
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modeling, synthesis and NMR characterization of novel chimera compounds targeting STAT3†
Silvia
Dell'Orto
a,
Daniela
Masciocchi
a,
Stefania
Villa
a,
Fiorella
Meneghetti
a,
Giuseppe
Celentano
a,
Daniela
Barlocco
a,
Diego
Colombo
b,
Laura
Legnani
*bc,
Lucio
Toma
c,
Yoon Jung
Jeon
d,
Byoung-Mog
Kwon
d,
Akira
Asai
e and
Arianna
Gelain
*a
aDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, Via L. Mangiagalli 25, 20133 Milano, Italy. E-mail: arianna.gelain@unimi.it; Fax: +39-02-503-19359; Tel: +39-02-503-19369
bDipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano, Via Saldini 50, 20133 Milano, Italy
cDipartimento di Chimica, Università degli Studi di Pavia, Via Taramelli 12, 27100 Pavia, Italy. Fax: +39-0382-98-7323; Tel: +39-0382-98-7311
dLaboratory of Chemical Biology and Genomics, Korea Research Institute of Bioscience & Biotechnology and Department of Biomolecular Science, Korea University of Science and Technology, Eoun-Dong, Yuseong-gu, DaeJeon 305-333, South Korea
eCenter for Drug Discovery, Graduate School of Pharmaceutical Sciences, University of Shizuoka, 52-1 Yada, Suruga-ku, Shizuoka, 422-8526, Japan
First published on 1st August 2014
Abstract
Signal Transducer and Activator of Transcription 3 (STAT3) is a cytoplasmic factor that mediates intracellular signaling commonly generated at cell surface receptors and transmits it to the nucleus. In previous research studies we synthesized several molecules inhibiting STAT3 and among them oxadiazole MD77 and pyridazinone I showed an interesting activity. For the first one, a direct inhibition mechanism was determined, while I interfered within the STAT3 pathway at a different level. In order to discover novel compounds possibly endowed with an improved activity, we decided to merge their scaffolds on the basis of their calculated conformational properties. Therefore we designed and synthesized the chimera diastereomers 3-oxo-N-(4-(trifluoromethyl)phenyl)-2,3,4,4a,5,6-hexahydrobenzo[h]cinnoline-6-carboxamide (1, 2) and 3-oxo-N-(4-(trifluoromethyl)phenyl)-2,3,5,6-tetrahydrobenzo[h]cinnoline-6-carboxamide (3) as racemate and their enantiomeric separation was performed. Modeling studies and theoretical prediction of [α]D values were carried out beside NMR studies which allowed us to assign 1 and 2 relative configurations.
Introduction
STATs (Signal Transducers and Activators of Transcription) are latent cytoplasmic proteins transducing signals through the cytoplasm and acting as transcription factors in the nucleus, regulating cell growth and survival.1 They are constituted by several structurally and functionally conserved domains: among them, the Src homology 2 (SH2) domain is essential for the STAT activation cascade. STAT3, a member of the STAT family, has been found constitutively activated in a wide variety of human solid and blood tumors and the suppression of deregulated STAT3 activation leads to cancer cell apoptosis with tumor regression, slightly affecting normal cells.2In previous research studies focused on the discovery of new inhibitors targeting aberrant STAT3 signaling, we described the oxadiazole MD77,3 a direct STAT3 SH2 domain inhibitor (IC50 value 17.7 μM, by Alpha-Screen based assay) and the pyridazinone I,4 exhibiting good inhibitory activity (46% STAT3 inhibition at 2 μM concentration, by dual luciferase assay), but unable to bind the SH2 domain (Chart 1).
 | ||
| Chart 1 | ||
Docking studies performed on MD77 showed that, in the best scored pose of the STAT3–MD77 complex, it maintained the same geometry shown to be preferred in water solution, established by molecular modeling calculations, and characterized by torsional angles τ1 − τ4 more or less deviated from planarity.3 In this pose MD77 showed a series of interactions with suitable functional groups in the SH2 domain involving its various molecular portions, among which the p-trifluoromethylphenyl moiety engaged in several hydrogen bonds. As far as pyridazinone I is concerned, an important requisite for inhibitory activity versus STAT3 seems to be the planar or almost planar arrangement of the polycyclic system, that presents only one geometry significantly populated, in complete agreement with that described in a previous paper and obtained with a molecular mechanics approach.5
The main degree of conformational freedom in I is represented by the N2 side chain, able to assume two almost isoenergetic orientations, that, however, does not seem to exert a significant effect on the activity.
Taking into account the biological results of these previously studied molecules, and their geometrical features determined by molecular modeling calculations, we have chemically merged their scaffolds aided by molecular modeling calculations in order to identify new molecules endowed with enhanced activity against STAT3. Their synthesis was planned to verify if the MD77p-trifluoromethylphenyl moiety, which plays a crucial role for the binding to the target, could address the pyridazinone nucleus in proximity of the dimerization interface. This approach started from the above stated consideration concerning the almost planar conformation of the tricyclic moiety. Therefore, the chimeras 1–3 (Chart 2) were designed introducing on the central ring of I the p-trifluoromethylphenyl moiety of MD77 in a position able to maintain its original orientation within the protein binding pocket.
 | ||
| Chart 2 | ||
Here we report the synthesis of 1–3, the complete exploration of their conformational space performed through theoretical calculations, the racemate separations, the NMR characterization as well as the results of the biological assays.
Chemistry
Starting from the commercially available α-phenylglutaric anhydride, by treatment with polyphosphoric acid, the racemic acid 4 was obtained (Scheme 1). The carboxylic group of 4 was condensed with 4-(trifluoromethyl)aniline, in the presence of the coupling agent TBTU (O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoborate), to obtain the amide 5. Aldol condensation between 5 and glyoxylic acid, followed by dehydration, led to the α,β-unsaturated acid 6. | ||
| Scheme 1 Reagents and conditions: (a) PPA, 100/110 °C, 30 min; (b) 4-(trifluoromethyl)aniline, CH2Cl2, TBTU, N-methlymorphine upto pH = 7, rt, 24 h; (c) CHO–COOH, NaOH, H2O, EtOH, rt, 1 h; (d) Zn, CH3COOH, H2O, rt, 20 min; (e) NH2NH2·H2O, EtOH, reflux, 2 h; (f) Sodium 3-nitrobenzenesulfonate, NaOH, H2O, reflux, 30 min. | ||
Reduction of the double bond by zinc in acetic acid gave a mixture of the two diastereoisomers 7 and 8, which were not separated and directly cyclized with hydrazine monohydrate to give the diastereoisomers 1 and 2 which were separated by flash chromatography. Dehydrogenation of the dihydropyridazinonic ring of 1 was carried out with sodium 3-nitrobenzenesulfonate to give 3 while 2 did not react under the same conditions. The relative configurations of 1 and 2 were assigned by means of NOESY experiments.
Finally, in order to perform the biological evaluation of the single enantiomeric entities, we separated the racemates 1, 2 and 3 by means of a chiral semi-preparative HPLC.
Computational studies
The modeling studies on the diastereoisomers 1 and 2, and on compound 3 were carried out with the Gaussian09 program package using the B3LYP exchange–correlation functional at the 6-311+G(d,p) level.6 The studies were performed on the stereoisomers 1a, 2a and 3a. The discussion is referred to them but it also applies to their enantiomers 1b, 2b and 3b, whose conformers are mirror images of those of 1a–3a. The degrees of conformational freedom were considered, namely inversion of the central hexacyclic ring and rotation along the single bond connecting the amide carbonyl group to this ring, with energy minimization and optimization of all the possible starting geometries. After the optimization in vacuo, a further optimization was performed at the same level as above using a polarizable continuum solvent model (PCM), choosing chloroform, dimethyl sulfoxide, or methanol as the solvent, for 1a, 2a and 3a, respectively. Fig. 1 shows the minimum energy conformations of each compound together with the corresponding relative energy and the percentage contribution to the overall population at 298 K. A very different conformational behaviour was found between the two diastereoisomers 1a and 2a. In fact, whereas in 1a the trifluoromethylphenylamido chain almost exclusively (99.9%) prefers the pseudo-axial orientation, in its diastereoisomer 2a the opposite pseudo-equatorial orientation is largely preferred (96.2%). Compound 3a closely resembles 1a in the preference for the pseudo-axial orientation, though in a lesser percentage (89.4%).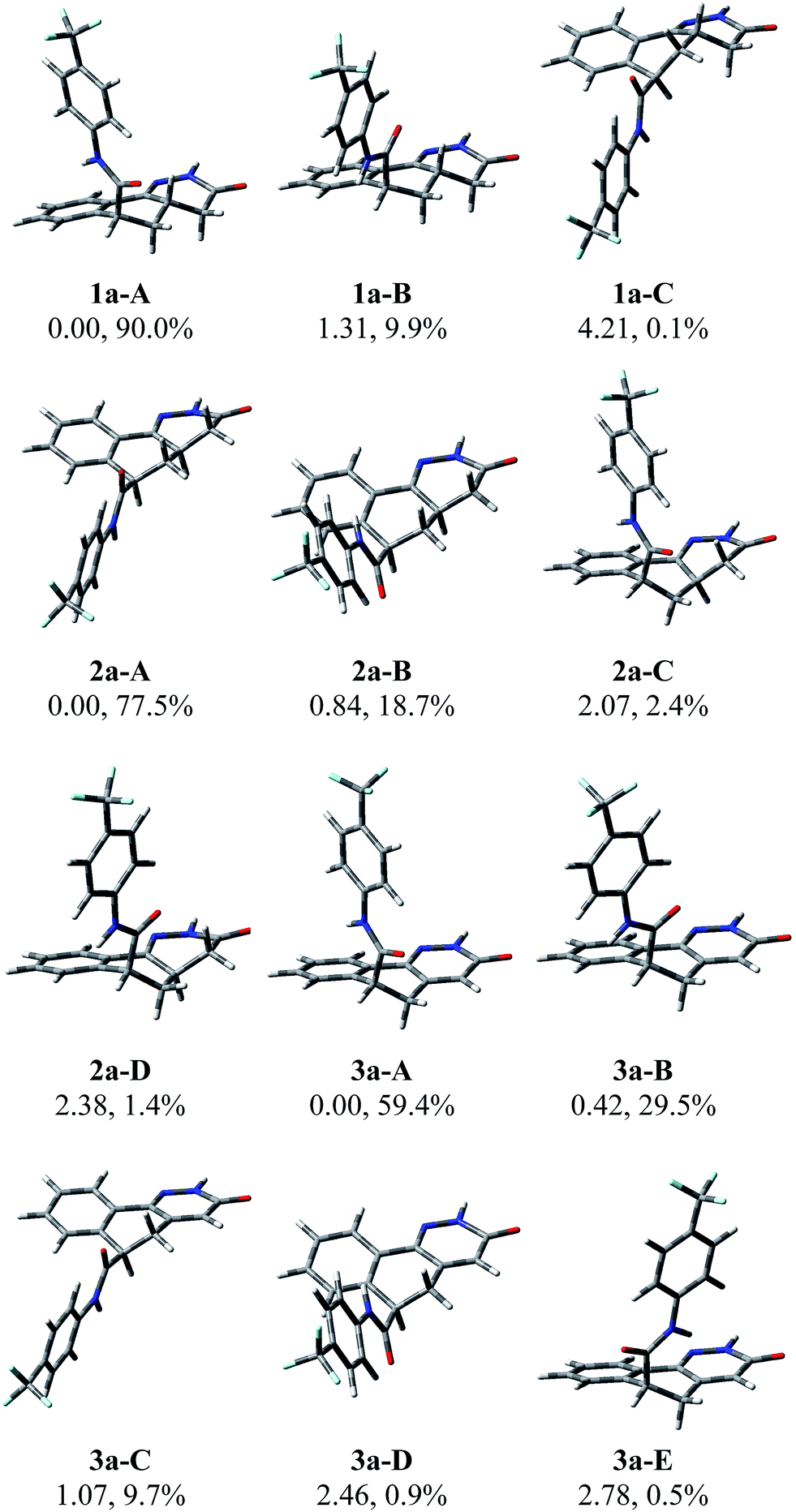 | ||
| Fig. 1 Three-dimensional plots of the populated conformers of compounds 1a (1a-A,B,C), 2a (2a-A,B,C,D) and 3a (3a-A,B,C,D,E) with their relative energy (kcal mol−1) and population percentages. | ||
Then, in an attempt to suggest the absolute configuration of these compounds, the specific optical rotation of each conformer of compounds 1a–3a was predicted using DFT calculations at the same level as above.7 The calculated [α]D for each conformer was weighted averaged on the basis of its percentage contribution to the overall population and yielded a positive value for 2a as well as for 3a ([α]D = +375 and +387, respectively) whereas the value obtained for 1a was too low for a reliable prediction. The largely positive values predicted for 2a and 3a, compared with the largely positive rotation values of the first eluted enantiomer of 2 and the second eluted enantiomer of 3 (see Experimental), allows us to suggest that the former has the (6R,4aR) configuration and the latter the (6R) configuration whereas no prediction seems possible for the enantiomers of 1.
1H-NMR studies
Racemic compounds 1, 2 and 3 were submitted to high field NMR spectroscopy (see Experimental) in order to get experimental data on their 3D structure. In particular for each compound 1H resonances were assigned and coupling constants measured. They are reported in Tables 1 and 2 together with the theoretical chemical shifts and coupling constants calculated for each optimized conformation of 1a–3a and averaged on the basis of the population percentages reported in Fig. 1.| H | 1 Exp.a | 1 Calcd | 2 Exp.b | 2 Calcd | 3 Exp.c | 3 Calcd |
|---|---|---|---|---|---|---|
| a CDCl3, 298 K. b DMSO-d6, 308 K. c CD3OD, 298 K. d The indexes ax and eq are referred to the most stable conformer of each compound. | ||||||
| 6 | 3.99 | 3.83 | 3.97 | 3.52 | 4.09 | 3.56 |
| 5axd | 1.89 | 1.71 | 1.95 | 2.07 | 3.20 | 2.93 |
| 5eqd | 2.84 | 2.85 | 2.29 | 2.08 | 3.26 | 3.47 |
| 4a | 3.01 | 2.80 | 2.98 | 2.69 | — | — |
| 4 | — | — | — | — | 6.83 | 6.78 |
| 4axd | 2.23 | 2.00 | 2.36 | 2.16 | — | — |
| 4eqd | 2.71 | 2.47 | 2.52 | 2.47 | — | — |
| J | 1 Exp.a | 1 Calcd | 2 Exp.b | 2 Calcd | 3 Exp.c | 3 Calcd |
|---|---|---|---|---|---|---|
| a CDCl3, 298 K. b DMSO-d6, 308 K. c CD3OD, 298 K. | ||||||
| 6,5ax | 4.8 | 5.1 | 12.5 | 10.2 | 5.4 | 6.1 |
| 6,5eq | 2.3 | 2.5 | 4.0 | 3.8 | 4.8 | 2.9 |
| 5ax,5eq | 13.0 | 11.8 | 12.5 | 11.6 | 16.4 | 14.5 |
| 5ax,4a | 13.0 | 11.1 | 12.5 | 10.4 | — | — |
| 5eq,4a | 5.0 | 4.8 | 4.0 | 4.9 | — | — |
| 5ax,4 | — | — | — | — | 1.8 | 2.2 |
| 5eq,4 | — | — | — | — | 1.0 | 0.5 |
| 4a,4ax | 16.0 | 13.4 | 16.0 | 13.5 | — | — |
| 4a,4eq | 6.5 | 6.1 | 6.6 | 6.1 | — | — |
| 4ax,4eq | 16.0 | 15.2 | 16.0 | 15.1 | — | — |
NOESY experiments were also performed to confirm the 1 and 2 relative configurations. In fact for the (6R,4aR)/(6S,4aS) configuration (Fig. 2B), a 2.6 Å calculated distance between the C-6 and C-4a protons should account for a NOE contact. On the contrary, this should not be observable for the (6R,4aS)/(6S,4aR) configuration (Fig. 2A) that showed a 3.8 Å calculated distance. Actually, the comparison of the NOESY spectrum of 1 with the corresponding spectrum of 2 (see Fig. S1a and S1b, ESI† section) clearly showed an intense cross peak only between H-6 and H-4a of 2 whose configuration was therefore ascertained to be (6R,4aR)/(6S,4aS). Consequently 1 resulted to be unambiguously the (6R,4aS)/(6S,4aR) isomer.
 | ||
| Fig. 2 Chemical structures of compounds 1a and 2a and their corresponding preferred conformers. In the 3D-plot of 2a a double arrow highlights the proximity of H-6 and H-4a that explains the NOESY observed cross-peak. | ||
Biological evaluation
The STAT3 inhibitory activity was evaluated at 10 μM and 50 μM concentrations through a modified procedure of dual luciferase assay8 in human colorectal carcinoma cells HCT-116, characterized by uncontrolled expression of STAT3. The results are expressed as percentage inhibition after 24 h treatment with the test compounds and Cryptotanshinone9 (a natural phenanthrene-quinone derivative) which was used as the control (Table 3).| Compound | % inh. [10 μM] | % inh. [50 μM] |
|---|---|---|
| 1 | −3 | 6 |
| (+)-1 | 9 | 35 |
| (−)-1 | 9 | 14 |
| 2 | −6 | 3 |
| (+)-2a | −8 | 12 |
| (−)-2b | 1 | 16 |
| 3 | 2 | 7 |
| (+)-3a | −2 | 24 |
| (−)-3b | 2 | 30 |
| Cryptotanshinone | 78 | 85 |
At the lower concentration none of the compounds showed any significant activity, while at 50 μM concentration the best derivative was (+)-1, which however was less active than cryptotanshinone (35% and 85%, respectively). Also, the AlphaScreen-based assay10 did not reveal any strong interaction of the chimeras with the STAT3 SH2 domain (% inhibition < 5), though they are incorporating the trifluoromethyl moiety that, in the case of MD77, cooperates in the binding to this domain.
Conclusion
During our studies, which aimed at the discovery of new potential STAT3 inhibitors, MD77 and I emerged as interesting molecules able to interfere in a different manner within the STAT3 pathway. These compounds were chemically merged leading to the chimeras1, 2 and 3, by utilizing the moieties identified as important for the STAT3 activity.The computational analysis on 1–3 revealed a very different conformational behavior of these compounds. In fact, while in 1 the trifluoromethylphenylamido chain almost exclusively prefers the pseudo-axial orientation, in 2 the opposite pseudo-equatorial orientation is largely preferred. Compound 3 closely resembles 1 in the preference for the pseudo-axial orientation, though in a lesser percentage. This geometrical diversity encouraged the synthetic efforts, in particular the orientation of the p-trifluoromethylphenyl group with respect to the tricyclic moiety in 2 that seems comparable with the orientation of the same group with respect to the chlorophenyl oxadiazole moiety in MD77. Thus, the chimeras were synthesized and their racemate resolved.
Then, high field NMR spectroscopy and NOESY experiments were performed to give experimental support to the calculations and unambiguously assign the 1 and 2 relative configurations. The vicinal J6,5ax coupling constant of 1 and 2 (Table 2) is in complete agreement with, respectively, an axial and equatorial orientation of the trifluoromethylphenylamido chain. The combination of the observed J6,5 and J5,4a in the two diastereoisomers is compatible with the assignment of the (6R,4aS)/(6S,4aR) relative configuration to 1 and the (6R,4aR)/(6S,4aS) to 2. Moreover, the intense cross-peak between H-6 and H-4a, observed only in the NOESY spectrum of 2, supports again its relative configuration, that makes these two hydrogens oriented on the same face of the molecule and relatively close (2.6 Å from the theoretical calculations) and not the diastereoisomeric (6R,4aS)/(6S,4aR) configuration, that makes them trans-oriented and more spaced (3.8 Å).
Finally, both the racemates and each single enantiomer of the new compounds were submitted to biological evaluation that did not bring about the identification of an advanced chemical lead, in spite of the presence in the molecules of moieties that have been shown suitable in other substrates. This indicates that the portions of MD77 not included in the chimera compounds seem to play a specific role that probably should not be neglected.
Experimental
General
Unless otherwise noted the materials were obtained from commercial suppliers (as starting reagents reported in Scheme 1) and used without purification. Commercial plates on aluminium-backed Silica Gel 60 plates (0.2 mm, Merck) were used for analytical TLC to follow the course of the reaction and to check the product purity. Silica gel 60 (Merck 40–63 μM) was used for flash chromatography to purify intermediates and final compounds. The purity of final compounds was determined by HPLC analysis and was ≥95%. Melting points were determined on a Buchi 510 capillary melting point apparatus and are uncorrected. 1H NMR spectra were recorded on a Bruker AVANCE-500 spectrometer operating at 500.13 MHz; the chemical shifts are reported as δ (ppm) using the solvent as the internal standard. The enantiomeric separations of compounds 1 and 3 were performed by a chiral semi-preparative HPLC Chiralcel ODH (5 μm, 250 × 4.6 mm). The enantiomeric separation of compound 2 was performed by a chiral semi-preparative HPLC Chiralpak AD (5 μm, 250 × 4.6 mm). Optical rotation values of the enantiomers were registered on a Perkin Elmer instrument (Mod 343) at 589 nm and 25 °C. The structures of all compounds were consistent with their analytical and spectroscopic data.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to afford the intermediate 5 (62% yield). 1H NMR (CD3OD) 2.43–2.48 (m, 2H, CH2), 2.64 (ddd, 1H, J = 17.5, J = 6.0 and J = 6.0 Hz, CH), 2.96 (ddd, 1H, J = 17.5, J = 14.0 and J = 6.2 Hz, CH), 4.13 (dd, 1H, J = 5.7 and J = 5.7 Hz, CH), 7.36 (dd, 1H, J = 7.50 and J = 1.0 Hz, ArH), 7.42 (ddd, 1H, J = 7.50, J = 7.50 and J = 1.0 Hz, ArH), 7.57 (ddd, 1H, J = 7.50, J = 7.50 and J = 1.5 Hz, ArH), 7.60 (d, 2H, J = 8.6 Hz, ArH), 7.80 (d, 2H, J = 8.6 Hz, ArH), 8.03 (dd, 1H, J = 7.50 and J = 1.5 Hz, ArH).
:1 with 0.02 mL of acetic acid) to provide compound 6 (47% yield). 1H NMR (CD3OD) 3.16 (ddd, 1H, J = 16.6, J = 4.8 and J = 2.7 Hz, CH), 4.13 (dd, 1H, J = 16.6 and J = 4.0 Hz, CH), 4.22 (dd, 1H, J = 4.8 and J = 4.0 Hz, CH), 6.91 (br s, 1H, =CH), 7.46 (dd, 1H, J = 7.50 and J = 1.0 Hz, ArH), 7.48 (ddd, 1H, J = 7.80, J = 7.50 and J = 1.1 Hz, ArH), 7.56 (d, 2H, J = 8.5 Hz, ArH), 7.61 (ddd, 1H, J = 7.50, J = 7.50 and J = 1.4 Hz, ArH), 7.70 (d, 2H, J = 8.5 Hz, ArH), 8.07 (dd, 1H, J = 7.8 and J = 1.4 Hz, ArH).
:2 = 8:2) were separated by flash chromatography (dichloromethane–methanol 9.8:0.2). Compound 1 (yellow solid): mp 230.2–232.4 °C. 1H NMR (CDCl3) 1.89 (ddd, 1H, J = 13.0, J = 13.0 and J = 4.8 Hz, CH), 2.23 (dd, 1H, J = 16.0 and J = 16.0 Hz, CH), 2.71 (dd, 1H, J = 16.0 and J = 6.5 Hz, CH), 2.84 (ddd, 1H, J = 13.0, J = 5.0 and J = 2.3 Hz, CH), 3.00 (dddd, J = 16.0, J = 13.0, J = 6.5 and J = 5.0 Hz, 1H, CH), 3.99 (dd, 1H, J = 4.8 and J = 2.3 Hz, CH), 7.10 (br s, 1H, NH), 7.34 (dd, 1H, J = 7.0 and J = 1.9 Hz, ArH), 7.42–7.52 (m, 6H, ArH), 8.23 (dd, 1H, J = 7.4 and J = 1.9 Hz, ArH), 8.52 (br s, 1H, NH). The enantiomeric separation of 1 was performed by a chiral semi-preparative HPLC: Chiralcel ODH (5 μm, 250 × 4.6 mm), hexane–ethanol (85:15), 0.8 mL min−1, UV 254 nm; tR(1) = 10.564 min (49.92%). (+)-1: [α]25D = +16 (c 0.0016, CH3OH); tR(2) = 12.503 min (50.08%) (−)-1: [α]25D = −16 (c 0.0016, CH3OH). Compound 2 (white solid): mp (277.1–279.8 °C). 1H NMR (DMSO-d6) 1.95 (ddd, 1H, J = 12.5, J = 12.5 and J = 12.5 Hz, 1H, CH), 2.29 (ddd, 1H, J = 12.5, J = 4.0 and J = 4.0 Hz, CH), 2.36 (dd, 1H, J = 16.0 and J = 16.0 Hz, CH), 2.52 (dd, 1H, J = 16.0 and J = 6.6 Hz, CH), 2.98 (dddd, J = 16.0, J = 12.5, J = 6.6 and J = 4.0 Hz, 1H, CH), 3.97 (dd, 1H, J = 12.5 and J = 4.0 Hz, CH), 7.16 (dd, 1H, J = 7.20 and J = 1.0 Hz, ArH), 7.34 (ddd, 1H, J = 7.80, J = 7.20 and J = 1.0 Hz, ArH), 7.37 (ddd, 1H, J = 7.20, J = 7.20 and J = 1.8 Hz, ArH), 7.73 (d, 2H, J = 8.5 Hz, ArH), 7.92 (d, 2H, J = 8.5 Hz, ArH), 8.08 (dd, 1H, J = 7.8 and J = 1.8 Hz, ArH). The enantiomeric separation of 2 was performed by a chiral semi-preparative HPLC: Chiralpak AD (5 μm, 250 × 4.6 mm), hexane–ethanol (85:15), 0.8 mL min−1, UV 254 nm; tR(1) = 14.970 min (49.68%). 2a: [α]25D = +264 (c 0.0012, DMSO); tR(2) = 19.781 min (50.32%). 2b: [α]25D = −287 (c 0.0012, DMSO).
:0.3) to afford 3 as a yellow solid (85% yield). Sublimation was performed at 333.4–338.7 °C. 1H NMR (CD3OD) 3.20 (ddd, 1H, J = 16.4, J = 5.4 and J = 1.8 Hz, CH), 3.26 (ddd, 1H, J = 16.4, J = 4.8 and J = 1.0 Hz, CH), 4.09 (dd, J = 5.4 and J = 4.8 Hz, 1H, CH), 6.83 (br s, 1H, CH), 7.38–7.44 (m, 3H, ArH), 7.57 (d, 2H, J = 8.5 Hz, ArH), 7.71 (d, 2H, J = 8.5 Hz, ArH), 8.14 (m, 1H, ArH). The enantiomeric separation of compound 3 was performed by a chiral semi-preparative HPLC: Chiralcel ODH (5 μm, 250 × 4.6 mm), hexane–ethanol (80:20), 0.8 mL min−1, UV 254 nm; tR(1) = 8.244 min (50.3%) 3b: [α]25D = −210 (c 0.0016, CH3OH); tR(2) = 8.968 min (49.7%) 3a: [α]25D = +194 (c 0.0016, CH3OH).
:3) to afford the intermediate 5 (62% yield). 1H NMR (CD3OD) 2.43–2.48 (m, 2H, CH2), 2.64 (ddd, 1H, J = 17.5, J = 6.0 and J = 6.0 Hz, CH), 2.96 (ddd, 1H, J = 17.5, J = 14.0 and J = 6.2 Hz, CH), 4.13 (dd, 1H, J = 5.7 and J = 5.7 Hz, CH), 7.36 (dd, 1H, J = 7.50 and J = 1.0 Hz, ArH), 7.42 (ddd, 1H, J = 7.50, J = 7.50 and J = 1.0 Hz, ArH), 7.57 (ddd, 1H, J = 7.50, J = 7.50 and J = 1.5 Hz, ArH), 7.60 (d, 2H, J = 8.6 Hz, ArH), 7.80 (d, 2H, J = 8.6 Hz, ArH), 8.03 (dd, 1H, J = 7.50 and J = 1.5 Hz, ArH).
:1 with 0.02 mL of acetic acid) to provide compound 6 (47% yield). 1H NMR (CD3OD) 3.16 (ddd, 1H, J = 16.6, J = 4.8 and J = 2.7 Hz, CH), 4.13 (dd, 1H, J = 16.6 and J = 4.0 Hz, CH), 4.22 (dd, 1H, J = 4.8 and J = 4.0 Hz, CH), 6.91 (br s, 1H, =CH), 7.46 (dd, 1H, J = 7.50 and J = 1.0 Hz, ArH), 7.48 (ddd, 1H, J = 7.80, J = 7.50 and J = 1.1 Hz, ArH), 7.56 (d, 2H, J = 8.5 Hz, ArH), 7.61 (ddd, 1H, J = 7.50, J = 7.50 and J = 1.4 Hz, ArH), 7.70 (d, 2H, J = 8.5 Hz, ArH), 8.07 (dd, 1H, J = 7.8 and J = 1.4 Hz, ArH).
:2 = 8:2) were separated by flash chromatography (dichloromethane–methanol 9.8:0.2). Compound 1 (yellow solid): mp 230.2–232.4 °C. 1H NMR (CDCl3) 1.89 (ddd, 1H, J = 13.0, J = 13.0 and J = 4.8 Hz, CH), 2.23 (dd, 1H, J = 16.0 and J = 16.0 Hz, CH), 2.71 (dd, 1H, J = 16.0 and J = 6.5 Hz, CH), 2.84 (ddd, 1H, J = 13.0, J = 5.0 and J = 2.3 Hz, CH), 3.00 (dddd, J = 16.0, J = 13.0, J = 6.5 and J = 5.0 Hz, 1H, CH), 3.99 (dd, 1H, J = 4.8 and J = 2.3 Hz, CH), 7.10 (br s, 1H, NH), 7.34 (dd, 1H, J = 7.0 and J = 1.9 Hz, ArH), 7.42–7.52 (m, 6H, ArH), 8.23 (dd, 1H, J = 7.4 and J = 1.9 Hz, ArH), 8.52 (br s, 1H, NH). The enantiomeric separation of 1 was performed by a chiral semi-preparative HPLC: Chiralcel ODH (5 μm, 250 × 4.6 mm), hexane–ethanol (85:15), 0.8 mL min−1, UV 254 nm; tR(1) = 10.564 min (49.92%). (+)-1: [α]25D = +16 (c 0.0016, CH3OH); tR(2) = 12.503 min (50.08%) (−)-1: [α]25D = −16 (c 0.0016, CH3OH). Compound 2 (white solid): mp (277.1–279.8 °C). 1H NMR (DMSO-d6) 1.95 (ddd, 1H, J = 12.5, J = 12.5 and J = 12.5 Hz, 1H, CH), 2.29 (ddd, 1H, J = 12.5, J = 4.0 and J = 4.0 Hz, CH), 2.36 (dd, 1H, J = 16.0 and J = 16.0 Hz, CH), 2.52 (dd, 1H, J = 16.0 and J = 6.6 Hz, CH), 2.98 (dddd, J = 16.0, J = 12.5, J = 6.6 and J = 4.0 Hz, 1H, CH), 3.97 (dd, 1H, J = 12.5 and J = 4.0 Hz, CH), 7.16 (dd, 1H, J = 7.20 and J = 1.0 Hz, ArH), 7.34 (ddd, 1H, J = 7.80, J = 7.20 and J = 1.0 Hz, ArH), 7.37 (ddd, 1H, J = 7.20, J = 7.20 and J = 1.8 Hz, ArH), 7.73 (d, 2H, J = 8.5 Hz, ArH), 7.92 (d, 2H, J = 8.5 Hz, ArH), 8.08 (dd, 1H, J = 7.8 and J = 1.8 Hz, ArH). The enantiomeric separation of 2 was performed by a chiral semi-preparative HPLC: Chiralpak AD (5 μm, 250 × 4.6 mm), hexane–ethanol (85:15), 0.8 mL min−1, UV 254 nm; tR(1) = 14.970 min (49.68%). 2a: [α]25D = +264 (c 0.0012, DMSO); tR(2) = 19.781 min (50.32%). 2b: [α]25D = −287 (c 0.0012, DMSO).
:0.3) to afford 3 as a yellow solid (85% yield). Sublimation was performed at 333.4–338.7 °C. 1H NMR (CD3OD) 3.20 (ddd, 1H, J = 16.4, J = 5.4 and J = 1.8 Hz, CH), 3.26 (ddd, 1H, J = 16.4, J = 4.8 and J = 1.0 Hz, CH), 4.09 (dd, J = 5.4 and J = 4.8 Hz, 1H, CH), 6.83 (br s, 1H, CH), 7.38–7.44 (m, 3H, ArH), 7.57 (d, 2H, J = 8.5 Hz, ArH), 7.71 (d, 2H, J = 8.5 Hz, ArH), 8.14 (m, 1H, ArH). The enantiomeric separation of compound 3 was performed by a chiral semi-preparative HPLC: Chiralcel ODH (5 μm, 250 × 4.6 mm), hexane–ethanol (80:20), 0.8 mL min−1, UV 254 nm; tR(1) = 8.244 min (50.3%) 3b: [α]25D = −210 (c 0.0016, CH3OH); tR(2) = 8.968 min (49.7%) 3a: [α]25D = +194 (c 0.0016, CH3OH).
NMR spectroscopy
NMR spectra were recorded at 298 K or 308 K (DMSO-d6 spectra) with a Bruker AVANCE-500 spectrometer operating at 500.13 MHz for 1H, using a 5 mm single pulsed field gradient (z-PFG) broadband reverse probe. Chemical shifts are reported on the δ (ppm) scale and are relative to chloroform (7.24 ppm), DMSO (2.51 ppm) or methanol (3.30 ppm) signals. Compounds (about 5 mg) were dissolved in 0.5 mL of CDCl3 (1), DMSO-d6 (2) or CD3OD (3) under N2, and their assignments were given by a combination of 1D and 2D COSY experiments, using standard Bruker pulse programs. Z-PFGs were used to obtain 1H–1H COSY spectra. The pulse width was 7.50 μs (90°) for 1H. Typically 32768 data points were collected for one-dimensional spectra. The spectral width was 11.45 ppm (5733 Hz) for 1H NMR (digital resolution: 0.17 Hz per point). 2D experimental parameters were as follows. For 1H–1H correlations: relaxation delay 2.0 s, 1024 × 1024 data point matrices (512 experiments to 1024 zero filling in F1, 1024 in F2), 2 or 16 transients in each experiment for COSY and NOESY respectively, spectral width 11.15 ppm (5580.37 Hz). The NOESY spectra were generated with a mixing time of 1.0 s and acquired in the TPPI mode. There were no significant differences in the results obtained at different mixing times (0.5–1.5 s). All 2D spectra were processed with the Bruker software package.
Computational methods
All calculations were performed using the Gaussian09 program package,6 at the B3LYP/6-311+G(d,p) level. Theoretical values of the [α]D were obtained at the same level of theory specifying appropriate keywords (polar and cphf) into the Gaussian input. GIAO NMR11 calculations were carried out at the B3LYP/6-311+G(d,p) level. The coupling constants were calculated with Ramsey's nonrelativistic approach12 and were weight-averaged on the basis of the population percentages.Biological assays
Acknowledgements
The authors acknowledge the Universities of Milan and Pavia, and CINECA for the allocation of computer time. This study was supported by funds from PRIN 2010-2011. B. M. Kwon was supported by the Bio & Medical Technology Development Program (2012M3A9C404877).Notes and references
- J. E. Darnell Jr, Science, 1997, 277, 1630–1635 CrossRef CAS.
- D. Masciocchi, A. Gelain, S. Villa, F. Meneghetti and D. Barlocco, Future Med. Chem., 2011, 3, 367–397 CrossRef PubMed.
- D. Masciocchi, S. Villa, F. Meneghetti, A. Pedretti, L. Legnani, L. Toma, B.-M. Kwon, S. Nakano, A. Asai and A. Gelain, MedChemComm, 2012, 3, 592–599 RSC.
- D. Masciocchi, A. Gelain, F. Meneghetti, A. Pedretti, G. Celentano, D. Barlocco, L. Legnani, L. Toma, B.-M. Kwon, A. Asai and S. Villa, MedChemComm, 2013, 4, 1181–1188 RSC.
- L. Toma, G. Cignarella, D. Barlocco and F. Ronchetti, J. Med. Chem., 1990, 33, 1591–1594 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. NakatsuJi, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. NakaJima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) P. J. Stephens, F. J. Devlin, J. R. Cheeseman, M. J. Frisch and C. Rosini, Org. Lett., 2002, 4, 4595–4598 CrossRef CAS PubMed; (b) B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS; (c) E. Giorgio, M. RoJe, K. Tanaka, Z. Hamersak, V. Sunjic, K. Nakanishi, C. Rosini and N. Berova, J. Org. Chem., 2005, 70, 6557–6563 CrossRef CAS PubMed; (d) A. C. Petrovic, J. He, P. L. Polavarapu, L. S. Xiao and D. W. Armstrong, Org. Biomol. Chem., 2005, 3, 1977–1981 RSC; (e) M. Zappalà, G. Postorino, N. Micale, S. Caccamese, N. Parrinello, G. Grazioso, G. Roda, F. S. Menniti, G. De Sarro and S. Grasso, J. Med. Chem., 2006, 49, 575–581 CrossRef PubMed.
- B. A. Sherf, S. L. Navarro, R. R. Hannah and K. V. Wood, Promega Notes Mag., 1996, 57, 2–8 Search PubMed.
- D.-S. Shin, H.-N. Kim, K. D. Shin, Y. J. Yoon, S.-J. Kim, D. C. Han and B.-M. Kwon, Cancer Res., 2009, 69, 193–202 CrossRef CAS PubMed.
- Y. Uehara, M. Mochizuki, K. Matsuno, T. Haino and A. Asai, Biochem. Biophys. Res. Commun., 2009, 380, 627–631 CrossRef CAS PubMed.
- (a) K. Wolinski, F. James, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS; (b) R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- T. Helgaker, M. Watson and N. C. Handy, J. Chem. Phys., 2000, 113, 9402–9409 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4md00177j |
| This journal is © The Royal Society of Chemistry 2014 |