 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ω-Heteroarylalkylcarbamates as inhibitors of fatty acid amide hydrolase (FAAH)†
Tobias
Terwege
,
Helmut
Dahlhaus
,
Walburga
Hanekamp
and
Matthias
Lehr
*
Institute of Pharmaceutical and Medicinal Chemistry, University of Münster, Corrensstrasse 48, D-48149 Münster, Germany. E-mail: lehrm@uni-muenster.de; Fax: +49 251 8332144; Tel: +49 251 8333331
First published on 16th May 2014
Abstract
Fatty acid amide hydrolase (FAAH) is a serine hydrolase that terminates the analgetic and anti-inflammatory effects of endocannabinoids such as anandamide. Herein we describe structure–activity relationship studies on a new series of ω-heteroarylalkylcarbamate inhibitors of FAAH. The most active compounds exhibit IC50 values in the low nanomolar range. Investigations on selectivity and metabolic stability of these inhibitors are also presented.
Anandamide is one principal endocannabinoid in the mammalian organism.1,2 It is formed “on demand” during several pathological disorders and mediates analgetic and anti-inflammatory effects by activation of the cannabinoid receptors CB1 and CB2. These two receptors are differentially distributed within the various cells.3–5 While the CB1 receptor is preferentially expressed in neuronal tissues, the CB2 receptor occurs mainly in cells of the immune system such as macrophages.
An important enzyme in the endocannabinoid metabolism is fatty acid amide hydrolase (FAAH), which rapidly cleaves the lipid mediator anandamide into arachidonic acid and ethanolamine.6–9 Inhibition of FAAH is supposed to potentiate the action of anandamide. Therefore, inhibitors of FAAH may represent new agents against pain and inflammation. In the past years many potent inhibitors of FAAH have been found.10–14 The majority of these compounds are substances, which form covalent binding interactions with the catalytic serine of the active site of FAAH such as carbamates like 1 (URB 597)15 and 2,16 activated ketone derivatives like the α-ketoheterocycle 3 (PHOP)17 and the propan-2-one 4,18 (Fig. 1), and urea derivatives like 5 (PF-04457845).19 The latter compound is being examined in clinical studies for fear response and cannabis withdrawl.20
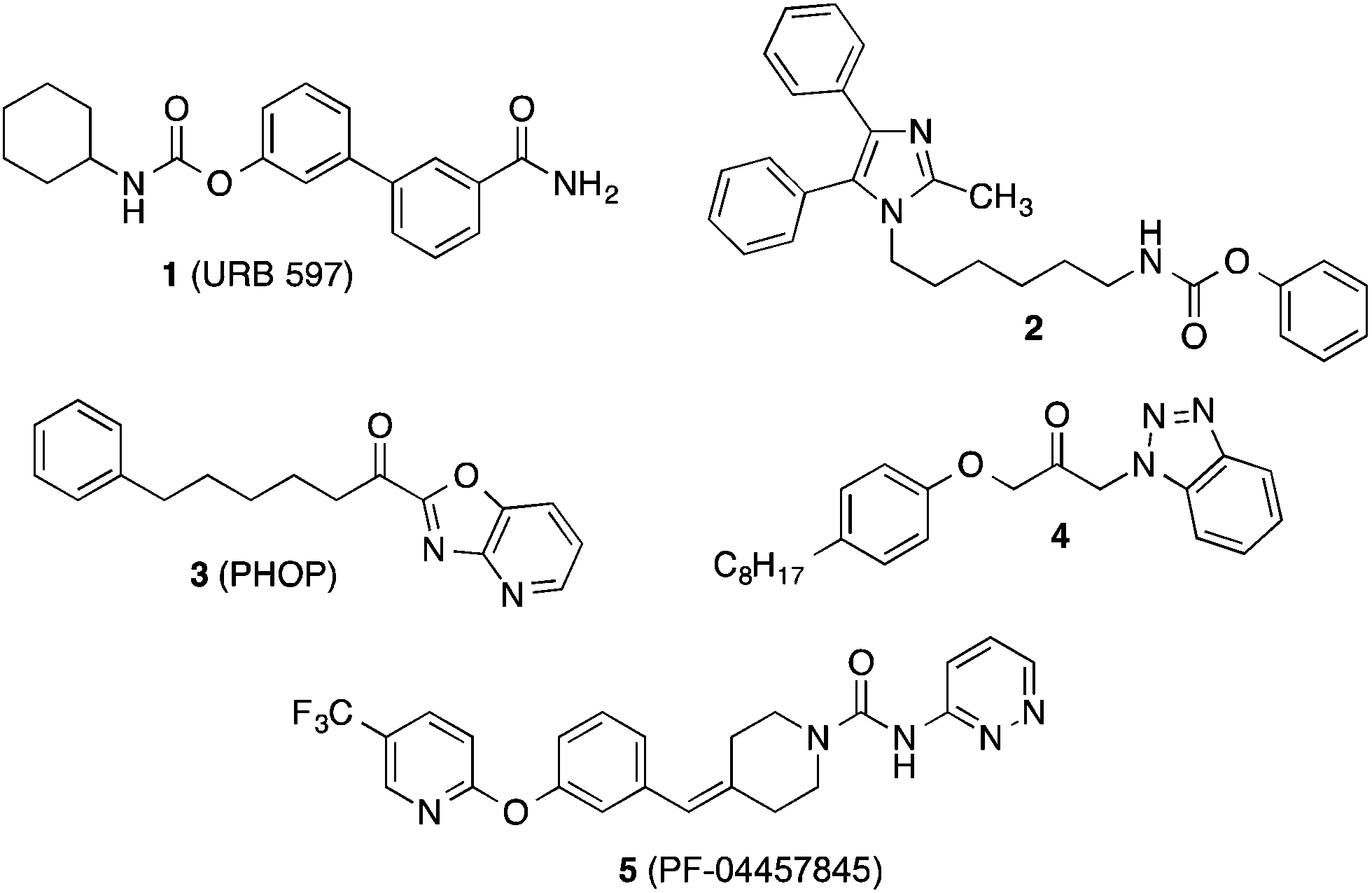 | ||
| Fig. 1 Structure of known inhibitors of FAAH. | ||
Several years ago we have described dual inhibitors of cyclooxygenase and 5-lipoxygenase, which contain a pyrrole scaffold with vicinal phenyl substituents (e.g.6)21 (Fig. 2) showing structural similarities with the heterocyclic part of the FAAH inhibitor 2.16 This initiated our interest in the question, in which way inhibitory potency of 2 is influenced when the 2-methyl-4,5-diphenylimidazole system of 2 is replaced by a 2,3-diphenyl-5-methylpyrrole substituent. Moreover, this pyrrole ring system offered the opportunity to introduce further substituents into the ring at the free carbon site, which could lead to additional binding interactions with FAAH. Therefore, we have synthesized the pyrrole analogue of 2 and used it as starting point for structure–activity relationship studies.
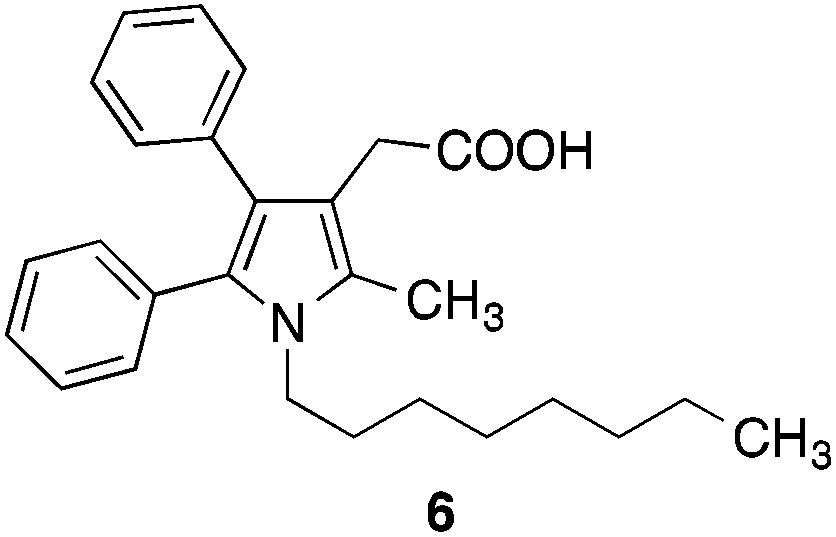 | ||
| Fig. 2 Structure of the dual cyclooxygenase–5-lipoxygenase inhibitor 6. | ||
The pyrrolyl-substituted N-alkylphenylcarbamates investigated were synthesized using one of the three routes outlined in Scheme 1. In the first instance, the appropriate nitrogen heterocycle was alkylated with a BOC-protected ω-bromoalkylamine followed by cleaving of the BOC group with trifluoroacetic acid and reaction of obtained amine with phenyl chloroformate. Alternatively, the heterocycle was alkylated with a dibromoalkane. The obtained bromoalkyl compound was treated with potassium phthalimide yielding a phthalimide-protected alkylamine. Removal of the protecting group was accomplished using hydrazine hydrate and the formed amine was directly reacted with phenyl chloroformate to yield the target compound. Instead, the phthalimide intermediate was directly prepared by reaction of the nitrogen heterocycle with ω-bromoalkyphthalimide as shown for the synthesis of 27 and 30.
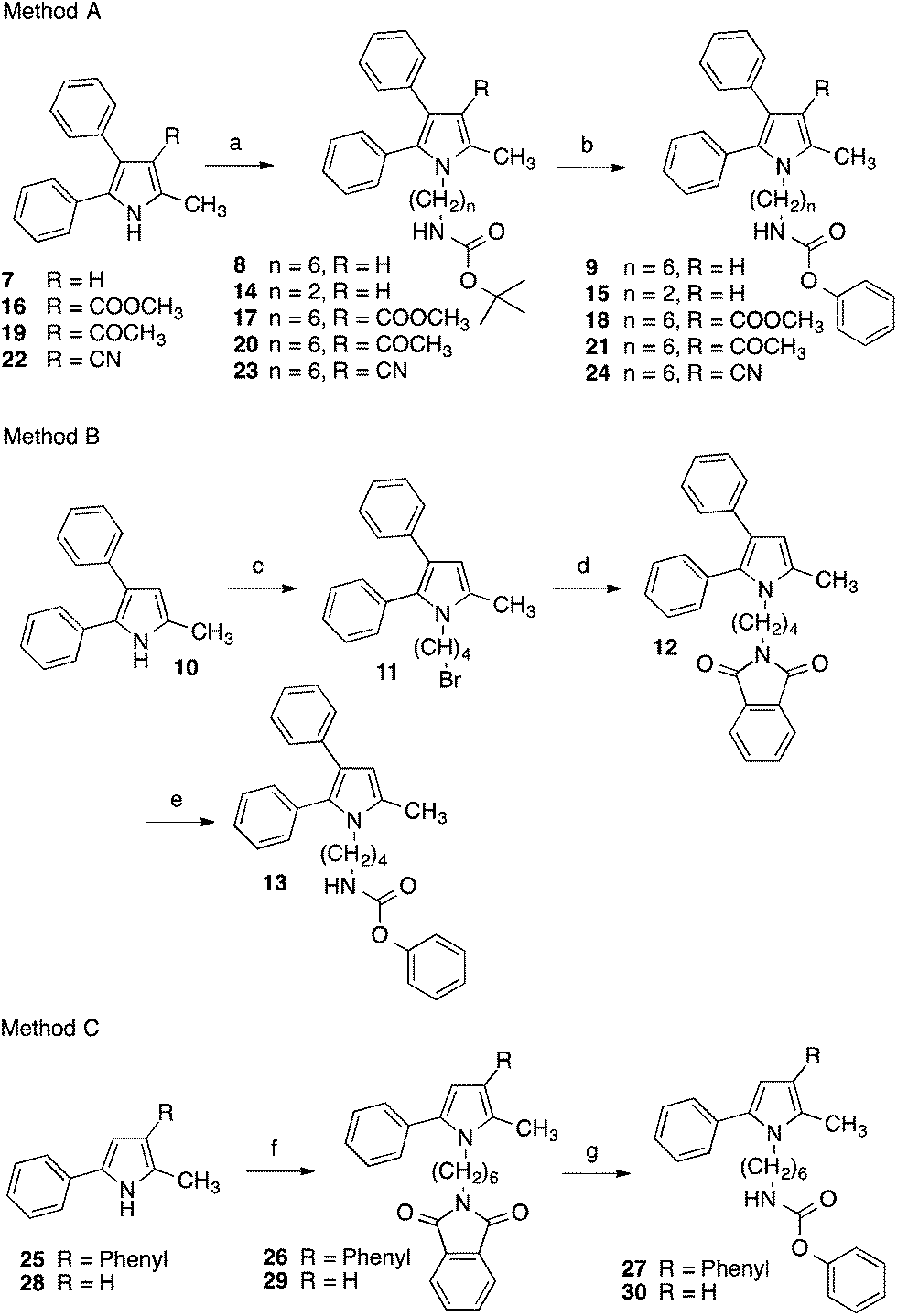 | ||
| Scheme 1 Reagents and conditions: (a) tert-butyl-N-(6-bromohexyl)carbamate–tert-butyl-N-(2-bromoethyl)carbamate, K-tert-butylate, DMSO, 60/80 °C; (b) (1) trifluoroacetic acid, CH2Cl2, 0 °C; (2) phenyl chloroformate, triethylamine, CH2Cl2, room temp.; (c) 1,4-dibromobutane, tetrabutylammonium bromide, toluene, 50% aqueous NaOH, 45 °C; (d) potassium phthalimide, DMF, 130 °C; (e) (1) hydrazine-hydrate, ethanol, reflux; (2) phenyl chloroformate, triethylamine, CH2Cl2, room temp.; (f) N-(6-bromohexyl)phthalimide, K-tert-butylate, DMSO, 50/70 °C; (g) (1) hydrazine-hydrate, ethanol, reflux; (2) phenyl chloroformate, triethylamine, CH2Cl2, 0 °C to room temp. | ||
The related derivatives with indole, indazole, imidazole and benzotriazole heterocycle were prepared similarly to Method C of Scheme 1 using K-tert-butylate–DMSO, NaH–DMSO, NaH–DMF, K2CO3–DMSO and K2CO3–acetonitrile, respectively, for the N-alkylation of the appropriate heterocycle with N-(ω-bromoalkyl)phthalimide.22
The indole derivatives with 3-pyridyl carbamate residues (52 and 54) were synthesized with the latter method utilizing 3-pyridyl chloroformate, synthesized from diphosgene and pyridin-3-ol, instead of phenyl chloroformate in the last step (Scheme 2).
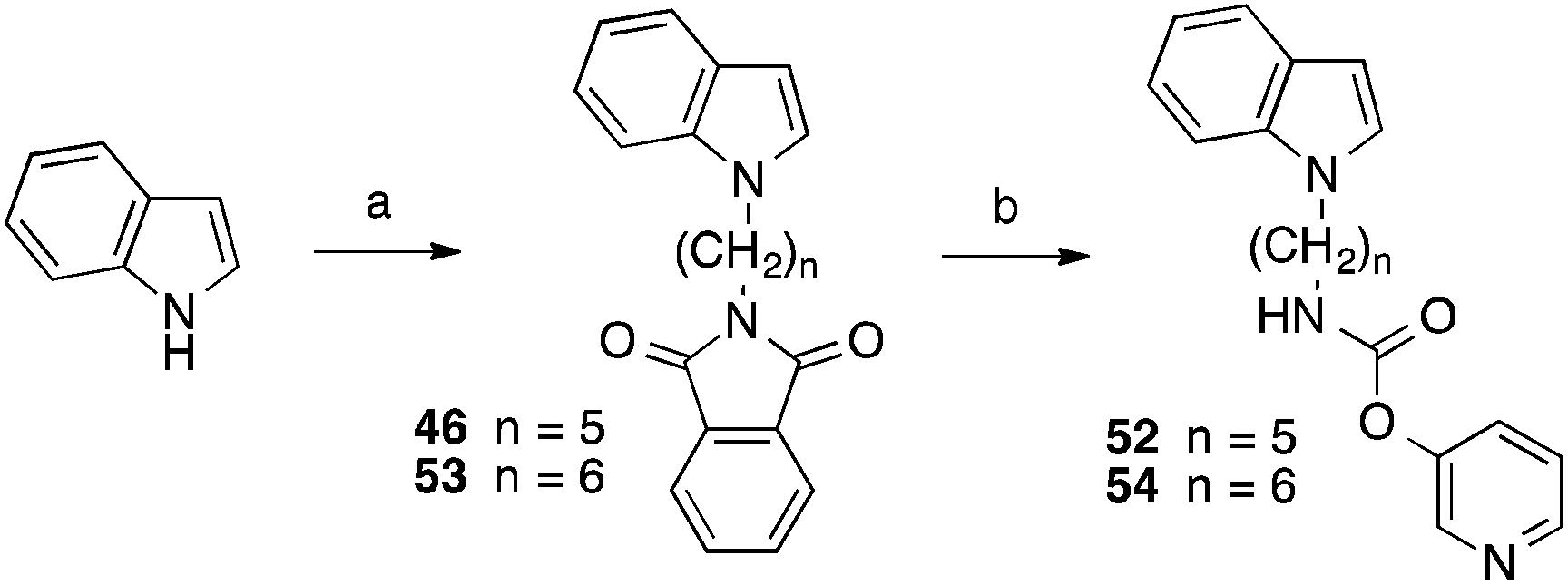 | ||
| Scheme 2 Reagents and conditions: (a) N-(ω-bromoalkyl)phthalimide, NaH, DMSO, 100 °C; (b) (1) hydrazine-hydrate, ethanol, reflux, 2 h; (2) trichloromethyl chloroformate, pyridin-3-ol, ethyl(diisopropyl)amine, CH2Cl2, THF, – 30 °C to room temp. | ||
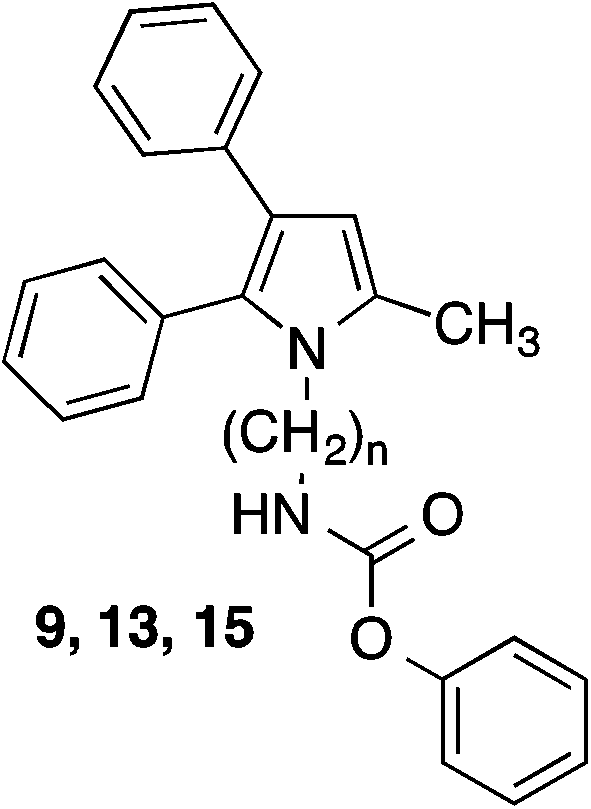
Inhibition of FAAH hydrolase by the test compounds was determined with an assay using microsomes from rat brain as enzyme source and N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide as fluorogenic substrate.23 Inhibitory potencies of the tested compounds were assessed by comparing the amount of 4-pyren-1-ylbutanoic acid released from the substrate in their absence and presence after an incubation time of 60 min by reversed-phase HPLC with fluorescence detection (see ESI†). With this assay for the diphenylimidazole 2 an IC50 of 0.35 μM was measured (Table 1). Replacement of the imidazole scaffold of 2 by a pyrrole (9) led to an about sevenfold loss of activity. Shortening of the alkyl chain connecting the carbamate group and the pyrrole heterocycle from six to four and two carbon atoms further decreased inhibitory potency. With an IC50 of 6.2 μM the butyl derivative 13 was only about half as active as the hexyl compound 9. The derivative with an ethyl linker (15) even was inactive at the highest test concentration of 10 μM. Because removing of the methyl group in position 2 of the imidazole ring of 2 did not change the activity of the compound,24 the impact of the corresponding methyl group of the pyrrole 9 on inhibitory potency was not examined.
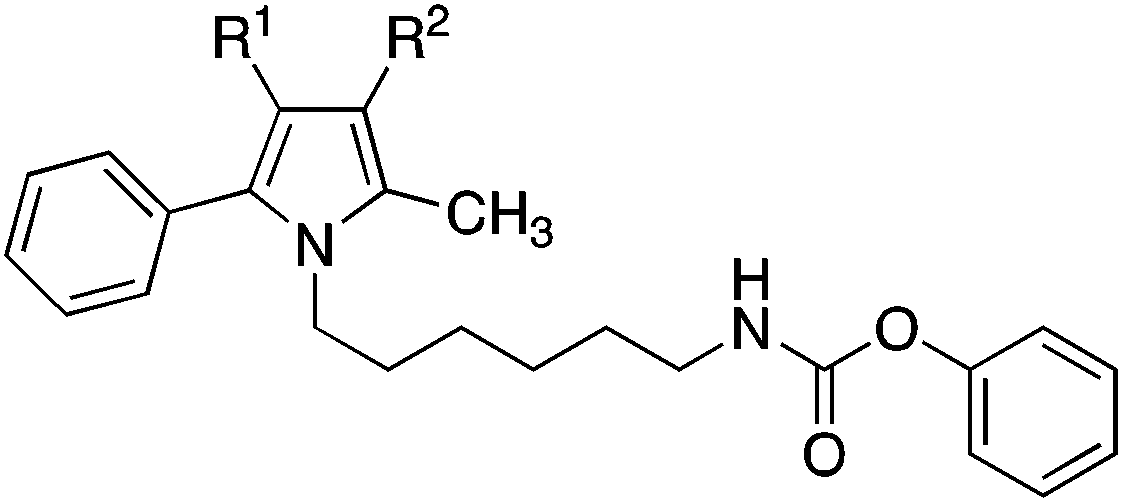
Introduction of polar ethoxycarbonyl, acetyl and cyano substituents at the free position of the pyrrole ring of 9 was also detrimental. The inhibition values of obtained compounds 18, 21 and 24 were in the magnitude of 10 μM (Table 2). Interestingly, moving the phenyl group from position 3 to position 4 of the pyrrole ring, to give compound 27, as well as completely removing the phenyl ring, to give compound 30, significantly increased FAAH inhibition. The activity of these two inhibitors was comparable to that of the diphenylimidazole 2.
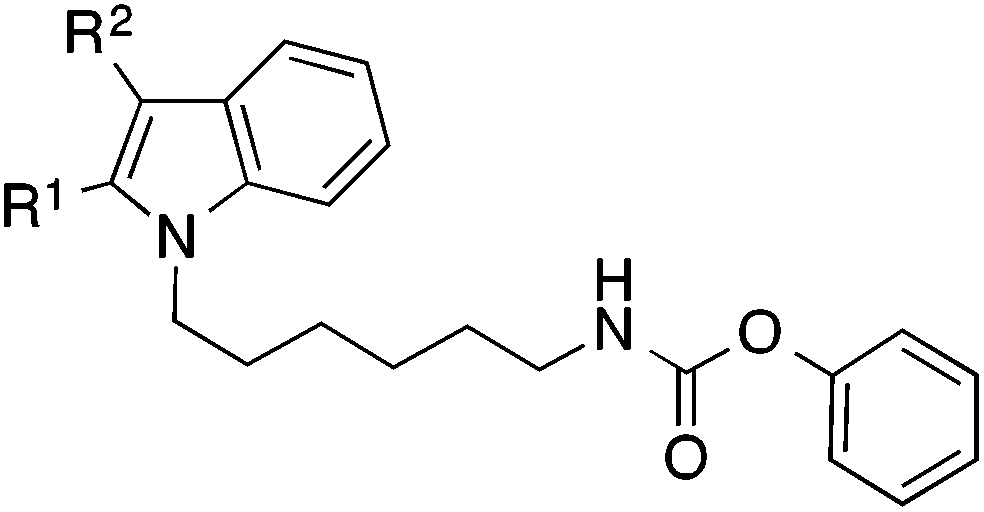
Similar results were obtained with analogous indole derivatives (Table 3). With IC50 values of about 5 μM the 2,3-diphenylindole 32 and the 3-phenylindole 34 possessed the lowest potency of the series. The 2-phenylindole 36 was about tenfold more active than the 3-phenylderivative 34. Thus, a phenyl substituent is much better tolerated in position 2 than in position 3 of the indole scaffold. The indole derivative 38, which has no phenyl-substituents in 2- and 3-position, possessed the highest activity (IC50 0.25 μM) showing that phenyl substituents are even unfavourable.
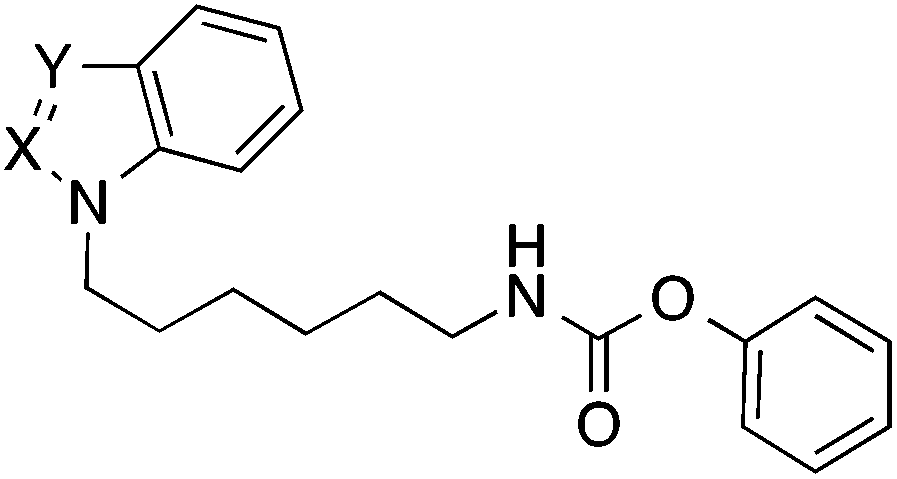
Next the effect of an introduction of nitrogen atoms in position 2 and 3 of the indole ring of 38 was studied. The indazole and benzotriazole derivatives 40 and 42 bearing a nitrogen in position 2 of the heterocycle were about three- to fourfold less active than the indole 38 (Table 4). In contrast, the imidazole derivative 44 possessing an additional nitrogen only in position 3 was equipotent to the indole 38.
Taken together, replacement of the diphenylpyrrole heterocycle from the starting compound 9 by an unsubstituted indole (38) led to an about tenfold increase of FAAH inhibitory potency. At this point we wanted to evaluate the role of the alkyl linker of 38 on activity in more detail. Both shortening and elongation of the hexyl chain by one carbon increased activity about twofold: the IC50 values of the pentyl and heptyl derivatives 47 and 49 were about 0.09 μM (Table 5). A more pronounced variation of the chain length to four (45) or eight (51) carbons resulted in a reduction of FAAH inhibition.
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | n | R | Inhibition of FAAH IC50a (μM) | Inhibition of MAGL IC50a (μM) | Stability in liver S9 fractionsb (%) | Stability in blood plasmac (%) |
| a Values are the means of at least two independent determinations, errors are within ± 20%. b Percent of parent remaining after incubation with rat liver S9 fractions for 30 min, in presence of the co-factor NADPH; values are means ± standard deviations of independent determinations (n = 3). c Percent of parent remaining after incubation with porcine blood plasma for 30 min; values are means ± standard deviations of independent determinations (n = 3). d n.a.: not active at 10 μM. e n.t.: not tested. | ||||||
| 45 | 4 | Phenyl | 0.96 | n.a.d | 53 ± 4.7 | 82 ± 9.0 |
| 47 | 5 | Phenyl | 0.094 | n.a.d | 52 ± 2.3 | 91 ± 6.5 |
| 38 | 6 | Phenyl | 0.25 | n.a.d | 50 ± 1.5 | 68 ± 7.8 |
| 49 | 7 | Phenyl | 0.090 | n.a.d | 59 ± 3.1 | 39 ± 7.4 |
| 51 | 8 | Phenyl | 0.24 | n.a.d | 49 ± 0.3 | 64 ± 4.2 |
| 52 | 5 | Pyridin-3-yl | 0.0036 | 0.22 | 43 ± 7.0 | 0 |
| 54 | 6 | Pyridin-3-yl | 0.0052 | 0.46 | 47 ± 13 | 0 |
| 1 (URB 597) | 0.060 | n.t.e | 13 ± 4.6 | 76 ± 12 | ||
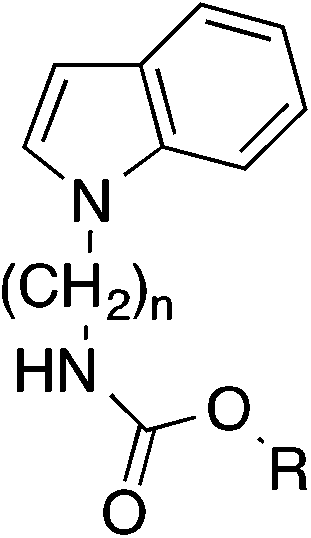
In literature a series of highly potent 3-pyridyl carbamate inhibitors of FAAH is described.25 Therefore, we finally wanted to investigate the effect of a replacement of the phenyl group of the carbamates 47 and 38 by a 3-pyridyl residue. This structural variation led to a drastic increase of activity (Table 5). With IC50 values of 0.0036 μM and 0.0052 μM obtained compounds 52 and 54 were about as active as the α-ketoheterocycle 3 (PHOP), which counts to the most active inhibitors of FAAH known today.
Besides FAAH, monoacylglycerol lipase (MAGL) is another important enzyme during the process of endocannabinoid inactivation. Just like FAAH, MAGL contains a catalytic serine in the active site. The main substrate of MAGL is 2-arachidonoyl glycerol. To get some information about the specificity of the developed substance class, some of the new FAAH inhibitors were also tested for MAGL inhibition.26 As shown in Table 5, the evaluated phenyl ω-(indol-1-yl)alkylcarbamates did not inhibit MAGL at the highest test concentration of 10 μM. Contrary, with IC50 values in the submicromolar range, the corresponding pyridyl carbamates 52 and 54 inhibited MAGL significantly. However, the inhibitory potency of these substances against FAAH is still 60- to 90-fold higher than against MAGL.
Another enzyme of the serine hydrolase family is cytosolic phospholipase A2α (cPLA2α), which catalyzes the first step of the so called arachidonic acid cascade by cleaving membrane phospholipids in arachidonic acid and lysophospholipids. At the highest test concentration of 10 μM cPLA2α was not inhibited27 by the indole compounds listed in Table 5. These results indicate that the inhibitors investigated display a certain specificity with regard to their ability to inhibit serine hydrolase enzymes.
Since carbamates are known to be susceptible to hydrolysis in biological environments28–30 leading to the inactivation of the compounds, we also measured the stability of some of the new substances in rat liver S9 fractions in presence of the co-factor NADPH31 and in porcine blood plasma (Table 5). After incubation in the liver S9 fractions for 30 min, still about 50% of the selected indole compounds were present in the incubation mixture. For comparison, the metabolism rate of the known FAAH inhibitor URB 597, which also exhibits a carbamate structure, was significantly higher. Here, only about 13% of the parent compound could be detected after the metabolic reactions. Incubation in blood plasma for 30 min revealed striking different results. The pyridin-3-yloxy carbamates 52 and 54 had totally disappeared at the end of the trial most likely due to an extensive hydrolysis or transcarbamoylation. The stability of the phenoxycarbamates depended on the chain length of the alkyl spacer. The pentyl-substituted compound 47 exhibited high stability, with 91% parent remaining after incubation. In contrast, only 39% of the heptyl derivative 49 could be detected under the same conditions.
Conclusions
In conclusion, starting from the lead compounds 2 and 9, respectively, a pronounced increase of FAAH inhibitory potency could be attained by only a few structural variations. The most active compounds synthesized (52 and 54) exhibited IC50 values in the low nanomolar range.Notes and references
- C. J. Fowler, Fundam. Clin. Pharmacol., 2006, 20, 549 CrossRef CAS PubMed.
- V. Di Marzo, Rev. Physiol. Biochem. Pharmacol., 2008, 160, 1 CAS.
- A. C. Howlett, F. Barth, T. I. Bonner, G. Cabral, P. Casellas, W. A. Devane, C. C. Felder, M. Herkenham, K. Mackie, B. R. Martin, R. Mechoulam and R. G. Pertwee, Pharmacol. Rev., 2002, 54, 161 CrossRef CAS.
- S. D. McAllister and M. Glass, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2002, 66, 161 CrossRef CAS PubMed.
- E. S. Graham, J. C. Ashton and M. Glass, Front. Biosci., 2009, 14, 944 CrossRef CAS PubMed.
- M. K. McKinney and B. F. Cravatt, Annu. Rev. Biochem., 2005, 74, 411 CrossRef CAS PubMed.
- G. Labar and C. Michaux, Chem. Biodiversity, 2007, 4, 1882 CAS.
- F. Fezza, C. De Simone, D. Amadio and M. Maccarrone, Subcell. Biochem., 2008, 49, 101 Search PubMed.
- K. Ahn, M. K. McKinney and B. F. Cravatt, Chem. Rev., 2008, 108, 1687 CrossRef CAS PubMed.
- S. M. Saario and J. T. Laitinen, Basic Clin. Pharmacol. Toxicol., 2007, 101, 287 CrossRef CAS PubMed.
- S. Vandevoorde, Curr. Top. Med. Chem., 2008, 8, 247 CrossRef CAS.
- K. Ahn, D. S. Johnson and B. F. Cravatt, Expert. Opin. Drug Discov., 2009, 4, 763 CrossRef CAS PubMed.
- H. Deng, Expert. Opin. Drug Discov., 2010, 5, 961 CrossRef CAS PubMed.
- I. K. Khanna and C. W. Alexander, CNS Neurol. Disord.: Drug Targets, 2011, 10, 545 CAS.
- S. Kathuria, S. Gaetani, D. Fegley, F. Valino, A. Duranti, A. Tontini, M. Mor, G. Tarzia, G. La Rana, A. Calignano, A. Giustino, M. Tattoli, M. Palmery, V. Cuomo and D. Piomelli, Nat. Med., 2002, 9, 76 CrossRef PubMed.
- S. Y. Sit, C. Conway, R. Bertekap, K. Xie, C. Bourin, K. Burris and H. Deng, Bioorg. Med. Chem. Lett., 2007, 17, 3287 CrossRef CAS PubMed.
- D. L. Boger, H. Sato, A. E. Lerner, M. P. Hedrick, R. A. Fecik, H. Miyauchi, G. D. Wilkie, B. J. Austin, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5044 CrossRef CAS.
- L. Forster, J. Ludwig, M. Kaptur, S. Bovens, A. Schulze Elfringhoff, A. Holtfrerich and M. Lehr, Bioorg. Med. Chem., 2010, 18, 945 CrossRef CAS PubMed.
- D. S. Johnson, C. Stiff, S. E. Lazerwith, S. R. Kesten, L. K. Fay, M. Morris, D. Beidler, M. B. Liimatta, S. E. Smith, D. T. Dudley, N. Sadagopan, S. N. Bhattachar, S. J. Kesten, T. K. Nomanbhoy, B. F. Cravatt and K. Ahn, ACS Med. Chem. Lett., 2011, 2, 91 CrossRef CAS PubMed.
- http://www.clinicaltrials.gov/ct2/results?term=PF-04457845 .
- G. Dannhardt and M. Lehr, Arch. Pharm., 1993, 326, 157 CrossRef CAS.
- M. Lehr and T. Terwege, DE102012018115, 2014.
- L. Forster, A. Schulze Elfringhoff and M. Lehr, Anal. Bioanal. Chem., 2009, 394, 1679 CrossRef CAS PubMed.
- T. Terwege, PhD thesis, University of Münster, 2011.
- T. Ishii, T. Sugane and J. Maeda, WO2006088075, 2006.
- A. Holtfrerich, T. Makharadze and M. Lehr, Anal. Biochem., 2010, 399, 218 CrossRef CAS PubMed.
- W. Hanekamp and M. Lehr, J. Chromatogr. B, 2012, 900, 7 CrossRef PubMed.
- F. Vacondio, C. Silva, A. Lodola, A. Fioni, S. Rivara, A. Duranti, A. Tontini, S. Sanchini, J. R. Clapper, D. Piomelli, M. Mor and G. Tarzia, ChemMedChem, 2009, 4, 1495 CrossRef CAS PubMed.
- F. Vacondio, C. Silva, A. Lodola, C. Carmi, S. Rivara, A. Duranti, A. Tontini, S. Sanchini, J. R. Clapper, D. Piomelli, G. Tarzia and M. Mor, Eur. J. Med. Chem., 2011, 46, 4466 CrossRef CAS PubMed.
- F. Vacondio, C. Silva, M. Mor and B. Testa, Drug. Metabol. Rev., 2010, 42, 551 CrossRef CAS PubMed.
- A. Holtfrerich, W. Hanekamp and M. Lehr, Eur. J. Med. Chem., 2013, 63, 64 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and analytical data of the test compounds; procedures for biological evaluation. See DOI: 10.1039/c4md00181h |
| This journal is © The Royal Society of Chemistry 2014 |