 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A red light-controlled synthetic gene expression switch for plant systems†
Konrad
Müller
a,
David
Siegel
a,
Fernando
Rodriguez Jahnke
ab,
Katrin
Gerrer
a,
Sabrina
Wend
ab,
Eva L.
Decker
a,
Ralf
Reski
abcd,
Wilfried
Weber
abce and
Matias D.
Zurbriggen
*ac
aFaculty of Biology, University of Freiburg, Schänzlestrasse 1, 79104 Freiburg, Germany. E-mail: matias.zurbriggen@biologie.uni-freiburg.de; Fax: +49 761 203 97660; Tel: +49 761 203 97656
bSGBM – Spemann Graduate School of Biology and Medicine, University of Freiburg, Albertstrasse 19A, 79104 Freiburg, Germany
cBIOSS Centre for Biological Signalling Studies, University of Freiburg, Schänzlestrasse 18, 79104 Freiburg, Germany
dFreiburg Institute for Advanced Studies (FRIAS), University of Freiburg, Albertstrasse 19, 79104 Freiburg, Germany
eFreiburg Centre for Biosystems Analysis (ZBSA), University of Freiburg, Habsburgerstrasse 49, 79104 Freiburg, Germany
First published on 14th January 2014
Abstract
On command control of gene expression in time and space is required for the comprehensive analysis of key plant cellular processes. Even though some chemical inducible systems showing satisfactory induction features have been developed, they are inherently limited in terms of spatiotemporal resolution and may be associated with toxic effects. We describe here the first synthetic light-inducible system for the targeted control of gene expression in plants. For this purpose, we applied an interdisciplinary synthetic biology approach comprising mammalian and plant cell systems to customize and optimize a split transcription factor based on the plant photoreceptor phytochrome B and one of its interacting factors (PIF6). Implementation of the system in transient assays in tobacco protoplasts resulted in strong (95-fold) induction in red light (660 nm) and could be instantaneously returned to the OFF state by subsequent illumination with far-red light (740 nm). Capitalizing on this toggle switch-like characteristic, we demonstrate that the system can be kept in the OFF state in the presence of 740 nm-supplemented white light, opening up perspectives for future application of the system in whole plants. Finally we demonstrate the system's applicability in basic research, by the light-controlled tuning of auxin signalling networks in N. tabacum protoplasts, as well as its biotechnological potential for the chemical-inducer free production of therapeutic proteins in the moss P. patens.
Introduction
The ability to control transgene expression in plant systems is essential for the analysis of complex regulatory systems and metabolic pathways, and in particular to study genes that have deleterious effects on plant growth and development and can therefore not be expressed constitutively.1,2 Furthermore, inducible transgene expression can be utilized for the production of therapeutic proteins in plant cell culture to confine protein production to growth phases with an optimal biosynthetic capacity.3 Consequently, several transgene expression systems have been developed that can be controlled by chemicals.1,3 These systems are regulated by antibiotics,4,5 steroids,6–8 insecticides,9,10 ethanol11 or copper.12 While some glucocorticoid-responsive tools suffer from toxic effects of the inducer,13,14 most of the chemically-inducible systems are orthogonal to plant metabolism and are characterized by good induction properties. However, these systems are controlled by small molecules and are subject to limitations that are inherent to these chemicals. Because of diffusion of the inducers, chemically-controlled systems have a poor temporal resolution of gene expression. Furthermore, many inducers are pharmacologically active. Therefore, their addition to a plant cell culture for the production of biopharmaceuticals is undesirable.In contrast to small chemical inducers, light at a cell compatible wavelength is not subject to regulatory restrictions in the bioproduction of proteins and can be delivered with high temporal resolution. Hence, several light-responsive gene-expression systems have recently been developed for mammalian systems that can be controlled by UVB,15,16 blue17–19 or red light.20 None of these systems have been transferred to plants yet, probably due to the fact that as opposed to mammalian cells, plants are not “blind” and require light to gather information from their surroundings and to harness their energy.
Here we describe the adaptation, optimization and implementation in plant settings of a red/far-red light-switchable transgene expression system that was developed in mammalian cells.20 The synthetic switch is based on the red/far-red light-dependent interaction of phytochrome B (PhyB)21,22 and the phytochrome-interacting factor 6 (PIF6) from A. thaliana that is integrated in a light-responsive split transcription factor (Fig. 1). On the one hand, PIF6 (amino acids 1–100) is fused to a DNA-binding domain (BD) that binds its operator site in the reporter construct upstream of a minimal promoter and the reporter gene. On the other hand, PhyB (amino acids 1–650) is linked to an activator domain (AD) and a nuclear localization sequence (NLS). In red light, PhyB-AD-NLS is recruited to PIF6 at the promoter site, switching the system to the ON state. Only upon absorption of a far-red photon the interaction between PhyB and PIF6 is terminated, resulting in a shut-off of gene expression.20 We reasoned that this system is suited for light-inducible gene expression in plants, because gene expression can not only be triggered by illumination with red (660 nm) light, but can also be actively terminated by far-red (740 nm) light. This may, in the future, facilitate the implementation of the system in whole plants, where transgene expression in light-grown plants can be repressed by supplementary far-red light illumination. On the other hand, the possibility to repress transgene expression in white light is not available for UVB or blue light-responsive systems that cannot be returned to the OFF state actively, but return to the inactive state passively in the dark with half-life times of several hours.23
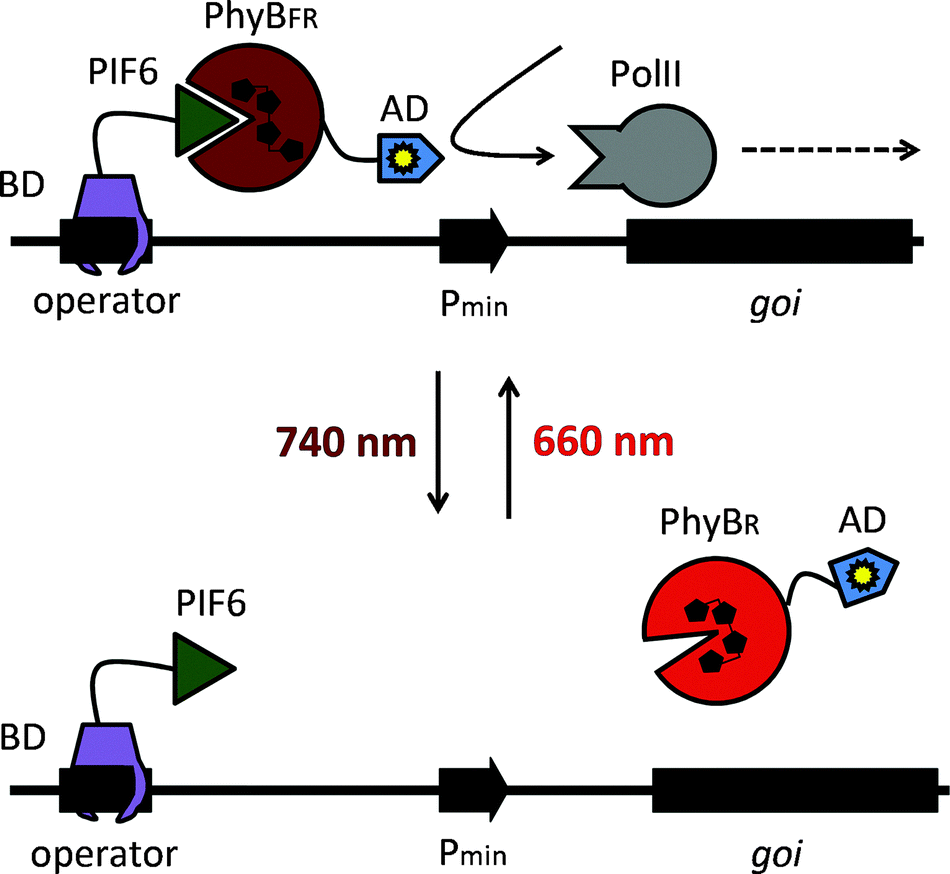 | ||
| Fig. 1 Molecular design of the red light-responsive gene expression system. Red light illumination converts PhyB into the active FR form (PhyBFR) and induces heterodimerization with PIF6 tethered via a DNA-binding domain (BD) to an operator site. The PhyB-fused activation domain (AD) recruits the transcription initiation complex and triggers activation of the minimal promoter (Pmin). Absorption of a far-red photon (740 nm) converts PhyB into the inactive R form (PhyBR) and triggers dissociation from PIF6, thereby resulting in de-activation of the target promoter and transcriptional silence. Adapted from ref. 20 with permission. | ||
Analogous to the mammalian cell lines and transient gene expression assays that are indispensable for the exploration of signalling processes, protoplast transient expression systems have been developed that offer a genetically accessible platform to dissect plant signal transduction pathways. Amongst other favorable biochemical, genetic and physiological characteristics, protoplasts retain the identity of the tissue they originate from and have been successfully applied to dissect various signalling pathways in plants.24 In light of these advantages, we decided to implement red light-controlled gene expression in N. tabacum leaf protoplasts.
In proof-of-principle applications to demonstrate the applicability of red light-controlled gene expression in the analysis of plant signalling and for the production of biopharmaceuticals, we then aimed to use red light to manipulate auxin signalling in tobacco protoplasts and to produce a therapeutic protein in the moss P. patens.
Results
Chemically-controlled gene expression in N. tabacum
The red light-responsive gene expression system for mammalian systems is based on the TetR protein for DNA-binding.20 To find a highly efficient DNA-binding protein to apply this system to plants, we first implemented and compared chemically-inducible systems, which respond to antibiotics that have been well-described and are widely used in mammalian cells and in N. tabacum protoplasts. To this end, we adapted a macrolide-responsive gene expression tool that is based on the macrolide repressor protein from E. coli (here referred to as E)25 to plant systems and compared it to tetracycline4 and pristinamycin-regulated5 systems. N. tabacum protoplasts were co-transformed with plasmids coding for the DNA-binding proteins fused to the Herpes simplex virus-derived VP16 transactivation domain and a nuclear localization sequence (NLS) and with reporter plasmids for firefly luciferase expression (Fig. 2A). Next, the protoplasts were incubated in the presence or absence of the regulating antibiotics at non-toxic concentrations5,26 (Fig. S1, ESI†) for 24 h. The macrolide-regulated transgene expression system showed higher reporter levels (245.5 [RLU] compared to 2.5 [RLU] (tetracycline-regulated) and 1.3 [RLU] (pristinamycin-regulated)) as well as a higher induction ratio of the unrepressed state compared to the repressed state (721-fold compared to 65-fold (tetracycline-regulated) and 52-fold (pristinamycin-regulated)) (Fig. 2B). In light of the superior performance of the E-based gene-regulation system in N. tabacum, we decided to modify the red light-regulated gene expression system with respect to the DNA binding protein by replacing TetR with the macrolide repressor protein E.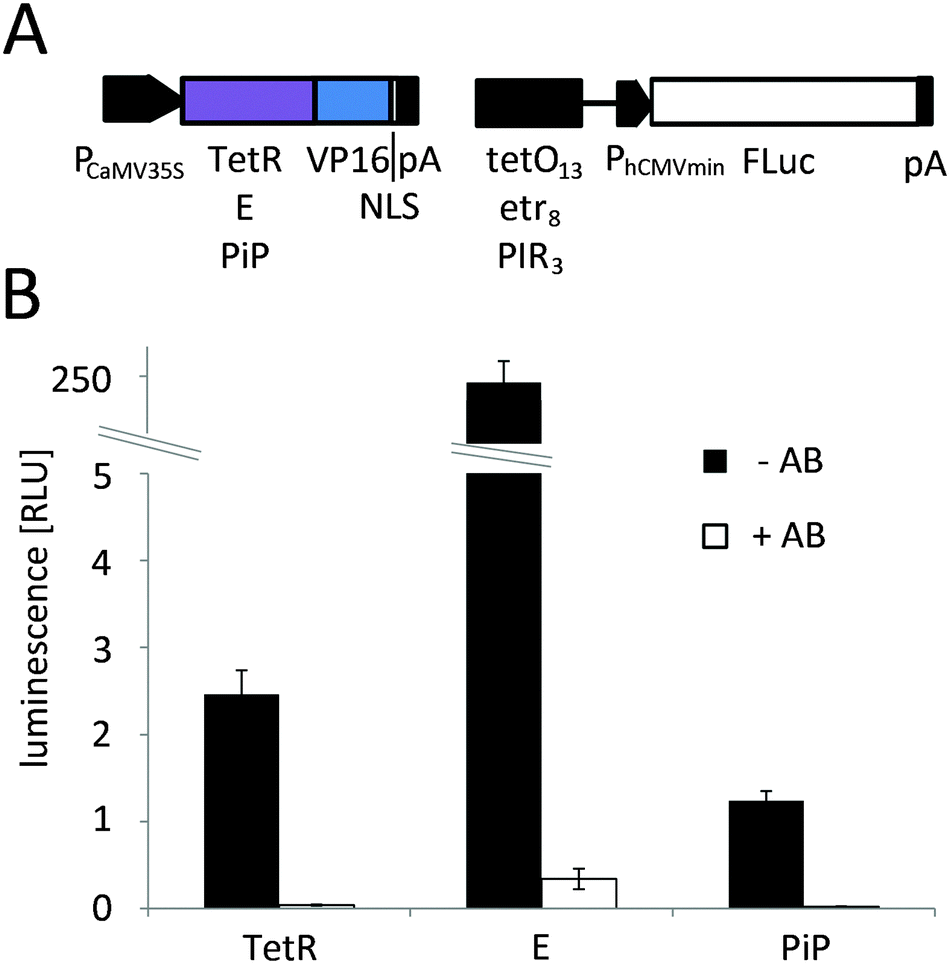 | ||
Fig. 2 Chemically-controlled gene expression in N. tabacum. (A) Configuration. For chemically-controlled gene expression a two vector system was employed. The first plasmid encoded a constitutively-expressed fusion protein of a DNA-binding protein and the Herpes simplex-derived VP16 transactivation domain under the control of the cauliflower mosaic virus 35S promoter (PCaMV35S). As binding proteins the tetracycline repressor protein (TetR, pMZ833), the macrolide repressor protein (E, pMZ824) or the pristinamycin repressor protein (PiP, pKM271) were used and the fusion proteins were targeted to the nucleus via a C-terminally fused nuclear localization sequence (NLS). The second plasmid coded for firefly luciferase (FLuc) under control of the human cytomegalovirus minimal promoter (PhCMVmin) that was positioned downstream of multimerized operator sequences for TetR (tetO13, pMZ802), E (etr8, pMZ836) or PiP (PIR3, pKM272). (B) Chemically controlled gene expression in N. tabacum-derived protoplasts. 125![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 protoplasts were transformed for tetracycline, clarithromycin or pristinamycin-controlled FLuc expression. After a 24 h incubation in the absence (−AB) or presence (+AB) of the regulating antibiotics the reporter luminescence was quantified. Data are means ± SEM (n = 12). 000 protoplasts were transformed for tetracycline, clarithromycin or pristinamycin-controlled FLuc expression. After a 24 h incubation in the absence (−AB) or presence (+AB) of the regulating antibiotics the reporter luminescence was quantified. Data are means ± SEM (n = 12). | ||
Red light-controlled gene expression in mammalian and N. tabacum cells
To adapt the red light-regulated gene expression system to plants, we followed a two-stage process. First, we capitalized on the established mammalian cell system to evaluate the functionality of the modified light-switch upon replacement of the TetR DNA-binding domain with the in-planta superior E DNA-binding protein. Next, we placed the system's components under the control of plant promoters and validated the system in N. tabacum-derived protoplasts.In the first step, CHO-K1 cells were transfected for red light-controlled expression of the reporter protein secreted alkaline phosphatase (SEAP) using TetR (pKM022/pKM006), E (pKM300/pKM082) or PIP (pKM301/pMF199) as the DNA-binding domains (Fig. 3A). 24 h after transfection, the medium was replaced with phycocyanobilin (PCB)-supplemented medium to provide the PhyB-chromophore absent in mammalian cells.27 After 1 h incubation in the dark for ligation of PCB to PhyB, the cells were illuminated with 660 nm or 740 nm light for 24 h before quantification of SEAP production. Total reporter levels were 1.8-fold and 2.5-fold higher for the PIP- and E-based systems compared to the original TetR-based light-inducible gene expression system (Fig. 3B). At the same time, the induction in 660 nm-illuminated compared to 740 nm-illuminated samples remained at comparable levels for all three systems.
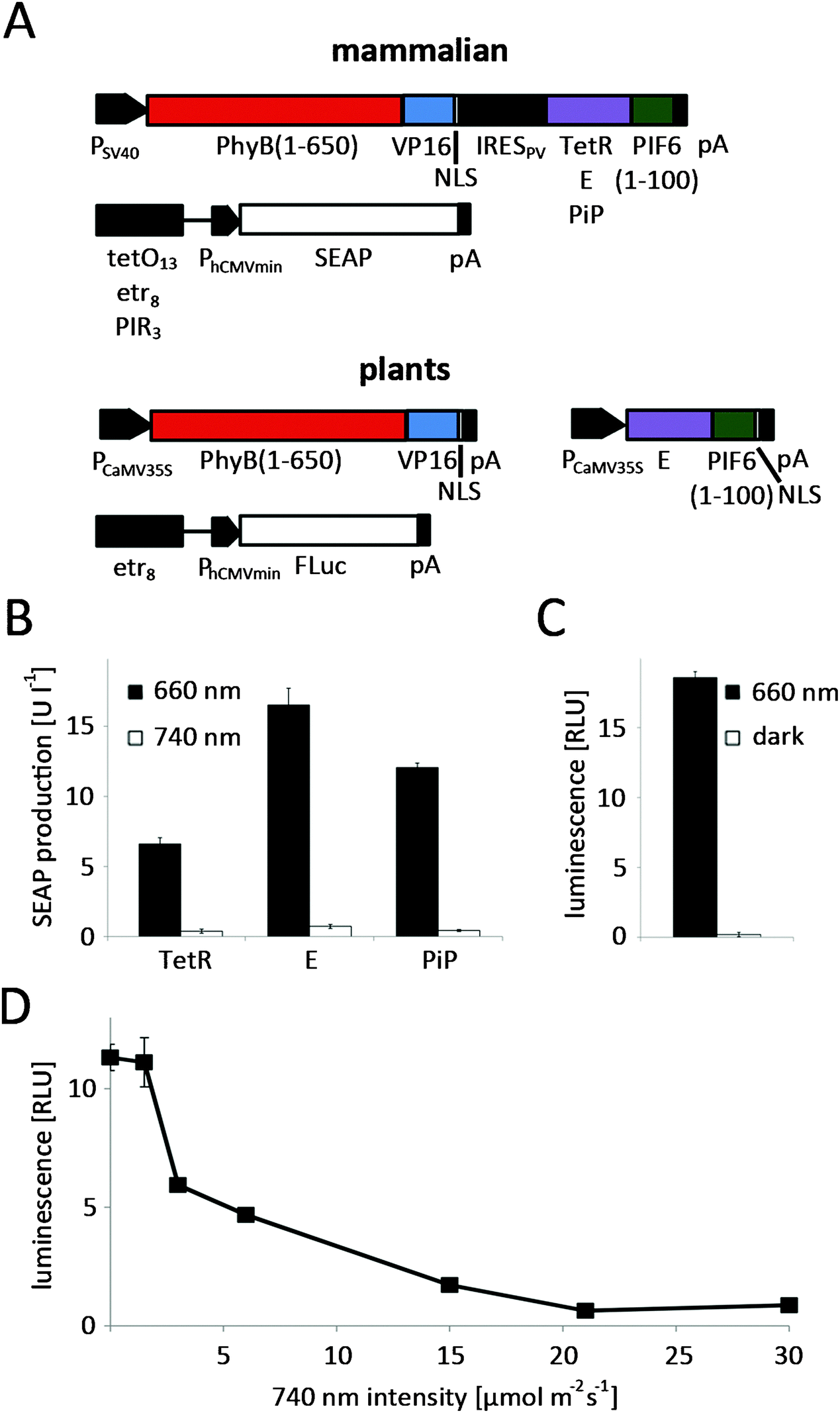 | ||
| Fig. 3 Red light-controlled gene expression in mammalian and in N. tabacum cells. (A) In mammalian cells, the red light-switchable split transcription factor was transcribed as a bicistronic expression unit under the control of the simian virus 40 promoter (PSV40). In the first cistron, the N-terminal fragment PhyB (PhyB(1-650)) was fused to VP16 and to an NLS. In the second cistron, the N-terminal 100 amino acids of PIF6 were fused to TetR (pKM022), E (pKM300) or PiP (pKM301). Translation of the second cistron was induced by a polioviral internal ribosome entry site (IRESPV). The response vectors comprised tetO13 (pKM006), etr8 (pKM082) or PIR3 (pMF199) fused to PhCMVmin and the reporter human placental secreted alkaline phosphatase (SEAP). For optimized red light-inducible gene expression in plants, the components of the split transcription factor PhyB(1-650)-VP16-NLS (pMZ827) and E-PIF6(1-100), which were enhanced by a C-terminal NLS (pMZ828), were under control of PCaMV35S. In the response construct FLuc expression was controlled by an etr8-PhCMVmin promoter (pMZ836). (B) Evaluation of different DNA binding proteins in CHO-K1 cells. 75000 CHO-K1 cells were transfected for red light-responsive SEAP production using TetR, E or PiP fusions of PIF6 in conjunction with PhyB-VP16-NLS and specific reporter plasmids. 24 h post transfection, the culture medium was replaced with phycocyanobilin (PCB)-supplemented medium. After 1 h incubation in the dark the cells were illuminated with 660 nm or 740 nm light for 24 h before SEAP quantification. (C) Red light-induced firefly luciferase expression in N. tabacum protoplasts. 125000 protoplasts were transformed for red light-responsive firefly luciferase production. Following incubation with 660 nm illumination or in the dark for 24 h, the luminescence was quantified. (D) Repression of transgene expression in white light by supplementary 740 nm illumination in N. tabacum protoplasts. 125000 N. tabacum protoplasts were first transformed for red light-regulated firefly luciferase production. Next, the protoplasts were illuminated with white light (1 μmol m−2 s−1) that was supplemented with 740 nm light of increasing intensities. 24 h after illumination start, the reporter luminescence was quantified. B, data are means ± SD (n = 4); C and D, data are means ± SEM (n = 12). | ||
Following the successful functional replacement of the DNA-binding domain, we proceeded to place the components of the red light-responsive transcription factor under the control of the cauliflower mosaic virus 35S promoter (PCaMV35S)28 for its application in plant cells. Next, N. tabacum-derived protoplasts were transformed with the red light responsive split transcription factor (pMZ827/pMZ828) along with a firefly luciferase reporter plasmid (pMZ836) (Fig. 3A). Because the natural PhyB chromophore phytochromobilin (PϕB) is synthesized by all plants,29,30 PCB-supplementation of the protoplast culture was not necessary. The protoplasts were either illuminated with activating 660 nm light for 24 h, or incubated in the dark immediately after the transformation. Quantification of luciferase luminescence revealed high expression levels in the 660 nm-illuminated samples, while expression in the dark-incubated protoplasts remained at basal levels, resulting in induction levels of 95-fold (Fig. 3C). For many applications it is essential to be able to grow plants in white light without activation of transgene expression. Therefore, we explored the possibility of preventing expression of a transgene under the control of the red light-inducible expression system by supplementing white light with inactivating 740 nm light. To this end, N. tabacum-derived protoplasts were transformed for red light-controlled firefly luciferase expression and incubated under white light (see spectrum in Fig. S2, ESI†) that was supplemented with 740 nm light of increasing intensities. Quantification of firefly luciferase luminescence revealed that a strong reduction of transgene expression by white light (1 μmol m−2 s−1) is possible by additional illumination with 3 μmol m−2 s−1 of 740 nm light. Complete repression of transgene expression to levels comparable to dark-incubated samples is achieved at 21 μmol m−2 s−1 740 nm light (Fig. 3D).
In mammalian cells, the red/far-red light controlled gene expression system is bistable – it remains in the ON state, when activating 660 nm illumination is followed by darkness and stays in the OFF state, when inactivating 740 nm illumination is succeeded by incubation in the dark.20 In plants, on the other hand, PhyB is known to return to the inactive R-form not only upon illumination with 740 nm light, but also in the dark. This process has been termed “dark reversion” and depends on several factors22 like pH, ionic strength, reducing agents, metal ions and phosphorylation.31 To gain a deeper insight into the kinetics of OFF-switching of gene expression by far-red light or in the dark in planta, N. tabacum-derived protoplasts were transformed for red light-responsive firefly luciferase expression and illuminated with 660 nm light for 8 h. Next, illumination with 660 nm light was either resumed, illumination was switched to 740 nm light or the protoplasts were moved to the dark. At the same time, control cells received clarithromycin that has been shown to induce the immediate dissociation of E from its operator sequence, resulting in an instantaneous shut-off of gene expression.25 The firefly luciferase reporter was followed over a time-course of 24 h by quantifying its luminescence (Fig. 4). Control cells for background reporter expression were incubated in the dark or under 740 nm light for the entire time course. Reporter expression increased after 4 h and continued to rise before reaching steady-state levels after 12 h of continuous 660 nm illumination. On the other hand, firefly luciferase luminescence from samples that received clarithromycin 8 h after illumination onset began to steadily decrease after 12 h and samples that were switched from 660 nm to 740 nm illumination after 8 h displayed the same time course of reporter expression as samples that received clarithromycin. This implies that transgene expression is instantaneously switched off upon illumination with 740 nm light. Samples that were transferred from 660 nm illumination to darkness after 8 h showed a decrease in the luminescence signal after 12 h as well, but this decrease was slightly delayed compared to samples that had been switched to 740 nm illumination. This suggests that dark reversion plays a significant role in red light-controlled gene expression in plants, but is slower than the active termination of gene expression by 740 nm illumination.
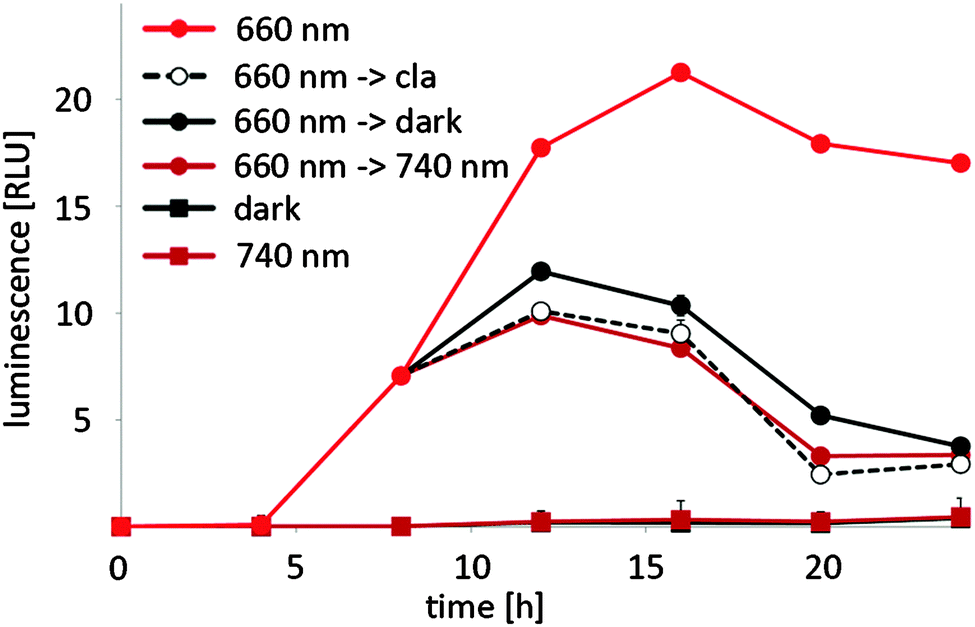 | ||
| Fig. 4 Switch-off kinetics of red light-regulated gene expression in N. tabacum. 125000 protoplasts were transformed for red light-responsive expression of firefly luciferase (pMZ827/pMZ828/pMZ836). After the transformation, the protoplasts were illuminated with 660 nm light for 8 h. Next, 660 nm illumination was continued, clarithromycin was added, and the cells were transferred to the dark or illumination was switched to 740 nm. Control cells were incubated in the dark or under 740 nm light for the entire experiment. Firefly luciferase luminescence was quantified at the indicated points in time. Data are means ± SEM (n = 12). | ||
Red light-controlled auxin signalling in N. tabacum
After implementation and characterization of the red light-responsive gene expression system in N. tabacum protoplasts, we sought to demonstrate its applicability in the investigation of plant signalling networks. To this end we chose to manipulate the auxin response, which plays a pivotal role in the regulation of plant growth and in developmental processes.32,33 The crux of auxin signalling is the auxin-dependent formation of a co-receptor complex between TIR1 (F-box protein component of an SCF E3 ubiquitin–ligase complex) and Aux/IAA family members (repressors of auxin-responsive genes) that triggers the de-repression of auxin-responsive genes via the degradation of Aux/IAA.34 We intended to target this switch point of auxin perception in a two-pronged approach. Firstly, we placed TIR1 under red light control to enhance the degradation of Aux/IAA. Secondly, we designed micro RNAs (miRNAs) targeting the TIR1 mRNA to reduce cellular TIR1-levels, resulting in reduced sensitivity to auxin and thus elevated Aux/IAA levels (Fig. 5A). To observe the effect of the light-regulated adjustment of TIR1-levels, we monitored the TIR1-dependent effect of auxin on a recently described, Aux/IAA-degradation-based ratiometric auxin sensor.35 To this end, N. tabacum-derived protoplasts were transformed for expression of an optimized quantitative auxin sensor construct (L2min17-Luc).35 After 24 h incubation in the dark, increasing amounts of auxin were added to the culture medium. Following 45 min incubation firefly luciferase and renilla luciferase luminescence was quantified. Due to increasing TIR1-mediated degradation of the Aux/IAA-coupled firefly luciferase, the ratio of firefly luciferase luminescence to renilla luciferase luminescence decreased from 4.1 in the absence of auxin to background levels at 10 nM auxin (Fig. 5B). In parallel, protoplasts were transformed for expression of the auxin sensor and red light controlled TIR1 (pMZ827/pMZ828/pMZ841) or for expression of the auxin sensor and red light-controlled miRNATIR1 (pMZ827/pMZ828/pMZ839). After transformation the protoplasts were incubated under 660 nm light or in the dark prior to auxin stimulation. Protoplasts that had been incubated in the dark displayed the same auxin-dependent decrease in Aux/IAA observed for the sensor alone, regardless of the deployed reporter. However, 660 nm-illuminated protoplasts transfected for red light-inducible miRNATIR1 expression showed increased Aux/IAA levels, as would be expected for the depletion of the cellular TIR1 pool resulting from the red light-induced knock-down of TIR1. On the other hand, the red light-induced expression of TIR1 triggered a decrease of Aux/IAA to background levels even without auxin supplementation (Fig. 5B).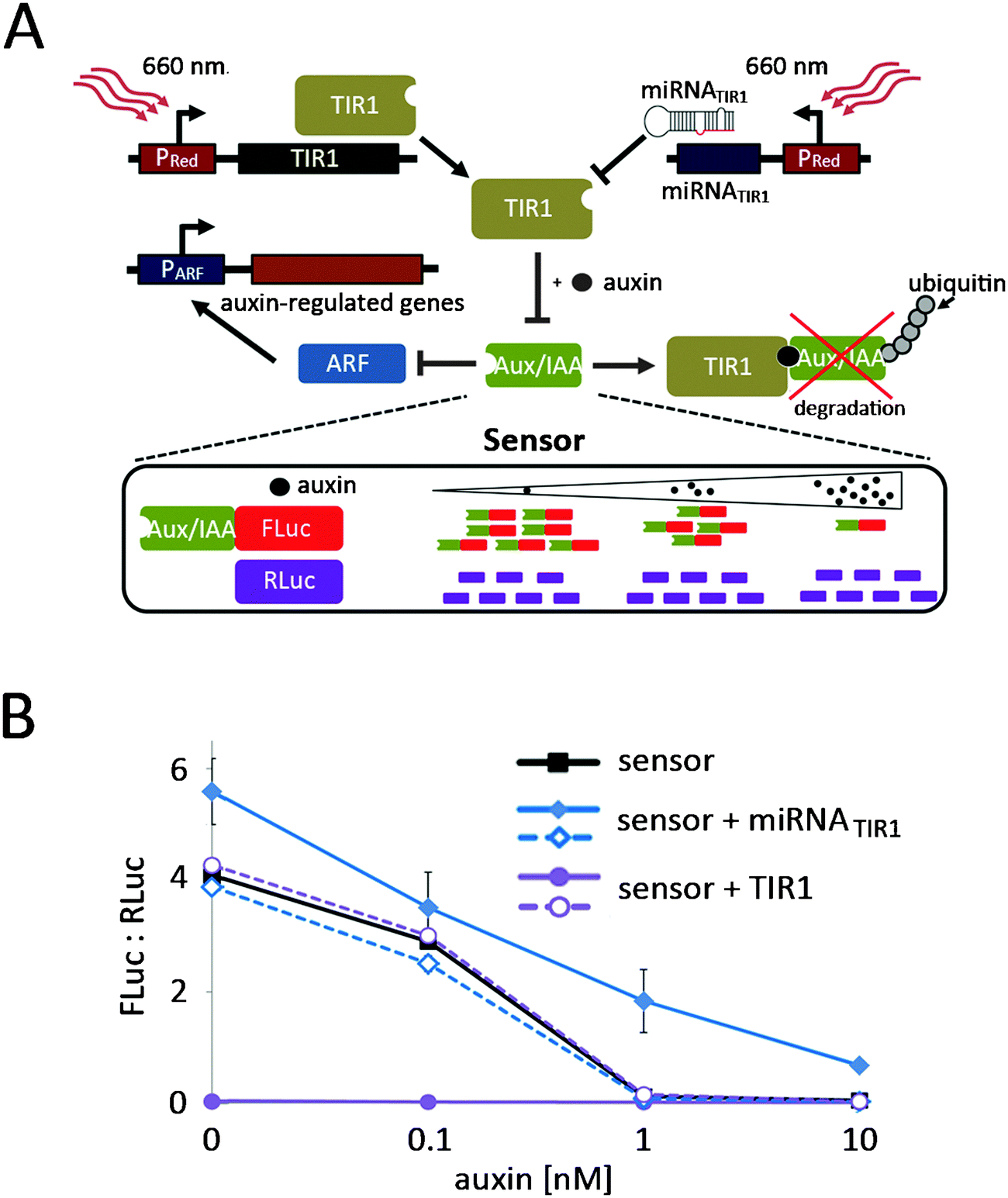 | ||
| Fig. 5 Red light-controlled tuning of auxin signalling in N. tabacum. (A) Principle of red light-controlled auxin signalling. TIR1 mediates auxin-induced degradation of Aux/IAA that in turn inhibits the expression of auxin-responsive genes via the repression of ARF. By red light-controlled expression of TIR1, Aux/IAA levels are further decreased, resulting in increased expression of auxin-regulated genes. On the other hand, the red light-induced expression of miRNATIR1 diminishes the cellular TIR1 pools, resulting in elevated Aux/IAA levels and strong repression of auxin signalling. The Aux/IAA-mediated signalling is monitored via a ratiometric sensor that is constituted by Aux/IAA-fused FLuc and RLuc that are produced in an equimolar ratio. An increase in auxin levels or the red light-induced expression of TIR1 results in a decrease in the Aux/IAA-FLuc pool, while the RLuc population is not affected. Therefore, the FLuc:RLuc ratio decreases. In the same way a decrease in auxin levels or the red light-induced knock-down of TIR1 will result in an increased FLuc:RLuc ratio. (B) Implementation of red light-controlled auxin signalling in N. tabacum cell culture. 125000 protoplasts were transformed with the auxin sensor alone, with the auxin sensor and a red light-controlled miRNATIR1 (pMZ827/pMZ828/pMZ839) or with the auxin and red light-controlled TIR1 (pMZ827/pMZ828/pMZ841). After transformation the protoplasts were either illuminated with 660 nm light (closed symbols) or incubated in the dark (open symbols). 24 h later increasing amounts of auxin were added to the protoplasts as indicated. After incubation for 45 min, the firefly luciferase and renilla luciferase luminescence was quantified. Data are means ± SEM (n = 3). | ||
These results illustrate the potential of the light-inducible expression system for the targeted analysis of complex plant signal sensing and transduction pathways.
Red light-triggered production of biopharmaceuticals in P. patens
To show the potential of the red light-inducible expression system not only in basic research, but also in biotechnology, we decided to apply it further to the production of biopharmaceuticals. Most biopharmaceuticals are currently produced in mammalian expression hosts, especially in Chinese hamster ovary (CHO) cells,36 to match their native glycosylation patterns. However, mammalian expression platforms are cost-intensive and are associated with the risk of contamination with human pathogens. Therefore, plants and in particular the moss P. patens have gained increasing attention as competitive production hosts for complex biopharmaceuticals.37–39P. patens is a versatile production platform,40 because it is mainly haploid, genetically accessible by highly efficient base-specific gene targeting, has been engineered for humanized glycosylation and proteins can be secreted in the culture medium.38,41,42 While several therapeutic proteins, like the vascular endothelial growth factor (VEGF),43 erythropoietin (EPO),44 monoclonal antibodies45 or factor H,46 have been produced in P. patens by batch fermentation, a chemical-inducer free inducible process would be highly desirable for the production of cytotoxic proteins that elude classical batch fermentation and to optimize the product yield by limiting protein production to growth phases with an optimal biosynthetic capacity.3Therefore, we tested the applicability of the red light-responsive gene expression system for P. patens. First, we confirmed that the clarithromycin-responsive gene expression system is functional in P. patens and observed excellent reporter expression when P. patens-derived protoplasts were co-transformed with E-VP16-NLS (pMZ824) and a firefly luciferase reporter (pMZ836) (Fig. 6A). Upon addition of 100 μg ml−1 clarithromycin, reporter expression was repressed to background levels, thus resulting in a 25-fold induction of the unrepressed state compared to the repressed state. Next, we tested red light-controlled gene expression by transformation of P. patens-derived protoplasts with the red light-responsive split transcription factor (pMZ827/pMZ828) and a luciferase reporter (pMZ836), followed by illumination with activating 660 nm light or incubation in the dark for 24 h. Quantification of firefly luciferase luminescence revealed a 26-fold induction of reporter expression in red light compared to the dark-control (Fig. 6B). Finally, we chose to place the human vascular endothelial growth factor (VEGF121) under red light control to demonstrate the suitability of the red light-controlled expression system for the production of biopharmaceuticals. VEGF is a small glycoprotein that plays an important role in angiogenesis47 and is being evaluated for applications in wound healing in diabetes.43 Illumination of P. patens-derived protoplasts transformed for red light-inducible VEGF121-production (pMZ827/pMZ828/pKM295) with 660 nm light for 40 h resulted in 616 pg ml−1 secreted VEGF (Fig. 6C). The lower induction ratio of the secreted protein VEGF (Fig. 6C) compared to light-induced expression of the cytoplasmatically-localized firefly luciferase (Fig. 6B) might be due to the delay in protein secretion upon illumination onset.
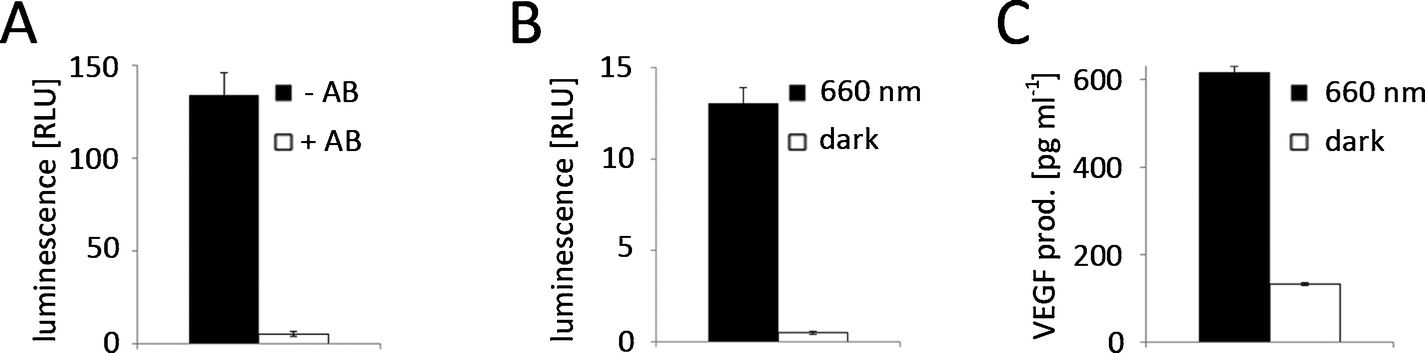 | ||
| Fig. 6 Inducible gene expression in P. patens. (A) Clarithromycin-regulated gene expression. 125000 protoplasts were transformed for clarithromycin (pMZ824/pMZ836)-controlled firefly luciferase expression. After a 24 h incubation in the absence (−AB) or presence (+AB) of 100 μg ml−1 clarithromycin the reporter luminescence was quantified. (B) Red light-inducible expression of firefly luciferase. 125000 protoplasts were transformed for red light-responsive firefly luciferase production (pMZ827/pMZ828/pMZ836). Following incubation under 660 nm illumination or in the dark for 24 h, the luminescence was quantified. (C) Production of VEGF121 in response to red light. 125000 protoplasts were transformed for red light-responsive VEGF production (pMZ827/pMZ828/pKM295). After illumination with 660 nm light for 40 h or incubation in the dark the VEGF production was quantified. Data are means ± SEM (n = 12). | ||
These results demonstrate the feasibility and the potential of red light-induced protein production in plants.
Discussion
Chemically-inducible gene expression systems have limitations regarding not only the temporal but also the spatial control of induction. Therefore, several light-responsive expression systems for bacteria,48–50 yeast18,51 and mammalian cells15,17,19,20 have been developed recently following synthetic biology principles.52 The idea of synthetic biology to create novel bio-molecular tools using engineering concepts is slowly gaining ground in plant systems.53,54 However, no light-controlled synthetic tools for plant systems have been reported to date, presumably because plants require light to sense their environment and as a source of energy. Consequently, it is impossible to keep light-responsive tools in the OFF state in-planta by ongoing incubation in the dark. We overcame this hitch by applying a phytochrome-based red light-responsive gene expression tool to plant systems. Phytochrome-based optogenetic tools are unique in that they can not only be activated by red light, but can also be rapidly returned to the OFF state by illumination with far-red light. This may, in the future, facilitate the implementation of the system in whole plants, where transgene expression in light-grown plants can be repressed by supplementary far-red light illumination. To activate gene expression with spatiotemporal precision, ambient light and far-red light illumination may then be terminated followed by the local activation of gene expression by spatially-defined illumination with activating red light (Fig. 7).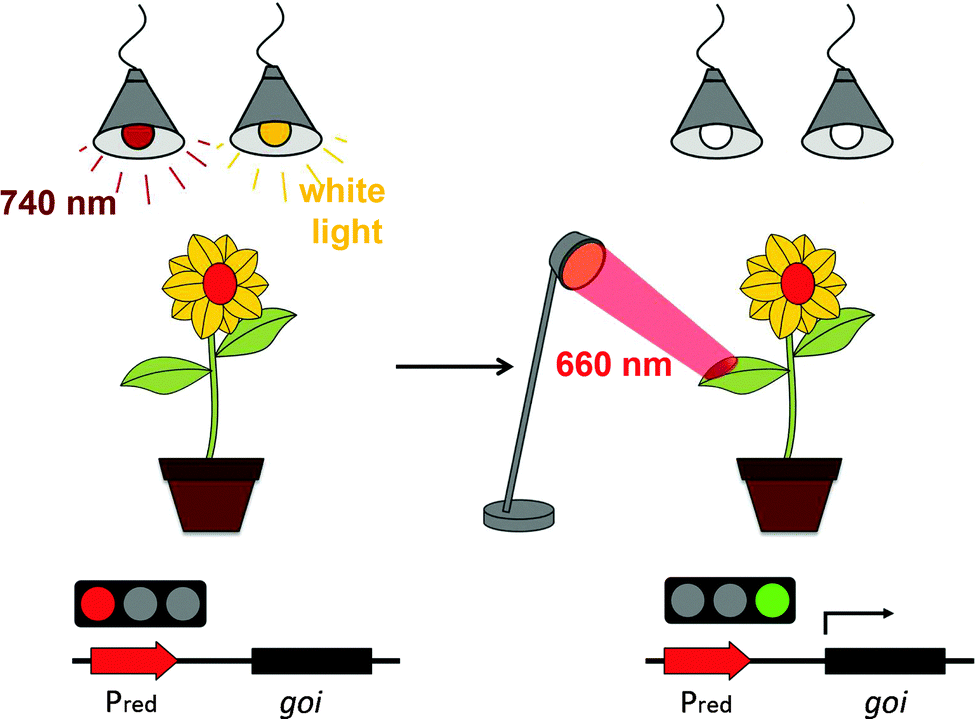 | ||
| Fig. 7 Layout of an experimental set-up for local red light-controlled gene expression in whole plants. To prevent activation of the red light-responsive gene switch in light-grown plants, supplementary 740 nm illumination is applied that constantly returns the system to the OFF state. For the spatiotemporal control of transgene expression, illumination with white light and supplementary 740 nm light is terminated and activating 660 nm light is applied locally. | ||
We applied a novel synergistic synthetic biology approach comprising mammalian cells and plant protoplasts. This strategy allowed a straightforward customization and optimization of the tool by profiting from the established experimental platform and optogenetic devices developed in mammalian cells. Furthermore, we developed a macrolide-responsive chemically-regulated expression system in the process of optimizing the red light-responsive gene expression system for plants. This system responds to the antibiotic clarithromycin and compares very favourably to existing chemically-controlled systems regarding expression strength and induction levels. Exposing the optimized red light-regulated gene expression system to red light resulted in an excellent induction of high reporter levels in N. tabacum-derived protoplasts and could be instantaneously switched off upon far-red light illumination. We took advantage of this feature and succeeded in keeping the system in the OFF state in the presence of white light by supplementation with inactivating far-red light.
We demonstrated the system's potential for the investigation of plant signalling by using red light to tune the auxin response. This approach may in future be adapted to whole plants to study auxin signalling with an unprecedented spatiotemporal resolution and can easily be adapted to the study of other signalling pathways and cellular processes. However, light does not merely act as an energy source for plants, but also constitutes an important environmental cue that controls various signalling pathways. Therefore, pleiotropic effects of illumination should be taken into consideration when the red light-responsive gene expression system is utilized to study signalling processes in plants. Beyond its application in basic research, we also highlighted the possibility to employ red light for the chemical inducer-free production of biopharmaceuticals in the biotechnologically relevant P. patens system.
It can be anticipated that this first optogenetic gene expression system for plants will, for some applications, initiate the replacement of complex equipment for the delivery of substances at specific points in time (e.g. microfluidic set-ups) by simple illumination.
In conclusion, the first red light-controlled gene expression system for plants can be expected to open new ways to study plant signalling processes with the spatiotemporal resolution of light and will, in addition, constitute a new tool for the inducible production of biopharmaceuticals.
Experimental
Expression vectors
The expression vectors and the detailed cloning strategies are described in Table S1 (ESI†).Mammalian cell culture and transfection
Chinese hamster ovary cells (CHO-K1, ATCC CCL 61) were cultivated in HTS medium (Cell Culture Technologies) supplemented with 10% fetal bovine serum (FBS) (PAN, cat. no. P30-3602, batch no. P101003TC) and 2 mM L-glutamine (Sigma). The medium was supplemented with 100 U ml−1 of penicillin and 0.1 mg ml−1 of streptomycin (PAN). Cells were transfected using a polyethyleneimine (PEI)-based method as described before.15 The bicistronic expression cassette for the split transcription factor (pKM022/pKM300/pKM301) was used in 2-fold excess (w:w) over the respective reporter plasmid (pKM006/pKM082/pMF199). After 24 h, the medium was replaced with fresh medium supplemented with 15 μM PCB (LivChem) from a 30 mM stock solution in DMSO. All experimental procedures after the addition of PCB were carried out under green LED light (522 nm). After 1 h cultivation in the dark, the cells were illuminated as indicated.Protoplast preparation and transformation
N. tabacum cultivation, protoplast isolation and polyethylene glycol-mediated transformation were performed as described before55 with minor variations: Enzymatic digestion of cut plant material was carried out with 0.5% cellulose Onozuka R10 and macerozyme R10 (Serva) in modified PIN solution (10 mM MES, 3.2 g l−1 Gamborg B5 basal salt powder with vitamins (EMELCA Bioscience), 0.38 M sucrose, adjusted to pH 5.8). After 16 h of incubation in the dark at 22 °C the incubation mixture was gently agitated and passed through a disposable 100 μm sieve. The protoplast solution was transferred into round-bottom falcon tubes and overlaid with 2 ml of 3 M solution (15 mM MgCl2, 5 mM MES, 0.465 M mannitol, adjusted to pH 5.8). After 30 min the protoplasts accumulating at the interphase were collected and transferred into a new tube with 10 ml of W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl, 5 mM glucose, adjusted to pH 5.8). After sedimentation at 80 g for 5 min the cells were re-suspended in 10 ml W5 and the cell density was determined. The cells were sedimented again and adjusted with 3 M solution to a density of 500000 ml−1. For the transformation, 5 μg of DNA in H2O was added to 100 μl protoplast solution in a round bottom falcon tube and incubated at room temperature for 5 min. Next, 100 μl PEG solution were added to the protoplasts in a dropwise manner and the falcon tube was gently tilted. After 8 min 1 ml, 2 ml, 3 ml and 4 ml of W5 were consecutively added to the tube. Next, the contents were mixed by gently tilting the tube and the cells were sedimented at 80 g for 5 min, re-suspended in 200 μl modified PCN (3.2 g l−1 Gamborg B5 basal salt powder with vitamins, 2 mg l−1 Ca-pantothenate, 0.2 mg l−1 biotin, 500 mg l−1 MgSO4·7H2O, 300 mg l−1 CaCl2·2H2O, 976 mg l−1 MES, 50 mg l−1 glutamine, 20 g l−1 sucrose, 80 g l−1 glucose, adjusted to pH 5.8). Immediately after transformation, the protoplasts were either illuminated with 660 nm light or incubated in the dark prior to reporter quantification.
P. patens was cultivated in liquid Knop medium (pH 4.5). The plants were cut and subcultured weekly as described before.56 Protoplast isolation and transformation was performed as detailed elsewhere.57,58 In brief, 300000 protoplasts were transformed and re-suspended in 1.2 ml regeneration medium. Finally, several transformation preparations were pooled and 125000 protoplasts transferred to each well of a 24-well plate in 500 μl regeneration medium. Immediately after transformation, the protoplasts were either illuminated with 660 nm light or incubated in the dark prior to reporter quantification.
Illumination and chemical inducers
Unless indicated, cells were illuminated with 660 nm (8 μmol m−2 s−1), 740 nm (20 μmol m−2 s−1) or white light (1 μmol m−2 s−1) from LED arrays.20 Light intensity was adjusted using neutral density filters (Schott) that were placed on top of the culture dishes. Regulating antibiotics were added to the culture medium where indicated: tetracycline (Sigma), 3 μg ml−1 from a 3 mg ml−1 stock in ethanol; clarithromycin (Sigma), 100 μg ml−1 from a 2 mg ml−1 stock in ethanol; pristinamycin (pyostacin pills, Aventis), 200 μg ml−1 from a 50 mg ml−1 stock in DMSO. Auxin (indole-3-acetic acid, Sigma) was added to the culture medium from a 200 mM stock in ethanol.Reporter gene assays
SEAP was quantified in the cell culture medium by a colorimetric assay as described before.59 Firefly luciferase and renilla luciferase luminescence was quantified in whole protoplasts as detailed elsewhere.35 VEGF was quantified in the cell culture medium using a human VEGF ELISA kit (PeproTech) according to the manufacturer's instructions.Acknowledgements
This work was supported by the Initiating and Networking Fund (IVF) of the Helmholtz Association within the Helmholtz Initiative on Synthetic Biology [SO-078]; the Baden-Württemberg Stiftung, under the programme ‘Internationale Spitzenforschung II’ [P-LS-SPII/2]; the European Research Council under the European Community's Seventh Framework Programme [FP7/2007-2013]/ERC [259043]-CompBioMat; the Excellence Initiative of the German Federal and State Governments [EXC 294-BIOSS, GSC 4-Spemann Graduate School]; the Alexander von Humboldt Foundation (research grant no. 1141629) and the Interreg IV Upper Rhine program project number A20.We thank Sophia L. Samodelov, Susanne Knall and Elke Wehinger for experimental assistance.
Notes and references
- M. Padidam, Curr. Opin. Plant Biol., 2003, 6, 169–177 CrossRef CAS.
- A. Junker and B. H. Junker, Methods Mol. Biol., 2012, 813, 343–358 CAS.
- G. Corrado and M. Karali, Biotechnol. Adv., 2009, 27, 733–743 CrossRef PubMed.
- P. Weinmann, M. Gossen, W. Hillen, H. Bujard and C. Gatz, Plant J., 1994, 5, 559–569 CAS.
- A. D. Frey, M. Rimann, J. E. Bailey, P. T. Kallio, C. J. Thompson and M. Fussenegger, Biotechnol. Bioeng., 2001, 74, 154–163 CrossRef CAS PubMed.
- T. Aoyama and N. H. Chua, Plant J., 1997, 11, 605–612 CAS.
- J. R. Zuo, Q. W. Niu and N. H. Chua, Plant J., 2000, 24, 265–273 CrossRef CAS.
- S. P. Zhang, L. L. Chen and S. A. Goff, Novartis Found. Symp., 2001, 236, 85–96 CAS.
- M. Padidam, M. Gore, D. L. Lu and O. Smirnova, Transgenic Res., 2003, 12, 101–109 CrossRef CAS.
- E. Unger, A. M. Cigan, M. Trimnell, R. J. Xu, T. Kendall, B. Roth and M. Albertsen, Transgenic Res., 2002, 11, 455–465 CrossRef CAS.
- B. Felenbok, J. Biotechnol., 1991, 17, 11–17 CrossRef CAS.
- V. L. Mett, L. P. Lochhead and P. H. Reynolds, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4567–4571 CrossRef CAS.
- H. G. Kang, Y. W. Fang and K. B. Singh, Plant J., 1999, 20, 127–133 CrossRef CAS.
- S. Amirsadeghi, A. E. McDonald and G. C. Vanlerberghe, Planta, 2007, 226, 453–463 CrossRef CAS PubMed.
- K. Muller, R. Engesser, S. Schulz, T. Steinberg, P. Tomakidi, C. C. Weber, R. Ulm, J. Timmer, M. D. Zurbriggen and W. Weber, Nucleic Acids Res., 2013, 41, e124 CrossRef PubMed.
- R. P. Crefcoeur, R. Yin, R. Ulm and T. D. Halazonetis, Nat. Commun., 2013, 4, 1779 CrossRef PubMed.
- X. Wang, X. Chen and Y. Yang, Nat. Methods, 2012, 9, 266–269 CrossRef CAS PubMed.
- M. J. Kennedy, R. M. Hughes, L. A. Peteya, J. W. Schwartz, M. D. Ehlers and C. L. Tucker, Nat. Methods, 2010, 7, 973–975 CrossRef CAS PubMed.
- S. Konermann, M. D. Brigham, A. E. Trevino, P. D. Hsu, M. Heidenreich, L. Cong, R. J. Platt, D. A. Scott, G. M. Church and F. Zhang, Nature, 2013, 500, 472–476 CAS.
- K. Muller, R. Engesser, S. Metzger, S. Schulz, M. M. Kampf, M. Busacker, T. Steinberg, P. Tomakidi, M. Ehrbar, F. Nagy, J. Timmer, M. D. Zubriggen and W. Weber, Nucleic Acids Res., 2013, 41, e77 CrossRef PubMed.
- I. Schepens, P. Duek and C. Fankhauser, Curr. Opin. Plant Biol., 2004, 7, 564–569 CrossRef CAS PubMed.
- N. C. Rockwell, Y. S. Su and J. C. Lagarias, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS PubMed.
- K. Muller and W. Weber, Mol. Biosyst., 2013, 9, 596–608 RSC.
- S. D. Yoo, Y. H. Cho and J. Sheen, Nat. Protocols, 2007, 2, 1565–1572 CAS.
- W. Weber, C. Fux, M. Daoud-el Baba, B. Keller, C. C. Weber, B. P. Kramer, C. Heinzen, D. Aubel, J. E. Bailey and M. Fussenegger, Nat. Biotechnol., 2002, 20, 901–907 CrossRef CAS PubMed.
- J. Love, G. C. Allen, C. Gatz and W. F. Thompson, J. Exp. Bot., 2002, 53, 1871–1877 CrossRef CAS PubMed.
- K. Muller, R. Engesser, J. Timmer, F. Nagy, M. D. Zurbriggen and W. Weber, Chem. Commun., 2013, 49, 8970–8972 RSC.
- J. T. Odell, F. Nagy and N. H. Chua, Nature, 1985, 313, 810–812 CrossRef CAS.
- R. Tanaka and A. Tanaka, Annu. Rev. Plant Biol., 2007, 58, 321–346 CrossRef CAS PubMed.
- R. J. Porra and L. H. Grimme, Int. J. Biochem., 1978, 9, 883–886 CrossRef CAS.
- M. Medzihradszky, J. Bindics, E. Adam, A. Viczian, E. Klement, S. Lorrain, P. Gyula, Z. Merai, C. Fankhauser, K. F. Medzihradszky, T. Kunkel, E. Schafer and F. Nagy, Plant Cell, 2013, 25, 535–544 CrossRef CAS PubMed.
- M. Kieffer, J. Neve and S. Kepinski, Curr. Opin. Plant Biol., 2010, 13, 12–20 CrossRef CAS PubMed.
- S. Depuydt and C. S. Hardtke, Curr. Biol., 2011, 21, R365–373 CrossRef CAS PubMed.
- L. I. Calderon Villalobos, S. Lee, C. De Oliveira, A. Ivetac, W. Brandt, L. Armitage, L. B. Sheard, X. Tan, G. Parry, H. Mao, N. Zheng, R. Napier, S. Kepinski and M. Estelle, Nat. Chem. Biol., 2012, 8, 477–485 CrossRef CAS PubMed.
- S. Wend, C. Dal Bosco, M. M. Kampf, F. Ren, K. Palme, W. Weber, A. Dovzhenko and M. D. Zurbriggen, Sci. Rep., 2013, 3, 2052 Search PubMed.
- G. Walsh, Nat. Biotechnol., 2010, 28, 917–924 CrossRef CAS PubMed.
- R. Fischer, E. Stoger, S. Schillberg, P. Christou and R. M. Twyman, Curr. Opin. Plant Biol., 2004, 7, 152–158 CrossRef CAS PubMed.
- E. L. Decker and R. Reski, Plant Cell Rep., 2012, 31, 453–460 CrossRef CAS PubMed.
- J. K. Ma, P. M. Drake and P. Christou, Nat. Rev. Genet., 2003, 4, 794–805 CrossRef CAS PubMed.
- M. Gitzinger, J. Parsons, R. Reski and M. Fussenegger, Plant Biotechnol. J., 2009, 7, 73–86 CrossRef CAS PubMed.
- J. Parsons, F. Altmann, M. Graf, J. Stadlmann, R. Reski and E. L. Decker, Sci. Rep., 2013, 3, 3019 Search PubMed.
- J. Parsons, F. Altmann, C. K. Arrenberg, A. Koprivova, A. K. Beike, C. Stemmer, G. Gorr, R. Reski and E. L. Decker, Plant Biotechnol. J., 2012, 10, 851–861 CrossRef CAS PubMed.
- A. Baur, R. Reski and G. Gorr, Plant Biotechnol. J., 2005, 3, 331–340 CrossRef CAS PubMed.
- A. Weise, F. Altmann, M. Rodriguez-Franco, E. R. Sjoberg, W. Baumer, H. Launhardt, M. Kietzmann and G. Gorr, Plant Biotechnol. J., 2007, 5, 389–401 CrossRef CAS PubMed.
- M. Schuster, W. Jost, G. C. Mudde, S. Wiederkum, C. Schwager, E. Janzek, F. Altmann, J. Stadlmann, C. Stemmer and G. Gorr, Biotechnol. J., 2007, 2, 700–708 CrossRef CAS PubMed.
- A. Buttner-Mainik, J. Parsons, H. Jerome, A. Hartmann, S. Lamer, A. Schaaf, A. Schlosser, P. F. Zipfel, R. Reski and E. L. Decker, Plant Biotechnol. J., 2011, 9, 373–383 CrossRef PubMed.
- N. Ferrara and T. Davis-Smyth, Endocr. Rev., 1997, 18, 4–25 CAS.
- A. Levskaya, A. A. Chevalier, J. J. Tabor, Z. B. Simpson, L. A. Lavery, M. Levy, E. A. Davidson, A. Scouras, A. D. Ellington, E. M. Marcotte and C. A. Voigt, Nature, 2005, 438, 441–442 CrossRef CAS PubMed.
- R. Ohlendorf, R. R. Vidavski, A. Eldar, K. Moffat and A. Moglich, J. Mol. Biol., 2012, 416, 534–542 CrossRef CAS PubMed.
- J. J. Tabor, A. Levskaya and C. A. Voigt, J. Mol. Biol., 2011, 405, 315–324 CrossRef CAS PubMed.
- S. Shimizu-Sato, E. Huq, J. M. Tepperman and P. H. Quail, Nat. Biotechnol., 2002, 20, 1041–1044 CrossRef CAS PubMed.
- A. S. Khalil and J. J. Collins, Nat. Rev. Genet., 2010, 11, 367–379 CrossRef CAS PubMed.
- M. D. Zurbriggen, A. Moor and W. Weber, J. Biotechnol., 2012, 160, 80–90 CrossRef CAS PubMed.
- J. V. Cabello, A. F. Lodeyro and M. D. Zurbriggen, Curr. Opin. Biotechnol., 2014, 26, 62–70 CrossRef PubMed.
- H. U. Koop, K. Steinmuller, H. Wagner, C. Rossler, C. Eibl and L. Sacher, Planta, 1996, 199, 193–201 CrossRef CAS.
- G. Schween, J. Schulte, R. Reski and A. Hohe, Bryologist, 2005, 108, 27–35 CrossRef.
- A. Hohe and R. Reski, Plant Sci., 2002, 163, 69–74 CrossRef CAS.
- R. Strepp, S. Scholz, S. Kruse, V. Speth and R. Reski, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4368–4373 CrossRef CAS.
- S. Schlatter, M. Rimann, J. Kelm and M. Fussenegger, Gene, 2002, 282, 19–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1 and S2. See DOI: 10.1039/c3mb70579j |
| This journal is © The Royal Society of Chemistry 2014 |