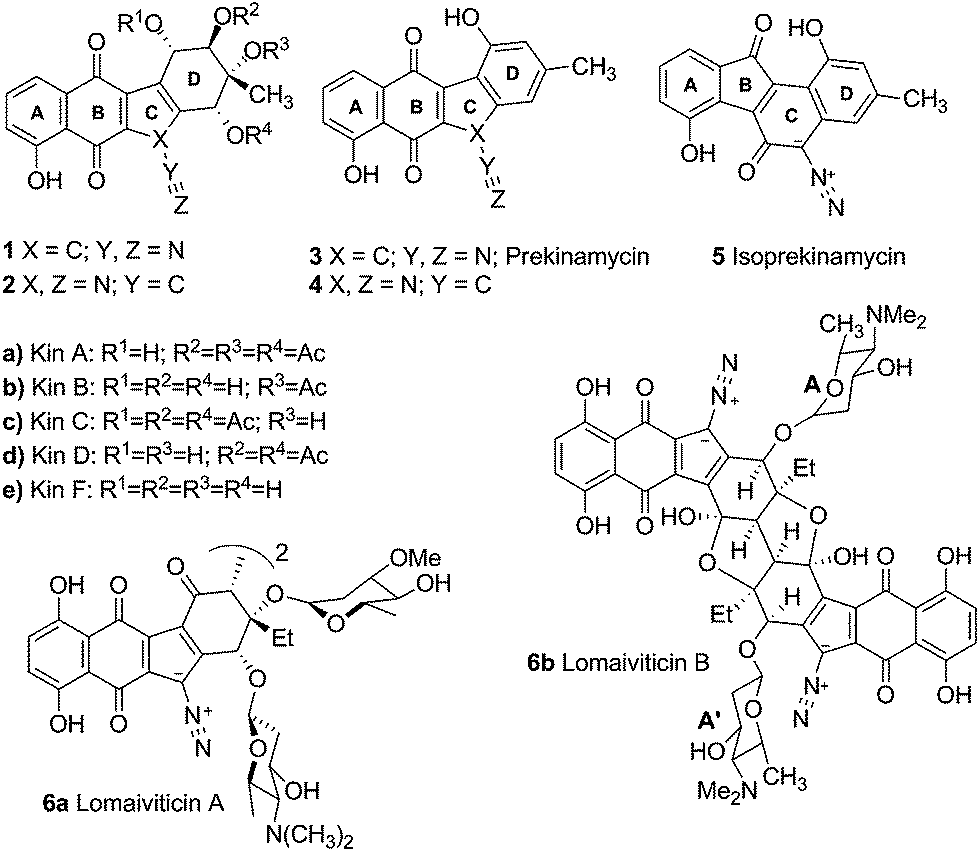 | ||
| Fig. 1 Representative members of the kinamycins and lomaiviticins. | ||
The structural novelty of the kinamycins has created interest in their mode of action (MOA), primarily in the context of their potential as anticancer reagents. The first indications of anticancer potential came from observations by the Omura group. Kinamycin C was reported to exhibit a significant survival effect at 0.1 mg kg−1 dosage i.p. in a mouse model of EHRLICH ascites carcinoma and at 1.0 mg kg−1 in a mouse model of sarcoma-180.2 Later, screening of kinamycin C versus the NIH panel of 60 cancer cell lines revealed potent in vitro cytotoxicity in the low to submicromolar IC50 range.9 The more recent report by He et al.10 of the very high cytotoxicity (nM to pM IC50 range) and DNA damaging activity of the lomaiviticins, which are glycosylated dimeric analogues of the kinamycins, has stimulated still more interest in the MOA of this class of antitumor antibiotics.11 The fact that the kinamycins incorporate a paraquinone functionality that is present in other anticancer agents (e.g. doxorubicin and daunorubicin12,13) has raised the possibility of a similarity in MOA that might involve intercalation of the quinone in dsDNA followed by bioreductive activation and DNA cleavage.
On the other hand, the presence of the relatively rare diazo functionality in the kinamycins raises other possibilities for the MOA. Evidence to indicate that the diazo group in these compounds is rather special can be found retrospectively by examination of the results of chemical studies reported by the Omura group, that were carried out at a time when the kinamycins were believed to be N-cyanobenzo[b]carbazoles and not diazobenzo[b]fluorenes. For example, the kinamycins were found to be stable to a number of acidic conditions such as 0.1 N HCl/methanol at room temperature2 and p-toluene sulfonic acid in refluxing acetone solution3 that would be expected to lead to C-protonation and decomposition of typical diazo groups.14 Furthermore, they were reported to be unstable under alkaline conditions. This property of being stable to acid but unstable to base is contrary to the known behaviour of diazo groups that are typically generated under basic conditions and decomposed under acidic conditions.14 On the other hand such behaviour is typical of aryl diazonium ions that are generated and stable under acidic conditions but are susceptible to decomposition under alkaline conditions.15
Several research groups have attempted to elucidate the reactivity of the diazo moiety in the context of biological activity. Jebaratnam and Arya demonstrated the DNA cleaving ability of the model compound 9-diazofluorene under oxidizing conditions with Cu(OAc)2.16 Our laboratory has shown previously that the diazo group of isoprekinamycin (5) is diazonium-like and hence should be activated toward attack by nucleophiles at the terminal nitrogen of the diazo group.17 We suggested that nucleophilic addition followed by homolysis generates a reactive radical, a reaction typical of aryl diazonium ions.15
Ar–N2+ + :Nu− → [Ar–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Nu] → Ar˙ + N2 + ˙Nu N–Nu] → Ar˙ + N2 + ˙Nu |
This reactivity is influenced by intramolecular H-bonding that enhances the diazonium ion character of the diazo group in isoprekinamycin and likewise in the kinamycins as indicated in Scheme 1.
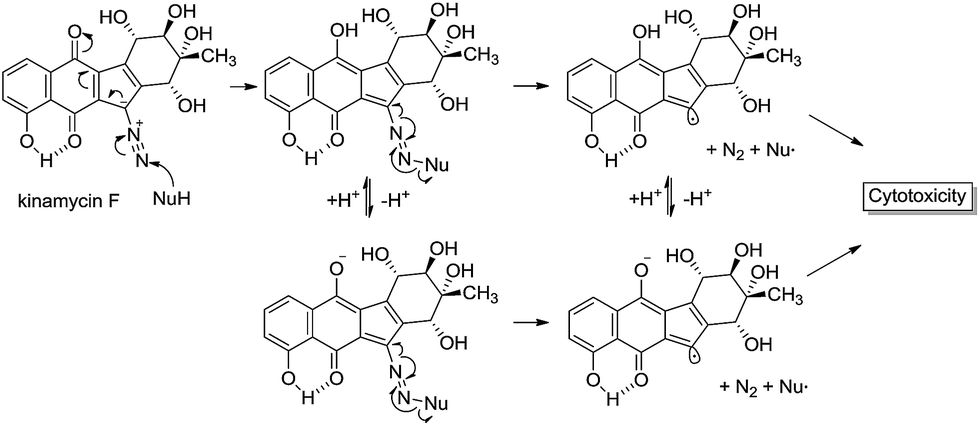 | ||
| Scheme 1 Possible interaction of kinamycin F (1e) with a biological nucleophile, potentially leading to cytotoxic events. | ||
We also pointed out that the electron affinity (EA) is enhanced by the same H-bonding interactions, potentially enhancing the ability of such systems to undergo activation by electron transfer processes in vivo.
Later, Feldman and Eastman studied prekinamycin derivatives in radical reactions with AIBN/(n-butyl)3SnH and suggested that the observed chemistry was analogous to a possible electron reduction to the semiquinone to afford, after nitrogen expulsion, an sp2 radical 7 (Fig. 2) that might be implicated in DNA damage via a number of potential mechanistic pathways.18,19 It was further postulated that an electrophilic o-quinone methide 8 was also generated under the same reducing conditions and might be trapped with biological nucleophiles as part of the MOA of the kinamycins.
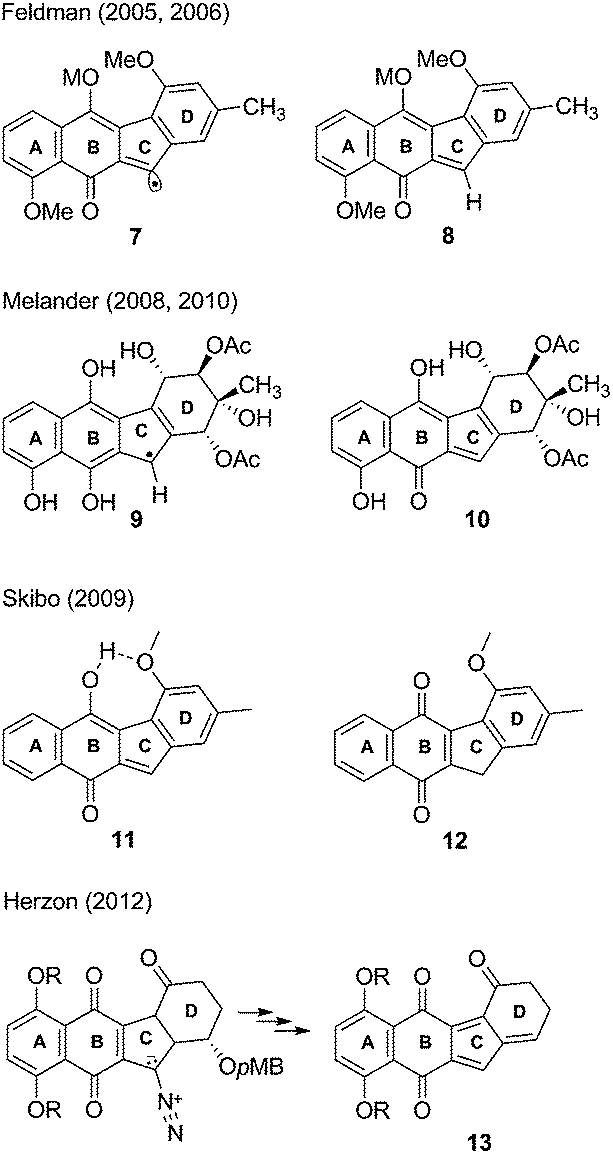 | ||
| Fig. 2 Possible reactive intermediates. | ||
More recently our focus has turned towards biological studies employing protein and cell culture techniques. We have demonstrated that kinamycins A and C (1a, 1c) are inhibitors, but not poisons, of topoisomerase IIα and are potent cell growth inhibitors.20 Furthermore, we have also shown that kinamycin F (1e), the deacetylated form of the kinamycins, is able to cleave DNA weakly on its own with increased cleavage observed in the presence of glutathione, albeit slowly under either condition.21 Isoprekinamycin (5), recently synthesized in our laboratory, was shown to cause significant growth inhibitory effects on CHO and K562 cells.22
In a series of studies using DNA-cleavage based assays, Melander and co-workers have suggested a two electron reduction of the quinone to the hydroquinone from which arise two potential pathways for DNA cleavage.23–25 The first pathway invokes (presumably with protonation occurring beforehand) a nucleophilic attack at the terminal nitrogen of the diazo moiety, furnishing a radical 9 which then can damage DNA. The second pathway involves protonation and subsequent loss of N2, providing the o-quinone methide 10 which may serve as a DNA alkylating agent.
Skibo and co-workers have carried out physical organic chemistry experiments aimed at identifying a possible MOA of prekinamycin.26 Prekinamycin and a number of analogues with the diazo carbon atom enriched with 13C were prepared and subjected to catalytic hydrogenation to mimic a two electron reduction that might occur in vivo. Skibo suggests that their observations are consistent with the formation of an o-quinone methide intermediate 11 upon a two electron reductive dediazoniation of prekinamycin. Furthermore, it was suggested that prekinamycin analogues that favour the ortho-quinone methide tautomer 11 over the quinone tautomer 12 are more likely to act as electrophiles with DNA bases as nucleophilic partners in conjugate addition reactions. Numerous examples are known of quinone methides alkylating DNA.27 The notion that kinamycins may generate quinone methides which may then go on to alkylate DNA has been known for some time,28 but still remains speculative.
Herzon and coworkers have recently described the total synthesis of (−)-kinamycin F29 and (−)-lomaiviticin aglycon.30 Subsequent studies using a pMB ether substituent at the C4 position of the D-ring was employed in “biological-like” conditions in efforts to gain insight into a MOA.31 When the analogue was subjected to mildly reducing conditions, an o-quinone methide was generated and subsequently underwent elimination of the pMB-alcohol to generate the corresponding acyl-fulvene 13. This acyl-fulvene underwent a 1,6-addition with methanol as the nucleophile, furnishing the methyl ether. Subsequent bioactivity studies (LC50) showed that the C4 pMB ether analogue was more active than the analogue that lacked such a group at C4. Acyl-fulvenes may indeed serve as cytotoxic agents but the structural dissimilarity of the compound used to generate 13 to that of the kinamycins suggests alternative MOAs. A very recent report from the Herzon group concludes that, in the case of the lomaiviticins, the potent cytotoxicity results from double strand cleavage of DNA that arises via a vinyl radical formed by a process analogous to that shown in Scheme 1 above.32
Our most recent studies have shown that kinamycin F selectively downregulates cyclin D3 in K562 leukemia cells at the transcription level and suggest that the kinamycins may not function simply by damaging DNA but might have a specific biological target associated with the induction of apoptosis.33 This study was the first to suggest a possible alternative MOA of the kinamycins that does not necessarily involve DNA cleavage. These observations have led us to consider the possibility that at least one of the modes of action of the kinamycins may involve a complementary fit to a specific biological receptor rather than the intrinsic chemical reactivity of the diazo moiety. This could conceivably be tested by abrogating the potential chemical reactivity of the diazo group while still keeping the overall size and electronic distribution intact. An isosteric-isoelectronic replacement that would also afford the vinylogous resonance of the lone pair at C11 with the quinone moiety would be the ideal candidate. All of these attributes are conveniently found within the N-cyano functionality as an isosteric-isoelectronic replacement of the diazo group (Fig. 3) and that bioactivity of the kinamycins may be approached from this yet to-be-addressed perspective. An initial assessment of the potential reactivity of 3 and 4 could be easily approached through theoretical calculations that might suggest an alternative MOA.
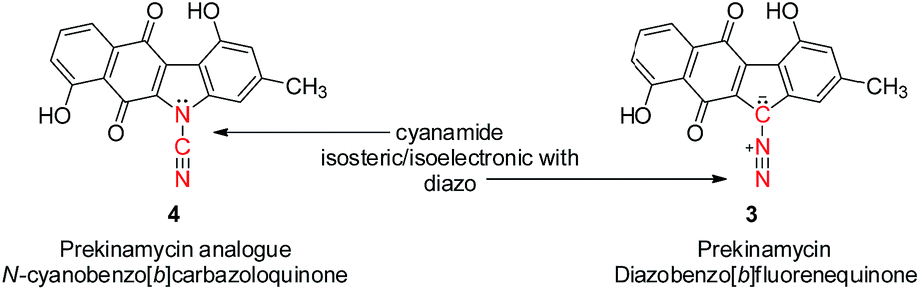 | ||
| Fig. 3 Prekinamycin 3 and the corresponding cyanamide analogue of prekinamycin 4. | ||
Replacing the diazo group with the cyanamide group would permit insight into the diazo group's role, if any, in the biological activity of the kinamycins. Thus, the primary motivation for the synthesis of 4 was to initiate biological studies not only to assess the importance of the diazo group, but also to evaluate benzo[b]carbazoloquinones in general which may be helpful in identifying the pharmacophore(s) responsible for the biological properties of the kinamycins and lomaiviticins. Ultimately, this information might facilitate drug design towards potential therapeutic agents.
Results and discussion
Theoretical studies
Quantum chemical calculations were undertaken on 4 and 3 at the DFT B3LYP 6-31G(d) level of theory. Fig. 4 illustrates the respective HOMOs and LUMOs of 4 and 3 and their corresponding energy levels. We have found that the HOMO of cyanamide 4 is predicted to be 9.9 kcal mol−1 lower in energy than that of prekinamycin (3). As well, inspection of the LUMOs reveals that the LUMO of 4 is 4.8 kcal mol−1 lower in energy than that of 3. In general, it is assumed that the EA is approximated by the negative reciprocal of a molecule's LUMO energy.34 From this perspective, 4 would be a better electron acceptor owing to its lower energy LUMO and hence higher EA. The Gibbs free energy was retrieved for 4 (−1102.150580 a.u.) and for 3 (−1102.130649 a.u.), indicating that the cyanamide analogue is thermodynamically more stable than prekinamycin by 12.5 kcal mol−1. These electronic properties suggest that there may be a difference by which these compounds elicit their MOA.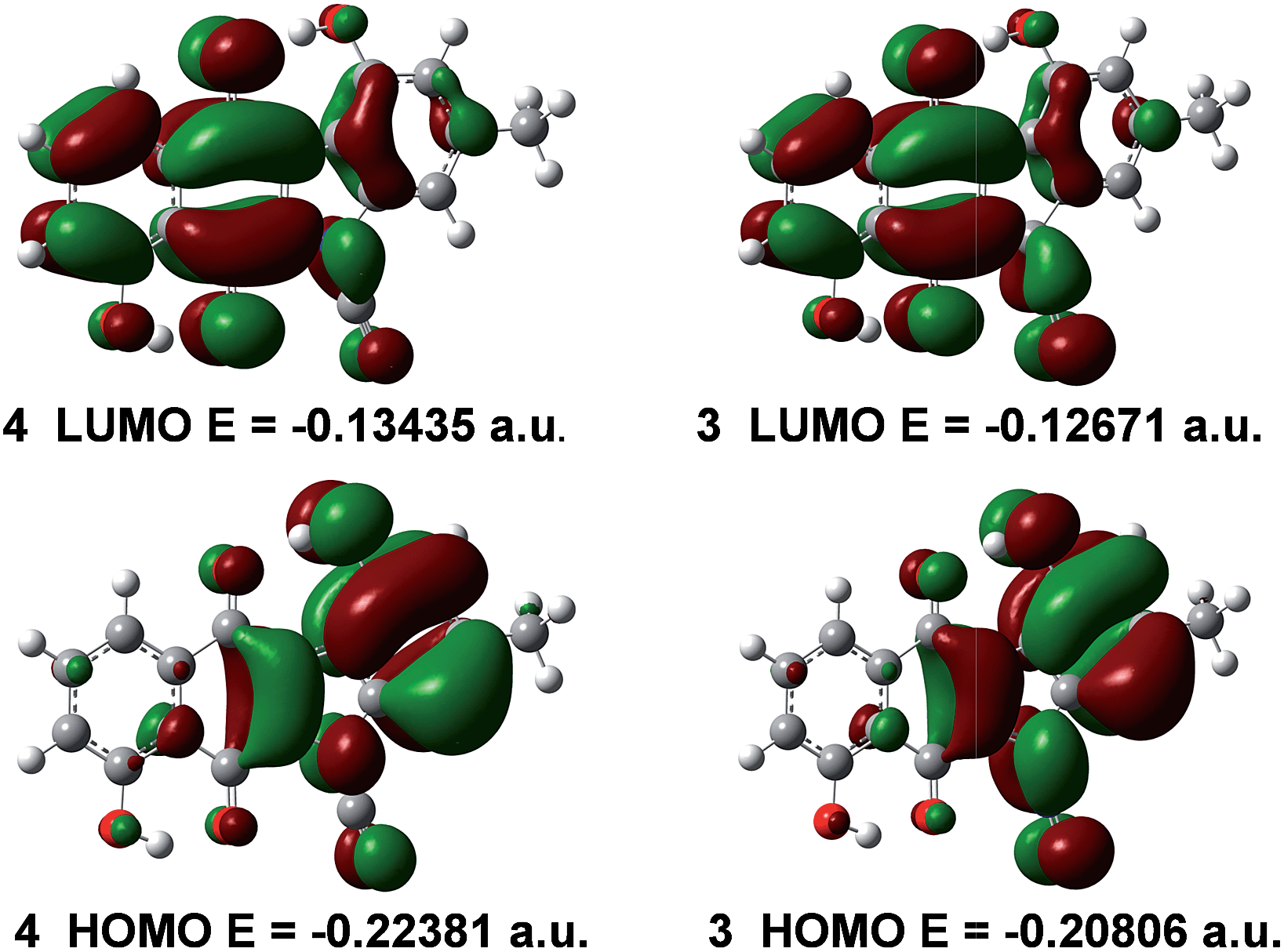 | ||
| Fig. 4 The HOMO, LUMO and their corresponding energy levels (eigenvalues) of cyanamide 4 and prekinamycin 3 generated at the DFT B3LYP 6-31 G(d) level of theory (1 a.u. = 627.5095 kcal mol−1). | ||
In order to probe the electronic differences between 4 and 3 further, an electrostatic potential energy (ESP)35 surface for both 4 and 3 was generated. Fig. 5 displays the ESP surface of 4 and 3, and for the most part, the gross molecular electron distribution is relatively similar with a few key differences.
 | ||
| Fig. 5 The electrostatic potential energy (ESP) surfaces of 4 and 3 generated at the DFT B3LYP 6-31G-(d) level of theory (isoval 0.0004) and range from ±30.8 kcal mol−1 for 4 and ±26.3 kcal mol−1 for 3. Regions coloured red indicate negative ESP values and regions coloured blue indicate positive values of the ESP with green being zero ESP. | ||
The terminal nitrogen of the cyano moiety of 4 possesses more electron density than that of the terminal nitrogen of the diazo group of 3 and intuitively, one would not expect the terminal nitrogen of the cyanamide group to be electrophilic. The lower electron density found at the terminal nitrogen of the diazo group of 3 fits well with the previous observations from this laboratory of the electrophilic character of the diazo group of the kinamycins.17,22,36 Also, the phenolic oxygens of prekinamycin display more electron density and, hence, more partial negative charge than that of the cyanamide analogue. This is a consequence of the increase in hydrogen bonding to the quinoid oxygens of the B-ring in 3 resulting from the greater need to delocalize partial negative charge from carbon to oxygen in 3 as compared to the delocalizaton of partial negative charge from nitrogen to oxygen in 4. The bond lengths in Fig. 6 clearly support the notion of stronger H-bonding to the quinoid oxygens in 3 than that of 4.
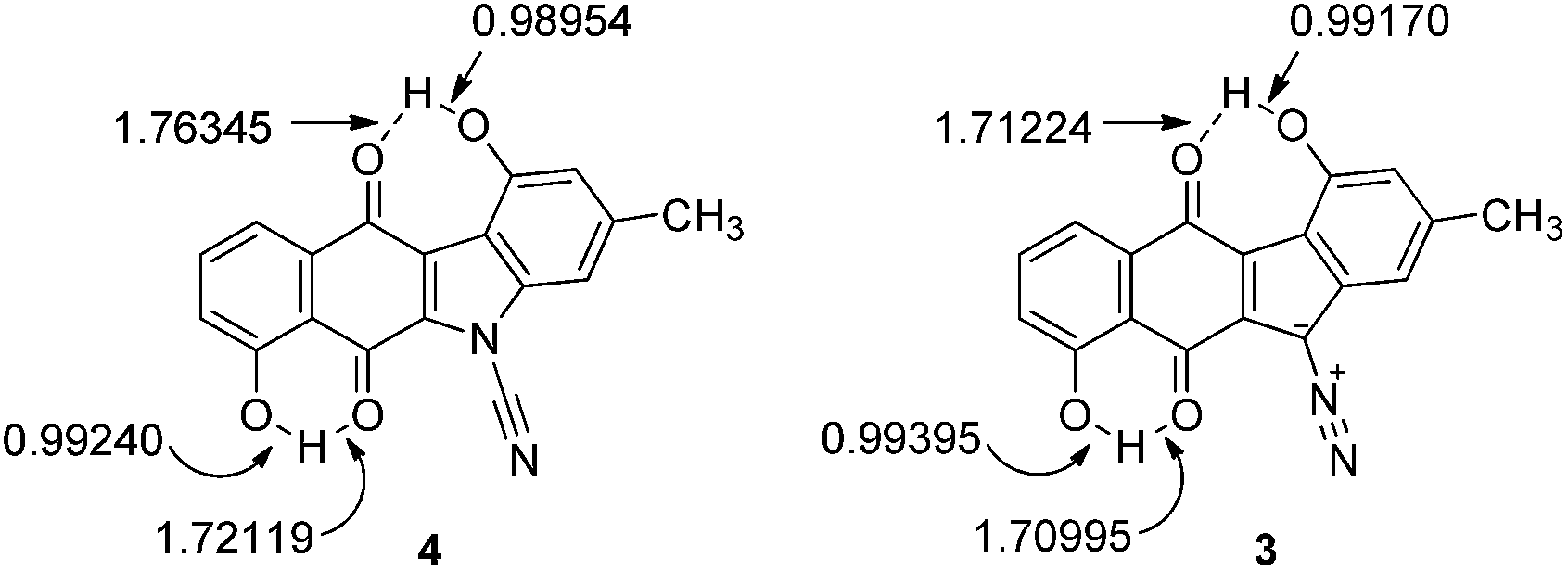 | ||
| Fig. 6 Bond lengths (Å) as observed in the minimized structures of 4 and 3 obtained at the DFT B3LYP 6-31G(d) level of theory. | ||
Overall, comparison of the calculated structures of 3 and 4 reveal similar spatial properties with small structural differences just described. Taken together, this would suggest that 3 and 4 may bind to the same receptor but do so with different affinities. An alternative explanation is that 3 and 4 may have different biological targets altogether. Biological testing of 3 and 4 under identical conditions would allow insight into these questions. To this end, the synthesis of the cyanamide analogue 4 was accomplished. Subsequent biological studies on the analogue 4, prekinamycin, kinamycin F and an earlier synthetic intermediate were performed and are described in the following sections.
Synthesis
The synthesis of 4 (Scheme 2) began with commercially available 1,5-dihydroxynaphthalene (14) which was protected as the O-methylated derivative (not shown).37 Using procedures reported by Rapoport,38 the formyl group was installed under Vilsmeier–Haack conditions and a subsequent Baeyer–Villiger oxidation provided formate 15 in 84% over 3 steps. Modifications39,40 to the Baeyer–Villiger oxidation conditions failed to increase either the purity or yield of the formate. Methanolysis, regioselective bromination and O-methylation furnished the trimethoxy-bromonaphthalene 16 in 49% yield over six steps and provided the key intermediate for the impending Buchwald–Hartwig cross-coupling.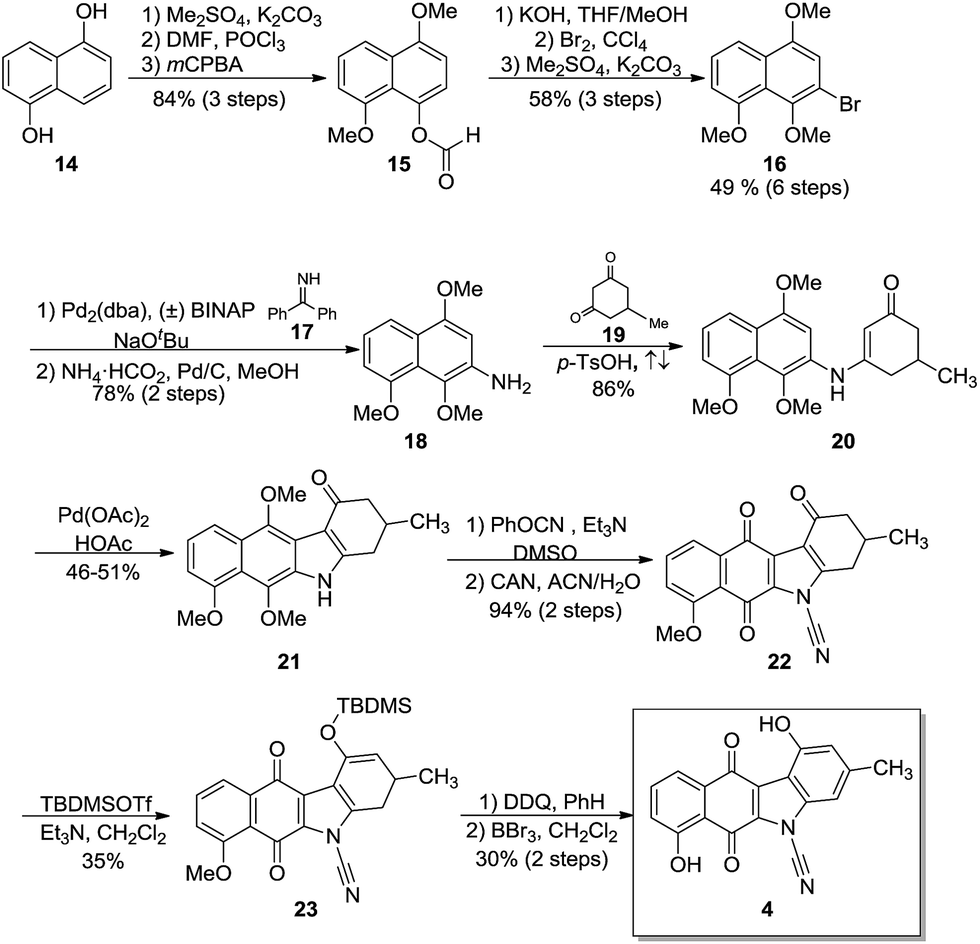 | ||
| Scheme 2 Synthesis of N-cyanobenzo[b]carbazoloquinone 4. | ||
Coupling of 16 with benzophenone imine (17) as described by Buchwald41 proceeded uneventfully and after hydrogenolysis, gave the requisite naphthylamine 18 in 78% yield over two steps. Condensation of 18 with the substituted cyclohexanedione 19 afforded anilinoketone 20 in 86% yield. Subsequent oxidative ring closure with palladium acetate using a modification of known procedures42,43 provided the tetracycle, the benzo[b]-tetrahydrocarbazole 21. Annulation typically gave yields of 27–36%. Several attempts were made to increase the yields by varying the time, temperature and the amount of oxidant. It was found that two equivalents of palladium acetate provided the best yields at 46–51%. N-Cyanation using phenyl cyanate44 and subsequent oxidation with CAN provided quinone 22 in 94% yield over 2 steps. Silylation45 of 22 using TBDMSOTf in dichloromethane provided the silyl enol ether 23 in 35% yield. Aromatization with DDQ in benzene and subsequent global deprotection with BBr3 furnished the target 4 in a 30% yield (2 steps) with an overall yield of 1.6% over fifteen steps.
Biological studies
The results from the testing of the growth inhibition of K562 cells (an erythroleukemic cell line) by two cyanamide analogues (4, 22) prepared in the present study as well as by prekinamycin (3) and kinamycin F (1e) are shown in Table 1. The details of the cellular inhibition assay as well as the preparation of kinamycin F have been described previously.21 The total synthesis of prekinamycin has been reported elsewhere.46|
|
|||
|---|---|---|---|
| IC50 (μM) | |||
| a Each IC50 value is the mean of three experiments performed on three separate days. | |||
| 22 | 4 | 3 | 1e |
| 46.6 ± 1.5 | 13.8 ± 4.4 | 13.5 ± 0.9 | 0.56 ± 0.12 |
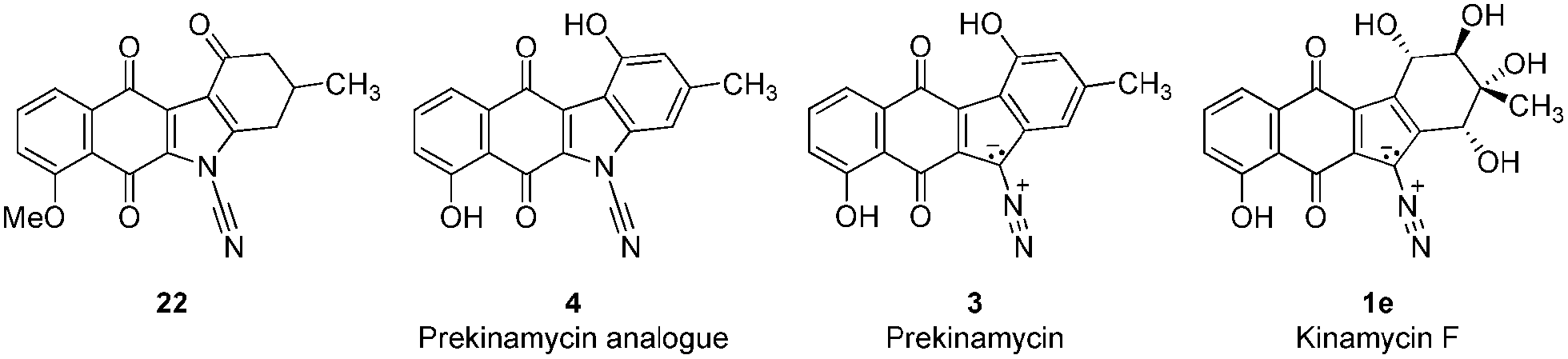
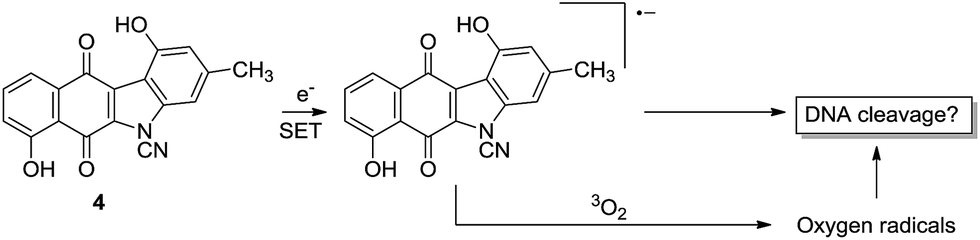
Inspection of Table 1 reveals that kinamycin F (1e) is the most active compound with an IC50 of 0.56 ± 0.12 μM. The cyanamide compound 22, an earlier synthetic precursor of 4, is the least bioactive with an IC50 of 46.6 ± 1.5 μM. We were interested in the biological testing of 22 as it presented as a crude monomeric model for the lomaiviticins. Further inspection of Table 1 shows that prekinamycin has an IC50 of 13.5 ± 0.9 μM and the cyanamide analogue has an IC50 of 13.8 ± 4.4 μM. The fact suggest that 4 and 3 possess comparable biological activity raises the question of what role the diazo function has with respect to mechanism of action and what the putative intracellular target(s) might be. These results have obvious implications in regard to the design of kinamycin analogues as well. We have suggested that the cytotoxicity of kinamycin F (1e) is due in part to the unique conformation of the polyhydroxylated D-ring, which has been shown to increase the diazonium ion character of the diazo group.36 The idea that the kinamycins may serve as biological electrophiles to generate reactive species is shown in Scheme 1. In this scenario, kinamycin F, the most bioactive form of the kinamycins, possessing an electrophilic diazo moiety, is attacked by a biological nucleophile at the terminal nitrogen, furnishing an azo adduct. Subsequently, it may be envisioned that in the biological milieu, the azo adduct, upon homolysis, produces molecular nitrogen and a reactive vinyl radical, that might lead to cell death via a number of potential pathways. On the other hand, if electron transfer is important, the lower LUMO for 4 would suggest a higher electron affinity (EA) and a greater tendency of 4 to accept an electron in a single electron transfer (SET). That is, 3 is more electrophilic but 4 has a higher electron affinity. This may suggest that compounds of the N-cyanocarbazole series may be more prone to the formation of free radicals including oxygen free radicals (i.e. S + e− → S]˙− + 3O2 → S + 3O2]˙− → ˙OH, ˙OOH, etc.) than are the kinamycins.
To investigate this issue, further ground state quantum chemical calculations on kinamycin F (1e) and its hypothetical cyanamide analogue 2e were pursued (Fig. 7). It was found that the cyanamide analogue of kinamycin F (2e) has a lower energy LUMO (by 5.5 kcal mol−1), while kinamycin F (1e) possesses a significantly larger coefficient at the terminal nitrogen than the corresponding cyanamide as shown in Fig. 7. Based on computed Gibbs free energies, the cyanamide 2e is predicted to be more stable than kinamycin F by 11.6 kcal mol−1. Taken together, these data parallel the same trend as found for comparisons of the electronic properties of 4 and 3. In this context, the results presented in Table 1 and Fig. 7 raise the question of which should be the more potent anti cancer agent: kinamycin F or its cyanamide congener.
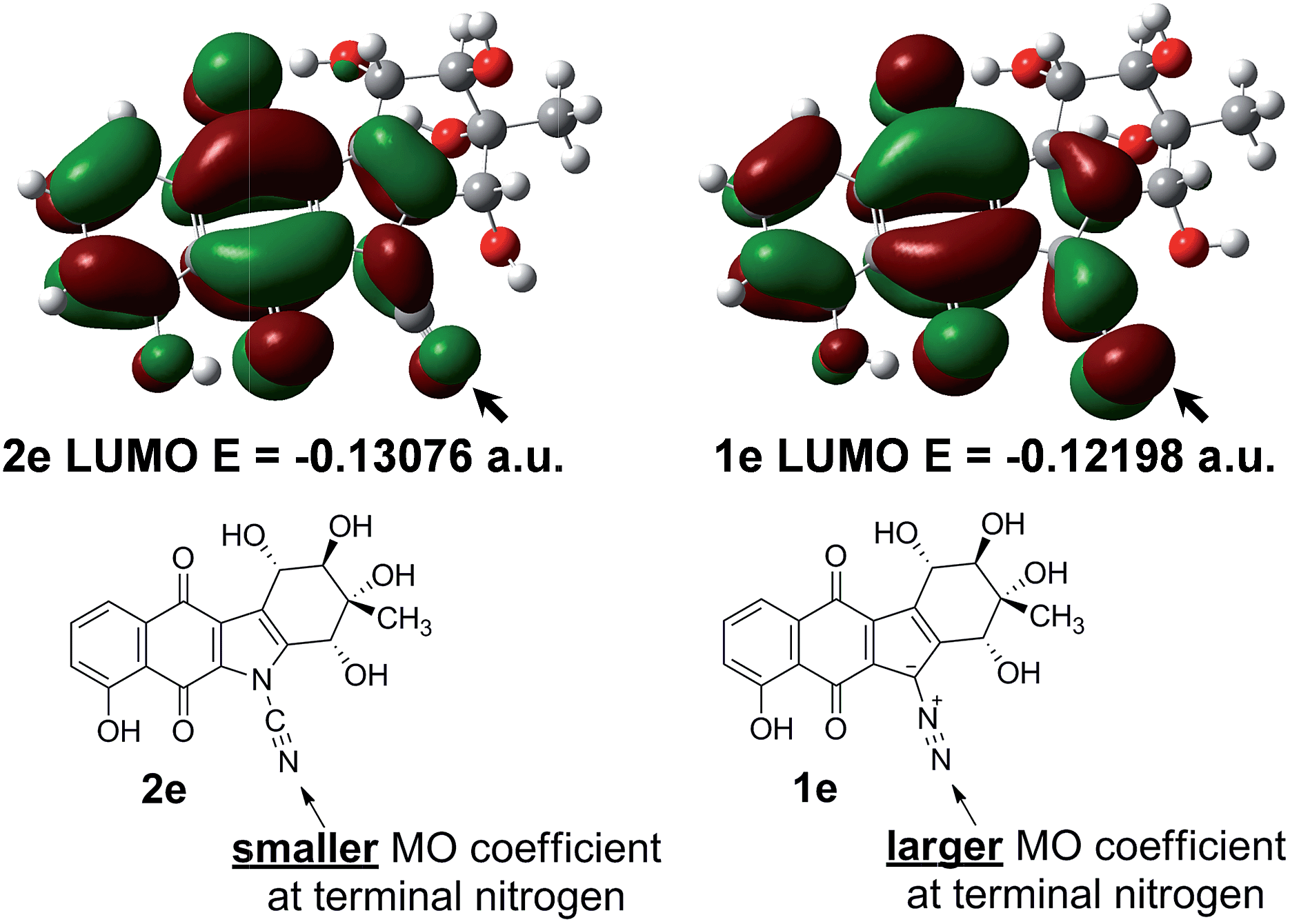 | ||
| Fig. 7 The LUMO and their corresponding energy levels (eigen-values) of kinamycin F 1e and its hypothetical cyanamide analogue 2e generated at the DFT B3LYP 6-31G(d) level (1 a.u. = 627.5095 kcal mol−1). | ||
It is possible that multiple interactions with various targets contribute to the activity of prekinamycin (3) and the cyanamide analogue 4. These include:
(1) Electron transfer to generate a radical anion which might then interact directly with DNA or first 3O2 leading to oxygen radicals which then go on to cleave DNA. This is possible for 4 and 3 but is more likely for 4 owing to its lower LUMO.
(2) Reaction with biological nucleophiles (DNA, protein). This applies for 3.
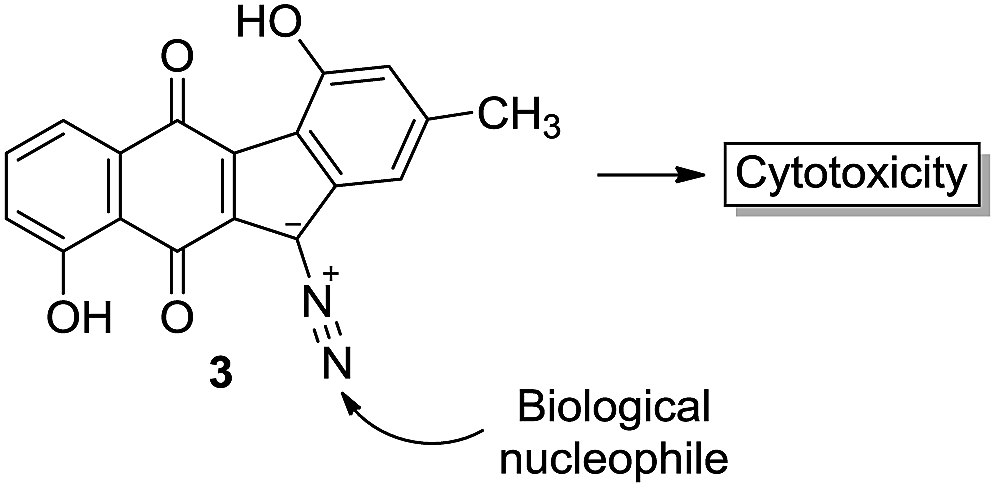
(3) Non-covalent binding to a biological receptor which is possible for both 4 and 3. In the case of kinamycin F, a recent study involving this laboratory has revealed that kinamycin F may be specifically targeting a protein involved in transcription of cyclin D3, one of a group of proteins involved in the cell cycle. The contribution of such an interaction to the overall MOA of kinamycin F is unlikely to be the consequence of a non-selective DNA cleavage event.33 A non-covalent or possibly a covalent binding to such a protein target by kinamycin F or one of its metabolites might explain the cytotoxicity since the diminution of cyclin D3 levels is known to induce apoptosis (programmed cell death). It is not yet known if similar effects on cyclin D3 levels are induced by prekinamycin, isoprekinamycin or the lomaiviticins.
One interpretation of the comparable cytotoxicity of 4 and 3 is that for 4, the electron transfer MOA makes a better contribution to the cytotoxicity effect, whereas for 3 the reaction with biological nucleophiles may be more important. Another interesting observation is the very low activity of the ketone 22. In the kinamycin series, the dimeric analogues called the lomaiviticins 6 possess such a keto group and are very active in DNA cleavage experiments under reducing conditions and are highly cytotoxic to cancer cells; more so than the monomeric kinamycins that lack the keto group. It is not clear, however, if the O-methyl group might play a role in the low activity of 22. This speculation is borne out by Herzon's recent observations in which kinamycin analogues bearing methoxy substituents displayed cytotoxicity that was substantially lower than values obtained from the corresponding analogues bearing hydroxyl groups.31
Conclusions
This study has provided some insight into the similarity and differences between the N-cyanobenzo[b]carbazole-based structure originally assigned to the kinamycins1 and the corrected (diazobenzo[b]fluorene-based) structure assigned more recently.6,7 We conclude that there is sufficient similarity between compounds possessing these core structures in regard to electronic structure and cell growth inhibition of K562 leukemia cells to warrant further study of N-cyanobenzo[b]carbazole-type analogues of the kinamycins. Synthetic efforts aimed at accessing such analogues are in progress.Acknowledgements
Mrs Valerie Goodfellow, Dr Nan Chen and Dr Otunola Adedayo are thanked for generating the sample of kinamycin F used in this study. This work was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (to G. I. D.), the Canadian Institutes of Health Research and a Canada Research Chair in Drug Development grants (to B.B.H.) and an American Chemical Society Petroleum Research Fund grant (to V.B.B.). An Ontario Graduate Scholarship (to G.L.A.) is gratefully acknowledged.Notes and references
- S. Ito, T. Matsuya, S. Omura, M. Ohtani and A. Nakagawa, J. Antibiot., 1970, 23, 315–317 CrossRef CAS.
- T. Hata, S. Omura, Y. Iwai, A. Nakagawa and M. Otani, J. Antibiot., 1971, 24, 353–359 CrossRef CAS.
- S. Omura, A. Nakagawa, H. Yamada, T. Hata, A. Furusaki and T. Watanabe, Chem. Pharm. Bull., 1971, 19, 2428–2430 CrossRef CAS.
- A. Furusaki, T. Watanabe, T. Hata, S. Omura, A. Nakagawa and M. Matsui, Isr. J. Chem., 1972, 10, 173–187 CrossRef CAS.
- S. Omura, A. Nakagawa, H. Yamada, T. Hata, A. Furusaki and T. Watanabe, Chem. Pharm. Bull., 1973, 21, 931–940 CrossRef CAS.
- A. M. Echevarren, N. Tamayo and M. C. Paredes, Tetrahedron Lett., 1993, 34, 4713 CrossRef.
- S. Mithani, G. Weeratunga, N. J. Taylor and G. I. Dmitrienko, J. Am. Chem. Soc., 1994, 116, 2209–2210 CrossRef CAS.
- S. J. Gould, N. Tamayo, C. R. Melville and M. C. Cone, J. Am. Chem. Soc., 1994, 116, 2207–2208 CrossRef CAS.
- See NCI138425 at http://www.dtp.nci.nih.gov.
- H. He, W. D. Ding, V. S. Bernan, A. D. Richardson, C. M. Ireland, M. Greenstein, G. A. Ellestad and G. T. Carter, J. Am. Chem. Soc., 2001, 123, 5362–5363 CrossRef CAS.
- S. B. Herzon and C. M. Woo, Nat. Prod. Rep., 2012, 29, 87–118 RSC.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS.
- M. Regitz and G. Maas, Diazo compounds: Properties and Synthesis, Academic Press, Orlando, 1986, p. 96 Search PubMed.
- H. Zollinger, Acc. Chem. Res., 1973, 6, 335–341 CrossRef CAS.
- D. P. Arya and D. J. Jebaratnam, J. Org. Chem., 1995, 60, 3268–3269 CrossRef CAS.
- R. S. Laufer and G. I. Dmitrienko, J. Am. Chem. Soc., 2002, 124, 1854–1855 CrossRef CAS PubMed.
- K. S. Feldman and K. J. Eastman, J. Am. Chem. Soc., 2006, 128, 12562–12573 CrossRef CAS PubMed.
- K. S. Feldman and K. J. Eastman, J. Am. Chem. Soc., 2005, 127, 15344–15345 CrossRef CAS PubMed.
- B. B. Hasinoff, X. Wu, J. C. Yalowich, V. Goodfellow, R. S. Laufer, O. Adedayo and G. I. Dmitrienko, Anti-Cancer Drugs, 2006, 17, 825–837 CrossRef CAS PubMed.
- K. A. O'Hara, X. Wu, D. Patel, H. Liang, J. C. Yalowich, N. Chen, V. Goodfellow, O. Adedayo, G. I. Dmitrienko and B. B. Hasinoff, Free Radical Biol. Med., 2007, 43, 1132–1144 CrossRef PubMed.
- W. Liu, M. Buck, N. Chen, M. Shang, N. J. Taylor, J. Asoud, X. Wu, B. B. Hasinoff and G. I. Dmitrienko, Org. Lett., 2007, 9, 2915–2918 CrossRef CAS PubMed.
- W. Zeng, T. E. Ballard, A. G. Tkachenko, V. A. Burns, D. L. Feldheim and C. Melander, Bioorg. Med. Chem. Lett., 2006, 16, 5148–5151 CrossRef CAS PubMed.
- T. E. Ballard and C. Melander, Tetrahedron Lett., 2008, 49, 3157–3161 CrossRef CAS PubMed.
- C. L. Heinecke and C. Melander, Tetrahedron Lett., 2010, 51, 1455–1458 CrossRef CAS PubMed.
- O. Khdour and E. B. Skibo, Org. Biomol. Chem., 2009, 7, 2140–2154 CAS.
- S. E. Rokita, Quinone methides, Wiley, Hoboken, N.J., 2009, p. 431 Search PubMed.
- H. W. Moore, Science, 1977, 197, 527–532 CAS.
- C. M. Woo, L. Lu, S. L. Gholap, D. R. Smith and S. B. Herzon, J. Am. Chem. Soc., 2010, 132, 2540–2541 CrossRef CAS PubMed.
- S. B. Herzon, L. Lu, C. M. Woo and S. L. Gholap, J. Am. Chem. Soc., 2011, 133, 7260–7263 CrossRef CAS PubMed.
- S. P. Mulcahy, C. M. Woo, W. D. Ding, G. A. Ellestad and S. B. Herzon, Chem. Sci., 2012, 3, 1070–1074 RSC.
- L. C. Colis, C. M. Woo, D. C. Hegan, Z. Li, P. M. Glazer and S. B. Herzon, Nat. Chem., 2014, 6, 504–510 CrossRef CAS PubMed.
- K. A. O'Hara, G. I. Dmitrienko and B. B. Hasinoff, Chem.-Biol. Interact., 2010, 184, 396–402 CrossRef PubMed.
- A. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, McGraw-Hill, New York, 1989, p. 127 Search PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science, Sausalito, Calif., 2006, pp. 14–15 Search PubMed.
- N. Chen, M. B. Carriere, R. S. Laufer, N. J. Taylor and G. I. Dmitrienko, Org. Lett., 2008, 10, 381–384 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1999, p. 23 Search PubMed.
- R. L. Hannan, R. B. Barber and H. Rapoport, J. Org. Chem., 1979, 44, 2153–2158 CrossRef CAS.
- R. W. Franck and R. B. Gupta, J. Org. Chem., 1985, 50, 4632–4635 CrossRef CAS.
- C. Bolchi, P. Catalano, L. Fumagalli, M. Gobbi, M. Pallavicini, A. Pedretti, L. Villa, G. Vistoli and E. Valoti, Bioorg. Med. Chem., 2004, 12, 4937–4951 CrossRef CAS PubMed.
- J. P. Wolfe, J. Ahman, J. P. Sadighi, R. A. Singer and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6367–6370 CrossRef CAS.
- B. Akermark, L. Eberson, E. Jonsson and E. Pettersson, J. Org. Chem., 1975, 40, 1365–1367 CrossRef CAS.
- H. J. Knolker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4427 CrossRef PubMed.
- R. E. Murray and G. Zweifel, Synthesis, 1980, 150–151 CrossRef CAS.
- L. N. Mander and S. P. Sethi, Tetrahedron Lett., 1984, 25, 5953–5956 CrossRef CAS.
- V. B. Birman, Z. F. Zhao and L. Guo, Org. Lett., 2007, 9, 1223–1225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational method, cell culture and growth inhibition assay details, experimental and synthetic procedures, full characterization data, 1H and 13C NMR spectra. See DOI: 10.1039/c4md00197d |
| This journal is © The Royal Society of Chemistry 2014 |