 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inhibitors of the human neuraminidase enzymes
Christopher W.
Cairo
*
Alberta Glycomics Centre, Department of Chemistry, University of Alberta, Edmonton Alberta, T6G 2G2, Canada. E-mail: ccairo@ualberta.ca; Fax: +1 780 492 8231; Tel: +1 780 492 0377
First published on 18th June 2014
Abstract
The human neuraminidase enzymes (hNEU) are a family of four isoenzymes that hydrolyze sialosides, including gangliosides and glycoproteins. These enzymes are proposed to play roles in several important signaling pathways and are implicated in diseases such as diabetes and cancer. Despite their importance, only a limited number of studies have sought to identify potent inhibitors for these enzymes. This review summarizes the substrate specificity and known inhibitors of the hNEU isoenzymes, as well as the emerging work on development of isoenzyme-specific inhibitors.
1. Introduction
The human neuraminidase enzymes (hNEU) are a family of glycosyl hydrolases (GH) that play critical roles in human health and disease. There are currently four known isoenzymes: NEU1, NEU2, NEU3, and NEU4. Individual isoenzymes have been found to vary in both subcellular and tissue localization.1–4 NEU1 is part of a membrane complex, and is found primarily in the lysosome and plasma membrane.5,6 Murine NEU1 has four sites of N-link glycosylation, several of which are critical for enzyme function.7 NEU2 has been found to be localized to the cytosol,8–10 while NEU3 and NEU4 are thought to be membrane-associated.11,12 The NEU4 isoenzyme is found in endoplasmic reticulum,13,14 mitochondrial,13,15 and lysosomal4 membranes and occurs as both a long and truncated form, which affect its subcellular localization.13,15The hNEU have been implicated in regulation of a variety of human pathologies. The identification of such a large number of hNEU isoenzymes may suggest that cells exploit these enzymes to remodel existing epitopes to rapidly generate modified glycan structures.16,17 Such a mechanism would avoid the time and energy required to biosynthesize a new glycan structure and may help explain the more rapid turnover of terminal carbohydrate residues in glycoproteins.18,19 Plasma-membrane associated forms of the enzyme can act on substrates found on other cells.20 The expression levels of hNEU isoenzymes are linked to malignancy.21,22 For example, NEU4 is overexpressed in neuroblastoma,23 and NEU1 expression has been linked to metastasis through regulation of integrin adhesion.24 hNEU have been linked to regulation of lipid metabolism, which may be relevant in cardiovascular disease.25 NEU1 has recently been linked to signaling by the insulin receptor in a model of diabetes.26 Several comprehensive reviews discuss the biology and links to disease for these isoenzymes in more detail.27–32
The family of hNEU are clearly important for a variety of biological mechanisms; however, few small molecule inhibitors of these enzymes have been reported. In contrast, a large body of research has been devoted to development of inhibitors for the viral neuraminidase enzymes (vNEU) as anti-influenza therapeutics.33 Importantly, compounds which target vNEU have not been found to be as active in the human enzymes.34 Thus, there remains a need for the development of compounds which target hNEU specifically. When considering the design of hNEU inhibitors, one should remember that certain functions of hNEU are essential for homeostasis. Genetic deficiency of the NEU1 isoenzyme results in sialidosis;35,36 thus, potential therapeutics must show specificity to avoid off-target or non-specific inhibition of undesired isoenzymes. The high homology within the enzyme family presents a challenging problem for the medicinal chemist, not only must potential therapeutics be active against the desired isoenzyme – but they must be highly selective. This review will summarize the current state of known small molecule inhibitors for the hNEU, as well as relevant molecular details regarding active-site structure and substrate specificity. Particular attention will be devoted to features and activities which are distinct among the isoenzymes.
2. Structure of hNEU
The hNEU are classified as family 33 glycosyl hydrolases (GH33) and cleave the terminal glycosidic linkage of sialic acid (exo-sialidases; EC 3.2.1.18).37 Alignment of the four human isoenzyme sequences shows the greatest similarity between NEU2 and NEU3. The NEU2 isoenzyme is the only hNEU studied by X-ray crystallography. Chavas et al. have reported co-crystals of the enzyme with 2-deoxy-2,3-didehydro-N-acetyl-neuraminic acid (DANA; 15),38 as well as viral neuraminidase enzyme (vNEU) inhibitors.39 The enzyme shows a 6-bladed β-propellar fold common to GH33 members which is conserved in many species.40 Unfortunately, there are currently limited structural data for the remaining hNEU members. This is likely due to the nature of the NEU1, NEU3, and NEU4 isoenzymes – each of these are either a component of a membrane complex or membrane-associated. The enzymes may contain post-translational modifications including phosphorylation4 and glycosylation7 that may be important for activity.In the absence of experimental protein structures, indirect methods of examining hNEU structure have been proposed. Magesh et al. first introduced homology models of the three remaining isoenzymes based on the reported structure of NEU2.41 Although NEU3 has been proposed to contain a transmembrane domain,2 further study has suggested that it is more likely a peripheral membrane-associated protein.12,42 A homology model of NEU3 has also been reported and validated using site-directed mutagenesis.43 A homology model of NEU4 was proposed to rationalize the binding of NEU4-selective inhibitors.44 The binding epitope of NEU3 has been examined using a trisaccharide analog of GM3.45
Some insight into the structure of hNEU can be gained from examining mutations of the enzymes. Naturally occurring mutations on NEU1 have been characterized in relation to sialidosis diseases.46 Mutagenesis of NEU1 has also revealed that N-link glycosylation of the protein is essential for its activity.7 The NEU2 enzyme has been studied by mutagenesis, confirming the catalytic pair as Y334 and E218.47,48 Site-directed mutagenesis of NEU3 was used to identify key active site residues, including the catalytic pair (Y370 and E225) and the arginine triad (R25, R245, R340).42,43 Deletion of a proline-rich segment of NEU4 has been found to alter protein–protein interactions and to alter substrate specificity.49
There are several key structural features of the active site of neuraminidase enzymes necessary for recognition of inhibitors across species.33,40,48 The C1 carboxylate of sialosides is recognized by a triad of arginine residues (Fig. 1). The catalytic residues of GH33 are a Tyr–Glu pair, with the Glu residue acting as a general acid–base and the Tyr-OH acting as a nucleophile.43,47,50 Finally an Asp residue is required as a general acid–base within the active site.48 The vNEU contain a flexible recognition site for the glycerol sidechain of the sialoside, and this has been exploited in the design of inhibitors.51 The recognition of the glycerol sidechain in hNEU appears to be distinct and relies on several contacts to Tyr residues.38,39
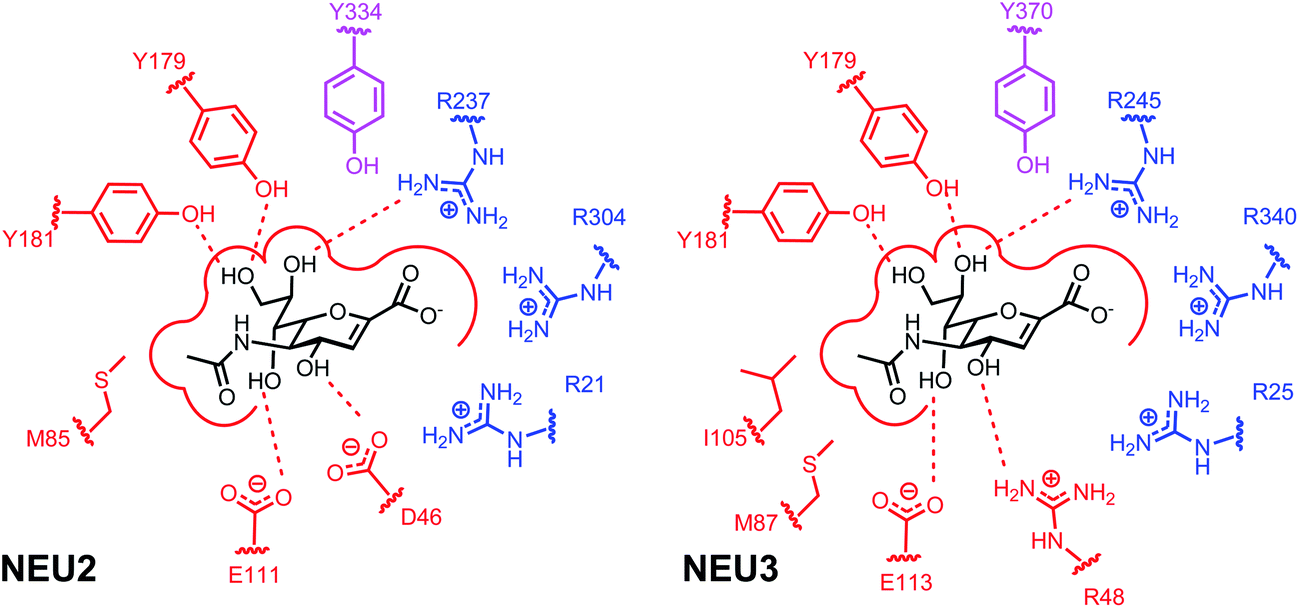 | ||
| Fig. 1 Binding site interactions of the hNEU enzymes NEU2 and NEU3. Schematics of the key interactions for DANA in the active site of NEU2 (left).38,47 The interactions proposed for the NEU3 active site are based on mutagenesis and homology modeling studies (right).43 Residues that form the arginine triad in both enzymes are colored in blue. The proposed Tyr nucleophile is shown in magenta for each binding site. The active site nucleophiles act as a catalytic pair with E218 (NEU2) and E225 (NEU3), not shown in this figure. | ||
3. Substrate recognition in hNEU
The hNEU are exo-sialidases which cleave α-linked N-acetyl-neuraminic acid (Neu5Ac); among these, NEU2 and NEU3 have been confirmed as retaining GH.43,52 Neu5Ac linkages in glycoproteins are typically α(2,6), though α(2,3) are possible, while α(2,3), α(2,6), and α(2,8) linkages are present in glycolipids (Fig. 2; 1–3).53 All four enzymes have acidic pH optima, typically found to be between pH 3.5 and 5.5.4,42,54 Reports differ on the activity of NEU2 in the presence of metal ions, although high concentrations of EDTA appear to reduce enzyme activity.52,55 The presence of Cu2+ ions is known to be inhibitory for NEU2,10,52,55 and NEU3;43 as a result, caution should be used when testing inhibitors that could contain this contaminant. Detection of neuraminidase activity often exploits the fluorogenic substrate 2′-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (4MU-NANA; 4).56,57 Alternative methods have been developed that detect the release of free sialic acid,58,59 or that use a coupled assay to detect the newly exposed terminal sugar after neuraminidase action.60 | ||
| Fig. 2 Substrates of the hNEU. (Sph = sphingolipid). | ||
There are few detailed studies of hNEU substrate specificity, and additional work is needed to develop the full picture of differences in substrate activity among the isoenzymes. Studies of enzyme specificity with synthetic analogs of sialosides are extremely valuable for the design of inhibitors or enzyme probes,61 and known substrate activities of each hNEU isoenzyme are briefly summarized below.
NEU1 targets glycoprotein substrates and has been shown to prefer α(2,3) over α(2,6) linkages (i.e.1vs.2).62 The enzyme has reduced activity for ganglioside substrates.4 NEU1 has been shown to target cell surface components such as the toll-like receptor,62 insulin receptor,26 and others.30
Synthetic substrates with variation in the Neu5Ac residue and its glycosidic linkage have been tested with NEU2.60 Similar to NEU1, NEU2 shows a strong preference for α(2,3) linkages. Testing with substrates containing N-glycolyl-neuraminic acid (Neu5Gc) residues (6) found that this substrate was preferred over the native Neu5Ac analog (5). Further modification of the Neu5Gc residue at the C9 position to an azido group also resulted in a substrate with improved activity (7).60 Modification at the C9 of Neu5Gc- or Neu5Ac-α(2,3) to O-methyl, -fluoro, or -deoxy analogs greatly reduced activity, suggesting that this position is critical for recognition. The NEU2 active site has been proposed to recognize portions of the glycan substrate distal to the Neu5Ac glycosidic linkage. Site directed mutagenesis and molecular modeling have implicated D358 and Y359 in the recognition of 6′-sialyllactose analogs,63 and Q112 and Q270 may interact with the Gal or Glc residues of sialyllactose substrates.47
Inactivating substrates of hNEU have been tested against the NEU2 isoenzyme. Kai et al. tested a 2-difluoromethylphenyl sialoside (12) against NEU2.64 They confirmed that the compound could inactivate the enzyme in vitro and used an azide tag to generate a library of potential inhibitors. The most active compound identified had a reported inhibition constant of 200 μM. Quinone methide based inactivators may have long lifetimes, and the rate of formation and consumption of the reactive intermediate may influence the specificity of these reagents.65 Another class of inactivators, 2-deoxy-2,3-difluoro-derviatives of Neu5Ac (13), have been tested against NEU2 but had weaker activity against the enzyme when compared to viral and bacterial enzymes.66 Crystallographic data on the inactivated complex between 2,3-difluoro sialic acid analogs and NEU2 has recently been reported.67
Fluorogenic or inactivating substrates for hNEU are potentially useful for sensitive detection and imaging of enzyme activity.61 Zamora et al. have tested a series of 4MU-NANA (4) derivatives against hNEU from cell lysates.68 Among the N5′-modified derivatives of 4, fluoromethyl and glycolyl derivatives showed improved activity. Notably, the activity of hNEU against the modified substrates was distinct from bacterial enzymes, raising the possibility of species- or isoenzyme-specific forms of 4MU-NANA. A fluorogenic benzothiazolylphenol-sialoside has been used to detect sialidase activity in mammalian tissues.69 Inactivating 2-deoxy-2,3-difluoro derivatives of Neu5Ac have been used as imaging probes for hNEU. Tsai et al. synthesized compound 14 (used as its per-acetylated form in cells) for labeling of NEU with an alkyne tag that could be easily visualized using Cu-catalyzed azide–alkyne cycloaddition (CuAAC).70 Direct labeling of all four hNEU isoenzymes and viral enzymes was observed by blotting. Fluorescent labeling of cells with these probes was also performed to visualize enzyme activity.
The NEU3 isoenzyme has been found to generally prefer glycolipid over glycoprotein substrates.3,4,71 The substrate specificity of NEU3 has been analyzed for gangliosides, demonstrating that the enzyme possesses both α(2,3) and α(2,8) sialidase activity.3,72,73 Additionally, the enzyme does not cleave α(2,3) linkages at internal sites, for example both GM1 and GM2 (which contain a single branched α(2,3)-Neu5Ac-[GlcNAc-β(1,4)]-Gal residue) are poor substrates.3,42,71 This observation has been rationalized using the homology model of NEU3.43 Furthermore, neoglycoproteins containing GD1a oligosaccharides are substrates of NEU3.74
The substrate activity of synthetic analogs of GM3 with NEU3 have been analyzed using a mass spectrometry-based kinetic assay.75 These studies revealed that the isoenzyme requires a hydrophobic aglycone; however, there is no requirement for diacyl or sphingosine-based lipids. Thus, the native GM3 substrate (8) can be simplified to an octyl-glycoside (9) providing identical substrate activity. The N5-azidoacetate (10) showed slightly improved activity over the native trisaccharide (9). NEU3 did not tolerate larger modifications at the N-acyl sidechain of Neu5Ac and showed minimal activity with triazole derivatives of 10. Interestingly, the C9-azido analog (11) retained significant substrate activity even after modification to a triazole. These findings suggest that modification of substrates at Neu5Ac may be better tolerated at C9 over N5.
NEU4 is able to hydrolyze both glycoprotein and glycolipid substrates.4,76 The short isoform of NEU4 has been shown to cleave the α(2,3) linkage of sialyl-Lewisa and sialyl-Lewisx antigens, which are critical for leukocyte adhesion mediated by E-selectin.77,78 NEU4 has also been shown to selectively cleave poly-sialic acids, containing α(2,8) linkages, in neuronal cells.79
4. Small molecule inhibitors of hNEU
Potent and selective small molecule inhibitors of hNEU will be valuable tools for research into the function of specific isoenzymes. Significant progress has been made in the design of inhibitors for glycosyl hydrolases.80 Neuraminidase inhibitors which target the vNEU enzymes have been extremely successful,33 but known vNEU inhibitors appear to have very weak activity against hNEU. Additionally, the hNEU system is complicated by the number of isoenzymes in the family which may impede the design of selective inhibitors. Studies of hNEU inhibitors are summarized below, paying particular attention to examples that provide isoenzyme selectivity.4.1. Non-carbohydrate inhibitors
Relatively few non-carbohydrate derived inhibitors have been tested against hNEU, and most of these have shown limited inhibitory activity. The cyclohexene-based oseltamivir and cyclopentane-derived peramivir are both potent vNEU inhibitors but show limited activity against hNEU.34,39 Derivatives of 2-fluorobenzoic acid were designed to inhibit hNEU; the most active of these had a Ki of ∼300 μM.81 A furan-2,5-dicarboxylic acid has been tested but was found to have weak inhibitory activity against NEU3 and NEU4.824.2. Neu5Ac- and DANA-based inhibitors of individual isoenzymes
The most commonly used pan-selective neuraminidase inhibitor is the 2-deoxy-2,3-didehydro-N-acetyl-neuraminic acid (DANA; 15) (Fig. 3). It is interesting to note that DANA can be synthesized by the NEU enzymes when Neu5Ac is present in high enough concentrations. The formation of DANA is likely the result of dehydration by Neu5Ac when bound in the active site.83 The hNEU enzymes are also capable of catalyzing this dehydration reaction.75,84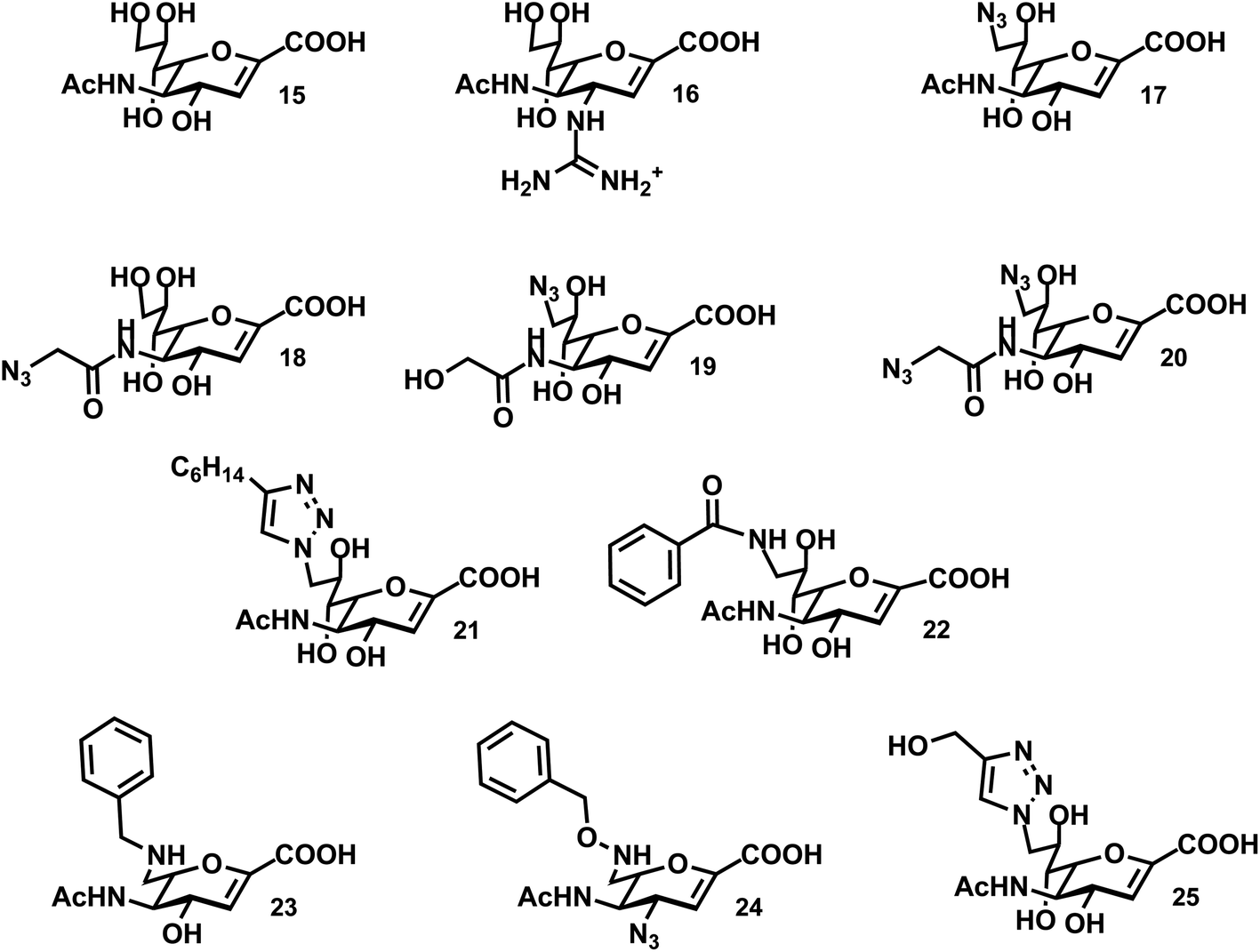 | ||
| Fig. 3 Inhibitors of the hNEU enzymes. | ||
Early work with purified hNEU isoenzymes confirmed that DANA (15) and C9-modified derivatives were active inhibitors.85 This was the first study to identify the low μM activity of these compounds; however, neither 15 nor 17 was found to have selectivity for isoenzymes of hNEU. Thioglycosides have been investigated as potential inhibitors of NEU2.52,63 The NEU2 enzyme shows a strong preference for α(2,3)-linked substrates,55,60 and a thioglycoside(methyl-2-deoxy-2-thio-Neu5Ac-α(2,3)-β-galactoside) exhibited mild inhibitory activity.52
Several reports have tested DANA-analogs for inhibition of the NEU2 isoenzyme. Li et al. found an IC50 of 18 ± 1 μM for 15 with a recombinant form of the enzyme.55 DANA itself was more potent as an inhibitor of α(2,3)-Neu5Ac substrates over α(2,6), and we will focus on results against this substrate. Modification of DANA at the N5 acyl group resulted in improved potency for compound 18 (IC50 of 8.3 ± 1 μM). The viral inhibitor, zanamivir (16), was also tested against NEU2 and was one of the most potent inhibitors in the study (IC50 of 5.3 ± 0.7 μM.) Khedri et al. explored modifications of DANA at C9 in combination with a modified N-acyl group.86 The N5-glycolyl-C9-azido analog (19) had relatively low potency. However, the N5-azidoacetate-C9-azido analog (20) showed improved activity as an inhibitor of NEU2 (IC50 of 13 ± 4 μM) over DANA (IC50 of 32 ± 6 μM). Other modifications at C9, including O-methyl and alkylamine derivatives showed reduced potency. DANA derivatives with the glycerol side chain replaced by a C6-ether showed moderate potency against NEU2, though these were all lower activity than the parent, 15.39
Testing of a small library of inhibitors against NEU3 was reported by Zou et al.87 Compounds included azide derivatives of DANA, which could be easily converted to triazole analogs.88 Modifications at both the N5 and C9 positions were used to explore the structure activity relationships of these compounds. In common with reports for NEU2, this study found that compound 18 had improved potency (IC50 of 21 ± 8 μM) relative to DANA (15; IC50 of 48 ± 5 μM).87 Modification at C9 to an azide, as in compound 17, resulted in decreased activity. However, conversion to a C9-triazole maintained activity when an alkyl group was included (21; IC50 of 20 ± 4 μM). Similar triazole modifications of 18 universally reduced potency, leading the authors to conclude that the active site of the enzyme could tolerate large modifications at C9 better than at N5. Similar observations were found in substrate studies of NEU3.75
4.3. Selectivity of inhibitors among the hNEU family
Recent studies have begun to realize isoenzyme-selective inhibitors of hNEU. It should be noted that even the well-known inhibitor, DANA (15), is typically found to have a range of activity among the hNEU isoenzymes that varies between 2 to 10-fold (comparing the most active to least active inhibitors).34,54,70,89 However, studies that have tested DANA against all hNEU typically find it to be most active against the NEU3 or NEU4 isoenzymes.54,70Testing of C9-amido derivatives of DANA has identified NEU1-selective inhibitors. Magesh et al. synthesized a library of compounds and tested these against lysates from hNEU transiently transfected cell lines.89 The design of the compounds tested was based on homology models of the four isoenzymes.41 Several compounds were selective inhibitors of NEU1, with the most potent compounds containing small alkyl or aryl groups (22; NEU1 IC50 13 μM). Notably, these compounds were significantly more active than the parent DANA (15) in the same assays. The NEU1-active inhibitors showed very little potency towards the other isoenzymes tested. Compound 22 showed minor activity for NEU3, with approximately 25-fold selectivity for NEU1 based on the reported IC50 data. These findings suggested the possibility that the binding pocket for the glycerol sidechain of Neu5Ac (C7–C9) may show variability among the isoenzymes that could be exploited for the design of selective inhibitors.
A recent study examined the activity of a library of DANA derivatives containing C4 and C7 modifications which allowed the identification of NEU2 and NEU3 selective inhibitors.54 This strategy was able to identify a 12-fold selective inhibitor for NEU2, 23 (IC50 86 ± 17 μM). Combining C4-azido and C7-benzyloxyamine modifications to the DANA scaffold generated a nearly 40-fold selective inhibitor for NEU3 (24; IC50 24 ± 2 μM, Ki 8 ± 1 μM).54 Albohy et al. identified oseltamivir analogs that were moderately active against NEU3 with partial selectivity for NEU3 over NEU4.90 Tsai et al. tested the activity of compound 14 against purified recombinant hNEU isoenzymes.70 They reported the compound was most active against NEU3 (IC50 5 ± 3 μM), although the selectivity was approximately 2-fold (based on the second most potent IC50 among hNEU).
The most potent and selective inhibitors for NEU4 were recently identified through only minor modifications of DANA.44 Albohy et al. tested a set of compounds against all four hNEU which had previously shown only moderate activity in assays of NEU3 alone.44 However, in testing against a panel of purified and recombinant hNEU, several of these compounds were found to have nanomolar activity against the NEU4 isoenzyme. The most potent compounds were C9-triazole derivatives of DANA containing an alkoxy group (25; Ki 30 ± 19 nM). Importantly, these compounds are the most potent inhibitors of any hNEU isoform reported to date and also exhibit unprecedented selectivity, with approximately 500-fold selectivity for NEU4 over all other isoenzymes. The activity of the compound was tested in assays using GM3 as a substrate to confirm that the activity was not an artifact of the 4MU-NANA-based assays used in screening.
4.4. Activity of NEU inhibitors across species
The hNEU enzymes share a great deal of homology with NEU of other species. Additionally, NEU enzymes of bacteria and viruses are important virulence factors and targets for the development of therapeutics.33,91 The active site topology and interactions between the enzyme and 15 are generally similar to the bacterial and viral enzyme. However, some notable differences are found in the interactions at the glycerol sidechain, and a dynamic loop containing D46 (NEU2).38,39Testing of clinically active vNEU inhibitors has shown that these compounds have weak to moderate potency for the hNEU. Hata et al. tested a panel including DANA (15), zanamivir (16), and oseltamivir with purified isoenzymes.34 Although zanamivir and oseltamivir are reported to have exceptional potency against vNEU,92 against hNEU only zanamivir shows low micromolar activity. This finding may be beneficial for the use of antivirals, as these compounds should have limited effects on native hNEU. Mutations of NEU2 have been found to alter its susceptibility to oseltamivir, which may be a concern for side effects in some individuals.93,94 In the case of oseltamivir, the glycerol sidechain is replaced by a 3-pentyl group. This feature of the inhibitor benefits activity against some viral enzymes due to a rearrangement in the active site.51 Other features of the viral active sites, such as the 150-loop,95 have also been used to target vNEU.96,97 However, compounds designed to exploit features such as the 150-loop of vNEU are not potent inhibitors of hNEU.90,96
The key structural differences between the active sites of vNEU and hNEU are the binding pockets that complex the glycerol side chain (C7–C9 pocket) and the C4 substituent (Fig. 1). The C7–C9 binding pocket of NEU2 features two Tyr residues (Y179, Y181) that complex O7 and O9.38 This is distinct from the Glu residue that forms H-bond contacts with this portion of DANA in vNEU.98 Additionally, computational studies have suggested that the O4-binding pocket residues are a key factor in the decreased activity for C4-amino/guanidino inhibitors for hNEU.99
Several examples confirm that hNEU have distinct requirements for inhibition relative to bacterial NEU enzymes. In a library of compounds with modified C7-substituents Zhang et al. found very little activity for the V. cholerae NEU, but many of these compounds were active against hNEU.54 Compound 20, while active against NEU2, was found to have much lower activity against the V. cholerae and C. perfringens neuraminidase enzymes. Interestingly, the C9-azido-N5-glycolyl analog of DANA (19) had the reverse selectivity, showing approximately 10-fold selectivity for V. cholerae neuraminidase over C. perfringens and NEU2.86 Difluoro inhibitors which target vNEU were found to have significantly lower potency against NEU2.66
5. Conclusions and future prospects
The hNEU enzyme family has begun to gain the attention of medicinal chemists as a potential target for the development of tools for glycobiology and as a source for therapeutics. A large body of work has focused on the study of hNEU enzyme function using genetic tools; however, the availability of small molecule inhibitors with known specificity for hNEU family members will provide for new approaches in this complex system. Consider that a family of enzymes with similar mechanisms may compensate for the deletion of a single isoenzyme. However, isoenzyme-selective inhibitors of sufficient potency could be used to dissect the contributions of individual enzymes. Significant progress has been made on the identification of inhibitors, yet there remains a distinct need for compounds that can target each of the hNEU isoenzymes with greater specificity and potency. The most useful compounds will have activity both in vitro and in vivo, but currently few in vivo studies for the inhibitors discussed here have been reported. Additionally, the most potent compounds currently available are based on the DANA scaffold, and organic scaffolds that are more synthetically accessible or which are conformationally restricted could be an important asset to the field.100Acknowledgements
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada. The author thanks M. Richards for helpful discussions and editorial assistance.References and notes
- E. Monti, A. Preti, B. Venerando and G. Borsani, Neurochem. Res., 2002, 27, 649–663 CrossRef CAS.
- E. Monti, M. T. Bassi, N. Papini, M. Riboni, M. Manzoni, B. Venerando, G. Croci, A. Preti, A. Ballabio, G. Tettamanti and G. Borsani, Biochem. J., 2000, 349, 343–351 CrossRef CAS.
- T. Wada, Y. Yoshikawa, S. Tokuyama, M. Kuwabara, H. Akita and T. Miyagi, Biochem. Biophys. Res. Commun., 1999, 261, 21–27 CrossRef CAS PubMed.
- V. Seyrantepe, K. Landry, S. Trudel, J. A. Hassan, C. R. Morales and A. V. Pshezhetsky, J. Biol. Chem., 2004, 279, 37021–37029 CrossRef CAS PubMed.
- K. E. Lukong, V. Seyrantepe, K. Landry, S. Trudel, A. Ahmad, W. A. Gahl, S. Lefrancois, C. R. Morales and A. V. Pshezhetsky, J. Biol. Chem., 2001, 276, 46172–46181 CrossRef CAS PubMed.
- M. V. Vinogradova, L. Michaud, A. V. Mezentsev, K. E. Lukong, M. El-Alfy, C. R. Morales, M. Potier and A. V. Pshezhetsky, Biochem. J., 1998, 330, 641–650 CAS.
- D. Wang, S. Zaitsev, G. Taylor, A. d'Azzo and E. Bonten, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 275–282 CrossRef CAS PubMed.
- E. Monti, A. Preti, E. Rossi, A. Ballabio and G. Borsani, Genomics, 1999, 57, 137–143 CrossRef CAS PubMed.
- T. Miyagi and S. Tsuiki, J. Biol. Chem., 1985, 260, 6710–6716 CAS.
- C. Tringali, N. Papini, P. Fusi, G. Croci, G. Borsani, A. Preti, P. Tortora, G. Tettamanti, B. Venerando and E. Monti, J. Biol. Chem., 2004, 279, 3169–3179 CrossRef CAS PubMed.
- Y. Wang, K. Yamaguchi, T. Wada, K. Hata, X. J. Zhao, T. Fujimoto and T. Miyagi, J. Biol. Chem., 2002, 277, 26252–26259 CrossRef CAS PubMed.
- G. Zanchetti, P. Colombi, M. Manzoni, L. Anastasia, L. Caimi, G. Borsani, B. Venerando, G. Tettamanti, A. Preti, E. Monti and R. Bresciani, Biochem. J., 2007, 408, 211–219 CrossRef CAS PubMed.
- K. Yamaguchi, K. Hata, K. Koseki, K. Shiozaki, H. Akita, T. Wada, S. Moriya and T. Miyagi, Biochem. J., 2005, 390, 85–93 CrossRef CAS PubMed.
- E. Monti, M. T. Bassi, R. Bresciani, S. Civini, G. L. Croci, N. Papini, M. Riboni, G. Zanchetti, A. Ballabio, A. Preti, G. Tettamanti, B. Venerando and G. Borsani, Genomics, 2004, 83, 445–453 CrossRef CAS PubMed.
- A. Bigi, L. Morosi, C. Pozzi, M. Forcella, G. Tettamanti, B. Venerando, E. Monti and P. Fusi, Glycobiology, 2010, 20, 148–157 CrossRef CAS PubMed.
- R. B. Parker and J. J. Kohler, ACS Chem. Biol., 2010, 5, 35–46 CrossRef CAS PubMed.
- S. Z. Gadhoum and R. Sackstein, Nat. Chem. Biol., 2008, 4, 751–757 CrossRef CAS PubMed.
- R. Tauber, C. S. Park, A. Becker, R. Geyer and W. Reutter, Eur. J. Biochem., 1989, 186, 55–62 CrossRef CAS PubMed.
- R. Tauber, C. S. Park and W. Reutter, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 4026–4029 CrossRef CAS.
- N. Papini, L. Anastasia, C. Tringali, G. Croci, R. Bresciani, K. Yamaguchi, T. Miyagi, A. Preti, A. Prinetti, S. Prioni, S. Sonnino, G. Tettamanti, B. Venerando and E. Monti, J. Biol. Chem., 2004, 279, 16989–16995 CrossRef CAS PubMed.
- T. Miyagi, T. Wada, K. Yamaguchi and K. Hata, Glycoconjugate J., 2003, 20, 189–198 CrossRef.
- T. Miyagi, T. Wada, K. Yamaguchi, K. Shiozaki, I. Sato, Y. Kakugawa, H. Yamanami and T. Fujiya, Proteomics, 2008, 8, 3303–3311 CrossRef CAS PubMed.
- C. Tringali, F. Cirillo, G. Lamorte, N. Papini, L. Anastasia, B. Lupo, I. Silvestri, G. Tettamanti and B. Venerando, Int. J. Cancer, 2012, 131, 1768–1778 CrossRef CAS PubMed.
- T. Uemura, K. Shiozaki, K. Yamaguchi, S. Miyazaki, S. Satomi, K. Kato, H. Sakuraba and T. Miyagi, Oncogene, 2009, 28, 1218–1229 CrossRef CAS PubMed.
- A. Yang, G. Gyulay, M. Mitchell, E. White, B. L. Trigatti and S. A. Igdoura, J. Lipid Res., 2012, 53, 2573–2585 CrossRef CAS PubMed.
- L. Dridi, V. Seyrantepe, A. Fougerat, X. Pan, É. Bonneil, P. Thibault, A. Moreau, G. A. Mitchell, N. Heveker, C. W. Cairo, T. Issad, A. Hinek and A. V. Pshezhetsky, Diabetes, 2013, 62, 2338–2346 CrossRef CAS PubMed.
- T. Miyagi and K. Yamaguchi, Glycobiology, 2012, 22, 880–896 CrossRef CAS PubMed.
- E. Monti, E. Bonten, A. D'Azzo, R. Bresciani, B. Venerando, G. Borsani, R. Schauer and G. Tettamanti, in Adv. Carbohydr. Chem. Biochem., Academic Press, 2010, vol. 64, pp. 403–479 Search PubMed.
- A. V. Pshezhetsky and L. I. Ashmarina, Biochemistry (Moscow), 2013, 78, 736–745 CrossRef CAS PubMed.
- A. V. Pshezhetsky and A. Hinek, Glycoconjugate J., 2011, 28, 441–452 CrossRef CAS PubMed.
- E. Monti and T. Miyagi, in Top. Curr. Chem., Springer, Berlin Heidelberg, 2012, ch. 328, pp. 1–26, DOI:10.1007/128_2012_328.
- T. Miyagi, K. Takahashi, K. Hata, K. Shiozaki and K. Yamaguchi, Glycoconjugate J., 2012, 29, 567–577 CrossRef CAS PubMed.
- M. von Itzstein, Nat. Rev. Drug Discovery, 2007, 6, 967–974 CrossRef CAS PubMed.
- K. Hata, K. Koseki, K. Yamaguchi, S. Moriya, Y. Suzuki, S. Yingsakmongkon, G. Hirai, M. Sodeoka, M. Von Itzstein and T. Miyagi, Antimicrob. Agents Chemother., 2008, 52, 3484–3491 CrossRef CAS PubMed.
- A. V. Pshezhetsky, C. Richard, L. Michaud, S. Igdoura, S. P. Wang, M. A. Elsliger, J. Y. Qu, D. Leclerc, R. Gravel, L. Dallaire and M. Potier, Nat. Genet., 1997, 15, 316–320 CrossRef CAS PubMed.
- E. J. Bonten, W. F. Arts, M. Beck, A. Covanis, M. A. Donati, R. Parini, E. Zammarchi and A. d'Azzo, Hum. Mol. Genet., 2000, 9, 2715–2725 CrossRef CAS PubMed.
- B. L. Cantarel, P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard and B. Henrissat, Nucleic Acids Res., 2009, 37, D233–D238 CrossRef CAS PubMed.
- L. M. G. Chavas, C. Tringali, P. Fusi, B. Venerando, G. Tettamanti, R. Kato, E. Monti and S. Wakatsuki, J. Biol. Chem., 2005, 280, 469–475 CrossRef CAS PubMed.
- L. M. G. Chavas, R. Kato, N. Suzuki, M. von Itzstein, M. C. Mann, R. J. Thomson, J. C. Dyason, J. McKimm-Breschkin, P. Fusi, C. Tringali, B. Venerando, G. Tettamanti, E. Monti and S. Wakatsuki, J. Med. Chem., 2010, 53, 2998–3002 CrossRef CAS PubMed.
- E. Giacopuzzi, R. Bresciani, R. Schauer, E. Monti and G. Borsani, PLoS One, 2012, 7, e44193 CAS.
- S. Magesh, T. Suzuki, T. Miyagi, H. Ishida and M. Kiso, J. Mol. Graphics Modell., 2006, 25, 196–207 CrossRef CAS PubMed.
- Y. Wang, K. Yamaguchi, Y. Shimada, X. J. Zhao and T. Miyagi, Eur. J. Biochem., 2001, 268, 2201–2208 CrossRef CAS.
- A. Albohy, M. D. Li, R. B. Zheng, C. Zou and C. W. Cairo, Glycobiology, 2010, 20, 1127–1138 CrossRef CAS PubMed.
- A. Albohy, Y. Zhang, V. Smutova, A. V. Pshezhetsky and C. W. Cairo, ACS Med. Chem. Lett., 2013, 4, 532–537 CrossRef CAS PubMed.
- A. Albohy, M. R. Richards, and C. W. Cairo, unpublished.
- V. Seyrantepe, H. Poupetova, R. Froissart, M. T. Zabot, I. Maire and A. V. Pshezhetsky, Hum. Mutat., 2003, 22, 343–352 CrossRef CAS PubMed.
- A. Mozzi, P. Mazzacuva, G. Zampella, M. E. Forcella, P. A. Fusi and E. Monti, Proteins: Struct., Funct., Bioinf., 2012, 80, 1123–1132 CrossRef CAS PubMed.
- A. Buschiazzo and P. M. Alzari, Curr. Opin. Chem. Biol., 2008, 12, 565–572 CrossRef CAS PubMed.
- A. Bigi, C. Tringali, M. Forcella, A. Mozzi, B. Venerando, E. Monti and P. Fusi, Glycobiology, 2013, 23, 1499–1509 CrossRef CAS PubMed.
- Y. Bourne and B. Henrissat, Curr. Opin. Struct. Biol., 2001, 11, 593–600 CrossRef CAS.
- P. J. Collins, L. F. Haire, Y. P. Lin, J. F. Liu, R. J. Russell, P. A. Walker, J. J. Skehel, S. R. Martin, A. J. Hay and S. J. Gamblin, Nature, 2008, 453, 1258–U1261 CrossRef CAS PubMed.
- S. Albouz-Abo, R. Turton, J. C. Wilson and M. von Itzstein, FEBS Lett., 2005, 579, 1034–1038 CrossRef CAS PubMed.
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2009 Search PubMed.
- Y. Zhang, A. Albohy, Y. Zou, V. Smutova, A. Pshezhetsky and C. W. Cairo, J. Med. Chem., 2013, 56, 2948–2958 CrossRef CAS PubMed.
- Y. Li, H. Cao, H. Yu, Y. Chen, K. Lau, J. Qu, V. Thon, G. Sugiarto and X. Chen, Mol. BioSyst., 2011, 7, 1060–1072 RSC.
- M. Potier, L. Mameli, M. Bélisle, L. Dallaire and S. B. Melançon, Anal. Biochem., 1979, 94, 287–296 CrossRef CAS.
- T. G. Warner and J. S. O'Brien, Biochemistry, 1979, 18, 2783–2787 CrossRef CAS.
- E. L. Romero, M. F. Pardo, S. Porro and S. Alonso, J. Biochem. Biophys. Methods, 1997, 35, 129–134 CrossRef CAS.
- L. R. A. Markely, B. T. Ong, K. M. Hoi, G. Teo, M. Y. Lu and D. I. C. Wang, Anal. Biochem., 2010, 407, 128–133 CrossRef CAS PubMed.
- Z. Khedri, M. M. Muthana, Y. Li, S. M. Muthana, H. Yu, H. Cao and X. Chen, Chem. Commun., 2012, 48, 3357–3359 RSC.
- C. W. Cairo, J. A. Key and C. M. Sadek, Curr. Opin. Chem. Biol., 2010, 14, 57–63 CrossRef CAS PubMed.
- S. R. Amith, P. Jayanth, S. Franchuk, T. Finlay, V. Seyrantepe, R. Beyaert, A. V. Pshezhetsky and M. R. Szewczuk, Cell. Signalling, 2010, 22, 314–324 CrossRef CAS PubMed.
- M. M. Rahman, S. Kitao, D. Tsuji, K. Suzuki, J.-I. Sakamoto, K. Matsuoka, F. Matsuzawa, S.-I. Aikawa and K. Itoh, Glycobiology, 2013, 23, 495–504 CrossRef CAS PubMed.
- H. Kai, H. Hinou and S.-I. Nishimura, Bioorg. Med. Chem., 2012, 20, 2739–2746 CrossRef CAS PubMed.
- D. H. Kwan, H.-M. Chen, K. Ratananikom, S. M. Hancock, Y. Watanabe, P. T. Kongsaeree, A. L. Samuels and S. G. Withers, Angew. Chem., Int. Ed., 2011, 50, 300–303 CrossRef CAS PubMed.
- J.-H. Kim, R. Resende, T. Wennekes, H.-M. Chen, N. Bance, S. Buchini, A. G. Watts, P. Pilling, V. A. Streltsov, M. Petric, R. Liggins, S. Barrett, J. L. McKimm-Breschkin, M. Niikura and S. G. Withers, Science, 2013, 340, 71–75 CrossRef CAS PubMed.
- S. Buchini, F.-X. Gallat, I. R. Greig, J.-H. Kim, S. Wakatsuki, L. M. G. Chavas and S. G. Withers, Angew. Chem., 2014, 126, 3450–3454 CrossRef.
- C. Y. Zamora, M. d'Alarcao and K. Kumar, Bioorg. Med. Chem. Lett., 2013, 23, 3406–3410 CrossRef CAS PubMed.
- A. Minami, T. Otsubo, D. Ieno, K. Ikeda, H. Kanazawa, K. Shimizu, K. Ohata, T. Yokochi, Y. Horii, H. Fukumoto, R. Taguchi, T. Takahashi, N. Oku and T. Suzuki, PLoS One, 2014, 9, e81941 Search PubMed.
- C.-S. Tsai, H.-Y. Yen, M.-I. Lin, T.-I. Tsai, S.-Y. Wang, W.-I. Huang, T.-L. Hsu, Y.-S. E. Cheng, J.-M. Fang and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2466–2471 CrossRef CAS PubMed.
- T. Miyagi, T. Wada, A. Iwamatsu, K. Hata, Y. Yoshikawa, S. Tokuyama and M. Sawada, J. Biol. Chem., 1999, 274, 5004–5011 CrossRef CAS PubMed.
- K. T. Ha, Y. C. Lee, S. H. Cho, J. K. Kim and C. H. Kim, Mol. Cells, 2004, 17, 267–273 CAS.
- Y. Azuma, H. Sato, K. Higai and K. Matsumoto, Biol. Pharm. Bull., 2007, 30, 1680–1684 CAS.
- J. Kopitz, C. Oehler and M. Cantz, FEBS Lett., 2001, 491, 233–236 CrossRef CAS.
- M. S. Sandbhor, N. Soya, A. Albohy, R. B. Zheng, J. Cartmell, D. R. Bundle, J. S. Klassen and C. W. Cairo, Biochemistry, 2011, 50, 6753–6762 CrossRef CAS PubMed.
- T. Hasegawa, N. Sugeno, A. Takeda, M. Matsuzaki-Kobayashi, A. Kikuchi, K. Furukawa, T. Miyagi and Y. Itoyama, FEBS Lett., 2007, 581, 406–412 CrossRef CAS PubMed.
- K. Shiozaki, K. Yamaguchi, K. Takahashi, S. Moriya and T. Miyagi, J. Biol. Chem., 2011, 286, 21052–21061 CrossRef CAS PubMed.
- R. Alon and S. Feigelson, Semin. Immunol., 2002, 14, 93–104 CrossRef CAS PubMed.
- K. Takahashi, J. Mitoma, M. Hosono, K. Shiozaki, C. Sato, K. Yamaguchi, K. Kitajima, H. Higashi, K. Nitta, H. Shima and T. Miyagi, J. Biol. Chem., 2012, 287, 14816–14826 CrossRef CAS PubMed.
- T. M. Gloster and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 683–694 CrossRef CAS PubMed.
- S. Magesh, V. Savita, S. Moriya, T. Suzuki, T. Miyagi, H. Ishida and M. Kiso, Bioorg. Med. Chem., 2009, 17, 4595–4603 CrossRef CAS PubMed.
- S. Magesh, S. Moriya, T. Suzuki, T. Miyagi, H. Ishida and M. Kiso, Med. Chem. Res., 2010, 19, 1273–1286 CrossRef CAS.
- W. P. Burmeister, B. Henrissat, C. Bosso, S. Cusack and R. W. H. Ruigrok, Structure, 1993, 1, 19–26 CrossRef CAS.
- J. P. Kamerling, J. F. G. Vliegenthart, R. Schauer, G. Strecker and J. Montreuil, Eur. J. Biochem., 1975, 56, 253–258 CrossRef CAS PubMed.
- T. G. Warner, A. Louie, M. Potier and A. Ribeiro, Carbohydr. Res., 1991, 215, 315–321 CrossRef CAS.
- Z. Khedri, Y. Li, H. Cao, J. Qu, H. Yu, M. M. Muthana and X. Chen, Org. Biomol. Chem., 2012, 10, 6112–6120 CAS.
- Y. Zou, A. Albohy, M. Sandbhor and C. W. Cairo, Bioorg. Med. Chem. Lett., 2010, 20, 7529–7533 CrossRef CAS PubMed.
- Y. Lu and J. Gervay-Hague, Carbohydr. Res., 2007, 342, 1636–1650 CrossRef CAS PubMed.
- S. Magesh, S. Moriya, T. Suzuki, T. Miyagi, H. Ishida and M. Kiso, Bioorg. Med. Chem. Lett., 2008, 18, 532–537 CrossRef CAS PubMed.
- A. Albohy, S. Mohan, R. B. Zheng, B. M. Pinto and C. W. Cairo, Bioorg. Med. Chem., 2011, 19, 2817–2822 CrossRef CAS PubMed.
- J. N. Varghese, Drug Dev. Res., 1999, 46, 176–196 CrossRef CAS.
- E. De Clercq, Nat. Rev. Drug Discovery, 2006, 5, 1015–1025 CrossRef CAS PubMed.
- C.-Y. Li, Q. Yu, Z.-Q. Ye, Y. Sun, Q. He, X.-M. Li, W. Zhang, J. Luo, X. Gu, X. Zheng and L. Wei, Cell Res., 2007, 17, 357–362 CrossRef CAS PubMed.
- M. Long, Cell Res., 2007, 17, 309–310 CrossRef CAS PubMed.
- R. J. Russell, L. F. Haire, D. J. Stevens, P. J. Collins, Y. P. Lin, G. M. Blackburn, A. J. Hay, S. J. Gamblin and J. J. Skehel, Nature, 2006, 443, 45–49 CrossRef CAS PubMed.
- S. Mohan, S. McAtamney, T. Haselhorst, M. von Itzstein and B. M. Pinto, J. Med. Chem., 2010, 53, 7377–7391 CrossRef CAS PubMed.
- S. Rudrawar, J. C. Dyason, M.-A. Rameix-Welti, F. J. Rose, P. S. Kerry, R. J. M. Russell, S. van der Werf, R. J. Thomson, N. Naffakh and M. von Itzstein, Nat. Commun., 2010, 1, 113 CrossRef PubMed.
- J. N. Varghese, J. L. McKimmbreschkin, J. B. Caldwell, A. A. Kortt and P. M. Colman, Proteins: Struct., Funct., Genet., 1992, 14, 327–332 CrossRef CAS PubMed.
- S. Hitaoka, H. Matoba, M. Harada, T. Yoshida, D. Tsuji, T. Hirokawa, K. Itoh and H. Chuman, J. Chem. Inf. Model., 2011, 51, 2706–2716 CrossRef CAS PubMed.
- M. G. Brant and J. E. Wulff, Org. Lett., 2012, 14, 5876–5879 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |