
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural mass spectrometry of tissue extracts to distinguish cancerous and non-cancerous breast diseases†
Kelly M.
Hines‡
abc,
Billy R.
Ballard
d,
Dana R.
Marshall
*cd and
John A.
McLean
*abc
aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA. E-mail: john.a.mclean@vanderbilt.edu; Fax: +1 615-343-1234; Tel: +1 615-322-1195
bVanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN 37232, USA
cVanderbilt Institute for Integrative Biosystems Research and Education, Vanderbilt University, Nashville, TN 37235, USA
dDepartment of Pathology, Anatomy and Cell Biology, Meharry Medical College, Nashville, TN 37208, USA. E-mail: dmarshall@mmc.edu; Fax: +1 615-327-6409; Tel: +1 615-327-6549
First published on 2nd September 2014
Abstract
Aberrant metabolism in breast cancer tumors has been widely studied by both targeted and untargeted analyses to characterize the affected metabolic pathways. In this work, we utilize ultra-performance liquid chromatography (UPLC) in tandem with ion mobility-mass spectrometry (IM-MS), which provides chromatographic, structural, and mass information, to characterize the aberrant metabolism associated with breast diseases such as cancer. In a double-blind analysis of matched control (n = 3) and disease tissues (n = 3), samples were homogenized, polar metabolites were extracted, and the extracts were characterized by UPLC-IM-MS/MS. Principle component analysis revealed a strong separation between disease tissues, with one diseased tissue clustering with the control tissues along PC1 and two others separated along PC2. Using post-ion mobility MS/MS spectra acquired by data-independent acquisition, the features giving rise to the observed grouping were determined to be biomolecules associated with aggressive breast cancer tumors, including glutathione, oxidized glutathione, thymosins β4 and β10, and choline-containing species. Pathology reports revealed the outlier of the disease tissues to be a benign fibroadenoma, whereas the other disease tissues represented highly metabolic benign and aggressive tumors. This IM-MS-based workflow bridges the transition from untargeted metabolomic profiling to tentative identifications of key descriptive molecular features using data acquired in one analysis, with additional experiments performed only for validation. The ability to resolve cancerous and non-cancerous tissues at the biomolecular level demonstrates UPLC-IM-MS/MS as a robust and sensitive platform for metabolomic profiling of tissues.
Introduction
Among women in the United States, breast cancer is the most prevalent type of invasive cancer, affecting 118.7 women per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in 2010, the most recent year for which statistics are available.1 Although it is currently the second leading cause of cancer deaths, the mortality due to breast cancer has steadily decreased from 26 women per 100000 in 2001 to 21.9 in 2010.1 The general decrease in breast cancer-related mortalities is in part due to the discovery of diagnostic and prognostic markers, and the subsequent development of targeted cancer therapies born of extensive genomic and proteomic profiling endeavors.2,3 Despite these advances, there still remain breast cancer subtypes, such as triple negative breast cancer (named so due to the absence of markers HER2, PR, and ER), which do not respond to the currently available targeted therapies.4,5
000 in 2010, the most recent year for which statistics are available.1 Although it is currently the second leading cause of cancer deaths, the mortality due to breast cancer has steadily decreased from 26 women per 100000 in 2001 to 21.9 in 2010.1 The general decrease in breast cancer-related mortalities is in part due to the discovery of diagnostic and prognostic markers, and the subsequent development of targeted cancer therapies born of extensive genomic and proteomic profiling endeavors.2,3 Despite these advances, there still remain breast cancer subtypes, such as triple negative breast cancer (named so due to the absence of markers HER2, PR, and ER), which do not respond to the currently available targeted therapies.4,5
Metabolomics approaches have also been applied in efforts to discover molecular differences between tumor and healthy cells. As the downstream endpoint of changes in the genome or proteome, metabolites best represent the cellular phenotype while also reflecting environmental influences.6,7 Cellular metabolism is significantly altered in the transformation of healthy cells into malignant cells, likely due to the rapid cellular proliferation in cancer.6 Affected pathways include glycolysis, oxidative phosphorylation, choline metabolism, and protection against reactive oxygen species, giving rise to a metabolic phenotype common to cancers in general.6,8,9 Among the most notable hypotheses of aberrant metabolism in cancer is the Warburg effect, which describes cancer cells' preferential use of glycolysis, typically an anaerobic process, to generate energy despite aerobic conditions amenable to oxidative phosphorylation.6,8–10 Thus, interrogation of the metabolic phenotype of cancer may provide targets for therapeutics designed to decrease tumor viability by disrupting the metabolic drivers specific to tumors.6
Among the tools typically used to perform cancer metabolomics studies are nuclear magnetic resonance (NMR) and mass spectrometry (MS).7–9 The former offers a number of tailored approaches, from proton (1H) and carbon (13C) NMR to phosphate (31P) NMR which may be used to monitor pathway-specific high-energy phosphate metabolites.11,12 Analysis of solid tissues is feasible with magnetic resonance spectroscopy (MRS) approaches such as high resolution magic angle spinning (HR-MAS).13,14 These techniques are nondestructive, which is an advantage when working with limited volumes of tissue; however, they are generally less sensitive than MS-based approaches.14,15 Chromatographic separations, such as gas chromatography (GC) and ultra-performance liquid chromatography (UPLC), are frequently combined with MS to increase peak capacity, reduce ionization suppression effects, and reduce mass spectral complexity. However, the derivatization necessary for GC-MS analyses can be challenging for untargeted analyses of complex biological samples, as there is no singular derivatization process which is applicable to all classes of biomolecular species due to their chemical diversity. MS-based cancer metabolomics are amenable to a variety of sample types including serum, plasma, urine, and tissues.15 These studies may be targeted or untargeted, where targeted approaches aim to measure a predefined subset of molecules based on their class or pathway and untargeted approaches aim to observe as many metabolites as possible without bias.16
Ion mobility-mass spectrometry (IM-MS) is a hybrid two-dimensional technique which combines the gas-phase structural separation of IM with mass-to-charge (m/z) separation of mass spectrometry. Briefly, IM separation occurs as ions travel through a drift cell containing a neutral buffer gas, such as helium or nitrogen, under the influence of a static or dynamic electric field. As the ions traverse the drift cell, they experience a number of collisions with the neutral buffer gas dependent on the collision cross section (CCS), or the effective ion surface area. This process results in a characteristic ion mobility drift time in the range of micro- to milliseconds, which can be used to calculate CCSs and determine coarse structural information.17
IM-MS-based analyses have been demonstrated for fields ranging from proteomics18,19 to systems biology.20,21 For metabolomics analyses of complex biological samples,22–25 IM-MS offers unique advantages relative to MS-only approaches. IM-MS platforms are highly flexible, allowing a range of pre-ionization separations, such as UPLC, to be combined in tandem with the IM-MS experiment. Relative to UPLC-MS approaches, UPLC-IM-MS provides even greater peak capacity without increasing the time of analysis due to the complementary time scales of minutes, milliseconds and microseconds for the UPLC, IM, and MS dimensions, respectively.18,19,26–28 For classes of biomolecules, such as lipids, proteins, and carbohydrates, a correlation is observed between mass and size, or m/z and CCS, based on their gas-phase packing efficiencies. In an IM-MS experiment, this correlation results in the separation of each biomolecular class into unique regions, or trendlines, in IM-MS conformation space.29–31 The separation of biomolecular classes in IM-MS conformation space enables a more holistic approach with minimal sample preparation for the analysis of complex biological samples, where multiple species of biomolecules may be present.32 Rather than performing sample purification strategies to isolate a particular class of biomolecules, the molecules of interest (e.g. metabolites) can be separated from other biomolecular classes in the IM dimension, effectively increasing the signal-to-noise (S/N). For metabolomics, this is particularly advantageous as isobaric species may be resolved in the IM dimension based on the differences in their gas phase structures. Lastly, data independent acquisition of MS/MS spectra post-ion mobility enables multiplexed fragmentation experiments to be performed nearly simultaneously, and minimizes the need to perform additional experiments to obtain fragmentation data.
Here, we describe an UPLC-IM-MS/MS approach to the characterization of the metabolites differentially expressed in breast diseases. Based on a previously demonstrated workflow for the analysis of diabetic wound fluid,32 disease (n = 3) and control (n = 3) breast tissues were homogenized, polar metabolites were extracted and characterized by UPLC-IM-MS/MS.33 Although it was known which tissues represented healthy and disease, the study was double-blind to the exact pathologies of the tissues. Principal component analysis (PCA) revealed unexpected grouping of the breast tissues. Features giving rise to this separation in the PCA were interrogated to determine tentative molecular identifications based on chromatographic retention time, accurate mass, drift time, and fragmentation analysis. While this study does not present enough statistical power to draw broad conclusions about the identified features, the general workflow highlights the benefits of incorporating IM-MS into untargeted metabolomics pipelines with particular emphasis on its utility in the identification process.
Results and discussion
The workflow demonstrated here for the UPLC-IM-MS/MS analysis of human tissues is based, in part, on a previously developed workflow to transition from wholly untargeted to targeted analyses of complex biological samples in the pursuit of identifying key biomolecular features distinguishing disease and healthy conditions.32 The general workflow demonstrated here is illustrated in Scheme 1, where this methodology has been adapted to include the additional online UPLC separation, methods for chromatographic peak picking and alignment, post-mobility data-independent acquisition of MS/MS spectra, and inclusion of MS/MS spectra, in addition to accurate mass, as a parameter for generating tentative identifications. | ||
| Scheme 1 Illustration of the workflow for the preparation and extraction of breast tissues, including the steps necessary to transition from a multidimensional dataset to the identification of statistically-prioritized molecular features. | ||
Data representative of the UPLC-IM-MS analysis of sample 4D is presented in Fig. 1. The IM-MS plot (Fig. 1(a)), shows the dimensions of m/z on the x-axis and drift time (ms) on the y-axis. Intensity, measured as counts, is depicted as a false color scale, where white indicates high intensity signals, blue represents low-to-medium intensity signals, and black indicates the absence of signals. A portion of the UPLC chromatogram from the analysis of 4D is presented in Fig. 1(b). User-defined regions of the multidimensional dataset may be selectively extracted to isolate species of interest and effectively increase the S/N by separating those species from chemical noise.32Fig. 1 demonstrates the extraction of two acetylated polypeptides, thymosins β4 and β10, in both the chromatographic (Fig. 1(b)) and drift time dimensions (Fig. 1(a)). The chromatographic peak containing thymosins β4 and β10, as indicated in Fig. 1(b) by the grey bar, may be extracted to yield the IM-MS plot containing the polypeptides and any co-eluting species. Likewise, a user defined area (outlined in white in Fig. 1(a)) of m/z-drift time space, or conformation space, containing the polypeptides may be extracted to provide an extracted ion chromatogram for thymosins β4 and β10. Extraction in both the chromatographic and ion mobility dimensions yields a mass spectrum (Fig. 1(c)) of the polypeptides (highlighted in grey), in which the isotopic distributions (inset) of the multi-charged species are sufficiently resolved to determine their charge state as 7+ with high S/N through this multistep filtering of chemical noise. This strategy may be applied in both the analysis of IM-MS and post-mobility-MS/MS spectra, where extraction in chromatographic and IM dimensions effectively increases confidence in tentative identifications by reducing isobaric interferences.31,32,34
 | ||
| Fig. 1 Data representative of the UPLC-IM-MS/MS characterization of breast tissue extract sample 4D. A user-defined region of conformation space, containing the polypeptides thymosin β4 and thymosin β10, is indicated on the IM-MS spectrum (a). Panel (b) represents a portion of the UPLC chromatogram from the same analysis, where the chromatographic peak in which the thymosin polypeptides eluted is highlighted by the grey bar. Extraction of these signals in both the drift time and chromatographic dimensions provides a mass spectrum (c) with improved S/N in which the isotopic distributions (inset, magnification of the area highlighted by grey bar) of the 7+ species are resolved. | ||
Although the presence or absence of molecular features may be visually observed from the IM-MS plots representing different disease statuses (i.e. disease vs. control), multivariate statistical analyses are required to detect more subtle variations in the expression of molecular features as a function of disease status. However, utilization of drift time in the initial peak picking and alignment remains a challenge for biostatistical tools. Therefore, peak picking and alignment of data resulting from the analysis of the breast tissue extracts was performed at the chromatographic level and the ion mobility dimension was returned to as an aid in generating or filtering putative identifications of statistically significant molecular features on the basis of post-mobility MS/MS.
Results from the statistical analysis of the breast tissue extract dataset are summarized in Fig. 2. As described above, samples were injected in three batches, where each batch contained one replicate injection per sample in a randomized order, and the set of replicates was bracketed by QC samples (for PCA with QC set, see ESI†). The PCA score plot (Fig. 2(a)) provides an overview of the samples. Grouping of the technical replicates for each sample, as seen in the score plot, indicates good reproducibility throughout the analysis. Samples forming a matched pair of disease and control tissues are identified with the dashed ellipses on the score plot (Fig. 2(a)). Generally, 4D and 6D separated from the other samples along principal component (PC) 1, but were separated from each other along PC2. Interestingly, 2D grouped with the control tissues. This suggested that although 2D represented a tissue affected by disease, this tissue was generally more metabolically normal than the pathologically abnormal and metabolically dysregulated tissues 4D and 6D. The initial hypothesis as to the location of 2D in the PCA score plot was that this tissue represented a benign breast disease, whereas 4D and 6D potentially represented more aggressive diseases.
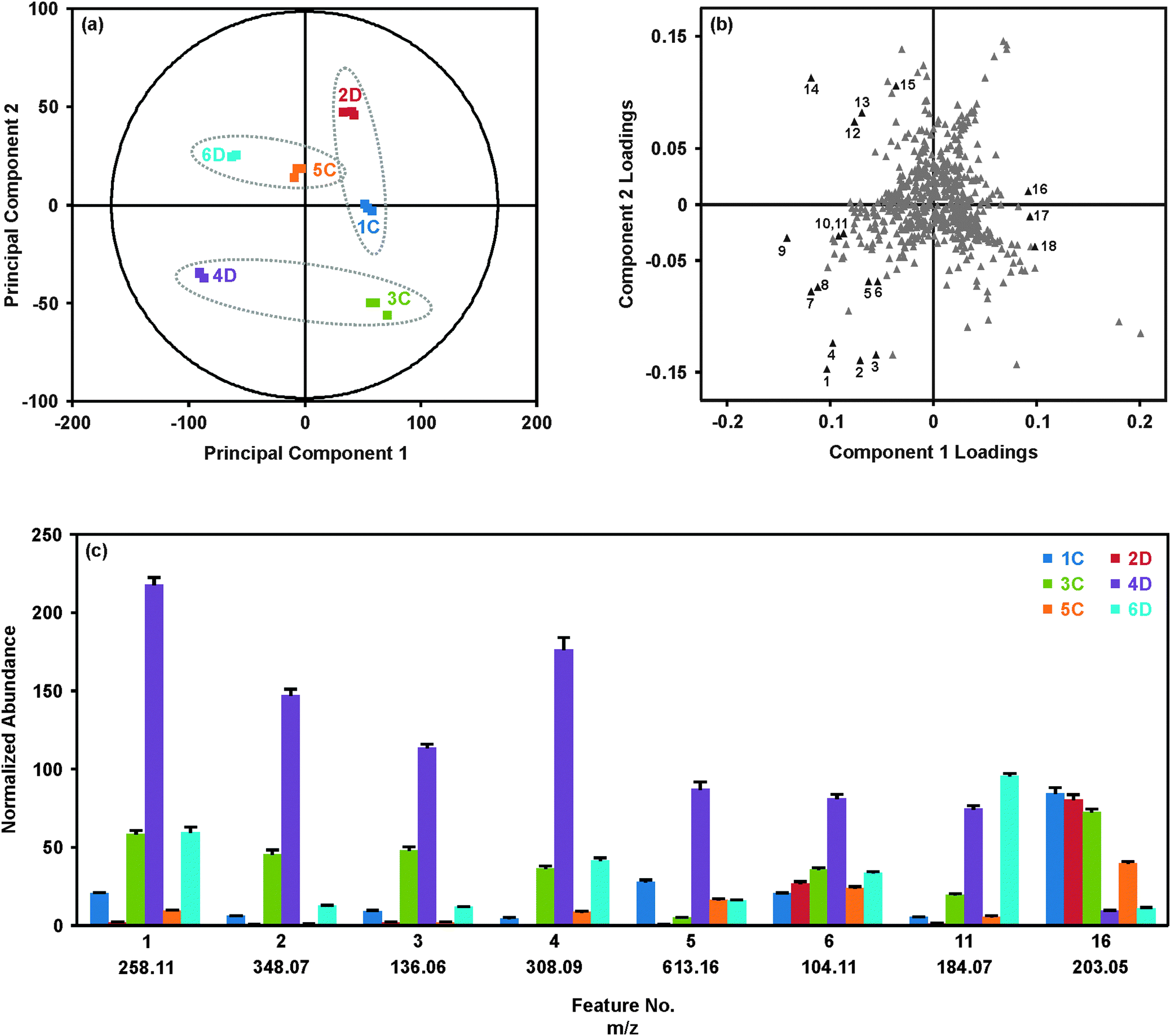 | ||
| Fig. 2 Summary of the multivariate statistical analysis of the breast tissue extract dataset. (a) Grouping of technical replicates and separation of biological replicates is shown by PCA of the UPLC-IM-MS/MS data. The plot contains three technical replicates for each sample, and each sample is represented by a different color. Matched control and disease tissues are indicated with ellipses. (b) Analysis of the corresponding loadings plot indicates the molecular features contributing to the separation of the samples along PC1 and PC2. Select features (in black) showing statistical significance are numbered. Average normalized abundances from three technical replicates for select features are shown in (c). The coloring scheme is the same as (a). Error bars indicate the standard deviations of the means. See Biostatistics in the Materials and Methods section for full description of the normalization method. | ||
The corresponding loadings plot (Fig. 2(b)) was investigated to determine the specific molecular features giving rise to the separations observed in the PCA score plot. In general, the loadings plot presents the coefficients or weights assigned to each variable in the process of generating the principal components. The values of the PC1 and PC2 loadings for a particular feature are representative of the correlation between the respective PC and the feature. For example, feature 16 in Fig. 2(b) has a loading of 0.09 for PC1 and a loading near zero (0.01) for PC2. This indicates that PC1 is highly correlated with feature 16 and PC2 is poorly correlated with feature 16. Similar to interpretation of the PCA score plot, features which group together in the loadings plot demonstrate similar behavior.
The loadings (Fig. 2(b)) and score plots (Fig. 2(a)) may be compared to understand the relationship between the molecular features, shown in the loadings plot, and the tissue samples, shown in the score plot. The histogram shown in Fig. 2(c) provides the average normalized abundances of several of the representative molecular features labeled in Fig. 2(b) across the six tissues. Looking at feature 16, a clear pattern emerges where the samples to the right of PC1 (2D, 1C and 3C) all have high abundances of feature 16 (m/z 203.05), while samples to the left of PC1 (4D and 6D) have significantly lower abundances. Sample 5C, which has a score near zero on PC1, has an intermediate abundance of m/z 203.05. This feature contributes significantly to the variability extracted by PC1, which generally separates the disease and control samples. Likewise, features 1–4 in the lower left quadrant of the loadings plot are all substantially more abundant in tissue 4D than the other tissues, giving rise to the location of 4D in the lower left quadrant of the PCA score plot. Features from Fig. 2(b) were selected to represent the molecules most strongly directing the loadings of samples 4D and 6D, and a few features which best represented the collective similarities of the control tissues and sample 2D. Pairwise comparisons between the matched disease-control samples were also performed via orthogonal partial least squares-discriminant analysis (OPLS-DA; see Fig. S3 to S6 in the ESI†) to maximize the group differences.35–38 Fold-changes for the data shown in Fig. 2(c) may be found in Table 1, along with the m/z and retention time associated with the feature.
| Feature | Experimental m/z | Retention time (min) | Tentative molecular identification | Mass accuracy (ppm) | p-Valuea (corrected) | Tissue 2Db,c | Tissue 4Db,d | Tissue 6Db,e |
|---|---|---|---|---|---|---|---|---|
| a Bonferroni corrected p-values from ANOVA, where p ≤ 0.05 determined significance. b Positive (+) and negative (−) symbols indicate directionality of fold-change, where (+) represents increase in tumor and (−) represents decrease in tumor relative to the respective matched control. N.S. indicates change is not significant (N.S.), as determined by p > 0.05 and fold-change ≤2. The p-values (shown in ESI) were calculated with the two-tailed Student's t-test with equal variance, and adjusted by the Bonferroni correction. p ≤ 0.05 and a fold-change ≥2 was used to determine significance. c Fold change and p-value calculated against the matched control, 1C. d Fold change and p-value calculated against the matched control, 3C. e Fold change and p-value calculated against the matched control, 5C. f MS/MS of adenosine 5′-monophosphate standard (shown in ESI) revealed presence of m/z 136.06 with no or little collision energy applied. Feature #3 may be adenine fragment of adenosine monophosphate (feature #2). “V” indicates features whose identifications where validated with standards. | ||||||||
| 1 | 258.11 | 1.51 | Glycerophosphocholine | 4.7 | 1.1 × 10−11 | −8.1 | +3.7 | +6.0 |
| 2 | 348.07 | 2.33 | Adenosine monophosphate (V) | 1.7 | 2.8 × 10−11 | −5.7 | +3.2 | +10.5 |
| 3 | 136.06 | 2.33 | Adeninef | 2.2 | 7.6 × 10−12 | −3.9 | +3.4 | +5.2 |
| 4 | 308.09 | 2.45 | Reduced glutathione (V) | 1.6 | 4.7 × 10−10 | N.S. | +4.8 | +4.6 |
| 5 | 613.16 | 3.76 | Oxidized glutathione (V) | 0.3 | 9.3 × 10−9 | −20.1 | +15.6 | N.S. |
| 6 | 104.11 | 1.51 | Choline | 1.0 | 8.3 × 10−9 | N.S. | +2.3 | N.S. |
| 7 | 705.94 | 4.56 | Thymosin β10, acetylated (7+) | 2.4 | 6.5 × 10−11 | N.S. | +22.1 | +2.3 |
| 8 | 823.44 | 4.56 | Thymosin β10, acetylated (6+) | 0.5 | 1.4 × 10−13 | −6.8 | +29.9 | +2.5 |
| 9 | 827.76 | 4.56 | Thymosin β4, acetylated (6+) | 3.4 | 9.9 × 10−9 | −11.8 | +10.4 | +2.0 |
| 10 | 152.06 | 2.26 | Guanine | 3.3 | 2.5 × 10−10 | N.S. | +17.1 | +3.5 |
| 11 | 184.07 | 1.51 | Phosphocholine | 5.4 | 9.3 × 10−13 | −3.5 | +3.7 | +5.8 |
| 12 | 137.05 | 3.93 | Hypoxanthine | 4.4 | 9.7 × 10−4 | N.S. | N.S. | N.S. |
| 13 | 246.17 | 5.01 | 2-Methylbutyroylcarnitine pivaloylcarnitine | 1.2 | 1.5 × 10−17 | N.S. | N.S. | +67.8 |
| 14 | 137.05 | 2.67 | Allopurinol | 3.6 | 3.8 × 10−9 | +3.3 | +3.5 | +2.6 |
| 15 | 152.06 | 3.93 | 2- or 8-hydroxyadenine | 3.9 | 1.3 × 10−6 | N.S. | +5.7 | N.S. |
| 16 | 203.05 | 1.45 | Monosaccharide [M + Na]+ | 1.5 | 9.7 × 10−9 | N.S. | −7.3 | −3.5 |
| 17 | 437.19 | 9.31 | Unknown | — | 8.0 × 10−6 | N.S. | −3.2 | N.S. |
| 18 | 387.19 | 9.14 | Unknown | — | 6.9 × 10−7 | N.S. | −7.0 | −2.1 |
The raw data was then revisited to determine tentative identities of the differentially abundant molecular features. Upon investigating feature 4, m/z 308.09, the extracted ion chromatogram indicated this species was found in two chromatographic peaks (retention times 2.47 and 3.78 min). While the peak at 2.47 min contained m/z 308.09 as the base ion, the chromatographic peak at 3.78 min contained a base ion of m/z 613.16. Closer inspection of the mass spectrum around m/z 308.1 revealed that there were overlapping isotopic envelopes present (Fig. 3(b)). Analysis of the IM-MS spectrum using the same approach described above revealed that there were in fact two species contributing to the spectrum in Fig. 3(b). In contrast to one dimensional MS analyses, these overlapping isotopic distributions can be easily resolved in the IM dimension as the separation occurs on the basis of size-to-charge, where the general trend is that higher charge state species have shorter drift times owing to greater mobilities at a given electric field strength.17,31,34 This can be seen in Fig. 3(a)–(d), where Fig. 3(b) represents the mass spectrum obtained from summing across all drift time as demonstrated with the white dashed lines in Fig. 3(a), mimicking a typical one dimensional MS analysis. Creating a user-defined area of drift time-m/z space by combining the vertical white dashed lines and the horizontal blue dashed lines allows the peak inside the blue box to be extracted away from the isobaric interference. This yields the spectrum presented in Fig. 3(c), from which it is evident that this species, m/z 308.09 is singly charged. Performing the same steps for the region outlined in Fig. 3(a) with green dashed lines provides a mass spectrum Fig. 3(d) of a doubly charged species, m/z 307.09. This corresponds to a molecular weight of 612.16, or [M + H]+ of 613.16, which is the same m/z value as feature 5 (Table 1). Database searches of the m/z and molecular weight values suggested that m/z 308.09 (feature 4) and m/z 307.09 (i.e. [M + H]+ 613.16, feature 5) were reduced and oxidized glutathione, respectively.
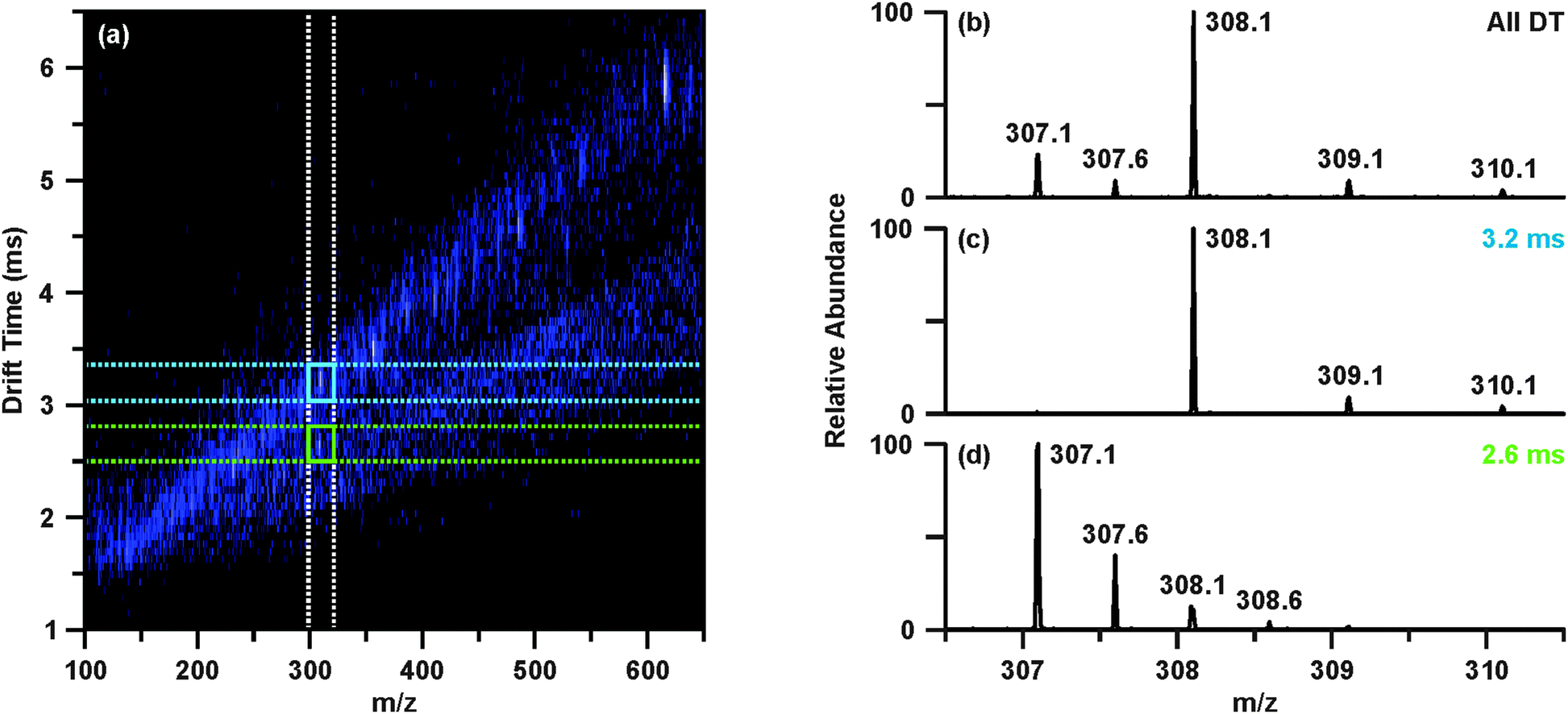 | ||
| Fig. 3 Demonstration of IM separation of isobaric overlapping isotopic distributions convoluting the mass spectrum around m/z 308.1 at retention time 3.78 min. (a) IM-MS plot with regions correlating to extracting the mobility separated isobaric ion. Extracting a defined window around m/z 308 across all drift times (outlined by the white dashed lines) yields the extracted mass spectrum (b). Combining the defined window about m/z 308 with a discrete window of drift times (blue dashed lines) allows the peak in the blue box to be extracted (c) away from the isobaric interference (green dashed lines, d). The end result is separate mobility-extracted mass spectra resolving the isotopic distributions to that of a singly-charged m/z 308.1 (feature 4) and doubly charged m/z 307.1 (feature 5). | ||
The post-mobility fragmentation spectra for m/z 308.09 and m/z 613.16 were studied to assess the plausibility of the potential identifications of reduced and oxidized glutathione as features 4 and 5, respectively. The chromatographic peak containing m/z 613.16 at 3.78 min was extracted to provide the IM-MS spectrum of the data independent MS/MS acquired post-mobility separation, shown in Fig. 4(a). Multiple regions of fragmentation were evident in the IM-MS spectrum, as outlined by the green and blue lines. As fragmentation occurs after mobility separation, the fragment ion should retain the drift time of the corresponding precursor ion, thus generating a horizontal line of precursor and fragment ions (regions outlined by green and blue lines).39 Effectively, this mobility organization of fragments circumvents complications which may arise from the presence of isobaric species and eliminates the requirement of mass selection prior to MS/MS experiments. Similarly, fragmentation which occurred prior to IM can be discerned from the post-mobility fragmentation as each ion, regardless of whether it is a precursor or fragment, will have a unique drift time. This may arise in situations where in-source fragmentation has occurred, and such a phenomenon is indicated by the white lines in Fig. 4(a). In order to obtain a true fragmentation spectrum of the intact molecular ion m/z 613.16, only a discrete window of drift time centered at the drift time of m/z 613.16 is extracted. As shown in Fig. 4(a) with the green lines, this area contains the mobility-organized fragments of m/z 613.16. Extracting this region provides the MS/MS spectrum shown in Fig. 4(b). Database searches and in silico fragmentation analysis of this spectrum supported the tentative identification of m/z 613.16 as oxidized glutathione. To validate this identification, a standard of oxidized glutathione was characterized by ESI-IM-MS/MS. The resulting extracted post-mobility MS/MS spectrum is shown in Fig. 4(c), and confirms the proposed identification as there is a peak-to-peak match between the experimental and standard. A similar process was performed to obtain the identifications shown in Table 1, and standards were used to validate IDs where possible (noted in Table 1).
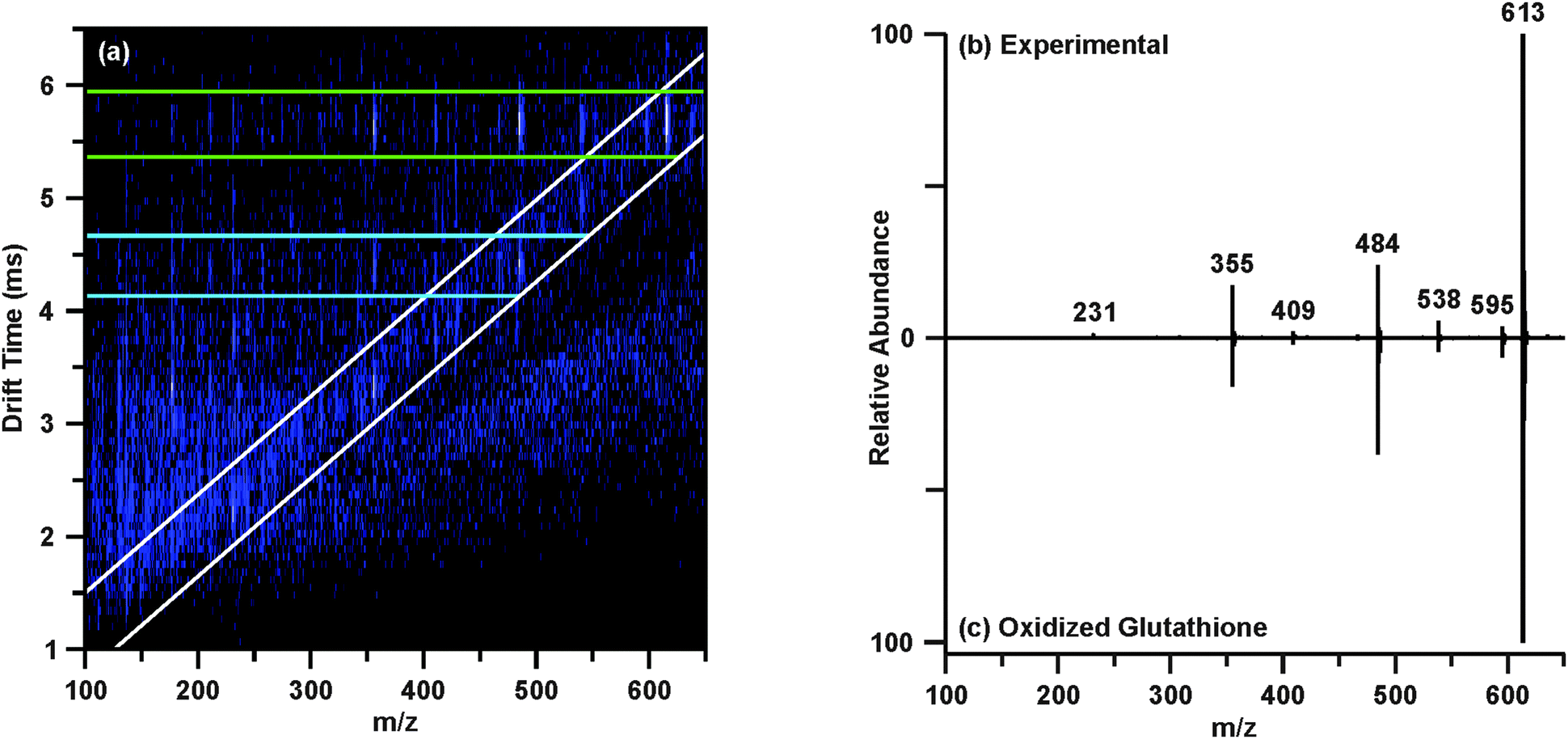 | ||
| Fig. 4 Interpretation of post-mobility MS/MS of m/z 613.16 (feature 5) at 3.78 min acquired by data-independent acquisition. (a) The IM-MS spectrum resulting from extraction of the chromatographic peak at 3.78 min reveals multiple regions of fragmentation. The region outlined in white is indicative of fragmentation which occurred prior to the mobility separation (e.g. in-source fragmentation), while the green region corresponds to post-mobility fragmentation of the intact molecular ion (m/z 613.16), and the blue region corresponds to post-mobility fragmentation of an in-source fragment ion (m/z 484) of m/z 613.16. Extracting the green region yields a true MS/MS spectrum of m/z 613.16 (b). MS/MS was performed on a standard of oxidized glutathione (c) to validate the tentative identification. | ||
Although tissue 4D is the only sample to reside in the lower left quadrant of the PCA (Fig. 2(a)), a number of the more significant molecular features (No. 1–11) are located in the corresponding quadrant of the loadings plot (Fig. 2(b)). Interestingly, many of these features were identified as biomolecular species previously demonstrated to be differentially expressed in the tumor environment. For example, thymosins β4 and β10 (features #7–9) are highly conserved, highly abundant polar polypeptides which are overexpressed in a number of tumor types, including breast cancer.40 A primary function of thymosin peptides is to bind G-actin, the primary component of the cellular cytoskeleton, and inhibit G-actin polymerization.40 As actin sequestration increases the motility of the cell, thymosin polypeptides have been suspected to play a key role in the processes of cell migration and tumor metastasis.40–42 Characterization of the effects of thymosin β4 overexpression on lung tumor metastasis revealed increases in tumor sizes, number of metastatic nodules, neoangiogenesis, and cell migration, strongly suggesting that thymosin β4 stimulates tumor metastasis.41 In the analysis described here, thymosin β4 and β10 were found to be increased greater than 10 and 20-fold (based on single peak in isotopic distribution of multiple charged species), respectively, between tissue 4D and its corresponding control 3C (p-values: β10 (#7), p = 7.3 × 10−7; β10 (#8), p = 2.9 × 10−7; β4 (#9), p = 1.5 × 10−5). A histogram demonstrating the differential expression of thymosins β4 and β10 across the disease and control breast tissues using summed peak areas for the whole isotopic envelope of the 6+ charge states can be found in the ESI.† Using the peak area results, fold-changes of 38 (p = 7.0 × 10−7) and 75 (p = 1.4 × 10−6) were observed between samples 3C and 4D for thymosins β4 and β10, respectively.
Three features contributing significantly to location of 4D within the PCA were identified as choline-containing metabolites: (#1) glycerophosphocholine, (#6) choline, and (#11) phosphocholine. Aberrant metabolism of choline phospholipids is a hallmark of cancer cells and tumors, including breast cancer, typically presenting in the form of increased levels of phosphocholine (PCho) and total choline-containing species (tCho; free choline (Cho) + PCho + glycerophosphocholine (GPC)) due to increased activity and/or expression of choline kinase, choline transporters and phospholipases.43–47 Generally, the extent to which levels of PCho and tCho are increased is reflective of the tumor aggressiveness or malignancy.12,45 From Fig. 2(c) and Table 1, it is evident that both 4D and 6D showed significant increases in GPC (4D: 3.7-fold, p = 1.4 × 10−6; 6D: 6.0-fold, p-value = 3.0 × 10−5) and PCho (4D: 3.7-fold, p = 3.3 × 10−6; 6D: 5.8-fold, p = 1.3 × 10−7) relative to the respective controls, suggesting both may represent types of breast tumors. Sample 2D, suspected to represent a non-cancerous breast disease, had significantly less GPC (8.1-fold decrease, p = 6.5 × 10−7) and PCho (3.5-fold decrease, p = 6.2 × 10−5) relative to the matched control.
Additionally, samples 4D and 6D presented higher levels of glutathione (#4; 4D: 4.8-fold, p = 1.3 × 10−5; 6D: 4.6-fold, p = 9.9 × 10−6) relative to the controls, while only 4D showed increased levels of oxidized glutathione (#5; 15.6-fold, p = 1.5 × 10−5; see Table 1 and Fig. 2(c)). Glutathione (GSH) is the primary intracellular thiol responsible for protection against free radicals.48 This detoxification may occur directly or in conjunction with the enzyme glutathione s-transferase, which conjugates electrophilic compounds to reducing sulfhydryl (–SH) group of glutathione's cysteine residue. Oxidized glutathione, or glutathione disulfide, is composed of two glutathione units linked through a disulfide bond between the cysteine residues. The relative abundances of reduced and oxidized glutathione (GSSG) in tissues have been examined as an indicator of the redox status of the tissue given the potential to detoxify to cancer drugs which work via oxidative damage.48–51 Previous studies of glutathione levels in breast tumors have found significantly elevated levels of reduced and oxidized glutathione in tumor tissues relative to matched peritumoral (i.e. control) tissues.48,49,51 However, it was observed in the tumor tissues that reduced glutathione levels were significantly greater than oxidized glutathione levels, representing an increase in the detoxification capacity of the tumors.51 Our results were consistent with these findings, where abundances of GSH were approximately 2-fold greater than GSSG in both 4D and 6D (Fig. 2(c) and ESI†).
For many of the tentatively identified features selectively highlighted in Fig. 2(c) and Table 1, a consistent pattern has been observed in which abundances are increased in 4D and 6D relative to their controls, while 2D demonstrates the opposite correlations despite also representing a tissue affected by disease. Our general hypothesis has thus been that 4D and 6D represented cancerous breast diseases, perhaps differing in malignancies, given the identities of the most significant molecular features and their known involvement in breast cancer. On the other hand, 2D has been suspected to represent a benign cancer or a non-cancerous breast disease. Examination of the pathology reports for 2D, 4D and 6D revealed our initial data-driven hypotheses to be generally accurate. Sample 2D was diagnosed as a fibroadenoma, a benign breast tumor most commonly diagnosed in patients in their early 20s.52 However, this sample did not represent a typical fibroadenoma diagnosis as the patient from whom this biopsy was taken was 47 years old. Sample 4D, as suspected, was diagnosed as an infiltrating ductal carcinoma of grade 3 and pathological state IIA which was found to be ER, HER2/NEU, and PR negative, often referred to as triple negative cancer. This particular type of breast cancer is challenging to treat as it does not respond to targeted therapies and is often associated with a shorter time between relapse and death.4 Lastly, the diagnosis of sample 6D was a pseudoangiomatous stromal hyperplasia, a benign breast tumor.53 Similar to triple negative tumors, pseudoangiomatous stromal hyperplasias are highly metabolic and perhaps this was the primary director for the separation of 4D and 6D away from the other tissues.
Conclusions
Metabolic profiling of breast tissues using the UPLC-IM-MS/MS-based platform described here was demonstrated to be a highly sensitive and selective technique for the differentiation of breast tissues representing a range of benign to cancerous breast diseases. The ion mobility aspect of the analysis provided an additional dimension of separation orthogonal to that provided in the chromatographic dimension. This enabled simultaneous isolation of features of interest in both IM and LC dimensions, improving confidence in locating features of interest while also increasing their signal-to-noise ratios. In the instance of co-eluting isobaric species, it was demonstrated that IM could effectively separate these species and eliminate the overlapping isotopic peaks which may confound accurate mass determination and subsequent identification. Data-independent tandem MS acquired post-mobility separation provided a means to distinguish fragmentation occurring prior to the mobility and collision cells from that of true collision induced dissociation, providing MS/MS spectra from which feature identifications could be made with high confidence.The molecular features giving rise to the distinction of cancerous and benign tissues from peritumoral control tissues included species previously well-known in the literature to be affected by the aberrant metabolism observed in breast cancer. These included choline, phosphocholine, glycerophosphocholine, glutathione, and oxidized glutathione. Similarly, our analysis revealed that the actin-sequestering polypeptides thymosin β4 and β10 were differentially expressed between disease and control tissues. A larger sample set including matched biological replicates for each type of breast cancer will be necessary for any conclusions to be drawn about the particular metabolites identified as statistically significant in this study. Power analysis based on the expression levels of choline indicates that a sample set of 18 age, gender and ethnicity matched tissues comprised of 3 triple negative tissues and 3 matched controls, 3 pseudoangiomatous hyperplasia tissues and 3 matched controls, and 3 fibroadenoma tissues and 3 matched controls would provide a statistical power near one for a two-tailed t-test where alpha is equal to 0.01. For the present work, the UPLC-IM-MS/MS platform provided a truly untargeted approach in which features from multiple classes of biomolecules could be utilized to differentiate tissues representing an array of breast diseases from cancerous to benign.
Materials and methods
Tissues
Six surgically resected fresh-frozen human tissues were selected from the Meharry Medical College Translational Pathology Shared Resource Core Facility to control for gender, age and ethnicity. Tissues were collected with informed consent (study approved by the Meharry Medical College Institutional Review Board #020715JEM133 13 and #060418SEA058 01). Matched controls were used when available, such that the control would be grossly normal peritumoral tissue from the same patient. The specific pathology of the disease tissues was withheld from the researchers to create a partially blinded experiment, however it was known which three of the six tissues represented a form of breast disease (C, control; D, disease; matched pairs indicated by consecutive numbering (i.e. 1C and 2D are a matched pair)). Tissues were stored at −80 °C.Sample preparation
The procedure for homogenization and extraction of the human breast tissues was adapted from the methods described by Want et al. for the extraction of polar metabolites from tissues for UPLC-MS analysis.33,54 Intact tissues were initially coarsely homogenized on ice in a dounce homogenizer, and 47 ± 4 mg (wet) of each tissue was transferred to an eppendorf tube. To each tissue, 1 mL of cold 1:1 methanol/water (v/v) (Chromasolv, Sigma-Aldrich, St. Louis, MO) was added and the samples were further homogenized on ice using a hand-held homogenizer with disposable plastic probes (Omni International, Kennesaw, GA). A fresh disposable probe was used for each sample. An additional 500 μL of cold 1:1 methanol/water was added to each sample for a total volume of 1.5 mL, and extraction was allowed to proceed overnight at −20 °C.
Samples were centrifuged (16500g for 5 min at 2 °C; Heraeus Fresco 21, Thermo Scientific) and the supernatants were transferred to fresh eppendorf tubes. For the UPLC-IM-MS/MS analysis, 750 μL of each supernatant was dried on a speed-vac and reconstituted with 200 μL of H2O. Protein precipitation was performed by adding cold (−20 °C) methanol in a 3:1 ratio, or 600 μL, to the samples on ice. Samples were then transferred to dry ice for 5 min, after which they were vortexed and centrifuged. Supernatants were transferred to fresh tubes, dried on a speed-vac, and then stored at −80 °C.
Samples were reconstituted with 500 μL of H2O with 0.1% formic acid (HPLC-grade, Fisher Scientific), vortexed, and centrifuged. From each sample, 250 μL was transferred to autosampler vials. A pooled quality control (QC) sample was prepared with 30 μL of each sample (taken from the 250 μL), for a total volume of 180 μL.
UPLC-IM-MS/MS
A nanoACQUITY (Waters Corporation, Milford, MA) UPLC system was used to perform chromatographic separations on an ACQUITY HSS C18 column (1.8 μm, 1.0 × 100 mm; Waters Corporation, Milford, MA). The autosampler and column temperatures were maintained at 4 and 40 °C, respectively, and an injection volume of 6 μL was used to overfill the 5 μL loop. Chromatographic separation was performed with a binary solvent system, where solvent A was 0.1% formic acid in water (Fisher Scientific) and solvent B was 0.1% formic acid in acetonitrile (Chromasolv, Sigma-Aldrich). Gradient conditions for the 25 min run with 60 μL min−1 flow rate were as follows: initial, 99% A; 1 min, 99% A; 6 min, 40% A; 16 min, 1% A; 18 min, 1% A; 19 min, 99% A; 21 min, 99% A.The UPLC was fluidly connected to a Synapt G2 ion mobility-mass spectrometer (Waters Corporation, Milford, MA) to perform IM-MS/MS detection. The traveling wave IM cell is pressurized with nitrogen gas and the MS is an orthogonal time-of-flight mass spectrometer (TOFMS) operated in the single stage reflectron configuration.34,55 The outflow from the chromatographic system was coupled to the instrument through the electrospray ionization (ESI) source. Conditions for positive mode ESI were as follows: capillary, 3 kV; sampling cone, 40 V; extraction cone, 7 V; source temperature, 80 °C; desolvation temperature, 150 °C; cone gas flow, 20 L h−1; desolvation gas flow, 300 L h−1. Mass calibration was performed with sodium formate in the range of m/z 50–1400, and a leucine enkephalin lockmass signal was continuously acquired throughout the MS acquisition for external mass correction of the data. IM separation was achieved with a traveling wave velocity of 650 m s−1 and height of 40 V. Data independent MS/MS by collision induced dissociation (CID) was performed in the transfer region with collision energies ramping between 10 and 30 V.
The sample queue was prepared to run one set of technical replicates (injections of a sample) in a randomized order, bracketed by QC samples. This was repeated two more times to provide three technical replicates for each sample and four QC injections through the queue to monitor performance.
Biostatistics
Data was first mass corrected using the continuously acquired lockmass signal, and the data was centroided during this process. The corrected data were then processed with ProteoWizard (version 3.0.4243) MSConvert to convert the .raw files to .mzXML files.56 The .mzXML files were processed with XCMS (Scripps, La Jolla, CA) to perform peak picking and peak alignment in the chromatographic domain.57,58 Briefly, the “matchedFilter” algorithm was used for peak picking and peak alignment, and retention time correction was performed with the “obiwarp” algorithm. Missing values were filled with the “fillPeaks” algorithm. Details of the XCMS processing are provided in the ESI.† The output from XCMS was normalized such that the sum of all intensities within a sample equaled 10000. This dataset was then imported into Extended Statistics (Umetrics) for visualization of multivariate statistical analyses. PCA was used to determine the quality of the dataset, in terms of the grouping of triplicate technical replicates and grouping of QC injections near the origin of the PCA plot. Model parameters (R2Y and Q2) for the PCA are provided in the ESI.† Analysis of the corresponding loadings plot was used to identify the features contributing to the score of each sample along PC1 and PC2 of the PCA plot. OPLS-DA S-plots were also generated in Extended Statistics to show the pairwise molecular differences between the matched disease and control samples. To determine significance between matched disease and control pairs, p-values were calculated with the Student's t-test for means (two-tailed, equal variance, α = 0.05) using the normalized aligned dataset and corrected by the Bonferroni method to account for multiple testing. ANOVA analyses were performed to determine the significance of the features when compared across all the samples, and the resulting p-values were Bonferroni corrected. For all p-values, p ≤ 0.05 was used as the threshold for significance.
Bioinformatics
For the statistically prioritized molecular features, values of m/z were retrieved from the lockmass-corrected .raw files to ensure the best mass accuracy. In addition, the IM-MS spectra containing the post-mobility data-independent MS/MS acquisitions (saved as function 2 of the data file) were accessed and mobility-organized fragmentation spectra for the features were extracted. This was performed by extracting a defined window of retention time containing the chromatographic peak in which the feature eluted, followed by extracting a defined window of drift time across all m/z which bracketed the drift time of the feature in DriftScope (for example, as indicated on Fig. 3 and 4). The extracted drift time peak was then exported to MassLynx, where all the scans across the peak were combined to generate the MS/MS spectrum. When possible, both accurate mass and fragmentation information were used to make tentative identifications from database searches. Databases used for generating tentative identifications by accurate mass included the Human Metabolite Database (HMDB, http://www.hmdb.ca), KEGG (http://www.kegg.com), and METLIN59–61 (http://metlin.scripps.edu). The METLIN MS/MS spectrum match feature or the MetFrag62 (http://msbi.ipb-halle.de/MetFrag/) in silico fragmentation tool were used to search the experimental fragmentation spectra peak lists, which were filtered by intensity to include only the top 30 peaks.Validation
Tentative identifications where validated with standards when possible. The experimental MS and MS/MS spectra, as well as the drift times, were compared against those of the standard. Standards of glutathione, oxidized glutathione, and adenosine 5′-monophosphate were purchased from Sigma-Aldrich (St. Louis, MO). Standards were prepared at concentrations from 1–3 μg mL−1 in 1:1 methanol/water with 0.1% formic acid. Each standard was directly infused into the ESI source of the Synapt G2 with an external syringe pump (10 μl min−1 flow rate) with ionization conditions identical to those described above. Post-mobility fragmentation was performed at collision energies between 10 and 30 V to approximate the conditions of the data-independent MS/MS acquisitions.
Acknowledgements
The authors would like to thank Purnima Ghose of the Meharry Medical College Translational Pathology Shared Resource Core Facility for assistance in the selection and descriptions of the tissues. The Translational Pathology Shared Resource Core Facility is funded by the U54/National Cancer Institute Grant (NIH/NCI 5U54CA163069-02). Financial support for this research was provided by funds to J.A.M. from the National Institutes of Health (UH2TR000491) and the Defence Threat Reduction Agency (HDTRA-09-1-00-13 and DTRA100271 A-5196), the Vanderbilt University College of Arts and Science, the Vanderbilt Institute for Chemical Biology, and the Vanderbilt Institute for Integrative Biosystems Research and Education.References
- U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999–2010 Incidence and Mortality Web-based Report; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute: Atlanta, 2013.
- R. Etzioni, N. Urban, S. Ramsey, M. McIntosh, S. Schwartz, B. Reid, J. Radich, G. Anderson and L. Hartwell, Nat. Rev. Cancer, 2003, 3, 243 CrossRef CAS PubMed.
- E. A. Vucic, K. L. Thu, K. Robison, L. A. Rybaczyk, R. Chari, C. E. Alvarez and W. L. Lam, Genome Res., 2012, 22, 188 CrossRef CAS PubMed.
- C. A. Hudis and L. Gianni, Oncologist, 2011, 16, 1 CrossRef PubMed.
- E. Senkus, F. Cardoso and O. Pagani, Cancer Treat. Rev., 2014, 40, 220 CrossRef PubMed.
- H. A. Coller, Am. J. Pathol., 2014, 184, 4 CrossRef PubMed.
- J. L. Spratlin, N. J. Serkova and S. G. Eckhardt, Clin. Cancer Res., 2009, 15, 431 CrossRef CAS PubMed.
- C. Oakman, L. Tenori, L. Biganzoli, L. Santarpia, S. Cappadona, C. Luchinat and A. Di Leo, Int. J. Biochem. Cell Biol., 2011, 43, 1010 CrossRef CAS PubMed.
- E. G. Armitage and C. Barbas, J. Pharm. Biomed. Anal., 2014, 87, 1 CrossRef CAS PubMed.
- M. G. V. Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029 CrossRef PubMed.
- J. L. Griffin and J. P. Shockcor, Nat. Rev. Cancer, 2004, 4, 551 CrossRef CAS PubMed.
- J. H. Chen, R. S. Mehta, H. M. Baek, K. Nie, H. Liu, M. Q. Lin, H. J. Yu, O. Nalcioglu and M. Y. Su, NMR Biomed., 2011, 24, 316 CrossRef CAS PubMed.
- T. F. Bathen, B. Sitter, T. E. Sjobakk, M.-B. Tessem and I. S. Gribbestad, Cancer Res., 2010, 70, 6692 CrossRef CAS PubMed.
- B. Sitter, T. F. Bathen, M.-B. Tessem and I. S. Gribbestad, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 239 CrossRef CAS PubMed.
- D. B. Liesenfeld, N. Habermann, R. W. Owen, A. Scalbert and C. M. Ulrich, Cancer Epidemiol., Biomarkers Prev., 2013, 22, 2182 CrossRef CAS PubMed.
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263 CrossRef CAS PubMed.
- K. M. Hines, J. R. Enders and J. A. McLean, in Encyclopedia of Analytical Chemistry, ed. R. A. Myers and D. C. Muddiman, John Wiley & Sons, 2012 Search PubMed.
- J. A. McLean, B. T. Ruotolo, K. J. Gillig and D. H. Russell, Int. J. Mass Spectrom., 2005, 240, 301 CrossRef CAS PubMed.
- S. J. Valentine, M. D. Plasencia, X. Liu, M. Krishnan, S. Naylor, H. R. Udseth, R. D. Smith and D. E. Clemmer, J. Proteome Res., 2006, 5, 2977 CrossRef CAS PubMed.
- J. R. Enders, C. C. Marasco, A. Kole, B. Nguyen, S. Sevugarajan, K. T. Seale, J. P. Wikswo and J. A. McLean, IET Syst. Biol., 2010, 4, 416 CrossRef CAS PubMed.
- J. R. Enders, C. R. Goodwin, C. C. Marasco, K. T. Seale, J. P. Wikswo and J. A. McLean, Current Trends in Mass Spectrom., 2011, 18 Search PubMed.
- P. Dwivedi, P. Wu, S. J. Klopsch, G. J. Puzon, L. Xun and H. H. Hill, Jr., Metabolomics, 2008, 4, 63 CrossRef CAS.
- J. C. May, C. R. Goodwin and J. A. McLean, in Encyclopedia of Drug Metabolism and Interactions, ed. A. V. Lyubimov, Wiley, 2012 Search PubMed.
- K. A. Kaplan, V. M. Chiu, P. A. Lukus, X. Zhang, W. F. Siems, J. O. Schenk and H. H. Hill, Jr., Anal. Bioanal. Chem., 2013, 405, 1959 CrossRef CAS PubMed.
- K. Kaplan, P. Dwivedi, S. Davidson, Q. Yang, P. Tso, W. Siems and H. H. Hill, Anal. Chem., 2009, 81, 7944 CrossRef CAS PubMed.
- B. T. Ruotolo, K. J. Gillig, E. G. Stone and D. H. Russell, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 782, 385 CrossRef CAS.
- B. T. Ruotolo, J. A. McLean, K. J. Gillig and D. H. Russell, J. Mass Spectrom., 2004, 39, 361 CrossRef CAS PubMed.
- X. Liu, M. Plasencia, S. Ragg, S. J. Valentine and D. E. Clemmer, Briefings Funct. Genomics Proteomics, 2004, 3, 177 CrossRef CAS PubMed.
- L. S. Fenn, M. Kliman, A. Mahsut, S. R. Zhao and J. A. McLean, Anal. Bioanal. Chem., 2009, 394, 235 CrossRef CAS PubMed.
- J. A. McLean, J. Am. Soc. Mass Spectrom., 2009, 20, 1775 CrossRef CAS PubMed.
- L. S. Fenn and J. A. McLean, Anal. Bioanal. Chem., 2008, 391, 905 CrossRef CAS PubMed.
- K. M. Hines, S. Ashfaq, J. M. Davidson, S. R. Opalenik, J. P. Wikswo and J. A. McLean, Anal. Chem., 2013, 85, 3651 CrossRef CAS PubMed.
- E. J. Want, P. Masson, F. Michopoulos, I. D. Wilson, G. Theodoridis, R. S. Plumb, J. Shockcor, N. Loftus, E. Holmes and J. K. Nicholson, Nat. Protoc., 2013, 8, 17 CrossRef CAS PubMed.
- S. D. Pringle, K. Giles, J. L. Wildgoose, J. P. Williams, S. E. Slade, K. Thalassinos, R. H. Bateman, M. T. Bowers and J. H. Scrivens, Int. J. Mass Spectrom., 2007, 261, 1 CrossRef CAS PubMed.
- D. I. Broadhurst and D. B. Kell, Metabolomics, 2006, 2, 171–196 CrossRef CAS.
- R. Goodacre, D. Broadhurst, A. K. Smilde, B. S. Kristal, J. D. Baker, R. Beger, C. Bessant, S. Connor, G. Capuani, A. Craig, T. Ebbels, D. B. Kell, C. Manetti, J. Newton, G. Paternostro, R. Somorjai, M. Sjostrom, J. Trygg and F. Wulfert, Metabolomics, 2007, 3, 231–241 CrossRef CAS.
- A. M. Evans, C. D. DeHaven, T. Barrett, M. Mitchell and E. Milgram, Anal. Chem., 2009, 81, 6656–6667 CrossRef CAS PubMed.
- P. Franceschi, U. Vrhovsek, F. Mattivi and R. Wehrens, Metabolic Biomarker Identification with Few Samples, Chemometrics in Practical Applications InTech, 2012 Search PubMed.
- C. S. Hoaglund-Hyzer, J. W. Li and D. E. Clemmer, Anal. Chem., 2000, 72, 2737 CrossRef CAS.
- T. Huff, C. S. G. Muller, A. M. Otto, R. Netzker and E. Hannappel, Int. J. Biochem. Cell Biol., 2001, 33, 205 CrossRef CAS.
- H. J. Cha, M. J. Jeong and H. K. Kleinman, J. Natl. Cancer Inst., 2003, 95, 1674 CrossRef CAS PubMed.
- A. E. Maelan, T. K. Rasmussen and L.-I. Larsson, Histochem. Cell Biol., 2007, 127, 109 CrossRef CAS PubMed.
- R. Katz-Brull, D. Seger, D. Rivenson-Segal, E. Rushkin and H. Degani, Cancer Res., 2002, 62, 1966 CAS.
- K. Glunde, C. Jie and Z. M. Bhujwalla, Cancer Res., 2004, 64, 4270 CrossRef CAS PubMed.
- K. Glunde, M. A. Jacobs and Z. M. Bhujwalla, Expert Rev. Mol. Diagn., 2006, 6, 821 CrossRef CAS PubMed.
- E. R. A. van Hove, T. R. Blackwell, I. Klinkert, G. B. Eijkel, R. M. A. Heeren and K. Glunde, Cancer Res., 2010, 70, 9012 CrossRef PubMed.
- K. Glunde, Z. M. Bhujwalla and S. M. Ronen, Nat. Rev. Cancer, 2011, 11, 835 CAS.
- M. P. Gamcsik, M. S. Kasibhatla, S. D. Teeter and O. M. Colvin, Biomarkers, 2012, 17, 671 CrossRef CAS PubMed.
- M. Perquin, T. Oster, A. Maul, N. Froment, M. Untereiner and D. Bagrel, Cancer Lett., 2000, 158, 7 CrossRef CAS.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191 CrossRef CAS.
- M. Perquin, T. Oster, A. Maul, N. Froment, M. Untereiner and D. Bagrel, J. Cancer Res. Clin. Oncol., 2001, 127, 368 CrossRef CAS.
- W. D. Dupont, D. L. Page, F. F. Parl, C. L. Vnencak-Jones, W. D. Plummer, M. S. Rados and P. A. Schuyler, N. Engl. J. Med., 1994, 331, 10 CrossRef CAS PubMed.
- M. Ferreira, C. T. Albarracin and E. Resetkova, Mod. Pathol., 2008, 21, 201 Search PubMed.
- P. Masson, A. C. Alves, T. M. D. Ebbels, J. K. Nicholson and E. J. Want, Anal. Chem., 2010, 82, 7779 CrossRef CAS PubMed.
- K. Giles, S. D. Pringle, K. R. Worthington, D. Little, J. L. Wildgoose and R. H. Bateman, Rapid Commun. Mass Spectrom., 2004, 18, 2401 CrossRef CAS PubMed.
- M. C. Chambers, B. Maclean, R. Burke, D. Amodei, D. L. Ruderman, S. Neumann, L. Gatto, B. Fischer, B. Pratt, J. Egertson, K. Hoff, D. Kessner, N. Tasman, N. Shulman, B. Frewen, T. A. Baker, M.-Y. Brusniak, C. Paulse, D. Creasy, L. Flashner, K. Kani, C. Moulding, S. L. Seymour, L. M. Nuwaysir, B. Lefebvre, F. Kuhlmann, J. Roark, P. Rainer, S. Detlev, T. Hemenway, A. Huhmer, J. Langridge, B. Connolly, T. Chadick, K. Holly, J. Eckels, E. W. Deutsch, R. L. Moritz, J. E. Katz, D. B. Agus, M. MacCoss, D. L. Tabb and P. Mallick, Nat. Biotechnol., 2012, 30, 918 CrossRef CAS PubMed.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779 CrossRef CAS PubMed.
- R. Tautenhahn, C. Bottcher and S. Neumann, BMC Bioinf., 2008, 9, 504 CrossRef PubMed.
- C. A. Smith, G. O'Maille, E. J. Want, C. Qin, S. A. Trauger, T. R. Brandon, D. E. Custodio, R. Abagyan and G. Siuzdak, Ther. Drug Monit., 2005, 27, 747 CrossRef CAS.
- R. Tautenhahn, K. Cho, W. Uritboonthai, Z. Zhu, G. J. Patti and G. Siuzdak, Nat. Biotechnol., 2012, 30, 826 CrossRef CAS PubMed.
- Z.-J. Zhu, A. W. Schultz, J. Wang, C. H. Johnson, S. M. Yannone, G. J. Patti and G. Siuzdak, Nat. Protoc., 2013, 8, 451 CrossRef CAS PubMed.
- S. Wolf, S. Schmidt, M. Mueller-Hannemann and S. Neumann, BMC Bioinf., 2010, 11, 148 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00250d |
| ‡ Current address: Mayo Clinic Metabolomics Resource Core, Mayo Clinic, Rochester, MN 55905, USA. |
| This journal is © The Royal Society of Chemistry 2014 |