 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bidirectional fluorescent two-hybrid system for monitoring protein–protein interactions†
Ida Karin
Nordgren
a and
Ali
Tavassoli
*ab
aChemistry, University of Southampton, Southampton, SO17 1BJ, UK
bFaculty of Medicine, University of Southampton, Southampton SO16 6YD, UK. E-mail: a.tavassoli@soton.ac.uk
First published on 29th November 2013
Abstract
Two-hybrid systems have been the cornerstone of research into protein–protein interactions, but these systems typically rely on life/death reporters that put additional selective pressure on the host organism, and potentially lead to false positives. Here we report a bidirectional fluorescence-based bacterial two-hybrid system that enables both the association and dissociation of a given protein–protein interaction to be monitored. The functionality of this system and its compatibility with FACS screening are demonstrated in the forward and reverse direction using known interacting protein-partners and their cyclic peptide inhibitors. The reported fluorescent two-hybrid system may be used in the forward direction for the identification of interacting protein partners, or as a reverse two-hybrid system for the high-throughput identification of protein–protein interaction inhibitors.
Introduction
Protein–protein interactions regulate the majority of cellular processes and are significant points of intervention for the development of therapeutics.1 Various two-hybrid technologies have been developed,2 with bacterial and yeast two-hybrid systems (THS)3–7 extensively used to identify, characterize and monitor interacting proteins.8–15 Each system has its advantages and disadvantages; bacterial THS eliminate the requirements for nuclear import and reduce false positives arising from complex formation and activation of transcription in the absence of a binding partner, whereas yeast THS allow the use of protein interactions that require post-translational modifications. The majority of two-hybrid systems rely on a life/death phenotype, which puts selective pressure on the host organism, potentially leading to false positives. This stress may be alleviated by a fluorescence reporter, which does not exert survival pressure on the host organism and therefore should reduce the number of isolated false positives. Bimolecular fluorescence complementation (BiFC) assays16,17 provide a viable alternative for monitoring protein–protein interactions using a fluorescent reporter,18–20 but these systems also bring potential complication in their use, as their expression in E. coli has been shown to be toxic due to the formation of insoluble products.21,22 Additionally, BiFC is not optimally suited to identification of protein–protein interaction inhibitors, as once assembled, the fluorescent reporter protein halves do not readily dissipate. Reverse two-hybrid systems (RTHS)23–25 are used to monitor for disruption of protein–protein interactions via life/death selections, and have been extensively used for the discovery of molecules that inhibit protein–protein interactions.12,15,26,27We have previously utilized a bacterial RTHS based on λ phage repressor proteins and two essential reporter genes; HIS3 (imidazole glycerol phosphate dehydratase) and KanR (aminoglycoside 3′-phosphotransferase for kanamycin resistance) inhibit growth on selective media lacking histidine and containing kanamycin.12,15 We have used this bacterial RTHS to identify and characterize interacting protein pairs,8,11,13 and in combination with a genetically-encoded cyclic peptide library of around a hundred million members28 for the rapid identification of inhibitors of a targeted protein–protein interaction.15,27,29,30 Aiming to eliminate the selective pressure exerted on the bacterial host by the reporter genes (HIS3 and KanR), and to enable automation of the screening process using fluorescence-activated cell sorting (FACS), we set out to develop a fluorescence-based two-hybrid system (FTHS) suitable for the identification of protein–protein interaction inhibitors. Here we report a bacterial FTHS that operates in both the forward and reverse direction, and may be used for the detection of protein–protein interactions, or for the identification of protein–protein interaction inhibitors.
Materials and methods
Construction of the FTHS reporter strain
The gene encoding tdTomato was amplified from pRSET-tdTomato31 with Deep Vent (New England Biolabs) using the primers listed in Table S1 (ESI†). The PCR product was ligated into the integration plasmid pAH15332via BamHI and EcoRI restriction endonuclease sites. The resulting plasmid was integrated onto the chromosome of the BW27786 strain of E. coli using the ϕ80 phage integrase encoded from the CRIM helper plasmid pAH123, as previously described.32 Plasmids encoding the targeted proteins (HIF-1α and HIF-1β, or P6 and UEV) as fusions with the bacteriophage 434 and chimeric P22 DNA binding domains were constructed in pTHCP14 as previously described.12,27 The region encoding the targeted fusion proteins was ligated into the integration plasmid pAH68 and incorporated onto the HK022 binding site on the chromosome of the above BW27786-derived FTHS reporter strain using a plasmid encoding the HK022 integrase,13,32 to give the HIF-1 and p6/UEV FTHS. A negative control FTHS encoding the repressor domains only (434 and P22) was also constructed.Characterizing the FTHS
The HIF-1 or p6/UEV FTHS was cultured overnight at 37 °C with gentamycin (12.5 μg mL−1) and ampicillin (50 μg mL−1), and subcultured into 1 mL of M9 media containing the same levels of antibiotics as the overnight culture, and varying concentrations of IPTG (0–100 μM). These mixtures were incubated with shaking for 24 hours at 30 °C, followed by 10-fold dilution with M9 media. Fluorescence of the resulting mixtures was assessed in black 96 well half-area microplates (Greiner Bio-One, USA), with excitation at 514 nm (10 nm bandwidth) and emission measured between 550 and 650 nm (5 nm bandwidth) using a Tecan Safire2 plate reader.Isolation of protein–protein interaction inhibitors by FACS
FACS readings and sorting were carried out using a Becton Dickinson FACSort mechanical cell sorter instrument with a 488 nm laser equipped with an orange (FL-2, 600 ± 30) filter, and Becton Dickinson CellQuest acquisition software. Analysis of FACS data was carried out using Windows Multiple Document Interface software (WinMDI). Cultures were prepared for measurements and sorting as above in M9 media. The number of events per second was kept to below 1000 by diluting the culture as required with M9 media to minimize inaccuracies in the measurements. SICLOPPS plasmids (with a p15A origin of replication, which results in ∼10 copies of SICLOPPS plasmid per cell)28 encoding cyclic peptide inhibitors of the p6/UEV (cyclo-SGWIYWNV),27 and HIF-1 (cyclo-CLLFVY)30 were transformed into their corresponding FTHS. A negative control SICLOPPS plasmid encoding cyclo-CAAAAA was transformed into both FTHS. The resulting cells were grown overnight in LB media supplemented with ampicillin (50 μg mL−1), gentamycin (12.5 μg mL−1) and chloramphenicol (35 μg mL−1), and subcultured into 1 mL of M9 media supplemented with IPTG (25 or 50 μM), L-arabinose (6.5 μM), ampicillin (50 μg mL−1), gentamycin (12.5 μg mL−1) and chloramphenicol (35 μg mL−1). These cultures were incubated with shaking for 24 hours at 30 °C and mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio with their equivalent FTHS with a plasmid encoding cyclo-CAAAAA and sorted by FACS. The output of each gate was collected for 2 minutes into 2× LB media (without antibiotics) and recovered by incubation with shaking at 37 °C for 1 hour 30 minutes. Ampicillin (50 μg mL−1), gentamycin (12.5 μg mL−1) and chloramphenicol (35 μg mL−1) were added to the recovery mixtures and these were incubated with shaking overnight at 37 °C. The SICLOPPS plasmids from these cultures were isolated (plasmid mini prep kit, QIAGEN) and analyzed by restriction digestion with PstI and ScaI restriction endonucleases (New England Biolabs).
:1 (v/v) ratio with their equivalent FTHS with a plasmid encoding cyclo-CAAAAA and sorted by FACS. The output of each gate was collected for 2 minutes into 2× LB media (without antibiotics) and recovered by incubation with shaking at 37 °C for 1 hour 30 minutes. Ampicillin (50 μg mL−1), gentamycin (12.5 μg mL−1) and chloramphenicol (35 μg mL−1) were added to the recovery mixtures and these were incubated with shaking overnight at 37 °C. The SICLOPPS plasmids from these cultures were isolated (plasmid mini prep kit, QIAGEN) and analyzed by restriction digestion with PstI and ScaI restriction endonucleases (New England Biolabs).
Results
Design and construction of the FTHS
Our FTHS monitors protein–protein interactions by utilizing repressor domains from the bacteriophage regulatory system,33 with the targeted proteins expressed as hybrid fusions of a 434, and chimeric 434.P22 repressor complex.10 The reporter construct contains the chimeric 434.P22 operators followed by the red fluorescent protein tdTomato31 (Fig. S1, ESI†), chosen from a series of fluorescent proteins for its fast maturation time and high visibility over E. coli autofluorescence (Fig. S2, ESI†).34The tdTomato reporter construct was integrated onto the Φ80 phage-attachment site on the chromosome of E. coli, using the Φ80 phage integrase.32 In the absence of a repressor complex, the fluorescence emission of the constitutively expressed tdTomato at 581 nm was readily detected in the FTHS strain using a fluorescence micro-plate reader (Fig. 1 and Fig. S3, ESI†). Expression of the targeted interacting proteins fused to the 434 and P22 repressor domains (induced by IPTG in the FTHS strain) would be expected to lead to the formation of a functional 434.P22 repressor that binds the chimeric 434.P22 operator upstream of tdTomato, and lead to the shutdown of the fluorescence signal (Fig. 1B), in an IPTG-dependent manner. If the targeted proteins do not interact, or in the presence of a protein–protein interaction inhibitor, the 434.P22 repressor will not form, allowing expression of the red fluorescence reporter gene (Fig. 1C).
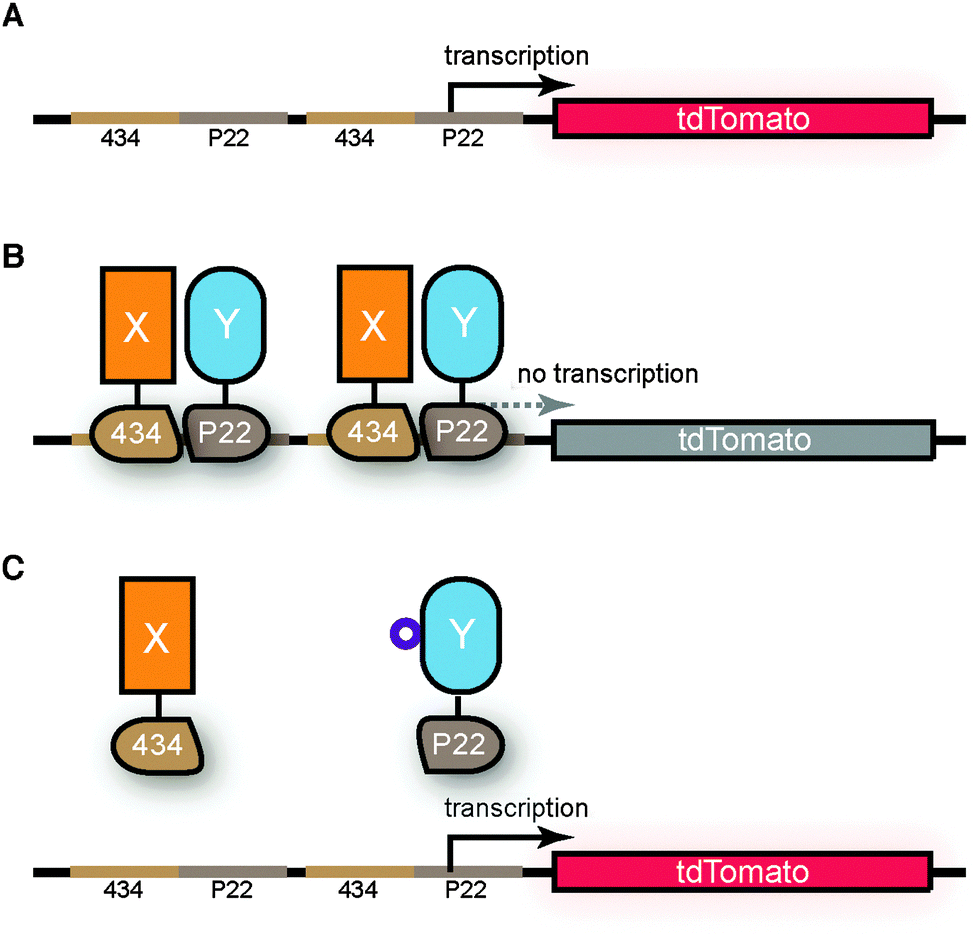 | ||
| Fig. 1 Monitoring protein–protein interactions with the FTHS (A) the tdTomato reporter construct integrated onto the chromosome of the E. coli host. (B) IPTG induces the expression of the targeted proteins (X and Y) and their interaction reconstitutes the functional repressor that prevents the transcription of the fluorescent reporter gene downstream. (C) In the presence of an inhibitor (purple circle, SICLOPPS cyclic peptide induced by arabinose), or if the targeted proteins do not interact, the 434.P22 repressor is not functional, allowing transcription of the fluorescent reporter gene tdTomato. | ||
Assessing the FTHS for identification of protein–protein interactions
To assess the function of the FTHS in the forward direction, we monitored the interaction of the α and β subunits of hypoxia inducible factor-1 (HIF-1), the primary sensor for cellular hypoxia,35 and p6 subunit of HIV Gag protein and the UEV domain of the human protein TSG101,27 an essential interaction for the budding of HIV from an infected host cell. These proteins were chosen as we have previously demonstrated their interaction within a bacterial THS,27,30 and because they represent two separate classes of protein–protein interactions; the HIF-1α/HIF-1β complex is formed by a large, multi-domain interaction interface,36–38 whereas p6 utilizes a tetrapeptide (PTAP) motif to bind into a groove in UEV.39 The above protein pairs were cloned into a vector that encodes the bacteriophage 434 repressor DNA binding domain, and a chimeric P22 variant of 434 (pTHCP14).12 Protein expression from this plasmid is regulated by IPTG via a tac promoter and a lacIq repressor. Increasing IPTG levels would therefore be expected to increase fusion protein levels and lead to a reduction in fluorescence. The target proteins were initially expressed in the FTHS from plasmid (pTHCP14); however, background expression (in the absence of IPTG) from the multiple plasmid copies in the cell was found to be sufficient to inhibit the fluorescence signal. To reduce the copy number, the constructs expressing the p6/UEV, HIF-1α/HIF-1β fusion proteins, and a control plasmid expressing the P22/434 repressors alone, were cloned into the chromosomal integration vector pAH68 and incorporated onto the chromosome of the bacterial FTHS host, via the HK022 phage attachment site.32 The ability of the FTHS to identify protein–protein interactions was assessed by measuring the IPTG-depended reduction in fluorescence; in the p6/UEV and HIF-1 FTHS, increased IPTG would be expected to result in upregulated expression of the fusion proteins, which dimerize to form a functional repressor and inhibit tdTomato expression (Fig. 1B). The P22/434 repressor domains alone do not interact, thus the fluorescence of the control strain expressing the blank repressors alone was not affected by increasing IPTG levels (Fig. 2A and D). In contrast, the p6/UEV (Fig. 2B and D) and HIF-1 (Fig. 2C and D) FTHS showed IPTG-dependent repression of tdTomato fluorescence, indicating the formation of a functional repressor capable of inhibiting the expression of tdTomato in each FTHS. | ||
| Fig. 2 IPTG-dependent repression of tdTomato fluorescence in FTHS (excitation at 514 nm). (A) The negative control FTHS strain expressing blank 434 and P22 repressors. (B) The P6/UEV FTHS. (C) The HIF-1 FTHS. (D) Relative change in fluorescence at 581 nm upon increasing IPTG levels for the control, p6/UEV and HIF-1 FTHS. | ||
Identification of protein–protein interaction inhibitors
We next sought to determine if our FTHS could be used to screen for inhibitors of a given protein–protein interaction. We used our previously reported27,30 cyclic peptide inhibitors of the p6/UEV and HIF-1α/HIF-1β protein–protein interactions in the p6/UEV FTHS and HIF-1 FTHS. These compounds were identified from genetically encoded SICLOPPS (split-intein circular ligation of peptides and proteins) libraries;28,40 the compound libraries are plasmid-encoded as SICLOPPS inteins that splices in E. coli to give cyclic peptides.The optimal filter for separation of cells with repressed tdTomato expression from those that express tdTomato was identified as that filtering for orange fluorescence (FL-2) at 600 ± 30 nm (Fig. S4, ESI†). A significant reduction in host size has been observed with DsRed expression in E. coli, a result of its aggregation in the cytoplasm and subsequent toxicity.22 To assess if the expression of tdTomato, or the repressor proteins results in a similar effect in our FTHS, we assessed the forward scatter (FSC) of the control FTHS (expressing tdTomato) and the p6/UEV and HIF-1 FTHS (repressed tdTomato expression). We did not observe significant difference in FSC in these systems (Fig. S5, ESI†), indicating that tdTomato expression in the FTHS is not significantly affecting host viability.
The SICLOPPS plasmid corresponding to our HIF-1 dimerization inhibitor (cyclo-CLLFVY)30 was transformed into the HIF-1 FTHS, and the plasmid encoding our p6/UEV dimerization inhibitor (cyclo-SGWIYWNV)27 was transformed into the p6/UEV strain. A SICLOPPS plasmid encoding cyclo-CAAAAA was constructed and transformed into the above FTHS as a negative control (the sequence was confirmed to not disrupt either protein–protein interaction using the conventional HIF-1 and p6/UEV RTHS). Inhibition of the targeted protein–protein interaction would be expected to restore tdTomato fluorescence to the host strain (Fig. 1C), whilst strains expressing the control peptide (cyclo-CAAAAA) would contain a functional repressor that inhibits expression of the fluorescent reporter (Fig. 1B). A series of trial FACS separations were carried out for the p6/UEV (Fig. 3A) and HIF-1 (Fig. 3B) FTHS, attempting to retrieve the plasmid encoding the inhibitor peptides from the negative control using the tdTomato reporter signal (Fig. 3C for p6/UEV FTHS and Fig. 3D for HIF-1 FTHS). Cells showing the lowest and highest tdTomato fluorescence were collected from gates M1 and M2 respectively (Fig. 3C and D); plasmids were isolated from each population and analyzed by restriction digestion. The plasmid encoding the negative control cyclic peptide contains a PstI restriction site (within the region encoding the cyclo-CAAAAA peptide), whereas the plasmids encoding cyclo-CLLFVY and cyclo-SGWIYWNV do not; all three plasmids contain a ScaI restriction site in their backbone. Restriction digestion with PstI and ScaI of the SICLOPPS plasmid encoding cyclo-CLLFVY therefore yields a 3939 bp fragment (lane 2, Fig. 3E), while the plasmid encoding cyclo-SGWIYWNV yields a 3945 bp fragment (lane 2, Fig. 3F), whereas the negative control plasmid (encoding CAAAAA) yields two fragments at 1462 bp and 2477 bp (lane 3, Fig. 3E and F). Restriction digestion of the plasmids isolated from the population of p6/UEV FTHS expressing tdTomato (corresponding to disruption of the p6/UEV interaction) from gate M2 (Fig. 3C) showed one band, corresponding to the plasmid encoding cyclo-SGWIYWNV (lane 6, Fig. 3E), thus indicating that the p6/UEV FTHS can be used for the identification of inhibitors of this protein–protein interaction. Similar results were obtained with plasmids from the HIF-1 FTHS isolated from gate M2 (Fig. 3D), showing one band corresponding to the plasmid encoding cyclo-CLLFVY (lane 6, Fig. 3F), again indicating that the HIF-1 FTHS may be used for the identification of HIF-1 inhibitors. Whilst the plasmids encoding the protein–protein interaction inhibitors were readily isolated from the inactive control using our FTHS, the opposite selection was not possible as plasmids encoding both the active and inactive peptides were present (lane 5, Fig. 3E and F) in the repressed population of both FTHS (not expressing tdTomato) isolated from gate M1 (Fig. 3C and D). The SICLOPPS construct is arabinose-induced, thus the inability to isolate inactive compounds may be due to the fact that arabinose induction is irregular and thus a portion of the SICLOPPS plasmid population will be unevenly induced.41 The FTHS may therefore be used for the identification of inhibitors of a given protein–protein interaction, but not as a negative screen (i.e. for compounds that do not affect a given protein–protein interaction); further experiments are necessary to probe the roots of this limitation.
 | ||
| Fig. 3 Trial selection of cyclic peptide protein–protein interaction inhibitors. (A) Density plot of the mixture of the p6/UEV inhibitor and control in the p6/UEV FTHS. (B) Density plot of the mixture of the HIF-1 inhibitor and control in the HIF-1 FTHS. (C) Histogram plot of inhibited p6/UEV FTHS, negative control FTHS, and trial separation; M1 and M2 represent the gates used for the separation. (D) Histogram plot of inhibited HIF-1 FTHS, negative control FTHS, and trial separation; M1 and M2 represent the gates used for the separation. (E) Restriction analysis of the separation of a mixture of plasmids encoding a p6/UEV inhibitor from a control plasmid. Lane 1 is ladder, lane 2 is restriction digestion of the plasmid encoding inhibitor, lane 3 is restriction digestion of a plasmid encoding control, lane 4 is restriction digestion of a mixture of plasmids encoding inhibitor and control, lane 5 is restriction digestion of plasmids from cells isolated by FACS from gate M1 in 3C, lane 6 is restriction digestion of plasmids from cells isolated by FACS from gate M2 in 3C. (F) Restriction analysis of the separation of a mixture of plasmids encoding a HIF-1 inhibitor from a control plasmid. Lane 1 is ladder, lane 2 is restriction digestion of a plasmid encoding inhibitor, lane 3 is restriction digestion of a plasmid encoding control, lane 4 is restriction digestion of a mixture of plasmids encoding inhibitor and control, lane 5 is restriction digestion of plasmids from cells isolated by FACS from gate M1 in 3D, lane 6 is restriction digestion of plasmids from cells isolated by FACS from gate M2 in 3D. | ||
Discussion
Two-hybrid technologies continue to expand and evolve, serving as a critical tool for deciphering and understanding protein interaction networks, as well as providing a platform for the identification of protein–protein interaction inhibitors.2 The FTHS detailed here operates in both the forward and reverse direction, and enables high throughput probing of protein–protein interactions without placing the host strain under life/death selective pressure. In the forward direction, the system may be used for the high-throughput identification of protein–protein interactions, for example assessing a cDNA library for binding partners of a given protein. The system was also demonstrated to function in the reverse direction for the identification of protein–protein interaction inhibitors; we illustrated this concept using SICLOPPS inhibitors, but it should be noted that any genetically encoded library could be used in conjunction with the reported FTHS.Alternatively, small molecule libraries may be screened for inhibitors of a given protein–protein interaction using the reported FTHS in a 96-well plate format. This approach would have the advantage of assessing membrane permeability of the potential inhibitor at the same time; compounds that do not cross the host's membrane will not inhibit the targeted interaction. Additionally, compounds that are toxic to the host would result in a decrease or loss of fluorescence, and so would not be isolated. It should be noted that the performance of the FTHS would vary to some degree for each protein–protein interaction examined, depending on the affinity of the targeted interacting proteins and the ability of the chosen library to disrupt them; thus functional parameters should be established for every constructed FTHS. We are currently using the FTHS reported here for the high-throughput identification of protein–protein interaction inhibitors.
Acknowledgements
We thank Professor Roger Tsien for plasmids encoding the fluorescent proteins used above, and Professor Barry Wanner for CRIM plasmids used for the chromosomal integration of expression and reporter constructs. This work was funded by Cancer Research UK (Career Establishment Award A10263 to A.T) and the Biotechnology and Biological Sciences Research Council (Doctoral Training Award for I.K.N. to A.T.).References
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS PubMed.
- B. Stynen, H. Tournu, J. Tavernier and P. Van Dijck, Microbiol. Mol. Biol. Rev., 2012, 76, 331–382 CrossRef CAS PubMed.
- C. T. Chien, P. L. Bartel, R. Sternglanz and S. Fields, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9578–9582 CrossRef CAS.
- M. Dmitrova, G. Younes-Cauet, P. Oertel-Buchheit, D. Porte, M. Schnarr and M. Granger-Schnarr, Mol. Gen. Genet., 1998, 257, 205–212 CrossRef CAS.
- S. L. Dove and A. Hochschild, Methods Mol. Biol., 2004, 261, 231–246 CAS.
- S. Fields and O. Song, Nature, 1989, 340, 245–246 CrossRef CAS PubMed.
- G. Karimova, J. Pidoux, A. Ullmann and D. Ladant, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5752–5756 CrossRef CAS.
- A. L. Andrews, I. K. Nordgren, G. Campbell-Harding, J. W. Holloway, S. T. Holgate, D. E. Davies and A. Tavassoli, Mol. BioSyst., 2013, 9, 3009–3014 RSC.
- C. T. Chien, P. L. Bartel, R. Sternglanz and S. Fields, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9578–9582 CrossRef CAS.
- G. Di Lallo, L. Castagnoli, P. Ghelardini and L. Paolozzi, Microbiology, 2001, 147, 1651–1656 CAS.
- A. L. Andrews, I. K. Nordgren, I. Kirby, J. W. Holloway, S. T. Holgate, D. E. Davies and A. Tavassoli, Biochem. Soc. Trans., 2009, 37, 873–876 CrossRef CAS PubMed.
- A. R. Horswill, S. N. Savinov and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15591–15596 CrossRef CAS PubMed.
- E. Miranda, F. Forafonov and A. Tavassoli, Mol. BioSyst., 2011, 7, 1042–1045 RSC.
- B. Suter, S. Kittanakom and I. Stagljar, Curr. Opin. Biotechnol., 2008, 19, 316–323 CrossRef CAS PubMed.
- A. Tavassoli and S. J. Benkovic, Angew. Chem., Int. Ed., 2005, 44, 2760–2763 CrossRef CAS PubMed.
- I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2000, 122, 5658–5659 CrossRef CAS.
- T. J. Magliery, C. G. Wilson, W. Pan, D. Mishler, I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2005, 127, 146–157 CrossRef CAS PubMed.
- C. D. Hu, Y. Chinenov and T. K. Kerppola, Mol. Cell, 2002, 9, 789–798 CrossRef CAS.
- C. D. Hu and T. K. Kerppola, Nat. Biotechnol., 2003, 21, 539–545 CrossRef CAS PubMed.
- T. K. Kerppola, Chem. Soc. Rev., 2009, 38, 2876–2886 RSC.
- C.-D. Hu, Y. Chinenov and T. K. Kerppola, Mol. Cell, 2002, 9, 789–798 CrossRef CAS.
- S. Jakobs, V. Subramaniam, A. Schonle, T. M. Jovin and S. W. Hell, FEBS Lett., 2000, 479, 131–135 CrossRef CAS.
- C. A. Leanna and M. Hannink, Nucleic Acids Res., 1996, 24, 3341–3347 CrossRef CAS PubMed.
- S. H. Park and R. T. Raines, Nat. Biotechnol., 2000, 18, 847–851 CrossRef CAS PubMed.
- M. Vidal, R. K. Brachmann, A. Fattaey, E. Harlow and J. D. Boeke, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10315–10320 CrossRef CAS.
- J. Huang and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13396–13401 CrossRef CAS.
- A. Tavassoli, Q. Lu, J. Gam, H. Pan, S. J. Benkovic and S. N. Cohen, ACS Chem. Biol., 2008, 3, 757–764 CrossRef CAS PubMed.
- A. Tavassoli and S. J. Benkovic, Nat. Protocols, 2007, 2, 1126–1133 CAS.
- C. N. Birts, S. K. Nijjar, C. A. Mardle, F. Hoakwie, P. J. Duriez, J. P. Blaydes and A. Tavassoli, Chem. Sci., 2013, 4, 3046–3057 RSC.
- E. Miranda, I. K. Nordgren, A. L. Male, C. E. Lawrence, F. Hoakwie, F. Cuda, W. Court, K. R. Fox, P. A. Townsend, G. K. Packham, S. A. Eccles and A. Tavassoli, J. Am. Chem. Soc., 2013, 135, 10418–10425 CrossRef CAS PubMed.
- N. C. Shaner, R. E. Campbell, P. A. Steinbach, B. N. Giepmans, A. E. Palmer and R. Y. Tsien, Nat. Biotechnol., 2004, 22, 1567–1572 CrossRef CAS PubMed.
- A. Haldimann and B. L. Wanner, J. Bacteriol., 2001, 183, 6384–6393 CrossRef CAS PubMed.
- J. C. Hu, E. K. O'Shea, P. S. Kim and R. T. Sauer, Science, 1990, 250, 1400–1403 CAS.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- I. K. Nordgren and A. Tavassoli, Chem. Soc. Rev., 2011, 40, 4307–4317 RSC.
- K. Gradin, J. McGuire, R. H. Wenger, I. Kvietikova, M. L. Fhitelaw, R. Toftgard, L. Tora, M. Gassmann and L. Poellinger, Mol. Cell. Biol., 1996, 16, 5221–5231 CAS.
- B. H. Jiang, E. Rue, G. L. Wang, R. Roe and G. L. Semenza, J. Biol. Chem., 1996, 271, 17771–17778 CrossRef CAS PubMed.
- P. J. Erbel, P. B. Card, O. Karakuzu, R. K. Bruick and K. H. Gardner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15504–15509 CrossRef CAS PubMed.
- O. Pornillos, S. L. Alam, D. R. Davis and W. I. Sundquist, Nat. Struct. Biol., 2002, 9, 812–817 CAS.
- C. P. Scott, E. Abel-Santos, M. Wall, D. C. Wahnon and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13638–13643 CrossRef CAS.
- A. Khlebnikov, K. A. Datsenko, T. Skaug, B. L. Wanner and J. D. Keasling, Microbiology, 2001, 147, 3241–3247 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mb70438f |
| This journal is © The Royal Society of Chemistry 2014 |