 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetabolism of a novel skepinone L-like p38 mitogen-activated protein kinase inhibitor
K.
Storch
,
M.
Gehringer
,
B.
Baur
and
S. A.
Laufer
*
Eberhard Karls University of Tuebingen, Institute of Pharmaceutical Sciences, Department of Pharmaceutical and Medicinal Chemistry, Auf der Morgenstelle 8, D-72076 Tuebingen, Germany. E-mail: stefan.laufer@uni-tuebingen.de
First published on 28th April 2014
Abstract
The p38 mitogen-activated protein kinase (MAPK) is a key mediator in cytokine-induced signaling events and is activated in response to a variety of extracellular stimuli such as stress factors, UV-light and inflammatory cytokines. Therefore, the p38 MAP kinase plays an integral role in disease states including oncogenesis, immune disorders and inflammatory processes. Recently a novel class of highly selective p38α inhibitors was described and characterized as tools and probes for in vivo studies. The objective of the current study was the preclinical characterization of 3-((2,4-difluorophenyl)amino)dibenzo[b,e]oxepin-11(6H)-one, a potent p38α MAP kinase inhibitor. In rat and human hepatic microsomal incubations, the examined compound is completely inactivated (concerning the inhibitory potency of the isolated p38α enzyme) by a CYP2B6 mediated phase 1 metabolism. The dehalogenation and subsequent hydroxylation in the para position of the 2,4-difluorophenyl residue was found to be the predominant transformation. The metabolite was detected in different quantities in both species. In a consecutive reaction the phase 1 metabolite conjugates with glucuronic acid in terms of a phase 2 metabolism. The responsible isoenzymes were identified to be UGT1A3, UGT1A9 and UGT1A10. In this reaction UGT1A10 is the predominant driver of the conversion. Similar to the phase 1 metabolite, the conjugate could also be found in different amounts in both examined species.
Introduction
The p38α kinase is the best-characterized member of the mitogen activated protein kinase (MAPK) family. It plays a central regulatory role in the production of pro-inflammatory cytokines like TNF-α, interleukin-1β, and interleukin-6β.1 The MAPK family is a group of serine/threonine kinases,2 which are activated by various extracellular stimuli, such as heat shock, osmotic shock or irradiation with UV light.3 Once being activated it mediates the signal transduction into the nucleus to turn on responsive genes. Diseases linked to this pathway are rheumatic arthritis,4,5 COPD, Crohn's disease6,7 or asthma.8,9 Consequently, p38 inhibitors have been considered as anti-TNF-α therapeutics that could potentially replace established anti-TNF antibodies or TNF receptors.Many pharmaceutical companies initiated p38 discovery programs,10,11 but despite the fact that a few candidates have reached phase III of clinical development, safety issues appear to be the main hurdle for a successful development. Especially when administered at high concentrations, many compounds showed toxic properties.12–14 Moreover kinase inhibitors are known to change their selectivity profile through metabolic transformations, adding an additional layer of complexity to toxicity prediction.15
There are four isoforms of p38 that demonstrate different patterns of tissue expression16–18 and activation.16 Several studies show that the α isoform is the major contributor to the role of p38 in inflammation.19–21 Besides the fact that there is a need for new potent p38 MAP kinase inhibitors showing high efficacy even at low dosage, ADMET investigations become more and more important.
As recently reported by our group,22 dibenzosuberones are highly selective and potent p38 MAP kinase inhibitors. A prominent example within this inhibitor class is skepinone-L, which has an outstanding potency in an isolated p38 enzyme assay as well as a human whole blood assay measuring the inhibition of TNF-α release. Furthermore, skepinone-L is hardly affected by metabolic conversion and displays favorable pharmacokinetic characteristics.
We also reported,23 that the metabolism of related dibenzosuberones gets suppressed by the introduction of hydrophilic side chains in position 9 of the A-ring. Besides increasing the hydrophilicity of the molecule, blocking the metabolically labile position of the central scaffold by introducing an ether bridge is the way of choice to prevent metabolic degradation. This concept led to another series of p38 inhibitors based on a dibenzo[b,e]oxepin-11(6H)-one core. Again hydrophilic side chains lead to increased metabolic stability. Based on the recently published results23 the present study determined that the main scaffold 3-((2,4-difluorophenyl)amino)dibenzo[b,e]oxepin-11(6H)-one is metabolized to a single metabolite bearing the corresponding hydroxyl function in para position of the 2,4-difluorophenyl moiety. The metabolism of this compound was simulated in rat liver microsomes as well as in human liver microsomes. Its phase 1 metabolism is exclusively mediated by CYP2B6. CYP2B6 is assumed to be expressed at only 3 to 5% of total P450s in human livers.24,25 Its expression and enzyme activity has been shown to vary approximately 100 fold in the human liver.26 Cytochrome 2B6 is also expressed in extrahepatic tissues such as brain, kidney, intestine,24 uterine endometrium,27 skin,28 and heart.29 It is one of the most polymorphic CYP genes in humans and currently 30 defined alleles with over 100 described polymorphisms are known.30,31 The polymorphic nature of the cytochrome P450 (CYP450) genes affects individual drug response and reactions to a big extent.32 Studies of human liver tissues have shown that females tend to express higher levels of CYP2B6 in liver than males.30 In an extension of the phase 1 metabolism study, incubations with glucuronosyltransferases (UGT) were performed. UGTs are responsible for glucuronidation, a major contributor to phase 2 metabolism.33 Incubations were again performed with rat and human liver microsomes as well as with different UGT isoenzymes. UGT1A3, 1A9 and 1A10 were identified to be involved in this reaction. Each of these three isoenzymes shows a genetic polymorphism in humans.34–37 Moreover UGT1A10 was identified as the key driver of the conversion.
Results
Metabolic stability of compound 1 in different species: male/female Sprague Dawley rat liver microsomes, and male/female human liver microsomes
The main metabolite of compound 1 was the corresponding 3-((2-fluoro-4-hydroxyphenyl)amino)dibenzo[b,e]oxepin-11(6H)-one. The introduction of a hydroxyl group in ortho position to the amino function (substituting the ortho-F) was not observed (Fig. 1).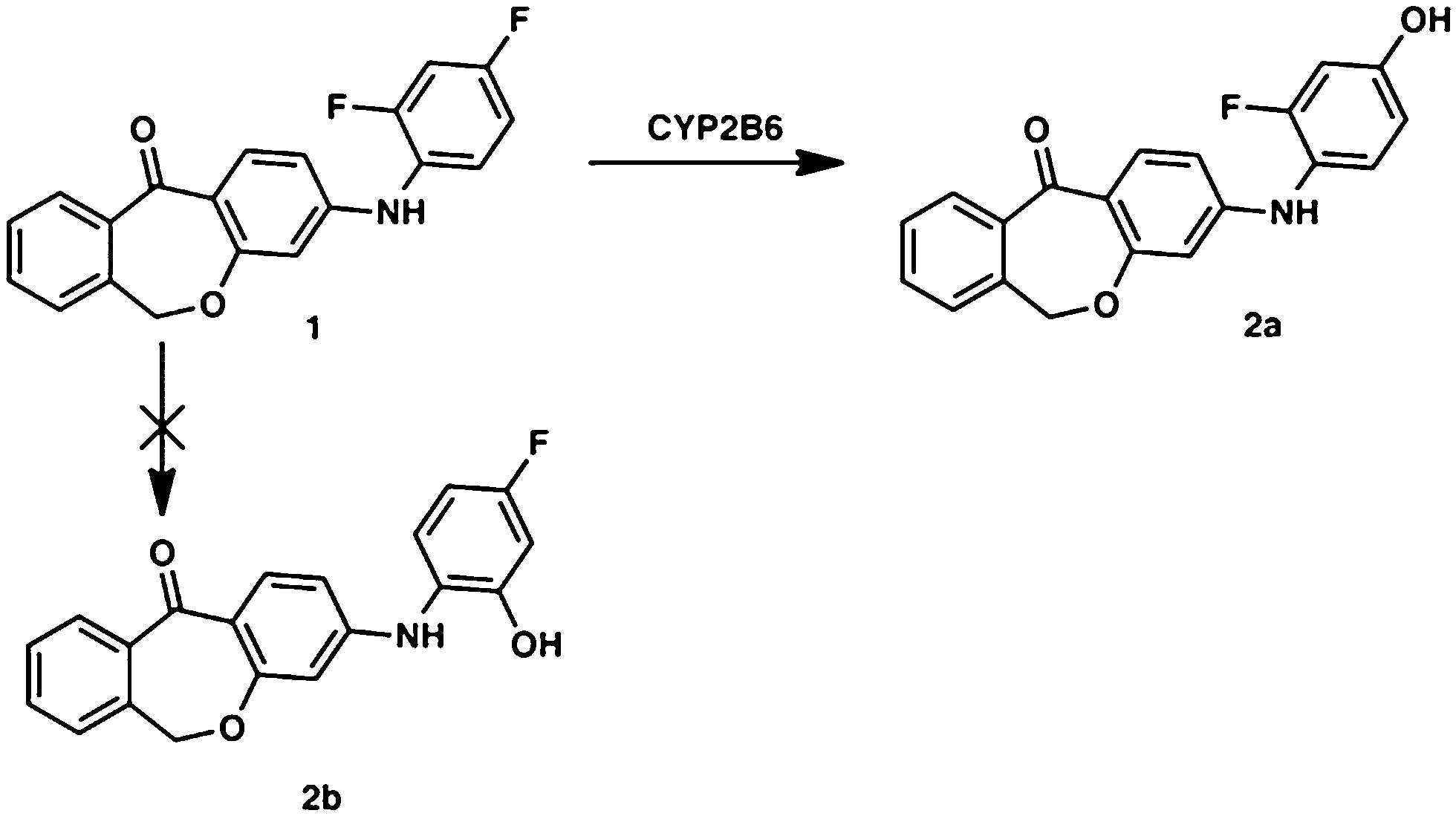 | ||
| Fig. 1 Metabolic profile of compound 1 with the enzyme involved in the biotransformation. The metabolic pathway was proven in rat and human hepatic microsomes after incubation for 180 min. | ||
This can primarily be attributed to the lower steric hindrance at the para position as well as to electronic factors. By analyzing both possible metabolites by HPLC, a retention time shift was observed between 2a and 2b (see Fig. 2). Although both compounds possess the same calculated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value, they differ significantly in lipophilicity. This observation can be explained by reduced solvation of 2b due to increased steric bulk around the hydroxyl group. Furthermore, a weak intramolecular hydrogen bond38 between the hydroxy and the diarylamino function can be formed in 2b, which partially masks the polar groups from hydrogen bonding with the solvent. Moreover, a conformational bias within 2b caused by this intramolecular hydrogen bond could further alter the lipophilicity of the molecule.
P value, they differ significantly in lipophilicity. This observation can be explained by reduced solvation of 2b due to increased steric bulk around the hydroxyl group. Furthermore, a weak intramolecular hydrogen bond38 between the hydroxy and the diarylamino function can be formed in 2b, which partially masks the polar groups from hydrogen bonding with the solvent. Moreover, a conformational bias within 2b caused by this intramolecular hydrogen bond could further alter the lipophilicity of the molecule.
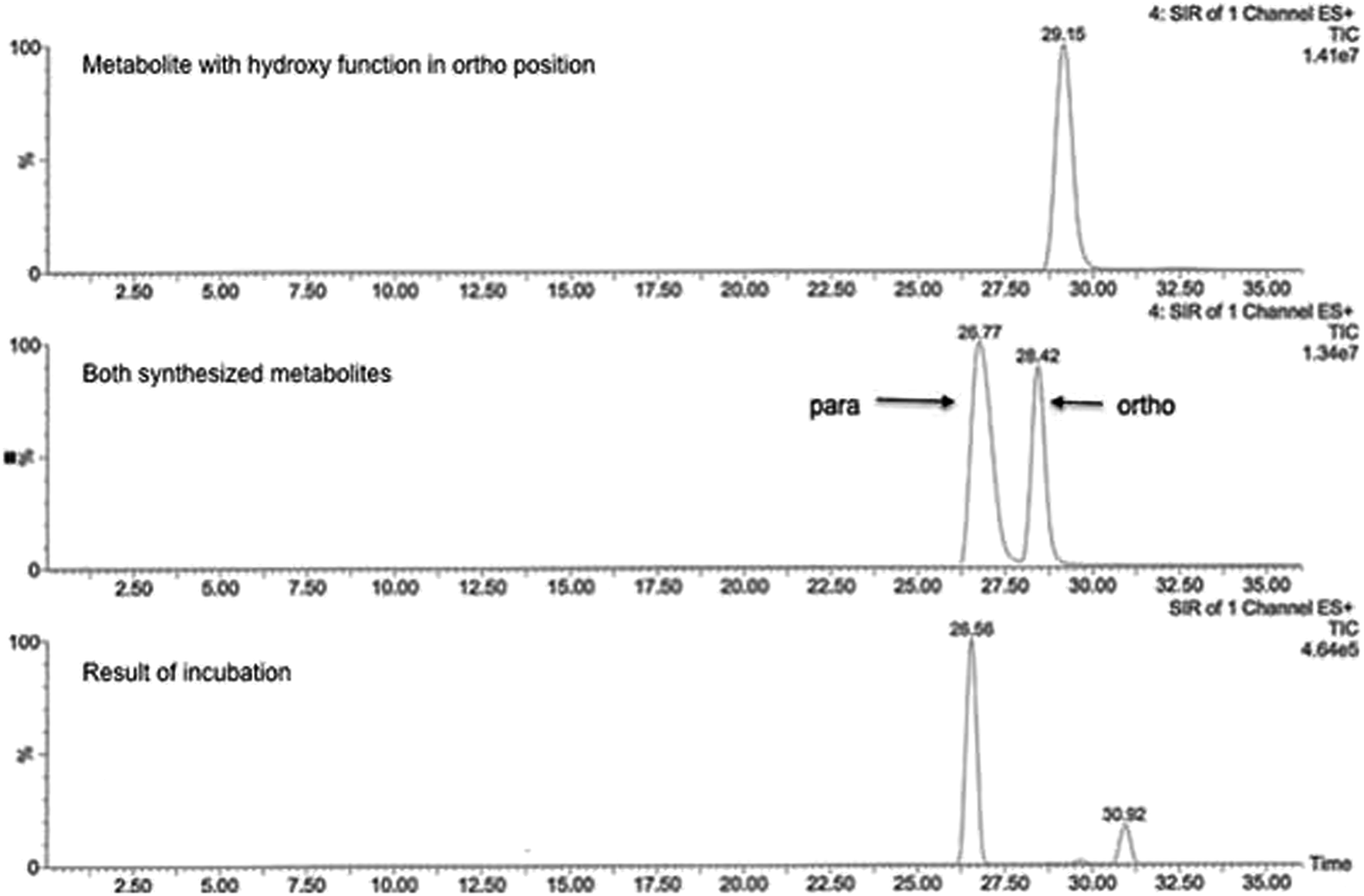 | ||
| Fig. 2 Representative chromatogram showing the feasibility of metabolite separation. The analysis was performed on RP-HPLC system with mass selective detection (positive ESI mode, SRM detection). | ||
MS/MS data did not provide any further information because both potential metabolites show an almost identical fragmentation pattern. Kinetic analysis demonstrated that the metabolite of compound 1 reached a concentration maximum between 100 and 120 min. The metabolite could be found in different amounts in both human and rat liver microsomes.
In vitro phase 1 metabolism in female and male rat and human liver microsomes
To examine gender-specific differences, incubations with male and female pooled RLMs as well as with male and female pooled HLMs were performed. The time profile of the RLMs showed clear-cut differences in the metabolite formation rates (Table 1). Male RLMs produced the metabolite faster, compared with female RLMs (Fig. 3). In HLMs, gender differences were much less distinct, especially when considering the higher P450 enzyme activity of female microsomes (see Table 1).30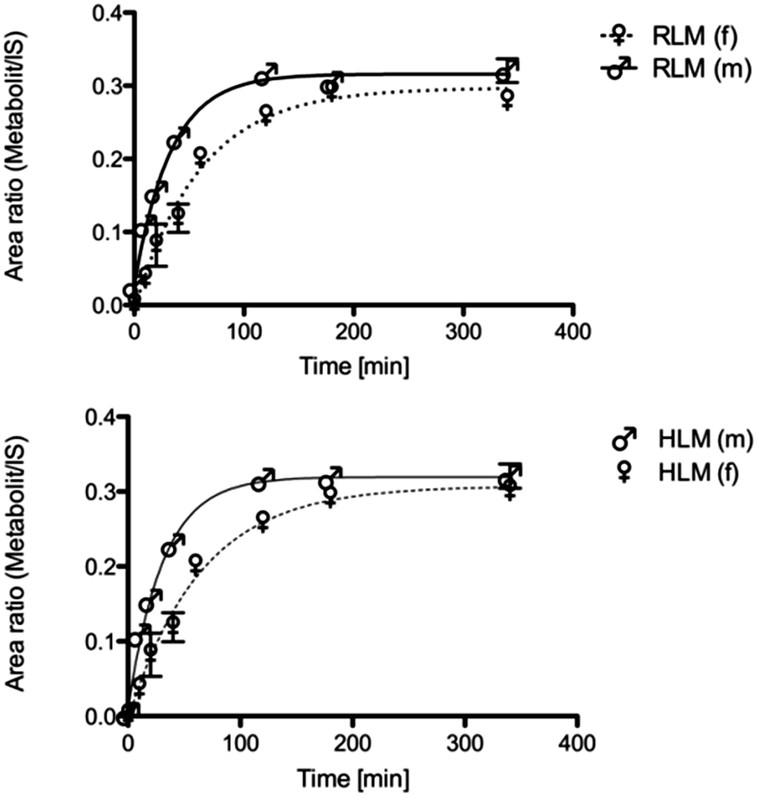 | ||
| Fig. 3 Metabolic conversion rate of male and female rat liver microsomes in the phase 1 reaction. | ||
In vitro phase 1 metabolism in baculovirus-expressed human P450 isoenzymes
The dehalogenation was catalyzed exclusively by CYP2B6, which was confirmed by incubation with several isolated P450 isoforms (CYP1A2, CYP2A6, CYP2B6, CYP3A4, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2J2, CYP4F12; see Table 2). Negative controls without NADPH and with cytochrome P450 reductase/b5 without P450 enzymes were carried out to exclude non-enzymatic metabolite formation.| Species | Gender specifity | K M [μM] | v max [nM min−1 per mg protein] | CLint [μL min−1 pmol P450] |
|---|---|---|---|---|
| Human | Male | 56.10 ± 12.46 | 3.015 ± 0.2955 | 20.19 |
| Human | Female | 58.91 ± 10.99 | 4.562 ± 0.3486 | 15 |
| Rat | Pooled | 63.99 ± 16.89 | 5.487 ± 0.7227 | 4.69 |
| rCYP2B6 | 14.14 ± 4.552 | 0.6293 ± 0.06293 | 12.21 |
Kinetics of phase 1 metabolism
The kinetic parameters KM and vmax were determined by incubating 1 (1–200 μM) with male and female HLM (0.19 mg mL−1), male/female RLM (0.15 mg mL−1) as well as with rCYP2B6 (0.1 mg mL−1). Calculated KM and vmax values are shown in Table 2. The respective Michaelis–Menten (M–M) plots of compound 1 can be found in Fig. 4. The M–M plots show a hyperbolic curve, indicating saturation of the metabolite formation over the substrate range used suggesting that the data obeyed M–M kinetics. M–M values did not differ significantly between male and female HLMs (see Table 2).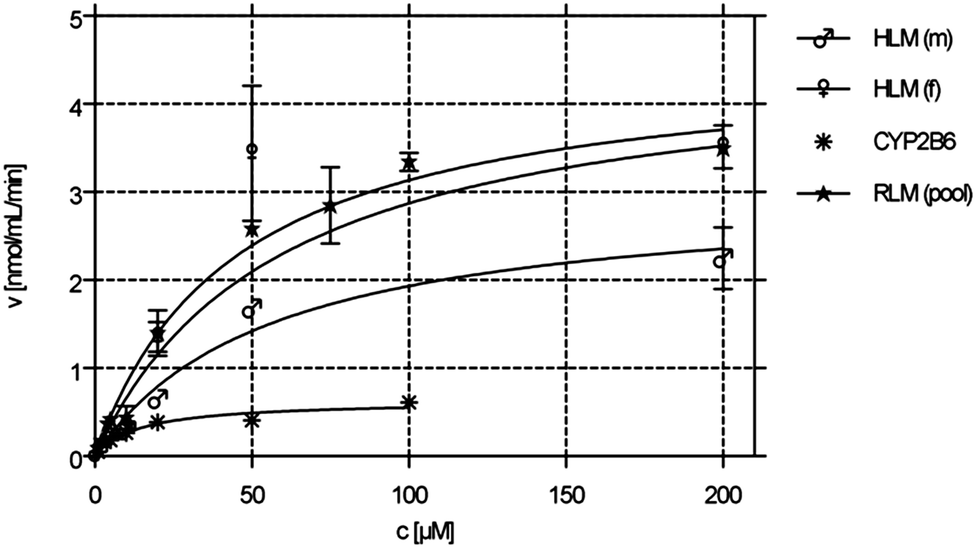 | ||
| Fig. 4 Michaelis–Menten kinetics (phase 1 metabolism) of male and female HLMs, pooled RLM as well as CYP2B6. | ||
In vitro phase 2 metabolism in baculovirus-expressed human UGT isoforms
In order to identify the isoenzymes responsible for the glucuronidation, incubations with a set of different UGT isoforms (UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A9, UGT1A10, UGT2B7, UGT2B15) were performed. The conjugation of 2a with glucuronic acid was mediated by UGT1A3, UGT1A9 and UGT1A10. The latter was identified to play a dominant role in the conversion (see Fig. 5). Incubations without UDPGA were used to rule out non-enzymatic conversion.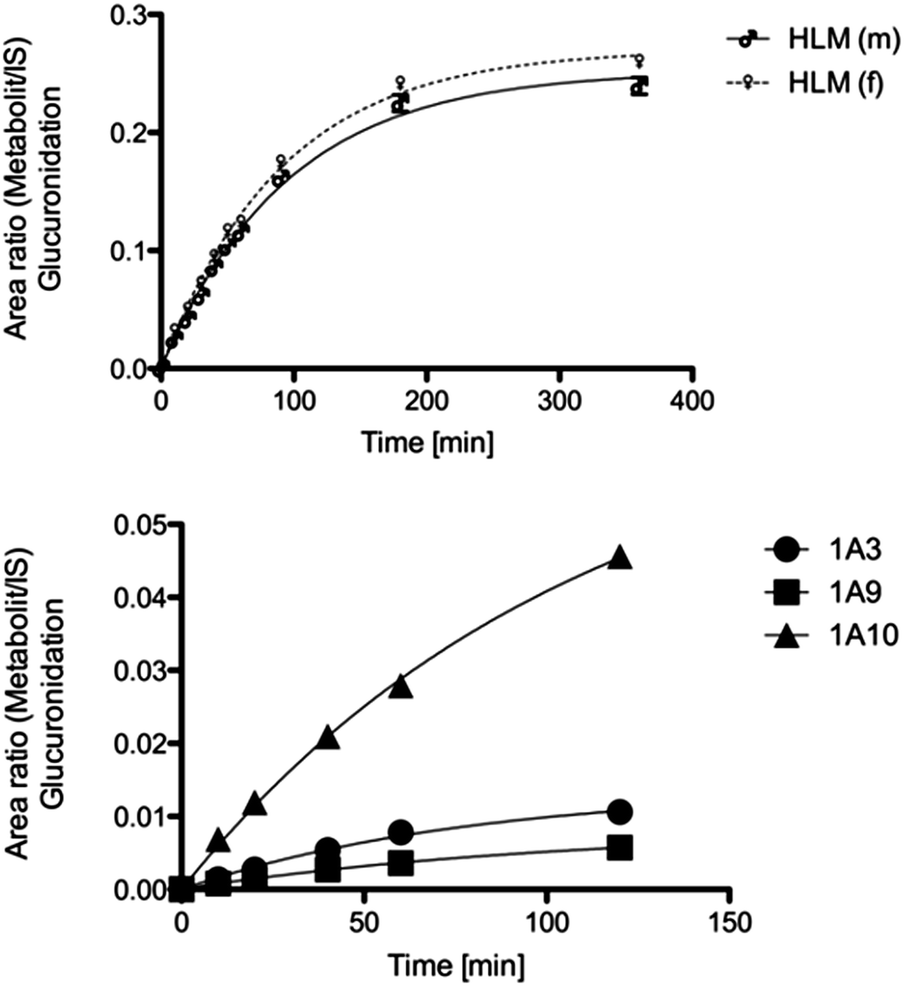 | ||
| Fig. 5 Metabolic conversion rate of male/female HLMs and recombinant isoenzymes in the phase 2 reaction. | ||
In vitro phase 2 metabolism in pooled rat and male/female human liver microsomes.
The time profile of the RLMs showed rapid conversion into the metabolite 3 (t1/2 = 9.218 ± 0.81 min). To examine gender-specific differences, incubations of male and female HLMs were compared. Female HLMs (t1/2 = 63.02 ± 3.47 min) and male HLMs (t1/2 = 65.68 ± 4.78 min) produce the metabolite with no significant difference in formation rates (Fig. 5 and Table 3).| Species | Gender specifity | t 1/2 [min] |
|---|---|---|
| Human | Male | 65.68 ± 4.78 |
| Female | 63.02 ± 3.47 | |
| Rat | Pool | 9.218 ± 0.81 |
| UGT1A3 | 54.92 ± 8.07 | |
| UGT1A9 | 74.67 ± 8.71 | |
| UGT1A10 | 75.36 ± 13.09 |
Discussion
The cytochrome P450 enzyme system plays a major role for the pharmacokinetics and toxicity of drugs. Knowledge about the dominant cytochrome P450 isoform responsible for the metabolism of a particular drug is valuable in predicting factors like efficacy or potential drug interactions.For this reason the aim of this study was to investigate the in vitro metabolism of 3-((2,4-difluorophenyl)amino)dibenzo[b,e]oxepin-11(6H)-one, a potent and selective skepinone-L like p38α MAPK inhibitor, by human and rat liver microsomes as cytochrome P-450 enzyme source. Isoenzyme CYP2B6 was identified as key driver in the phase 1 metabolism of 1. The value of vmax fits into the range between 1 and 15 nmol of product per minute per milligram of microsomal protein, which is in the typically observed range for organic substrates metabolized by cytochrome P-450 enzymes.39,40 CYP2B6 is mediating the dehalogenation and hydroxylation of the fluorine atom in the para position of the difluorphenyl moiety. Through this phase 1 metabolism, compound 1 undergoes a complete inactivation with regard to the inhibition of the isolated p38α enzyme (0.234 μM for substance 1 and 3.293 μM for substance 2b, respectively). The introduction of a hydroxyl group opens up the opportunity for phase 2 reactions. Phase 2 metabolism was investigated by incubations with uridine 5′-diphosphoglucuronic acid trisodium salt, which revealed a conjugation with glucuronic acid. The conversion was mediated by UGT1A3, UGT1A9 and UGT1A10 with a preference for the latter.
Experimental
Reagents
1: 3-((2,4-difluorophenyl)amino)dibenzo[b,e]oxepin-11(6H)-one, 2a: 3-((2-fluoro-4-hydroxyphenyl)amino)dibenzo[b,e]oxepin-11(6H)-one, 2b: 3-((2-hydroxy-4-fluorophenyl)amino)dibenzo[b,e]oxepin-11(6H)-one, the internal standard skepinone-L (3) (R)-2-((2,4-difluorophenyl)amino)-7-(2,3-dihydroxypropoxy)-10,11-dihydro-5H-dibenzo[a,d][7]annulen-5-one22 and the corresponding phase 2 metabolite were prepared at the Department of Pharmaceutical and Medicinal Chemistry, University of Tuebingen (Tuebingen, Germany). The corresponding chemical structures are shown in Fig. 6. Acetonitrile (ACN; LC-MS Grade) and methanol (LiChrosolv hypergrade) were purchased from VWR (Darmstadt, Germany). Formic acid (FA) p.a., NADP+ sodium salt, Trizma base and Trizma HCl, magnesium chloride hexahydrate, glucose 6-phosphate sodium salt, and glucose 6-phosphate dehydrogenase from yeast, uridine 5′-diphosphoglucuronic acid trisodium salt, D-saccharic acid 1,4-lactone monohydrate and alamethicin from Trichoderma viride were purchased from Sigma-Aldrich Chemie GmbH (Munich, Germany). Water was obtained from an in-house bi-distillation device (Department of Pharmaceutical and Medicinal Chemistry, University of Tuebingen, Germany).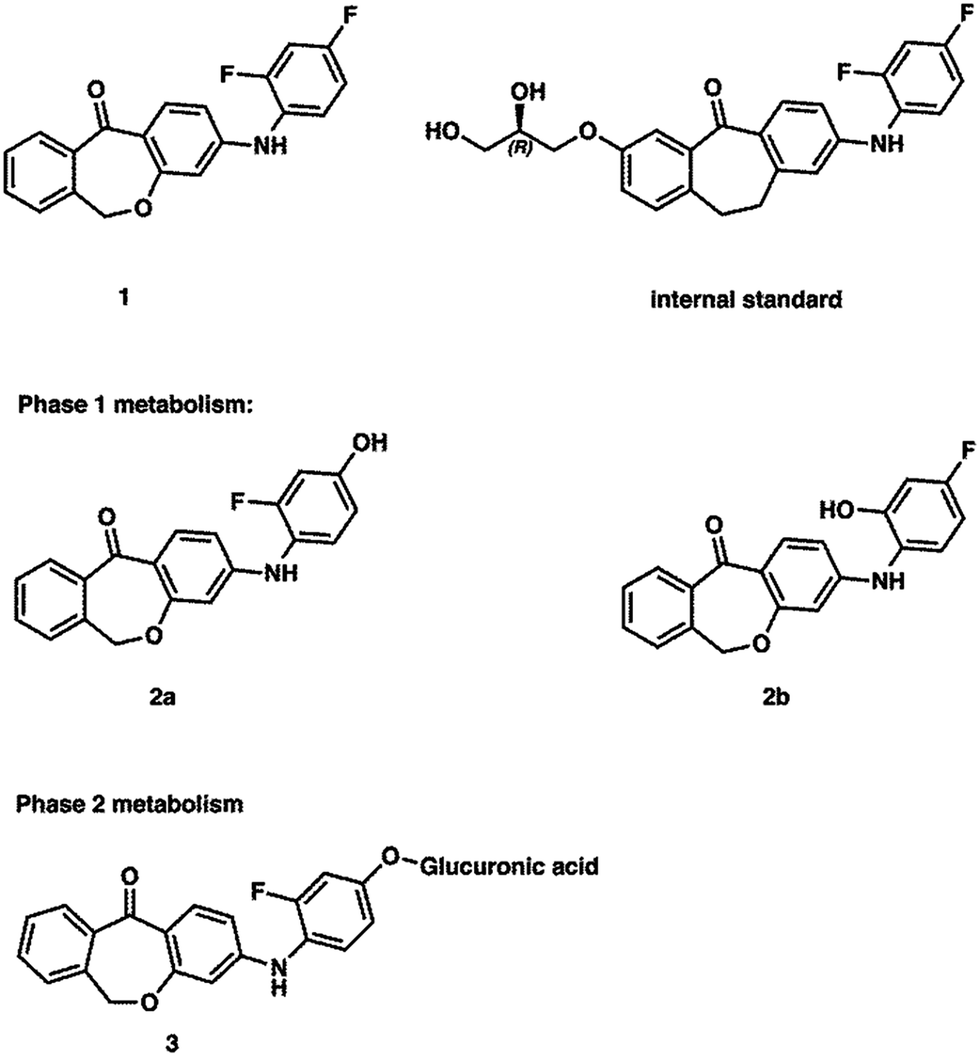 | ||
| Fig. 6 Chemical structures of all compounds with corresponding IUPAC nomenclature. 1: 3-((2,4-difluorophenyl)amino)dibenzo[b,e]oxepin-11(6H)-one; 2a: 3-((2-fluoro-4-hydroxyphenyl)amino)dibenzo[b,e]oxepin-11(6H)-one; 2b: 3-((4-fluoro-2-hydroxyphenyl)amino)dibenzo[b,e]oxepin-11(6H)-one; 3: (2R,3R,4R,5S,6R)-6-(3-fluoro-4-((11-oxo-6,11-dihydrodibenzo[b,e]oxepin-3-yl)amino)phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid; internal standard: (R)-2-((2,4-difluorophenyl)amino)-7-(2,3-dihydroxypropoxy)-10,11-dihydro-5H-dibenzo[a,d][7]annulen-5-one. | ||
In vitro metabolism studies (phase 1)
Pooled male and female liver microsomes of rats (RLMs) and humans (HLMs) were purchased from Life Technologies GmbH (Darmstadt, Germany). To compare the rates of substrate degradation, the microsomal protein content was arbitrarily standardized to 1 mg mL−1. All incubations were performed under the same conditions with respect to their protein content.All incubations (final total volume 1050 μL) were made in the presence of an NADPH-regenerating system consisting of 5 mM glucose 6-phosphate, 5 U mL−1 glucose 6-phosphate dehydrogenase, and 1 mM NADP+. The substrate (10 μM), NADPH regenerating system, and 3.8 mM MgCl2x6·H2O in 0.1 M Tris buffer (pH 7.4 at RT) were pre-incubated for 5 min in a shaking heating block at 37 °C, 550 rpm, and the reaction was started by addition of the respective microsomes. To follow the course of metabolism, 100 μL aliquots were withdrawn at different time points (0, 10, 20, 40, 60, 120 and 180 min) and transferred into an ice-cooled vial containing 100 μL internal Standard (100 μg mL−13 in acetonitrile, final concentration of 50 μg mL−1). The samples were vortexed for 15 s and centrifuged (19800 relative centrifugal force/4 °C per 10 min). The supernatant was directly used for analysis. All incubations were conducted in triplicate; average mean values of these incubations are shown in the figures. In all incubations a limit of 1% organic solvent was not exceeded.41,42
In vitro metabolism in baculovirus-insect cell expressed human P450 isoenzymes
All isoenzymes (CYP1A2, CYP2A6, CYP2B6, CYP3A4, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2J2, CYP4F12) were purchased from BD Biosciences (Heidelberg, Germany). The final enzyme concentration was 100 pmol mL−1 in all incubations. Incubations were conducted according to the liver microsome procedure.Proof of linearity for protein content and time of incubation
1 was incubated for 90 min with the same protein content in each of the different species' liver microsomes. Five aliquots were drawn at time points 0, 10, 30, 60, and 90 min as described above. The concentration of 1 was set to 100% at t = 0 min. The logarithm of the percentage remaining versus time was fitted by least-squares linear regression of at least four time points. Goodness of fit was assessed by calculation of the correlation coefficient (R2) and visual examination of the data.Determination of kinetic parameters for the dehalogenation of 1 to 2
K m and vmax were determined in pooled female and male human liver microsomes, using baculovirus cDNA-expressed isoenzymes (Supersomes) of CYP2B6 with P450 reductase. The substrate concentrations ranged from 0.1 μM to 50 μM. The protein content and time linearity were proven in the preceding analysis. The protein content determined was 0.03 mg mL−1 for HLMs and the cytochrome P450 content was 0.5 pmol mL−1 for CYP2B6. Incubations were conducted in a total volume of 250 μL for an incubation time of 10 min, and were performed in triplicate for the liver microsomes and CYP2B6. Data reported are averages ± standard errors of the determinations. Estimation of the enzyme kinetic parameters was made by fitting to the Michaelis–Menten equation by best-fit nonlinear regression using the Enzyme Kinetics Module of the GraphPad Prism software, version 5.0a for Mac OsX (GraphPad Software, San Diego, California, USA, http://www.graphpad.com). Goodness of fit was assessed by calculation of the correlation coefficient (R2) and visual examination of the data.In vitro metabolism studies (phase 2)
Phase 2 incubations were performed in analogous manner to phase 1 incubations. The NADPH regenerating system was replaced with uridine 5′-diphosphoglucuronic acid trisodium salt (100 mM), D-saccharic acid 1,4-lactone monohydrate (100 mM)43 and alamethicin (100 mM).44 The workup was done as described in the section above.Screening of metabolites by LC-MS/MS analysis
Metabolite formation was analyzed with a Jasco (Groβ-Umstadt, Germany) HPLC system consisting of a pump (PU-1580) and an autosampler from CTCAnalytics (HTSPal; Zwingen, Switzerland). The chromatographic separation was performed on a waters symmetry C18 column (150× 4.6 mm; 5 μm) with a pre-column made of the same material. The injection volume was 30 μL. A binary gradient of 30 min with solvent A (H2O/ACN/FA; 90/10/0.1%) and solvent B (ACN/0.1% FA) was set to a flow rate of 300 μL min−1. The initial composition of 0% B was held for 3 min, followed by a linear gradient up to 95% B in 20 min, holding for 5 min, changing to 0% B in 1.5 min, and re-equilibrating at the end. The detection was performed on a Micromass Quattro micro triple quadrupole mass spectrometer (Waters GmbH, Eschbronn, Germany) in the electrospray ionization-positive SRM mode. Spray voltage was set to 2.9 kV and the heated capillary operated at 350 °C. The desolvation gas flow worked at 200 L h−1. Eluates from the first 15 min and the last 4 min were diverted to waste in order to prevent contamination of the source due to matrix components. Fig. 7 shows a representative HPLC chromatogram of this metabolism assay. The retention times for compound 1 and the developing metabolite were 30.92 and 26.51 min, respectively, in a 33 min HPLC run (Fig. 7). Metabolites were also identified by direct injection of the according reference material. Substrates and metabolites were quantified by internal standards as well as with calibration curves constructed from known concentrations of reference material.
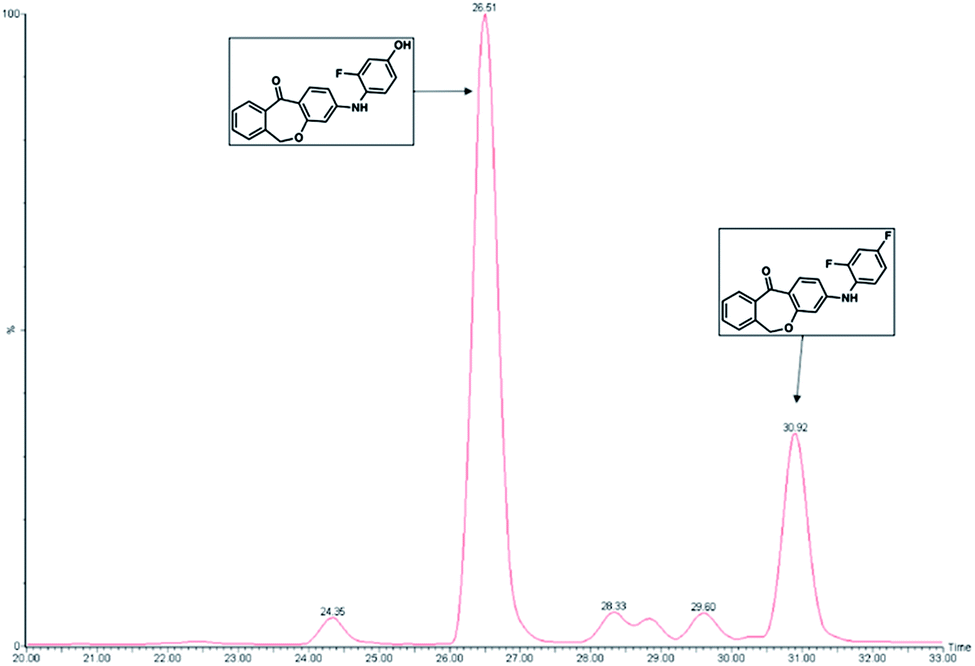 | ||
| Fig. 7 A representative HPLC chromatogram of phase 1 incubations showing the only metabolite detected in RLMs. Compound 1 (10 μM) was incubated with 1 mg mL−1 RLMs for 120 min. 1 and the corresponding metabolite peaks were detected at retention times of 30.92 and 26.51 min, respectively, in a 33 min HPLC run. The peaks in front of the metabolite and between the two observed peaks were determined to be impurities stemming from the RLMs. | ||
Data analysis
Enzyme kinetic parameters KM and vmax were determined using nonlinear regression analysis with GraphPad Prism software, version 5.0a for Mac OsX (GraphPad Software, San Diego, California, USA, http://www.graphpad.com). The results are expressed as the mean value of three experiments (±SEM). The concentrations were determined in μmol L−1.Metabolite formation was analyzed using a one-phase association equation. The half-time was computed as ln(2)/K, where K is the rate constant, expressed in reciprocals of time units.
Biological testing
The effect of both synthesized compounds on p38α inhibition was tested in an isolated p38α MAP kinase assay. In our system45 the tested compound competes with adenosine triphosphate (ATP) for the ATP binding site in the catalytic domain. The phosphorylation of activating transcription factor-2 (ATF-2) is determined with an anti-phospho-ATF-2 antibody. The degree of phosphorylation inversely correlates with the inhibitory activity of the tested compounds46 and was measured using an enzyme-linked immunosorbent assay (ELISA).Experimental procedures
All commercially available reagents and solvents were used without further purification. 1H NMR (200/400 MHz) and 13C NMR (50/100 MHz) spectra were recorded on a Bruker Advance 200/400 NMR spectrometer. Chemical shifts (δ) are reported in ppm relative to TMS and spectra were calibrated using the solvent resonance. Flash chromatography was performed using an Interchim PuriFlash 430 automated flash chromatography system with self-packed columns containing Davisil LC60A 20-45 micron silica from Grace Davison. The purity of the final compounds was determined by HPLC (Hewlett Packard HP 1090 Series II) LC equipped with a UV diode array detector (DAD, detection at 254 nm). The chromatographic separation was performed on a Phenomenex Luna 5u C8 column (150 mm × 4.6 mm, 5 μm) at 35 °C oven temperature, employing a gradient of 0.01 M KH2PO4 (pH 2.3) and methanol as solvent system with a flow rate of 1.0 mL min−1. EI mass spectra were measured on a Hewlett Packard HP 6890 Series GC-MS-system equipped with a HP-5MS capillary column (0.25 EM film thickness 30 m × 0.25 mm) and a Hewlett Packard HP 5973 Mass selective detector (70 eV). FAB (fast atom bombardment) data were obtained from the department for mass spectrometry, Institute of Organic Chemistry, Eberhard-Karls-University Tuebingen. All compounds were >95% pure.
× 100 mL) and the combined organic layers dried over sodium sulfate. After solvent evaporation, the product was purified by column chromatography (silica gel, gradient 100% petroleum ether to 100% ethyl acetate) to obtain a colorless oil (1.90 g, 12.0 mmol, 94.3%). EI-MS: m/z 201.1 [M]+, HPLC: 100% (254 nm), 1H NMR (200 MHz, chloroform-d) δH 7.99–7.81 (1H, m), 7.13–6.98 (1H, m), 6.88–6.67 (1H, m), 5.30 (2H, s), 3.54 (3H, s). 13C NMR (50 MHz, chloroform-d) δC 165.47 (d, J1 = 255.6 Hz), 152.74 (d, J3 = 11.5 Hz), 128.72 (d, J4 = 40.6 Hz), 127.73 (d, J3 = 10.9 Hz), 108.73 (d, J2 = 23.5 Hz), 104.96 (d, J2 = 26.7 Hz), 95.56, 56.94.
:1). The product was obtained as a yellowish foam (134 mg, 353 μmol, 86.2%). FAB-MS: m/z 380.2 [M + H]+, HPLC: 100% (254 nm), 1H NMR (200 MHz, chloroform-d) δH 8.18 (1H, d, J3 = 8.9 Hz), 7.94 (1H, dd, J3,4 = 7.3, 1.5 Hz), 7.47 (2H, pd, J3,4 = 7.4, 1.6 Hz), 7.37–7.17 (2H, m), 6.96 (1H, dd, J3,4 = 10.1, 2.8 Hz), 6.68 (2H, m), 6.54 (1H, d, J4 = 2.3 Hz), 6.26 (1H, s), 5.32–4.97 (4H, m), 3.45 (3H, s). 13C NMR (50 MHz, chloroform-d) δC 188.64, 163.40, 159.17 (d, J1 = 242.9 Hz), 150.92, 149.70 (d, J3 = 10.3 Hz), 140.70, 135.65, 134.14, 132.25, 129.67, 129.17, 127.68, 126.18 (d, J4 = 3.3 Hz), 122.03 (d, J3 = 9.6 Hz), 118.04, 110.83, 108.35 (d, J2 = 22.3 Hz), 103.65 (d, J2 = 26.8 Hz), 103.01, 95.32, 73.71, 56.50.
30 mL). The combined organic layers were dried over sodium sulfate, the solvent was evaporated and the product purified by gradient column chromatography (silica gel, gradient 100% petroleum ether to 100% ethyl acetate). A yellow foam was obtained (15.4 mg, 49.5 μmol, 60.1%). ESI-MS: m/z 335.1 [M + H]+, HPLC: 97.4% (254 nm), 1H NMR (200 MHz, chloroform-d) δH 8.13 (1H, d, J3 = 8.9 Hz), 7.91 (1H, dd, J3,4 = 7.4, 1.6 Hz), 7.64–7.37 (2H, m), 7.30 (1H, dd, J3,4 = 7.1, 1.5 Hz), 7.14 (1H, dd, J3,4 = 8.7, 5.9 Hz), 6.76 (1H, dd, J3,4 = 9.6, 2.7 Hz), 6.70–6.42 (3H, m), 6.20 (1H, d, J4 = 2.0 Hz), 5.71 (1H, s), 5.10 (2H, s). 13C NMR (50 MHz, chloroform-d) δC 189.24, 163.55, 159.42, 153.22 (d, J3 = 12.6 Hz), 152.78, 140.64, 135.62, 134.40, 132.47, 129.68, 129.30, 127.82 (d, J3 = 10.0 Hz), 127.78, 120.43 (d, J1 = 219.5 Hz), 110.26, 107.88 (d, J2 = 22.8 Hz), 103.81 (d, J2 = 25.7 Hz), 103.04, 73.79.
Conclusions
Both activity and selectivity of protein kinase inhibitors might be altered by metabolism. The metabolites of oxepine analogs of skepinone-type p38 MAPK-inhibitors are inactive for both p38 and a broad range of other kinases as well (Novartis kinase Panel, data not shown). The metabolic lability of aromatic fluorine residues is another major finding. 2,4-Difluorophenyl residues were hydroxylated to 2-fluoro-4-hydroxyphenyl derivates by CYP2B6.Acknowledgements
Dr Benjamin Baur and Matthias Gehringer from University of Tuebingen, Department of Pharmaceutical and Medicinal Chemistry designed and executed the synthesis of both possible hydroxyl metabolites. Prof. Dr Stefan Laufer supervised the project and was decisively involved in the initial hypothesis generation as well as in all data discussions and analysis. Kirsten Storch planed, established and accomplished the experimental setup. Moreover she generated/analyzed the data and wrote the manuscript. We thank Novartis Pharma AG (Jörg Trappe, Peter Drückes and Jürgen Köppler) for screening in the Novartis Kinase Panel.Notes and references
- J. Lisnock, A. Tebben, B. Frantz, E. A. O'Neill, G. Croft, S. J. O'Keefe, B. Li, C. Hacker, S. De Laszlo, A. Smith, B. Libby, N. Liverton, J. Hermes and P. LoGrasso, Biochemistry, 1998, 37, 16573–16581 CrossRef CAS PubMed.
- K. F. Chung, Chest, 2011, 139, 1470–1479 CrossRef CAS PubMed.
- N. C. Newton and C. P. Decicco, J. Med. Chem., 1999, 42, 2295–2314 CrossRef PubMed.
- F. Berenbaum, Curr. Opin. Rheumatol., 2004, 16, 616–622 CrossRef.
- C. Peifer, G. Wagner and S. Laufer, Curr. Top. Med. Chem., 2006, 6, 113–149 CrossRef CAS.
- Y. J. Feng and Y. Y. Li, J. Dig. Dis., 2011, 12, 327–332 CrossRef CAS PubMed.
- D. Hommes, B. Van Den Blink, T. Plasse, J. Bartelsman, C. Xu, B. Macpherson, G. Tytgat, M. Peppelenbosch and S. Van Deventer, Gastroenterol, 2002, 122, 7–14 CrossRef CAS PubMed.
- R. Newton and N. Holden, BioDrugs, 2003, 17, 113–129 CrossRef CAS PubMed.
- P. J. Barnes, Discov. Med., 2004, 4, 421–426 Search PubMed.
- J. Hynes Jr and K. Leftheri, Curr. Top. Med. Chem., 2005, 5, 967–985 CrossRef.
- S. R. Natarajan and J. B. Doherty, Curr. Top. Med. Chem., 2005, 5, 987–1003 CrossRef CAS.
- S. B. Cohen, T.-T. Cheng, V. Chindalore, N. Damjanov, R. Burgos-Vargas, P. DeLora, K. Zimany, H. Travers and J. P. Caulfield, Arthritis Rheuma, 2009, 60, 335–344 CrossRef CAS PubMed.
- R. J. Hill, K. Dabbagh, D. Phippard, C. Li, R. T. Suttmann, M. Welch, E. Papp, K. W. Song, K.-C. Chang, D. Leaffer, Y.-N. Kim, R. T. Roberts, T. S. Zabka, D. Aud, J. Dal Porto, A. M. Manning, S. L. Peng, D. M. Goldstein and B. R. Wong, J. Pharmacol. Exp. Ther., 2008, 327, 610–619 CrossRef CAS PubMed.
- C. Dominguez, D. A. Powers and N. Tamayo, Curr. Opin. Drug Discovery Dev., 2005, 8, 421–430 CAS.
- M. W. Karaman, S. Herrgard, D. K. Treiber, P. Gallant, C. E. Atteridge, B. T. Campbell, K. W. Chan, P. Ciceri, M. I. Davis, P. T. Edeen, R. Faraoni, M. Floyd, J. P. Hunt, D. J. Lockhart, Z. V. Milanov, M. J. Morrison, G. Pallares, H. K. Patel, S. Pritchard, L. M. Wodicka and P. P. Zarrinkar, Nat. Biotechnol., 2008, 26, 127–132 CrossRef CAS PubMed.
- G. Alonso, C. Ambrosino, M. Jones and A. R. Nebreda, J. Biol. Chem., 2000, 275, 40641–40648 CrossRef CAS PubMed.
- N. W. Court, C. G. dos Remedios, J. Cordell and M. A. Bogoyevitch, J. Mol. Cell. Cardiol., 2002, 34, 413–426 CrossRef CAS PubMed.
- K. K. Hale, D. Trollinger, M. Rihanek and C. L. Manthey, J. Immunol., 1999, 162, 4246–4252 CAS.
- J. S. Mudgett, J. Ding, L. Guh-Siesel, N. A. Chartrain, L. Yang, S. Gopal and M. M. Shen, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10454–10459 CrossRef CAS PubMed.
- M. Allen, L. Svensson, M. Roach, J. Hambor, J. McNeish and C. A. Gabel, J. Exp. Med., 2000, 191, 859–870 CrossRef CAS.
- V. A. Beardmore, H. J. Hinton, C. Eftychi, M. Apostolaki, M. Armaka, J. Darragh, J. McIlrath, J. M. Carr, L. J. Armit, C. Clacher, L. Malone, G. Kollias and J. S. C. Arthur, Mol. Cell. Biol., 2005, 25, 10454–10464 CrossRef CAS PubMed.
- S. C. Koeberle, J. Romir, S. Fischer, A. Koeberle, V. Schattel, W. Albrecht, C. Gruetter, O. Werz, D. Rauh, T. Stehle and S. A. Laufer, Nat. Chem. Biol., 2012, 8, 141–143 CrossRef CAS PubMed.
- B. Baur, K. Storch, K. E. Martz, M. I. Goettert, A. Richters, D. Rauh and S. A. Laufer, J. Med. Chem., 2013, 56, 8561–8578 CrossRef CAS PubMed.
- L. Gervot, B. Rochat, J. C. Gautier, F. Bohnenstengel, H. Kroemer, V. De Berardinis, H. Martin, P. Beaune and I. De Waziers, Pharmacogenetics, 1999, 9, 295–306 CrossRef CAS PubMed.
- T. Lang, K. Klein, J. Fischer, A. K. Nussler, P. Neuhaus, U. Hofmann, M. Eichelbaum, M. Schwab and U. M. Zanger, Pharmacogenetics, 2001, 11, 399–415 CrossRef CAS PubMed.
- S. R. Faucette, R. L. Hawke, E. L. Lecluyse, S. S. Shord, B. Yan, R. M. Laethem and C. M. Lindley, Drug Metab. Dispos., 2000, 28, 1222–1230 CAS.
- J. Hukkanen, M. Mantyla, L. Kangas, P. Wirta, J. Hakkola, P. Paakki, S. Evisalmi, O. Pelkonen and H. Raunio, BMC Pharmacol. Toxicol., 1998, 82, 93–97 CrossRef CAS.
- A. Janmohamed, C. T. Dolphin, I. R. Phillips and E. A. Shephard, Biochem. Pharmacol., 2001, 62, 777–786 CrossRef CAS.
- T. Thum and J. Borlak, Lancet, 2000, 355, 979–983 CrossRef CAS.
- V. Lamba, J. Lamba, K. Yasuda, S. Strom, J. Davila, M. L. Hancock, J. D. Fackenthal, P. K. Rogan, B. Ring, A. Wrighton Steven and G. Schuetz Erin, J. Pharmacol. Exp. Ther., 2003, 307, 906–922 CrossRef CAS PubMed.
- J. Solus, B. J. Arietta, J. R. Harris, D. P. Sexton, J. Q. Steward, C. McMunn, P. Ihrie, J. M. Mehall, T. L. Edwards and E. P. Dawson, Pharmacogenomics, 2004, 5, 895–931 CrossRef CAS PubMed.
- M. Ingelman-Sundberg, S. C. Sim, A. Gomez and C. Rodriguez-Antona, Pharmacol. Ther., 2007, 116, 496–526 CrossRef CAS PubMed.
- P. Jancova, P. Anzenbacher and E. Anzenbacherova, Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia, 2010, vol. 154, pp. 103–116 Search PubMed.
- C. Guillemette, Pharmacogenomics J., 2003, 3, 136–158 CrossRef CAS PubMed.
- A. Mori, Y. Maruo, M. Iwai, H. Sato and Y. Takeuchi, Drug Metab. Dispos., 2005, 33, 672–675 CrossRef CAS PubMed.
- H. Jinno, M. Saeki, Y. Saito, T. Tanaka-Kagawa, N. Hanioka, K. Sai, N. Kaniwa, M. Ando, K. Shirao, H. Minami, A. Ohtsu, T. Yoshida, N. Saijo, S. Ozawa and J.-I. Sawada, J. Pharmacol. Exp. Ther., 2003, 306, 688–693 CrossRef CAS PubMed.
- A. Iida, S. Saito, A. Sekine, C. Mishima, Y. Kitamura, K. Kondo, S. Harigae, S. Osawa and Y. Nakamura, J. Hum. Genet., 2002, 47, 505–510 CrossRef CAS PubMed.
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601–2611 CrossRef CAS PubMed.
- C. Sams, G. D. Loizou, J. Cocker and M. S. Lennard, Toxicol. Lett., 2004, 147, 253–260 CrossRef CAS PubMed.
- Y. Lin, P. Lu, C. Tang, Q. Mei, G. Sandig, A. D. Rodrigues, T. H. Rushmore and M. Shou, Drug Metab. Dispos., 2001, 29, 368–374 CAS.
- N. Chauret, A. Gauthier and D. A. Nicoll-Griffith, Drug Metab. Dispos., 1998, 26, 1–4 CAS.
- W. F. Busby Jr, J. M. Ackermann and C. L. Crespi, Drug Metab. Dispos., 1999, 27, 246–249 CAS.
- L. Oleson and M. H. Court, J. Pharm. Pharmacol., 2008, 60, 1175–1182 CrossRef CAS PubMed.
- T. Pallmann, PhD thesis, Universität Marburg, 2007.
- S. Laufer and S. Linsenmaier, J. Biomol. Screening, 2006, 11, 528–536 CrossRef CAS PubMed.
- M. Goettert, R. Graeser and S. A. Laufer, Anal. Biochem., 2010, 406, 233–234 CrossRef CAS PubMed.
- S. A. Laufer, G. M. Ahrens, S. C. Karcher, J. S. Hering and R. Niess, J. Med. Chem., 2006, 49, 7912–7915 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |