 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformational analysis of peramivir reveals critical differences between free and enzyme-bound states†
Michele R.
Richards
b,
Michael G.
Brant
a,
Martin J.
Boulanger
c,
Christopher W.
Cairo
b and
Jeremy E.
Wulff
*a
aDepartment of Chemistry, University of Victoria, Victoria British Columbia V8W 3V6, Canada. E-mail: wulff@uvic.ca
bAlberta Glycomics Centre, Department of Chemistry, University of Alberta, Edmonton Alberta T6G 2G2, Canada
cDepartment of Biochemistry and Microbiology, University of Victoria, Victoria British Columbia V8W 3V6, Canada
First published on 19th June 2014
Abstract
Peramivir is a potent inhibitor of influenza neuraminidase, and is used clinically to treat influenza infections. The substantial potency of peramivir for its target suggests that similar structures might be useful as lead compounds for designing inhibitors of related viral, mammalian, or bacterial neuraminidases. At the same time, the large number of rotatable bonds in peramivir's structure led us to consider the conformational flexibility for the drug, since a more flexible scaffold might be a disadvantage in cases where isoenzyme selectivity is required. An examination of previously published X-ray data for the free and bound states of the drug, together with solution-phase NMR, conformational analysis, and DFT calculations leads us to conclude that peramivir undergoes a substantial conformational shift upon binding to the neuraminidase active site. Peramivir's previously unrecognized conformational flexibility may be a liability for peramivir itself, or for future applications of the underlying cyclopentane scaffold. Our analysis finds a consensus among enzyme-bound conformations of the inhibitor, and suggests that favoring this conformation could be used to develop inhibitors with greater potency or isoform selectivity.
Introduction
Pathogens including viruses, bacteria, and parasites have developed sophisticated mechanisms to engage surface structures on host cells as a crucial step towards establishing infection. Sialic acid (or N-acetyl neuraminic acid, Neu5Ac) residues on host cells frequently underpin the host–pathogen interaction,1 and enzymes that cleave these residues (sialidases or neuraminidases) are often identified as critical to the progression of the disease. Inhibitors of neuraminidase enzymes therefore have emerged as high-value drug candidates.2The value of this approach is particularly evident in the influenza field, where three such inhibitors – oseltamivir (Tamiflu™),3 zanamivir (Relenza™),4 and peramivir (PeramiFlu™ or Rapiacta™)5,6 – are used clinically to control the spread and symptoms of both seasonal and pandemic flu (see Fig. 1 for structures). At the same time, bacterial neuraminidases have long been appreciated as virulence factors for pneumonia7 and cholera8 (as well as several other pathogenic bacteria), while the related hemagglutinin–neuraminidase fusion protein is now seen as a therapeutic target for the prevention of parainfluenza-induced croup and bronchiolitis.9,10 Perhaps most interestingly, isoenzyme-specific inhibitors of mammalian neuraminidase enzymes11 may have value as anti-cancer agents (in that the mamalian NEU3 enzyme is highly expressed in several solid tumors)12 or as preservative agents for blood platelets.13,14
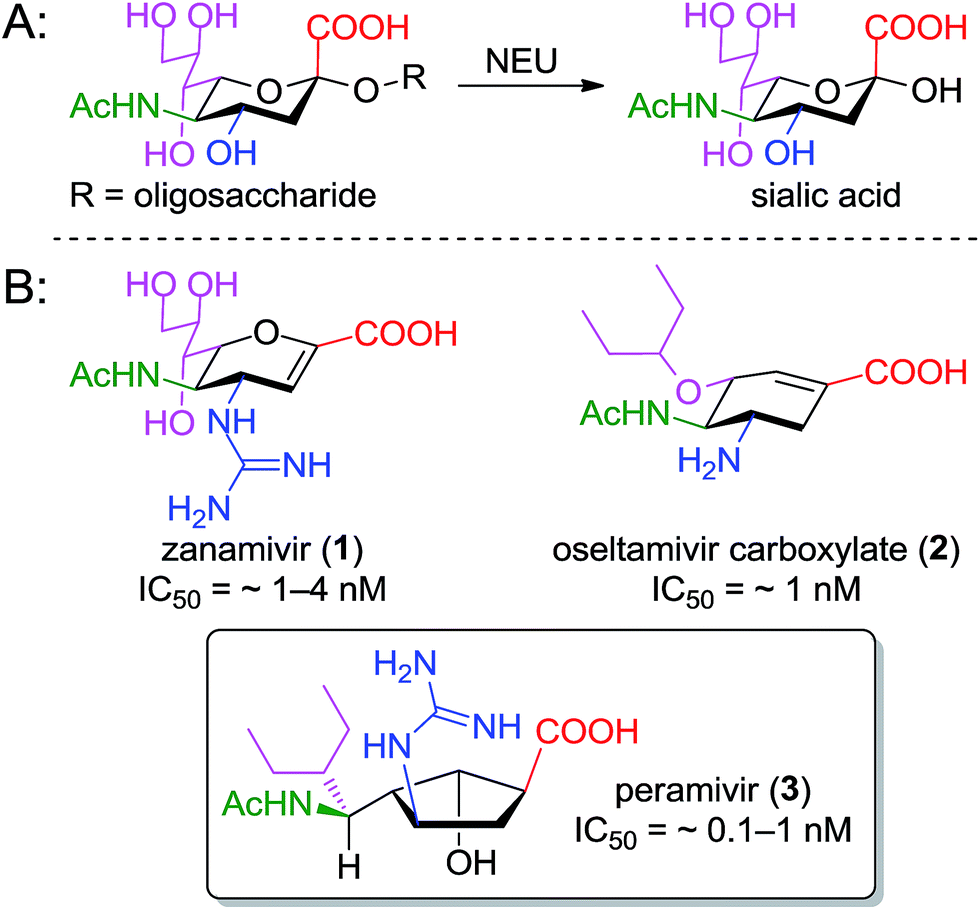 | ||
| Fig. 1 Function and inhibition of the neuraminidase enzyme; (A) glycosidase reaction catalyzed by neuraminidase; (B) structures of clinically used inhibitors of the influenza enzyme; colors indicate related functional groups. | ||
The neuraminidase enzymes have broadly similar active sites across species, suggesting that lessons learned over two decades of structure–function studies targeting the influenza enzyme could profitably be applied to the development of inhibitors for new targets. To design new inhibitors, one must be cognizant of the role played by the underlying cyclic scaffold (i.e. the dihydropyran of zanamivir, the cyclohexene of oseltamivir, or the cyclopentane of peramivir). In each case, the scaffold serves to position functional groups into orientations that match the boat-shaped conformation adopted by sialic acid substrates upon binding to neuraminidase.19 Scaffolds that predispose these groups to their optimal presentation in the binding site should lead to improved potency.
In the cases of zanamivir and oseltamivir, it is well understood that the presence of the alkene in the central core serves to favor a twist-boat conformation, resulting in improved binding of the substituents to the appropriate subpockets of the neuraminidase active site. For this reason, the enzyme-bound15,20 and unbound16 geometries of oseltamivir and zanamivir are structurally conserved (see Fig. 2).
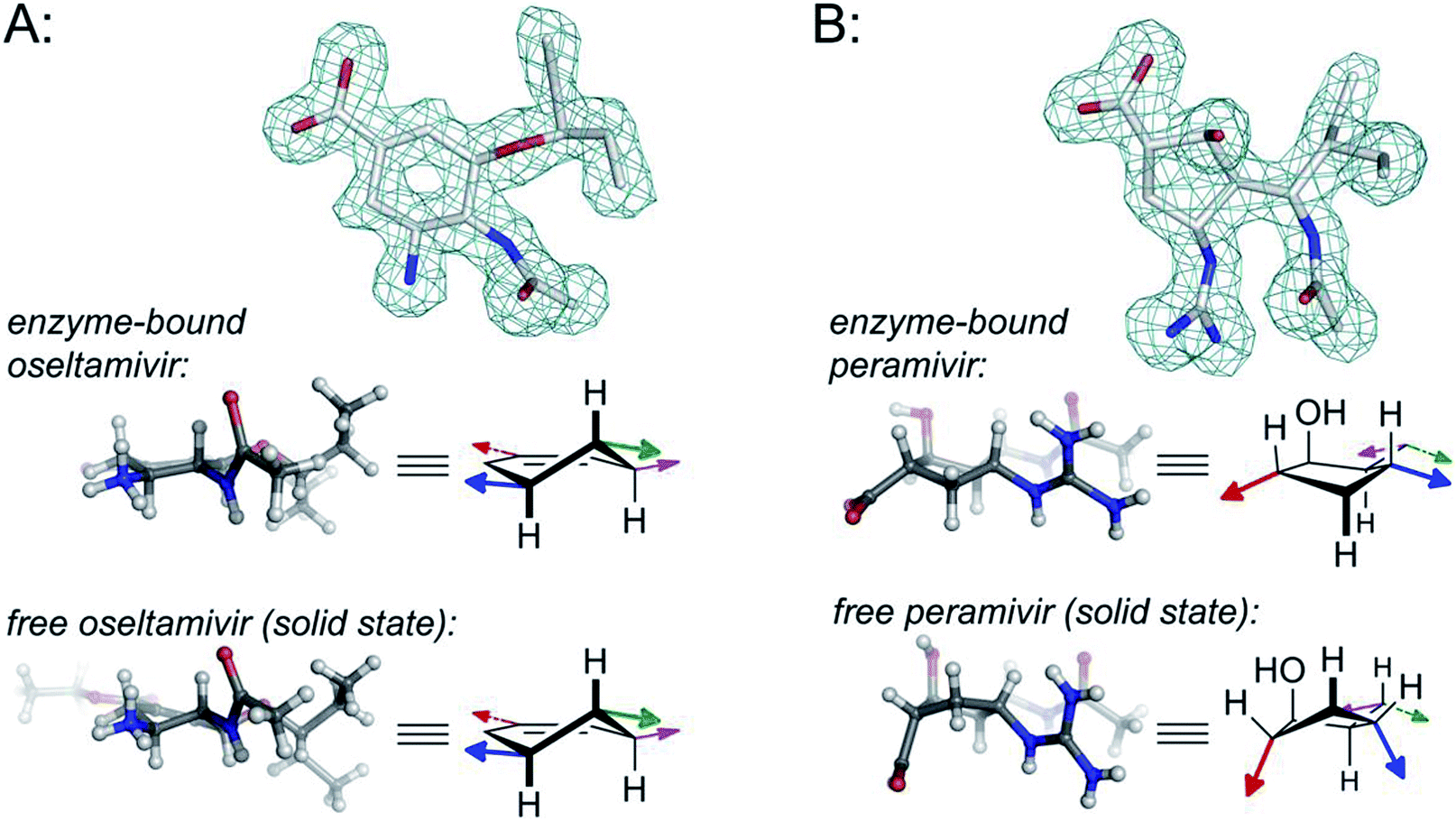 | ||
| Fig. 2 Comparison of structural properties for enzyme-bound and free oseltamivir and peramivir; (A) oseltamivir carboxylate bound in the enzyme active site of H1N1 neuraminidase (from PDB 3TI6)15versus small-molecule X-ray data for oseltamivir (as the ethyl ester prodrug);16 (B) peramivir bound in the enzyme active site of H1N9 neuraminidase (from PDB 1L7F);17versus small-molecule X-ray data for peramivir.18 Colored arrows on the structural drawings correspond to the orientation of functional groups using colors from Fig. 1. Wireframe models correspond to omit maps from the PDB structures. For more details, along with additional data for zanamivir and for peramivir bound to an influenza B neuraminidase, see Fig. S1–S4 in the ESI.† | ||
The structure of peramivir (BCX-1812) is notably different from oseltavimir and zanamivir (Fig. 1), and it is not immediately apparent how the underlying cyclopentane ring favors the presentation of its sidechains to the neuraminidase binding site. The binding of peramivir has typically been examined within co-crystal structures in the neuraminidase active site. However, to the best of our knowledge, no studies have determined or compared the solution conformation of peramivir to its enzyme-bound conformer. As part of our ongoing efforts to explore structure-activity relationships for peramivir,21 and to design rigidified analogues as inhibitors of several neuraminidase targets,22 we sought to obtain experimental evidence of the conformational preferences of peramivir in the absence of its enzyme target. These data would provide insight into the energetic changes required for binding as critical information for the design of new, more potent inhibitors.
In this report, we describe solution 1H NMR data which was used in combination with molecular modeling to rigorously determine the conformational preferences of peramivir in aqueous solution. We then compare the conformers of peramivir observed in single crystals and in the active site of neuraminidase by X-ray diffraction (XRD). We find a remarkable range of conformations for peramivir, which vary significantly between the free and bound states. Taken together, these data suggest that the ground state conformation for peramivir is not optimal for binding the enzyme target. While these observations may have implications for the treatment of influenza infections with peramivir (in that such a conformationally-flexible drug as peramivir may be expected to have a greater number of off-target binding partners), their chief value is in guiding the development of new drugs for neuraminidase targets in other diseases, where isoform selectivity will be of increased importance.
Results and discussion
Conformational analysis of peramivir in solution
We obtained 1D (1H, 13C, nOe) and 2D (COSY, NOESY) NMR spectra of peramivir in D2O at 500 and 700 MHz. The 1D 1H NMR spectrum was analyzed by fitting to obtain precise coupling constants (see Table 1 and ESI†). The simulated spectrum shows excellent agreement with the experimental data (RMSD = 0.003 Hz).| 1H | δ(1H) |
|---|---|
| a Measured at 700 MHz, in D2O at 27 °C. | |
| H1 | 2.73 ppm (ddd, J = 8.6, 3.9, 1.3 Hz) |
| H2 | 4.38 ppm (ddd, J = 4.6, 1.3, 0.7 Hz) |
| H3 | 2.23 ppm (ddd, J = 10.9, 9.1, 4.6 Hz) |
| H4 | 3.85 ppm (ddd, J = 9.1, 9.0, 5.7 Hz) |
| H5α | 1.82 ppm (dddd, J = 14.1, 5.7, 3.9, 0.7 Hz) |
| H5β | 2.56 ppm (ddd, J = 14.1, 9.0, 8.6 Hz) |
| H3′ | 4.37 ppm (dd, J = 10.9, 2.2 Hz) |
| Ac | 1.97 ppm (s) |
| 3-Pentyl substituent | 1.53–1.40 ppm (3H, m) |
| 1.08–0.96 ppm (2H, m) | |
| 0.95 ppm (3H, t, J = 7.2 Hz) | |
| 0.89 ppm (3H, t, J = 7.2 Hz) | |
Qualitative interpretation of the scalar coupling values provides an initial view of the peramivir solution conformation (see Fig. 3). The large coupling constant between H3′ and H3 (10.9 Hz) indicates that these protons are close to an anti-orientation. Furthermore, the observation of a somewhat smaller coupling (9.1 Hz) between H3 and H4, and a strong nOe (4.4%) between H3′ and H4 suggest an anti/anti geometry between H4, H3 and H3′. The signals corresponding to H5α and H5β could be readily distinguished by the presence of a 1.4% nOe between H3 and the more upfield of the two signals (1.82 ppm) in the 1H NMR spectrum. Since the configuration at C3 is known to be R, this allowed us to assign this signal to H5α, and to assign the corresponding signal at 2.56 ppm to H5β. Significantly, H5β was observed to have large coupling constants to H1 (8.6 Hz) and H4 (9.0 Hz), while H5α maintains a much smaller coupling to both H1 (3.9 Hz) and H4 (5.7 Hz). The values for H5β are close to the maximum for syn protons,23 and indicate exceptionally small values for φH1,H5β and φH4,H5β. The smaller couplings of H5α with H1 and H4 are also consistent with a nearly eclipsing orientation of H5β with both H1 and H4, since this would necessarily provide φH1,H5α and φH4,H5α values of close to 110°. Further evidence for the solution-phase conformation of peramivir was obtained from an analysis of nOe interactions between H1 and H4. These protons gave only a very small nOe (0.6%), suggesting that the ring pucker of peramivir substantially increased the distance between these 1,3-related protons on the same face of the cycopentane scaffold. Finally, a small W coupling was observed between H5α and H2 (−0.7 Hz).24
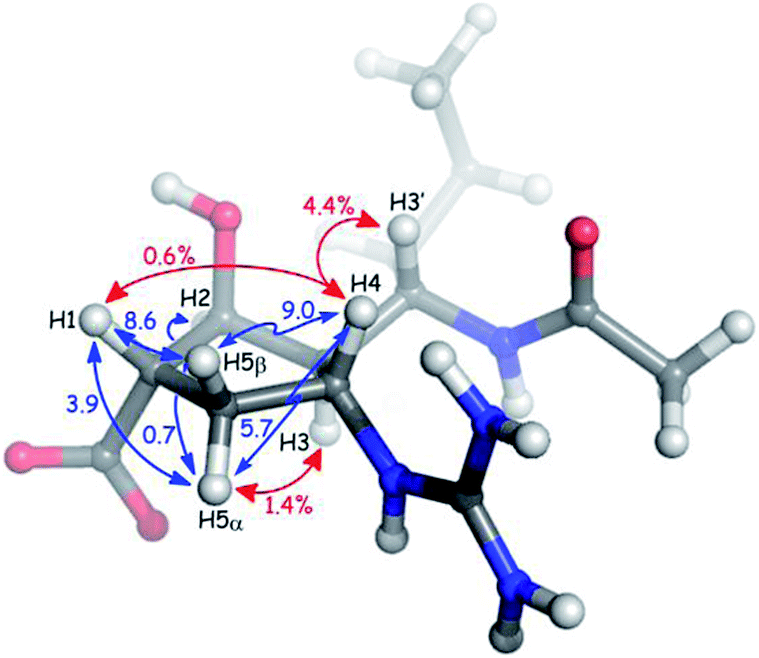 | ||
| Fig. 3 Key nOe interactions (red) and coupling constants (blue) observed for peramivir in D2O. The neuraminidase–peramivir co-crystal structures examined have an average P value of 20°. | ||
Molecular modelling of peramivir
To develop a quantitative model of the peramivir solution-state conformation, we employed molecular modeling in conjunction with the high resolution NMR data already obtained for the inhibitor (vide supra). The conformational flexibility of five membered rings is well known, and has most effectively been analyzed using a combination of molecular dynamics and spectroscopy.25,26 A convenient method for describing ring pucker, and the approach we have used here, is to use the pseudorotational phase angle (P). For a 5-membered ring, P is defined by the following equation:where τ0–τ4 represent the five torsion angles within the ring.27 This method has previously been applied to both cyclopentane and furanoside scaffolds.25,27,28
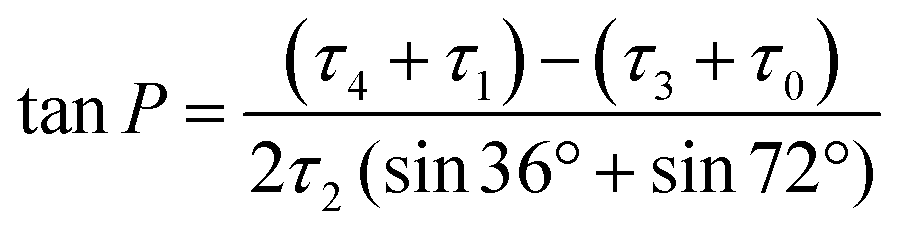
The NMR spectrum of peramivir represents an equilibrium population of solution conformations. We simulated this range of conformations using molecular dynamics (MD) of peramivir in explicit water using AMBER.29 The distribution is relatively narrow and centered around −30°, giving a ring conformation of E3/2T3 (Fig. 4). To identify a representative conformation, we used Chimera30 to determine clusters in the MD trajectory. The top cluster contained 30% of the simulation, and we chose this cluster as most representative of this population and used it for further comparisons to spectroscopic data. From within this top 30% cluster, 200 conformations were chosen for calculation of the expected 1H–1H scalar couplings using density functional theory (DFT).31,32 The calculated coupling constants were in excellent agreement with our experimentally determined values (see ESI†).
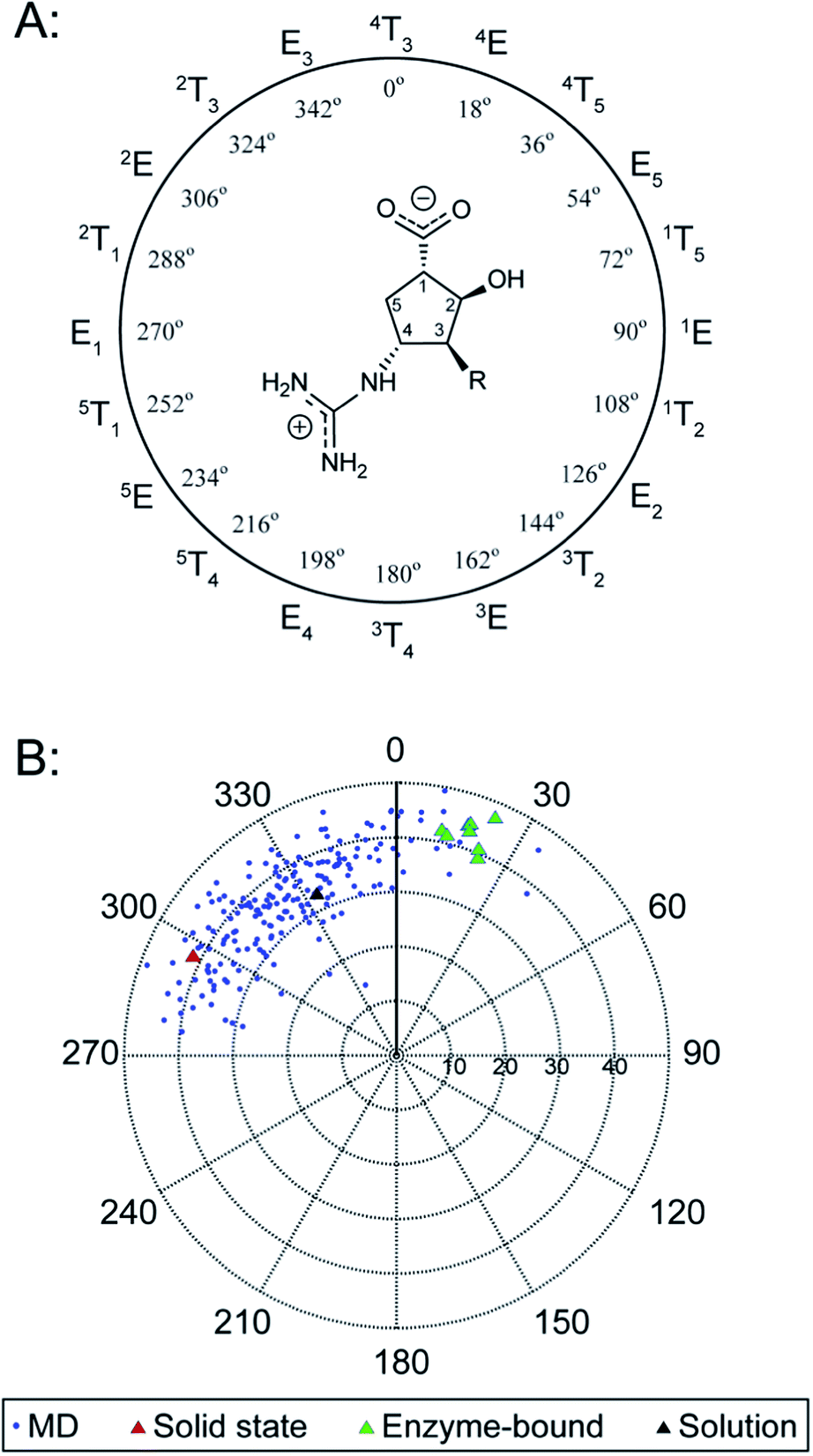 | ||
| Fig. 4 Ring pucker of peramivir varies dramatically over solid-state and solution conditions. The neuraminidase peramivir is a potent inhibitor of influenza neuraminidase, and is used clinically to treat influenza infections. peramivir co-crystals structures examined have an average P value of 20°. The single crystal conformers are found at an average of 296°. The average solution conformation from MD lies at 333°. The pseudorotational phase angle, P, is plotted versus the extent of ring pucker (ϕm). | ||
Inspection of the solution model shows φH1,H5β and φH1,H5α at 9° and 110° respectively, consistent with the large (8.6 Hz) and small (3.9 Hz) coupling constants between these groups determined by NMR. The H3 and H3′ protons are found in an anti-orientation (179°), as are H3 and H4 (159°). The interatomic distance between H1 and H4 was 3.6 Å, and that of H3′ and H4 was 2.1 Å, consistent with the much stronger nOe measurements found for the latter interaction. The φH1,H2 was found to be 92°, consistent with the small coupling observed for these two protons (1.3 Hz); and φH2,H3 was 42°, consistent with the 4.6 Hz coupling observed.
Single crystal conformation of peramivir
We next searched for peramivir structures in the Cambridge Structural Database, and were pleased to find that the structure of peramivir trihydrate had been solved by Keller and Krämer in 2007.18 The Keller–Krämer structure contains four molecules of peramivir in slightly different conformations, together with twelve partially disordered water molecules in the unit cell. The four conformations of peramivir visible in the Keller–Krämer crystal all have fairly similar geometries (see Fig. 5A for an overlay), with an average pseudorotational phase angle of 296° (Fig. 4), shifting the ring pucker closer to 2T1/2E.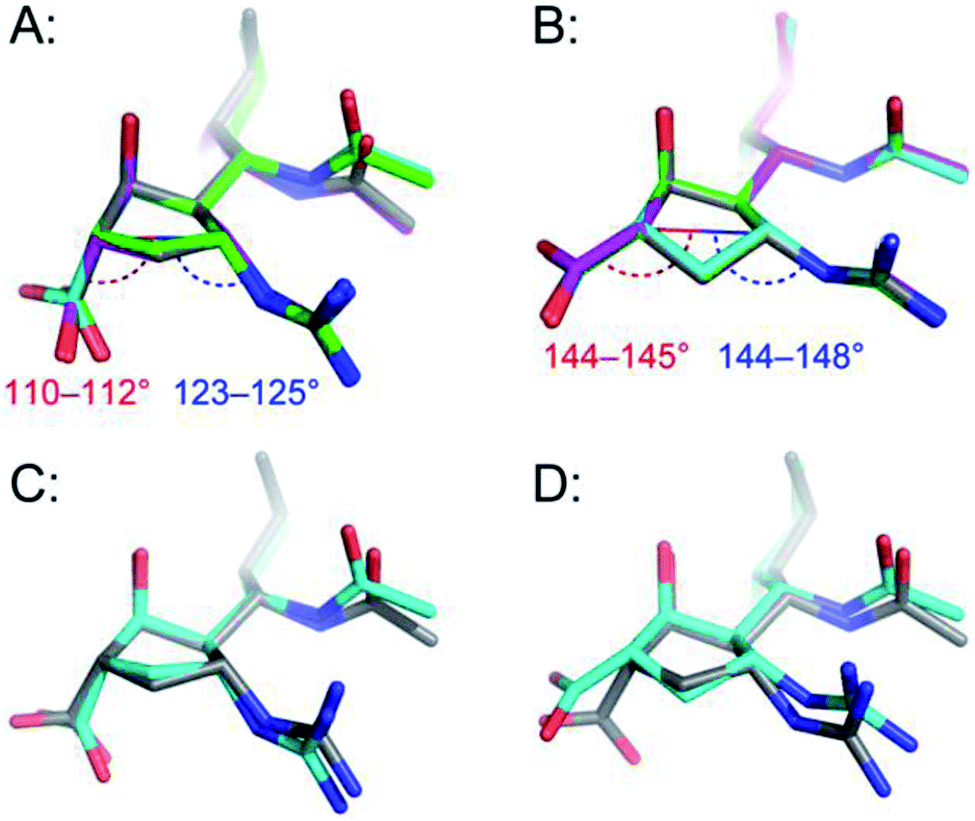 | ||
| Fig. 5 Comparison of peramivir conformations from the solid state with aqueous bound- and unbound-conformations; (A) the four conformations of unbound peramivir from the Keller–Krämer X-ray, (individually colored grey, cyan, green and magenta); (B) the enzyme-bound structure of peramivir in the active site of influenza A neuraminidase, from PDB 2HTU (grey), 1L7F (cyan), 1L7G (green), and 1L7H (magenta); (C) overlay of the average MD structure (grey) with the structure of peramivir in the solid state (cyan); (D) overlay of the average MD structure (grey) with the structure of peramivir in the active site (1L7F, cyan). Hydrogen atoms are hidden for clarity. Numerical values indicate vectors of projection for carboxylate and guanidinium functions. | ||
As illustrated in Fig. 5A, the most noticeable consequence of this altered ring geometry is to shift the C1 carboxylate to a pseudoaxial orientation. Nonetheless, the solid-state and solution-state (unbound) conformations for peramivir remained relatively close to one another (see Fig. 5C for an overlay; RMSD = 0.49 Å). In particular, the vectors at which the carboxylate and guanidinium functions project from the central cyclopentane core are highly conserved between the solution- and solid-state structures.
Enzyme-bound conformations of peramivir
Peramivir has been crystallized in complex with neuraminidase from influenza A,17,33,34 influenza B,35 and the human neuraminidase enzyme, NEU2 (hNEU2).36 Analysis of the conformation of peramivir found in these co-crystals revealed significant conformational differences from the solution and single crystal conformations discussed above. We first analyzed the pseudorotational phase angles of all peramivir conformations obtained from neuraminidase co-crystals found in the PDB. In cases where multiple forms of peramivir were observed in the unit cell, their P values were averaged. A plot of the P values for all co-crystals is shown in Fig. 4B, along with those of the solution and single crystal. Although neuraminidase enzymes from multiple species are included, the ring pucker of bound conformations of peramivir are tightly clustered close to 4E. The result of this ring conformation is to place the C1 carboxylate at a more pseudoequatorial orientation than is found in either the solution or single crystal structures, as illustrated by the overlay of an enzyme-bound conformation (1L7F) with the solution conformation in Fig. 5D (RMSD = 0.62 Å). Indeed, evaluation of the bound complexes reveals that this orientation is critical for optimal engagement with the S1 and S2 subpockets of the neuraminidase active site.22 It is clear from a comparison of the overlays in Fig. 5C and D – as well as the ring-pucker data in Fig. 4 – that the conformations favored by peramivir in both the solution and solid state leave these functional groups incorrectly positioned for binding in the neuraminidase active site.It is notable that the enzyme-bound orientation forces the carboxylate and guanidinium substituents into pronounced pseudo-equatorial orientations, while the hydroxyl group is nearly coplanar to the locations of the hydrogen atoms on the carbons bearing these substituents (Fig. 2B). This conformation would be disfavored in free solution, since it would result in significant unfavourable diaxial interactions. In order to quantify the energetic penalty that would result from such a conformation (in the absence of the enzyme host), we extracted the bound ligand from a peramivir–neuraminidase complex (PDB 2HTU), populated the structure with hydrogen atoms, and performed a minimization by DFT methods (B3LYP/6-31G*). An identical minimization was performed on the Keller–Krämer structure. Our results (see ESI† for details) indicate that the conformation found in the solid state structure is favored by approximately 20 kJ mol−1 in the gas phase, relative to that of the bound conformation.
Examination of the populations found in the MD simulations of peramivir in solution should provide further insight into the likely energy differences between these conformers. We binned the number of frames observed for each of the three conformers indicated in Fig. 4B (−180° to 47°, solid state; −45° to −6°, solution; −3° to 71°, enzyme-bound). This analysis gives relative populations of 36%, 55%, and 9% for the solid state, solution, and enzyme-bound conformers in solution (or a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:1). These populations support the conclusion that the solution conformation is more stable than the solid state by 1.1 kJ mol−1, and that the enzyme bound conformation is approximately 4.5 kJ mol−1 higher in energy than the solution conformation.
:6:1). These populations support the conclusion that the solution conformation is more stable than the solid state by 1.1 kJ mol−1, and that the enzyme bound conformation is approximately 4.5 kJ mol−1 higher in energy than the solution conformation.
Conclusions
The data presented here make a striking case for the previously unrecognized conformational flexibility of peramivir. Our findings support a wide range of ring conformations for the peramivir cyclopentane structure depending on its environment. Although the peramivir core would appear to be too crowded to allow for significant flexibility (with four of five ring positions being substituted, and with the C4 group containing a branched structure), our data unequivocally establish that there are significant differences in the conformational preference of peramivir in the solid state, solution phase, and in all examined enzyme-bound conformations.The differences in the free and enzyme-bound solid state structures for peramivir are striking (e.g. compare the bound and unbound structures in Fig. 2B). Particularly notable is the number of eclipsed or nearly-eclipsed interactions on the cyclopentane ring in the bound structure. This would seem to suggest that in order to bind the neuraminidase target, peramivir must undergo an unfavorable conformational change. Indeed, this large conformational shift, which is not required for oseltamivir or zanamivir, may explain the very slow kon and koff rates reported for peramivir relative to the rates of other neuraminidase inhibitors.37
Among the three clinically used inhibitors (Fig. 1), peramivir is the most potent in vitro inhibitor of influenza neuraminidase, indicating that it is able to overcome the apparent energetic penalties associated with any structural reorganization necessary for binding. At the same time, this degree of conformational flexibility may contribute to off-target interactions for the drug itself, or may be a liability for researchers wanting to create peramivir-like inhibitors of non-influenza neuraminidases.
In the case of hNEU (NEU1-NEU4), isoenzyme selectivity of inhibitors is essential11 and may be difficult to rationally design.38 Inhibition of hNEU by peramivir has only been reported for the NEU2 isoenzyme, and its activity is relatively modest (Ki = 330 μM).36 Chavas et al. suggest that peramivir has reduced contacts in the active site of NEU2 as compared to other inhibitors, although the conformation adopted by the inhibitor is the same as in other neuraminidase active sites.
Our determination of the solution conformation of peramivir, when taken together with existing crystallography data, identifies a substantial conformational change required for peramivir's inhibitory activity. We conclude that inhibitors such as peramivir have a conformational bias which must be overcome in order to bind to the neuraminidase active site. As a result, structural modifications of peramivir that predispose it towards the active conformation may reduce the energetic penalty required for binding, thus increasing potency even further. Additionally, these results suggest that the identification of conformationally restricted scaffolds compatible with the neuraminidase active site will be valuable tools for the design of future inhibitors.
Acknowledgements
We would like to thank the Cancer Research Society of Canada for operating funds (Operating Grant #17396), as well as the Michael Smith Foundation for Health Research and the Canada Research Chairs program for salary support to J.W. Access to high-field NMR facilities was provided by the Alberta Glycomics Centre and the Department of Chemistry at the University of Alberta.Notes and references
- G. Reuter and H. J. Gabius, Biol. Chem. Hoppe-Seyler, 1996, 377, 325–342 CAS.
- M. von Itzstein, Nat. Rev. Drug Discovery, 2007, 6, 967–974 CrossRef CAS PubMed.
- C. U. Kim, W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690 CrossRef CAS.
- M. von Itzstein, W.-Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. V. Phan, M. L. Smythe, H. F. White, S. W. Oliver, P. M. Colman, J. N. Varghese, D. M. Ryan, J. M. Woods, R. C. Bethell, V. J. Hotham, J. M. Cameron and C. R. Penn, Nature, 1993, 363, 418–423 CrossRef CAS PubMed.
- Y. S. Babu, P. Chand, S. Bantia, P. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, A. J. Elliott, C. D. Parker, S. L. Ananth, L. L. Horn, G. W. Laver and J. A. Montgomery, J. Med. Chem., 2000, 43, 3482–3486 CrossRef CAS.
- D. Birnkrant and E. Cox, N. Engl. J. Med., 2009, 361, 2204–2207 CrossRef CAS PubMed.
- G. Xu, M. J. Kiefel, J. C. Wilson, P. W. Andrew, M. R. Oggioni and G. L. Taylor, J. Am. Chem. Soc., 2011, 133, 1718–1721 CrossRef CAS PubMed.
- J. E. Galen, J. M. Ketley, A. Fasano, S. H. Richardson, S. S. Wasserman and J. B. Kaper, Infect. Immun., 1992, 60, 406–415 CAS.
- K. J. Henrickson, Clin. Microbiol. Rev., 2003, 16, 242–264 CrossRef CAS.
- M. Winger and M. von Itzstein, J. Am. Chem. Soc., 2012, 134, 18447–18452 CrossRef CAS PubMed.
- Y. Zhang, A. Albohy, Y. Zou, V. Smutova, A. V. Pshezhetsky and C. W. Cairo, J. Med. Chem., 2013, 56, 2948–2958 CrossRef CAS PubMed.
- T. Miyagi, T. Wada, K. Yamaguchi and K. Hata, Glycoconjugate J., 2003, 20, 189–198 CrossRef.
- K. M. Hoffmeister, E. C. Josefsson, N. A. Isaac, H. Clausen, J. H. Hartwig and T. P. Stossel, Science, 2003, 301, 1531–1534 CrossRef CAS PubMed.
- A. V. Pshezhetsky and A. Hinek, Glycoconjugate J., 2011, 28, 441–452 CrossRef CAS PubMed.
- C. J. Vavricka, Q. Li, Y. Wu, J. Qi, M. Wang, Y. Liu, F. Gao, J. Liu, E. Feng, J. He, J. Wang, H. Liu, H. Jiang and G. F. Gao, PLoS Pathog., 2011, 7, e1002249 CAS.
- P. Naumov, N. Yasuda, W. M. Rabeh and J. Bernstein, Chem. Commun., 2013, 49, 1948–1950 RSC.
- B. J. Smith, J. L. McKimm-Breshkin, M. McDonald, R. T. Fernley, J. N. Varghese and P. M. Colman, J. Med. Chem., 2002, 45, 2207–2212 CrossRef CAS PubMed.
- E. Keller and V. Kraemer, Z. Naturforsch., 2007, 62b, 983–987 Search PubMed.
- V. c. Spiwok and I. Tvaroška, J. Phys. Chem. B, 2009, 113, 9589–9594 CrossRef CAS PubMed.
- X. Xu, X. Zhu, R. A. Dwek, J. Stevens and I. A. Wilson, J. Virol., 2008, 82, 10493–10501 CrossRef CAS PubMed.
- C. M. Bromba, J. W. Mason, M. G. Brant, T. Chan, M. D. Lunke, M. Petric, M. J. Boulanger and J. E. Wulff, Bioorg. Med. Chem. Lett., 2011, 21, 7137–7141 CrossRef CAS PubMed.
- M. G. Brant and J. E. Wulff, Org. Lett., 2012, 14, 5876–5879 CrossRef CAS PubMed.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- L. Pogliani, M. Ellenberger and J. Valat, Org. Magn. Reson., 1975, 7, 61–71 CrossRef CAS.
- M. R. Richards and T. L. Lowary, ChemBioChem, 2009, 10, 1920–1938 CrossRef CAS PubMed.
- H. A. Taha, M. R. Richards and T. L. Lowary, Chem. Rev., 2012, 113, 1851–1876 CrossRef PubMed.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS.
- J. E. Kilpatrick, K. S. Pitzer and R. Spitzer, J. Am. Chem. Soc., 1947, 69, 2483–2488 CrossRef CAS.
- D. A. Case, T. Darden, T. E. Cheatham III, C. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, D. A. Pearlman and M. Crowley, AMBER 9, University of California, San Francisco, 2006 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- T. Bally and P. R. Rablen, J. Org. Chem., 2011, 76, 4818–4830 CrossRef CAS PubMed.
- H. A. Taha, P.-N. Roy and T. L. Lowary, J. Chem. Theory Comput., 2011, 7, 420–432 CrossRef CAS.
- R. J. Russell, L. F. Haire, D. J. Stevens, P. J. Collins, Y. P. Lin, G. M. Blackburn, A. J. Hay, S. J. Gamblin and J. J. Skehel, Nature, 2006, 443, 45–49 CrossRef CAS PubMed.
- Y. Wu, Y. Bi, C. J. Vavricka, X. Sun, Y. Zhang, F. Gao, M. Zhao, H. Xiao, C. Qin, J. He, W. Liu, J. Yan, J. Qi and G. F. Gao, Cell Res., 2013, 23, 1347–1355 CrossRef CAS PubMed.
- A. J. Oakley, S. Barrett, T. S. Peat, J. Newman, V. A. Streltsov, L. Waddington, T. Saito, M. Tashiro and J. L. McKimm-Breschkin, J. Med. Chem., 2010, 53, 6421–6431 CrossRef CAS PubMed.
- L. M. G. Chavas, R. Kato, N. Suzuki, M. von Itzstein, M. C. Mann, R. J. Thomson, J. C. Dyason, J. McKimm-Breschkin, P. Fusi, C. Tringali, B. Venerando, G. Tettamanti, E. Monti and S. Wakatsuki, J. Med. Chem., 2010, 53, 2998–3002 CrossRef CAS PubMed.
- S. Bantia, C. S. Arnold, C. D. Parker, R. Upshaw and P. Chand, Antiviral Res., 2006, 69, 39–45 CrossRef CAS PubMed.
- A. Albohy, Y. Zhang, V. Smutova, A. V. Pshezhetsky and C. W. Cairo, ACS Med. Chem. Lett., 2013, 4, 532–537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional figures illustrating the binding of oseltamivir, zanamivir, and peramivir in the active site of neuraminidase A and B enzymes, comparisons of calculated and experimental NMR data, population analysis and calculation data. See DOI: 10.1039/c4md00168k |
| This journal is © The Royal Society of Chemistry 2014 |