 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fragment growing to retain or alter the selectivity of anchored kinase hinge-binding fragments†
Charlotte E.
Allen
,
Amanda J.
Welford
,
Thomas P.
Matthews
,
John J.
Caldwell
and
Ian
Collins
*
Cancer Research UK Cancer Therapeutics Unit, The Institute of Cancer Research, 15 Cotswold Road, Sutton, Surrey SM2 5NG, UK. E-mail: ian.collins@icr.ac.uk
First published on 16th December 2013
Abstract
The activity patterns of kinase hinge-binding fragments can be retained or redirected in fragment growing strategies. Targeting conserved kinase features preserved the selectivity pattern of a PKB hinge-binding fragment over a 5000-fold increase in potency, while late-stage modification of a CHK1 hinge-binding fragment substantially changed the pattern.
Introduction
Many ATP-competitive protein kinase inhibitors are approved as anticancer drugs and the discovery of new kinase inhibitors remains an area of intense focus in oncology and other therapeutic areas.1 Successful strategies for achieving selective inhibition of specific enzymes in the kinase superfamily include targeting non-conserved residues in the ATP-binding site of catalytically active kinases, or binding to inactive conformations of the enzymes.2 Inhibitors that bind allosterically at sites remote from the ATP-binding site, or displace the peptide substrate, may also achieve high selectivity.3,4 Promiscuous kinase inhibitors may present an increased risk of toxicities,5 but it is also recognized that molecules inhibiting broader, well-defined, groups of kinases may give better efficacy against deregulated signalling pathways and could forestall the development of resistance in cancer cells.6,7 While it is common practise to measure kinase inhibitor selectivity profiles in lead discovery8 and to seek selectivity for specific targets, strategies to design and maintain well-defined poly-pharmacology are less developed.9–11Fragment-based drug discovery has proved highly effective for generating new ATP-competitive kinase inhibitors, particularly starting with anchoring fragments that mimic the binding of the adenine moiety of the cofactor and growing these into the ATP-binding site.12,13 It was recently shown that, despite binding to a highly conserved protein backbone, hinge-binding fragments can have distinct kinase selectivity profiles.14 However, there is little data on how easily fragment selectivity patterns can be maintained during elaboration to potent inhibitors.
We have previously applied distinct fragment-growing strategies, guided by protein crystallography, to discover selective inhibitors of PKB15,16 and CHK117,18 starting from hinge-binding fragments which showed some common features in their interactions with the kinases, but led to very different lead profiles. In each case, multiple crystallographic structures confirmed the hinge-binding elements were stably anchored throughout the optimisations. Although developed to produce inhibitors of specific target kinases, these compound series provided an opportunity to examine, how broader protein kinase selectivity profiles evolve during fragment elaboration and to determine when the selectivity profiles became predictive of the ultimate leads.
Methods
Compounds 1–13 (Fig. 1) from three distinct fragment-to-lead series of protein kinase inhibitors were prepared according to published procedures.15–18 The compounds were chosen to exemplify major functional group additions or structural modifications during the fragment elaboration, or where order-of-magnitude increases in on-target potency were observed, and where a consistent binding mode of the hinge-binding component of the ligands to the target kinase had been confirmed by crystallography.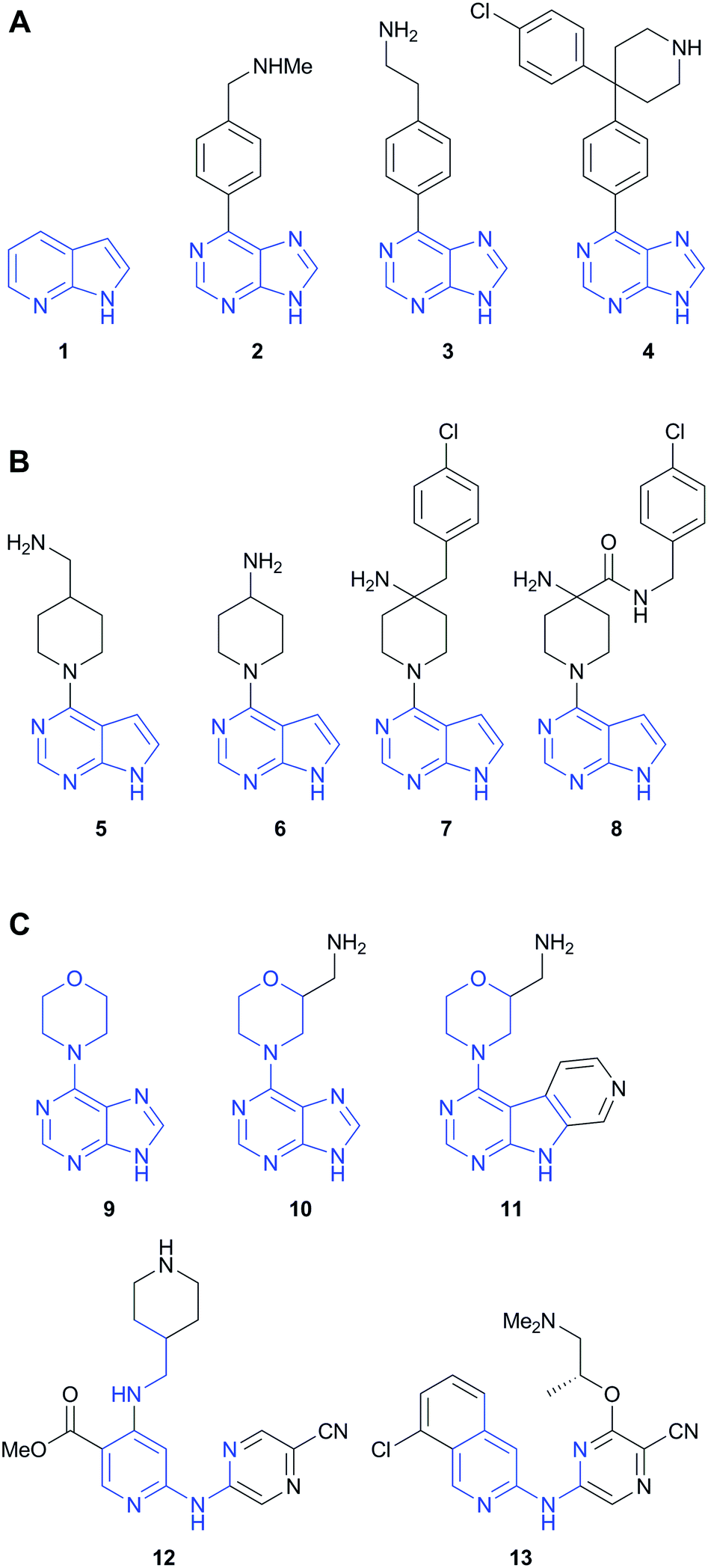 | ||
| Fig. 1 Three fragment-to-lead series of kinase inhibitors. (A and B) PKB inhibitors developed from 7-azaindole (1); (C) CHK1 inhibitors developed from 4-(9H-purin-6-yl)morpholine (9). The heavy atom footprint of the fragment preserved in the elaborated molecules is indicated in blue. | ||
Compounds were tested for inhibition of 91 protein kinases representing the major kinome sub-families using a microfluidic mobility-shift peptide phosphorylation assay19 (Table S1, ESI†). The assay was effective for assessing nanomolar potent inhibitors, as well as low molecular weight fragments at high micromolar concentrations, which has been highlighted as an area of caution for some biochemical assay formats.14 An ATP concentration equal to the Km,ATP for each individual kinase was used in the assays, so that the measured % inhibitions consistently reflected affinity for the binding site.2 The compounds were tested in duplicate at a concentration of 10× the IC50 at the target kinase, in order to capture a defined selectivity relative to the target potency as it increased during the elaboration. Measuring the inhibition at a constant multiple of the target kinase IC50 takes account of the limits on the dynamic range of single-concentration profiling, where assays may be unresponsive to further concentration decreases or increases once <10% or >90% inhibition are reached. The determination of percentage inhibition values at a single test concentration is an established screening approach for kinase selectivity profiling.14,20,21
Selectivity profiles (Table S2, ESI†) were plotted as the mean percentage inhibition of each kinase at the test concentration for each compound, as described in a recent seminal study of fragment selectivity.14 Selectivity indices (S),22 with cut-off points of either 70% (S(70%)) or 30% (S(30%)) inhibition were also calculated. Although the S index does not capture the pattern of a selectivity profile, it can describe general levels of promiscuity,2 in this case at the level of 10-fold selectivity relative to the target kinase level, and is appropriate for use with single-concentration screening data.
Results and discussion
The three previously reported inhibitor series (1 → 4, 1 → 5 → 8 and 9 → 13) were developed independently from two hinge-binding fragments, which were elaborated using structure based design (Fig. 1). For the PKB inhibitors 1–8, 7-azaindole (1) was identified through a virtual screen of low molecular weight fragments (MW < 250 Da) against the structure of PKB, followed by validation in biochemical assay and X-ray crystallography with a PKA–PKB chimeric protein.15 For the CHK1 inhibitors 9–13, the morpholine purine fragment (9) was identified by virtual screening of a compound set containing some larger fragments (8–24 non-hydrogen atoms), and hits were validated by biochemical assay and X-ray crystallography with CHK1.17 Importantly for the present study, multiple crystal structures were determined during these separate fragment elaborations, which showed the hinge-binder fragments and their variants remained anchored in equivalent positions for the sequences studied (Fig. 2).15–18,23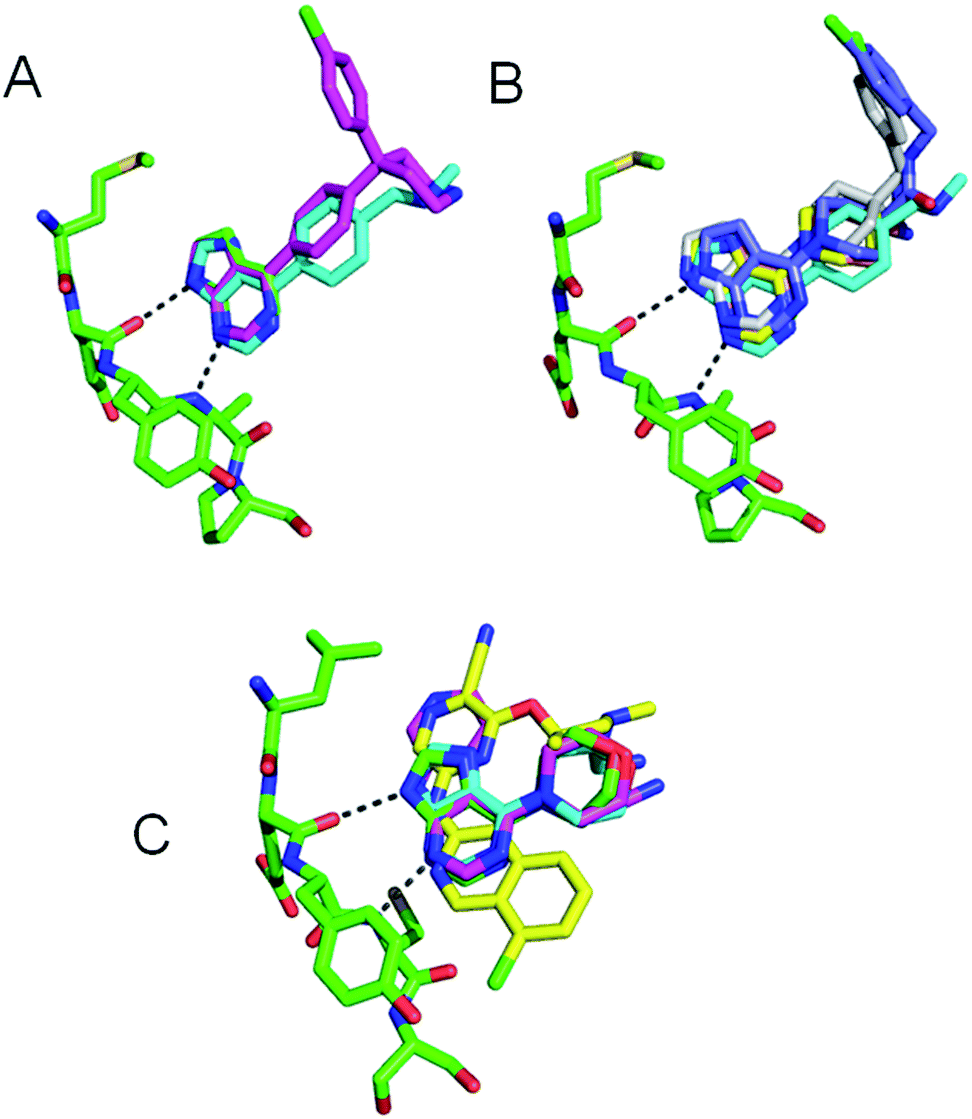 | ||
| Fig. 2 (A) Overlay of 1 (green, 2UVX), 2 (cyan, 2UVY), 4 (magenta, 2UWO) bound to PKB–PKA chimera; (B) overlay of 1 (green, 2UVX), 5 (cyan, 2VNW), 6 (yellow, 2VNY), 7 (grey, 2VO6) bound to bound to PKB–PKA chimera and 8 (blue, 2X39) bound to PKBβ; (C) overlay of 9 (green, 2WMU), 10 (cyan, 2WMV), 11 (magenta, 2YM6), 13 (yellow, 2YM8) bound to CHK1. Kinase hinge regions (GK to GK + 4, green) and hydrogen bonds from fragments 1 or 9 (dashed lines) are shown. | ||
The PKB inhibitors in this series were known to be active on all PKB isoforms and the highly homologous PKBα isoform (AKT1) was included in the screen. 7-Azaindole 1, tested at 1 mM, showed inhibition of members of all the kinase subfamilies in the panel, but with less activity for CMGC kinases (Fig. 3A). This has been observed previously and may reflect reduced hydrophobicity of the ATP binding site in CMGC kinases rendering it less suited to binding heteroaromatic fragments.14 Promiscuous activity can be due to aggregation of poorly soluble compounds when tested at high concentrations.24 The fragments 1 and 9 in this study were identified in screening cascades that contained a counter screen to exclude aggregating compounds, and the activity profiles are unlikely to be due to non-specific effects even when measured at high concentrations.15,17 The introduction of new polar interactions in the binding site led to >10-fold increases in PKBβ potency for amines 2 and 3 (Table 1), but the activity pattern seen for fragment 1 and the S-indices were maintained, showing that affinity was increased generally across the kinase panel (Fig. 3A and Table 1). The introduction of a 4-chlorophenyl substituent in compound 4 increased the potency for PKBβ and a moderate increase in S index was observed. Correspondingly, the activity profile of 4 showed more selectivity against TK family enzymes compared to 1–3, but retained a general similarity to 1–3 across the remainder of the panel. Thus many of the features of the kinase activity profile of the hinge-binding fragment 1 were maintained during a 5000-fold increase in inhibition of the target kinase in the elaboration to 4.
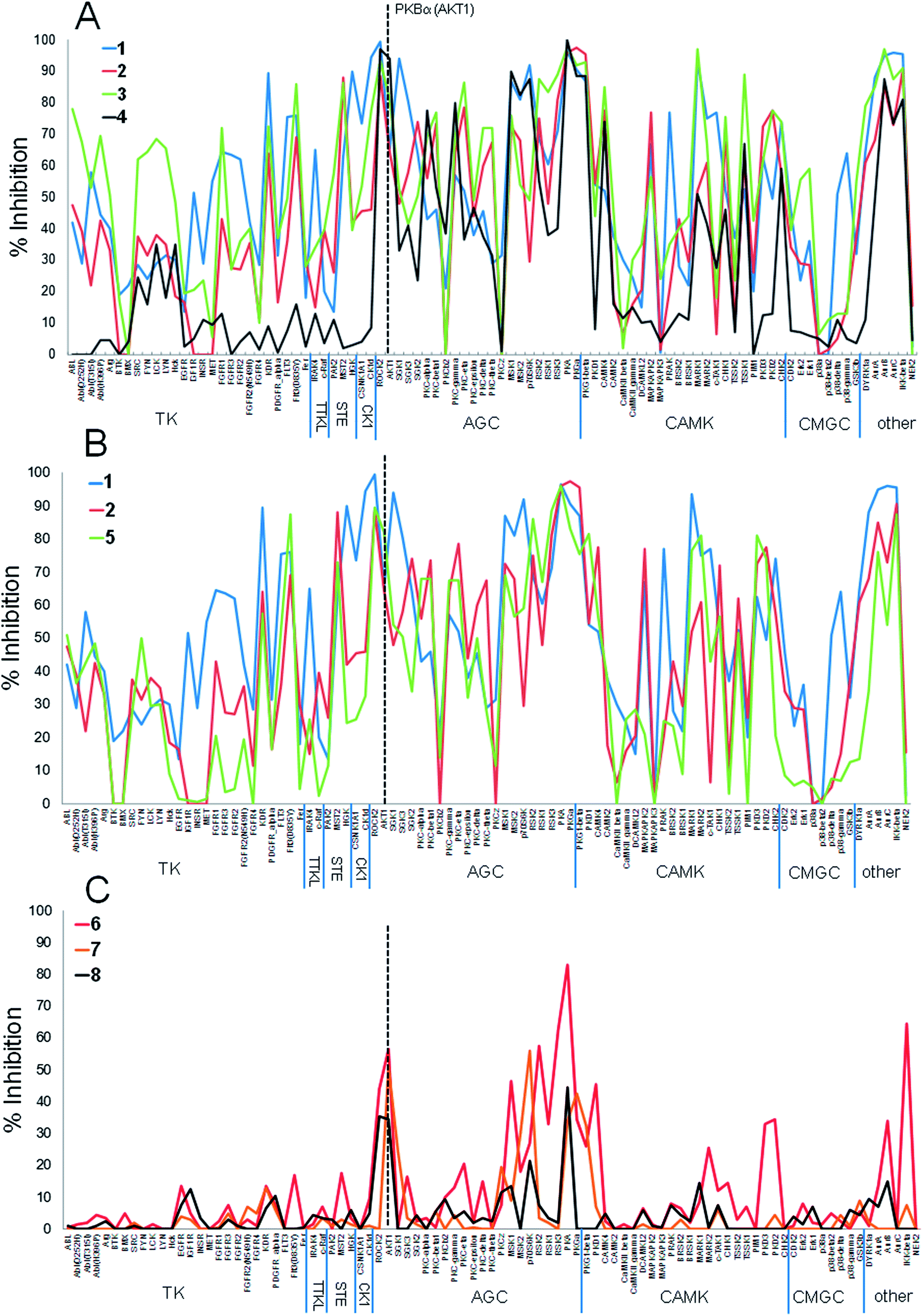 | ||
| Fig. 3 Percentage inhibition (mean of n = 2) at a single concentration (ca. 10× target kinase IC50, see Table 1) of 91 kinases by (A) PKBβ inhibitors 1–4; (B) PKBβ inhibitors 1, 2, 5; (C) PKBβ inhibitors 6–8. PKB (AKT1 isoform) is indicated by dashed line. | ||
| MW | PKBβ IC50a (μM) | CHK1 IC50b (μM) | Test conc. (μM) | S (70%) | S (30%) | |
|---|---|---|---|---|---|---|
| a Data from ref. 15 and 16. b Data from ref. 17 and 18. c Calculated from the mean of duplicate percentage inhibitions for each kinase at the concentrations specified. d Not calculated as highest inhibitions <70%. | ||||||
| 1 | 118 | >100 | — | 1000 | 0.27 | 0.73 |
| 2 | 239 | 6.9 | — | 70 | 0.21 | 0.62 |
| 3 | 239 | 9.5 | — | 95 | 0.36 | 0.78 |
| 4 | 390 | 0.02 | — | 0.2 | 0.17 | 0.35 |
| 5 | 231 | 0.77 | — | 8 | 0.17 | 0.50 |
| 6 | 217 | 0.18 | — | 2 | 0.01 | 0.14 |
| 7 | 342 | 0.006 | — | 0.06 | —d | 0.07 |
| 8 | 385 | 0.002 | — | 0.02 | —d | 0.03 |
| 9 | 205 | — | 42 | 420 | 0.55 | 0.89 |
| 10 | 234 | — | 9.2 | 90 | 0.30 | 0.63 |
| 11 | 284 | — | 0.88 | 9 | 0.14 | 0.48 |
| 12 | 367 | — | 0.036 | 0.35 | 0.01 | 0.05 |
| 13 | 382 | — | 0.013 | 0.1 | 0.01 | 0.04 |
Replacing the phenyl linker of 2 with the piperidine 5 generally reproduced the promiscuous inhibition seen for fragment 1 across the 91 kinases (Fig. 3B). However, modification of 5 to the 4-aminopiperidine 6 dramatically changed the selectivity profile from that of the hinge binder fragment 1, such that 6 significantly inhibited only 6 of the 23 AGC kinases in the panel as well as IKKb and PKD1 when tested at 10× its PKBβ IC50 (Fig. 3C), reflected in the decreased S-indices. We had previously shown the 4-aminopiperidine 6 was selective for PKBβ over the closely related kinase PKA due to interactions of the basic amine with an acidic residue and a favourable close approach to the residue Met 173 in the floor of the binding pocket of PKB.16 The current results show that these selectivity determinants confer more generally increased specificity for PKBβ in a molecule (6, MW = 217) that is still a fragment.
Crystallography confirmed that the lipophilic substituents added in 7 and 8 interacted with similar residues to that of 4 (Fig. 2B).16,23 The selectivity profiles and S-indices of 7 and 8 closely resembled those of the fragment 6. As with the elaboration of 1 to 4, the addition of P-loop targeting elements in the sequence from 6 to 8 did not modify the selectivity pattern conferred by 6, and suggests fragment growing along this vector could be a generally useful strategy for preserving hinge-binding fragment selectivity patterns while increasing affinity. However, the contrasting behaviour of the closely related compounds 5 and 6 suggests that hinge-binding fragment selectivity patterns can also be over-ruled by targeting specific structural determinants, and that this can be achieved with minimal changes to the structure of the fragment.
The two examples above involved a high degree of conservation of the initial hinge-binding fragment in the elaborated structures (Fig. 1). We also studied the changes in patterns of kinase inhibition when greater modification of the hinge-binding group was carried out in the evolution of the CHK1 inhibitors 9–13 (Fig. 4). The purine 9 showed indiscriminate activity against the kinome sub-families present in the screening panel, and was more promiscuous than 1 as measured by the S-indices (Table 1). The amine substituent of 10 was previously shown to introduce new interactions in the ribose pocket of CHK1.17 This led to an increase in potency but did not substantially change the activity profile in the kinase panel when tested at 10× the CHK1 IC50, with small changes in the S indices (Table 1). Although there was a decrease in inhibition of the TK sub-family, the inhibition profile largely remained consistent with the initial fragment. Extension of the hinge-binding fragment to the tricycle 11 introduced a water-mediated interaction between the pyridine nitrogen and Asp 148 in CHK118 while maintaining the hinge-binding pose. This again increased potency against CHK1, with a further small increase in selectivity against the 91-kinase panel.
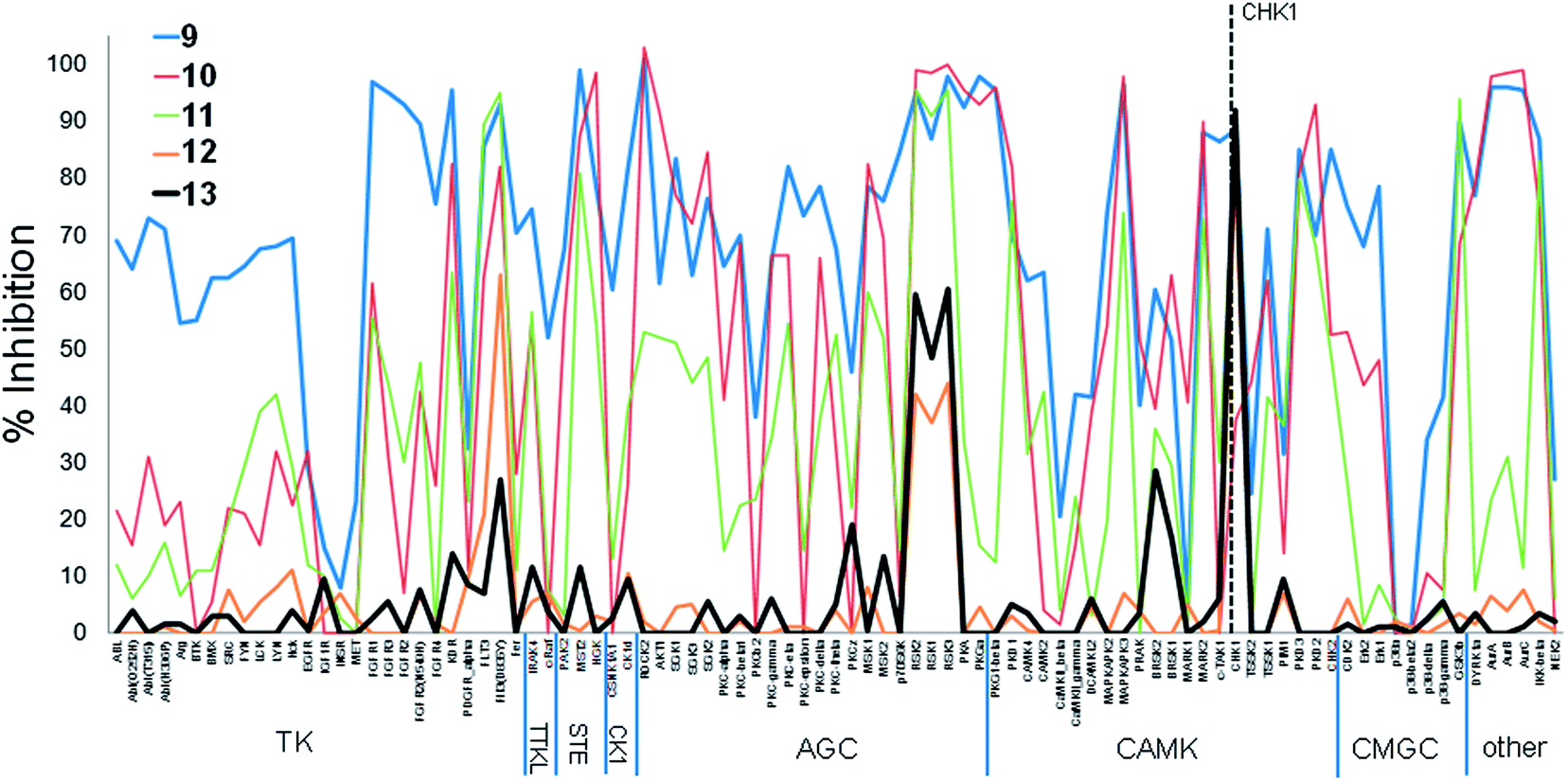 | ||
| Fig. 4 Percentage inhibition (mean of n = 2) at a single concentration (ca. 10× target kinase IC50, see Table 1) of 91 kinases by CHK1 inhibitors 9–13. CHK1 is indicated by dashed line. | ||
A more dramatic change in selectivity was observed on elaboration of 11 to the ring-opened analogue 12, as shown by the changes in the S indices. Compound 12 showed a more specific profile than 9–11, and this was maintained on elaboration to 13 despite a distinct structural change in the basic amine substituent probing the ribose pocket. Compounds 11–13 all showed water-mediated interactions to polar residues in the CHK1 interior pocket. The analogues 12 and 13, although maintaining equivalent hydrogen bonding contacts to the kinase hinge, also interacted with the conserved lysine in the ATP-site, and displayed their constituent rings in a more ‘opened out’ conformation compared to 9–11. Both of these factors may contribute to the change in activity profile during the fragment growing.
Conclusions
We have shown that the pattern of kinase inhibition of an anchoring hinge binding fragment can be maintained during a 5000-fold increase in affinity for a target kinase (1 → 4) when the hinge-binding architecture remains constant in the compound and elaboration occurs along the well-defined ATP-mimetic vectors for type-I kinase inhibitors.2 Since differences in kinase inhibitory profiles are observed between structurally distinct hinge binding fragments, we speculate it should be feasible to select a kinase selectivity profile at the fragment level and maintain that pattern of activities during potency optimisation through fragment-growing. The structurally similar components that bind the ATP cofactor in the active conformation of protein kinases are productive areas to exploit for additional potency without introducing new selectivity determinants, although this may also limit the options to add new functionality to modulate other properties. Provided appropriate selectivity can be introduced to the fragments, it may therefore be possible to apply fragment-based approaches to develop inhibitors with defined protein kinase poly-pharmacology.We have also shown that, starting from the same fragment (1), a minimal change in the structure while still in fragment chemical space, altered the trajectory of the optimisation to give more specific inhibitors (1 → 8) than suggested by the activities of 1. The structural change associated with the more specific activity pattern was minimal (a methylene deletion; 5 → 6), and reflected the targeting of a specific residue in the ribose binding pocket of PKB. This illustrates the benefit of ‘fragment-optimisation’ early in the elaboration. Once established, the new selectivity pattern was carried through further potency optimisation (6 → 8) when groups targeting common features of the ATP site were added, as suggested above. The sensitivity of the inhibitor activity pattern to small structural changes while still in fragment chemical space may confound a strategy of early selection of poly-pharmacology, and underlines the importance of structural biology in understanding the interactions of the fragments and elaborated compounds with the targets.
A fragment-morphing step during the elaboration of CHK1 inhibitors (9 → 13) significantly changed the pattern of activities shown by the inhibitors. In the example studied here, established structural features specific to the CHK1 active site were deliberately targeted to confer high selectivity. This experience suggests that the inhibitory pattern of a hinge-binding fragment is likely to survive only conservative fragment-morphing or scaffold-hopping strategies. The selectivity of the lead 13 illustrates how promiscuous fragments such as 9 can be optimised for potency and selectivity by targeting specific structural determinants in individual kinases, although in this example this was achieved through significant modification of the hinge-binding elements.
Early and extensive profiling of kinase selectivity is now well established in pharmaceutical hit-to-lead projects.2,8 Our data suggest that broad kinase selectivity screens of fragments could be predictive of the lead, provided strategies to conserve the profile are followed in the elaboration, avoiding introducing new interactions with target-specific residues. Conversely, the initial fragment selectivity patterns are unlikely to reflect those of developed leads if the fragment does not already encode the anticipated target-specific interactions.
Conflicts of interest
The Institute of Cancer Research has a commercial interest in PKB and CHK1 inhibitors. Authors who are, or have been, employed by The Institute of Cancer Research are subject to a ‘Rewards to Inventors Scheme’ which may reward contributors to a programme that is licensed. The authors have, or have had, direct or indirect commercial interactions with Astex Pharmaceuticals, Sareum Ltd, AstraZeneca Plc.Acknowledgements
This work was supported by Cancer Research UK grants C309/A11566, C19524/A8027 (studentship to A.J.W.) and The Institute of Cancer Research (studentship to C.E.A.). We acknowledge provision of consumables under the ‘Reward to Discover’ program from Caliper Life Science.References
- D. Fabbro, S. W. Cowan-Jacob, H. Mobtz and G. Martiny-Baron, Methods Mol. Biol., 2012, 795, 1 CAS.
- L. A. Smyth and I. Collins, J. Chem. Biol., 2009, 2, 131 CrossRef PubMed.
- Z. Fang, C. Grutter and D. Rauh, ACS Chem. Biol., 2013, 18, 58 CrossRef PubMed.
- K. C. Han, S. Y. Kim and E. G. Yang, Curr. Pharm. Des., 2012, 18, 2875 CrossRef CAS.
- C. A. Dasanau, P. Padmanabhan, B. A. Clark, 3rd and C. Do, Expert Opin. Drug Saf., 2012, 11, 445 CrossRef PubMed.
- R. Morphy, J. Med. Chem., 2010, 53, 1413 CrossRef CAS PubMed.
- Z. A. Knight, H. Lin and K. M. Shokat, Nat. Rev. Cancer, 2010, 10, 130 CrossRef CAS PubMed.
- P. Bamborough, Expert Opin. Drug Discovery, 2012, 7, 1053 CAS.
- B. Apsel, J. A. Blair, B. Gonzalez, T. M. Nazif, M. E. Feldman, B. Aizenstein, R. Hoffman, R. L. Williams, K. M. Shokat and Z. A. Knight, Nat. Chem. Biol., 2008, 4, 691 CrossRef CAS PubMed.
- A. C. Dar, T. K. Das, K. M. Shokat and R. L. Cagan, Nature, 2012, 486, 80 CrossRef CAS PubMed.
- T. Anastassiadis, K. C. Duong-Ly, S. W. Deacon, A. Lafontant, H. Ma, K. Devarajan, R. L. Dunbrack Jr, J. Wu and J. R. Peterson, J. Biol. Chem., 2013, 288, 28068 CrossRef CAS PubMed.
- G. Chessari and A. G. Woodhead, Drug Discovery Today, 2009, 14, 668 CrossRef CAS PubMed.
- T. G. Davies, S. J. Woodhead and I. Collins, Curr. Top. Med. Chem., 2009, 9, 1705 CrossRef CAS.
- P. Bamborough, M. J. Brown, J. Christopher, C. Chung and G. W. Mellor, J. Med. Chem., 2011, 54, 5131 CrossRef CAS PubMed.
- A. Donald, T. McHardy, M. G. Rowlands, L. K. Hunter, T. G. Davies, V. Berdini, R. G. Boyle, G. W. Aherne, M. D. Garrett and I. Collins, J. Med. Chem., 2007, 50, 2289 CrossRef CAS PubMed.
- J. J. Caldwell, T. G. Davies, A. Donald, T. McHardy, M. G. Rowlands, G. W. Aherne, L. K. Hunter, K. Taylor, R. Ruddle, F. I. Raynaud, M. Verdonk, P. Workman, M. D. Garrett and I. Collins, J. Med. Chem., 2008, 51, 2147 CrossRef CAS PubMed.
- T. P. Matthews, S. Klair, S. Burns, K. Boxall, M. Cherry, M. Fisher, I. M. Westwood, M. I. Walton, T. McHardy, K. J. Cheung, R. van Montfort, D. Williams, G. W. Aherne, M. D. Garrett, J. Reader and I. Collins, J. Med. Chem., 2009, 52, 4810 CrossRef CAS PubMed.
- J. C. Reader, T. P. Matthews, S. Klair, K. J. Cheung, J. Scanlon, N. Proisy, G. Addison, J. Ellard, N. Piton, S. Taylor, M. Cherry, M. Fisher, K. Boxall, S. Burns, M. I. Walton, I. M. Westwood, A. Hayes, P. Eve, M. Valenti, A. de Haven Brandon, G. Box, R. L. M. van Montfort, D. H. Williams, G. W. Aherne, F. I. Raynaud, S. A. Eccles, M. D. Garrett and I. Collins, J. Med. Chem., 2011, 54, 8328 CrossRef CAS PubMed.
- L. J. Blackwell, S. Birkos, R. Hallam, G. Van De Carr, J. Arroway, C. M. Suto and W. P. Janzen, in High Throughput Screening: Methods and Protocols, ed. W. P. Janzen, 2009, pp. 225–237 Search PubMed.
- A. Card, C. Caldwell, H. Min, B. Lokchander, X. Hualin, S. Sciabola, A. V. Kamath, S. L. Clugston, W. R. Tschantz, W. Leyu and D. J. Moshinsky, J. Biomol. Screen., 2009, 14, 31 CrossRef CAS PubMed.
- C. E. Allen, C. L. Chow, J. J. Caldwell, I. M. Westwood, R. L. van Montfort and I. Collins, Bioorg. Med. Chem., 2013, 21, 5707 CrossRef CAS PubMed.
- M. W. Karaman, S. Herrgard, D. K. Treiber, P. Gallant, C. E. Atteridge, B. T. Campbell, K. W. Chan, P. Ciceri, M. I. Davis, P. T. Edeen, R. Faraoni, M. Floyd, J. P. Hunt, D. J. Lockhart, Z. V. Milanov, M. J. Morrison, G. Pallares, H. K. Patel, S. Pritchard, L. M. Wodicka and P. P. Zarrinkar, Nat. Biotechnol., 2008, 26, 127 CrossRef CAS PubMed.
- T. McHardy, J. J. Caldwell, K. M. Cheung, L. J. Hunter, K. Taylor, M. Rowlands, R. Ruddle, A. Henley, A. Brandon, M. Valenti, T. G. Davies, L. Fazal, L. Seavers, F. I. Raynaud, S. Eccles, G. W. Aherne, M. D. Garrett and I. Collins, J. Med. Chem., 2010, 53, 2239 CrossRef CAS PubMed.
- S. L. McGovern, B. T. Helfand, B. Feng and B. K. Shoichet, J. Med. Chem., 2003, 46, 4265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of kinases tested; experimental protocol for kinase inhibition assays; data for inhibition of kinases by 1–13. See DOI: 10.1039/c3md00308f |
| This journal is © The Royal Society of Chemistry 2014 |