DOI:
10.1039/C3MB70370C
(Paper)
Mol. BioSyst., 2014,
10, 54-64
On the catalytic mechanism of polysaccharide lyases: evidence of His and Tyr involvement in heparin lysis by heparinase I and the role of Ca2+†‡
Received
28th August 2013
, Accepted 29th October 2013
First published on 31st October 2013
Abstract
The structurally diverse polysaccharide lyase enzymes are distributed from plants to animals but share common catalytic mechanisms. One, heparinase I (F. heparinum), is employed in the production of the major anticoagulant drug, low molecular weight heparin, and is a mainstay of cell surface proteoglycan analysis. We demonstrate that heparinase I specificity and efficiency depend on the cationic form of the substrate. Ca2+–heparin, in which α-L-iduronate-2-O-sulfate residues adopt 1C4 conformation preferentially, is a substrate, while Na+–heparin is an inhibitor. His and Tyr residues are identified in the catalytic step and a model based on molecular dynamics and docking is proposed, in which deprotonated His203 initiates β-elimination by abstracting the C5 proton of the α-L-iduonate-2-O-sulfate residue in the substrate, and protonated Tyr357 provides the donor to the hexosamine leaving group.
Introduction
Polysaccharide lyases are a widely distributed but structurally diverse class of enzymes, which have been divided into 21 families but, within these, only 6 general folds and 2 principal modes of action are evident.1 They employ β-elimination to cleave polysaccharides containing an oxidized hexose sugar residue (uronic acid) at the C-4 position, to generate an unsaturated non-reducing terminal and a fresh reducing end.2 Polysaccharide lyases are of considerable biological significance, and are distributed throughout the animal and plant kingdoms, as well as in prokaryotes. They are utilized for activities as diverse as the preparation of food stabilizers such as pectins, the degradation of biomass, re-modeling of cell wall structure and the production of pharmaceuticals. Understanding the detailed mechanisms of these enzymes is of considerable importance if they are to be controlled and effectively exploited. Heparinase I (EC 4.2.2.7) is a polysaccharide lyase isolated from Flavobacterium heparinum3–6 that cleaves heparin primarily at α-D-glucosamine N- and 6-sulfated (1 → 4) α-L-iduronate 2-sulfate7–10 by a β-eliminative process. Heparinase enzymes were first described by Korn & Payza in 195610 and the products of heparin degradation by Dietrich et al.7,11 Heparinase I is widely employed in the study of both heparin and heparan sulfate structure, which are important clinical anticoagulants and key regulators of cellular communication respectively. Heparinase I is also employed in the manufacture of low molecular weight heparin, a major anticoagulant drug, as well as in medical devices, including filters designed to promote the deliberate breakdown of heparin during surgical procedures.8 The mechanism of heparin degradation has not been elucidated fully but such information will be important if full control of its activity is to be achieved. A hypothesis is that the mechanism is initiated by base-catalyzed abstraction of the proton at C5 of the uronate resulting in a direct E2 eliminative cleavage. The enzyme breaks the oxygen–aglycone bond, with the loss of water, leading to production of an unsaturated uronic acid (C4–C5) on the non-reducing end and a hemiacetal (hexosamine) on the reducing end side of the cleaved glycosidic bond.3,12 Previous results have shown that bivalent cations can activate heparinase I11,13 and Ca2+, Zn2+, Mg2+ and Mn2+ bind specifically to α-L-iduronate residues in heparin driving the ring conformation to the 1C4 form,14–16 which is characterized by an antiperiplanar disposition between the proton at C5 and the oxygen–aglycone bond of C4, favorable to an E2 eliminative process. Employing evidence from site-directed mutagenesis and thiol-reactive probes, it has been hypothesized that the thiol group of Cys135 is negatively charged and is surrounded by a positively charged environment, serving as an important nucleophile, which could initiate the β-elimination reaction in the active site.17 Moreover, a catalytic role was assigned to His203; this residue being implicated as a second acid catalyst serving to protonate the leaving hexosamine.18,19 The heparinase I active site contains two Cardin–Weintraub heparin binding consensus motifs20 and heparinase I also has two calcium-binding motifs that may bridge heparin to heparinase I through Ca2+ in a ternary complex during catalysis.21 The structure of heparinase I from Bacteroides thetaiotaomicron has been determined by X-ray diffraction analysis of crystals obtained both in a free state and complexed with heparin fragments.22 These authors assigned the role of a base to His151 and that of an acid to Tyr357, with Gln149, Arg83, Lys353 and Lys81 residues maintaining a conformation favorable to β-elimination.22 Furthermore, Ca2+ seems to organize the amino acid residues involved in substrate binding.22 Although the β-elimination reaction is well understood3,12 and several potential catalytic amino acid residues at the heparinase I active site have been proposed,18–21 there remains no established mechanism for heparinase I acting on heparin. This work aimed to investigate the molecular mechanism by which heparinase I from F. heparinum depolymerizes heparin. The effects of Ca2+ ions, temperature, heparin structure, pH, solvents and 1H/2H isotopes were studied on substrate binding and catalysis by kinetics and conformational analysis, and following simulation and docking experiments, the prototropic groups responsible for the enzyme activity have been proposed.
Results and discussion
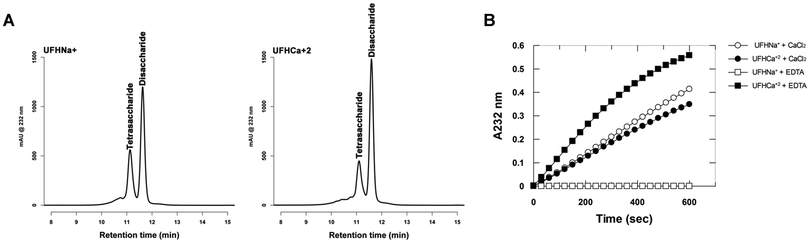
Fig. 1 shows analysis of the disaccharide products resulting from degradation of heparin by heparinase I under conditions in which the cationic form of the heparin substrate prevailing in the medium has been controlled. Whether Ca2+ or Na+ ions were present in the active site of the enzyme could not be established formally at the outset, although it is highly probable that Ca2+ ions were bound, following exposure of the enzyme to Ca2+ during its preparation. Comparison of the products of digestion of heparin–Na+ in the presence, or absence of Ca2+ ions (achieved by the addition of EDTA) in the media (Fig. 1A), reveals that Ca2+ is sequestered from the medium to achieve effective catalysis, and there is only very weak residual catalysis when Ca2+ ions are excluded from the medium. Hence, heparin–Na+ is not a substrate for heparinase I in the absence of Ca2+ ions. Once Ca2+ ions are bound to the heparin substrate, however, EDTA cannot sequester them (Fig. 1B) and the ratios of the principal disaccharide products of digestion of hep–Ca2+ are also unaffected by the addition of Ca2+. Thus, the binding of Ca2+ to the heparin substrate prior to enzymatic action determines the products of digestion, independently of the presence of Ca2+ in the digestion buffer. Comparison of the results of digestion under the two sets of conditions also reveals that the presence of Ca2+ bound to heparin prior to its addition to the medium (comparing UFH-Na+ plus CaCl2 in Fig. 1A to UFH-Ca2+ plus CaCl2 in Fig. 1B) alters the ratio of the products of digestion compared to when heparin has not bound Ca2+ and it needs to be sequestered from the medium, a higher proportion of ΔUA2S-GlcNS,6S to ΔUA2S-GlcNS being produced. This suggests that the location of the Ca2+ ions in the enzyme–substrate complex in the two cases is different and could result in distinct modes of binding in the two cases. Combined with the results concerning the rate of substrate digestion shown in Fig. 2A and B, in which higher ratios of disaccharide to tetrasaccharide products were observed in the presence of pre-bound Ca2+ ions, it can be concluded that the most efficient conversion of substrate to disaccharide products occurs when Ca2+ ions are pre-loaded onto the heparin substrate, rather than having to be sequestered from the medium.
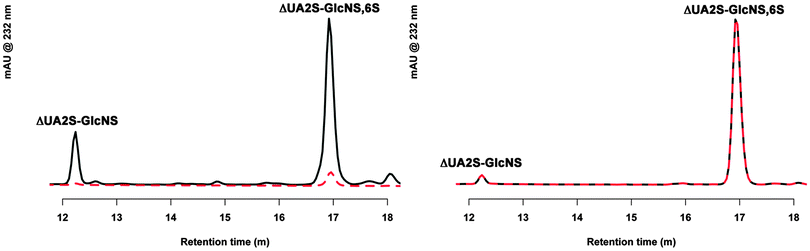 |
| | Fig. 1 Ca2+ modulates heparin digestion, influencing both the extent and the products of heparinase 1 action. (A) Disaccharide degradation products identified by HPLC when heparin is in the Na+ form in the presence of Ca2+ (black line) and absence, with EDTA (dotted red line). (B) Disaccharide degradation products identified by HPLC when heparin is in the Ca2+ form with Ca2+ added (black line) and with EDTA added (dotted red line). | |
 |
| | Fig. 2 (A) Proportion of heparin products released by heparinase I when heparin is saturated with Na+ (UFH-Na+; 1.8 disaccharides/tetrasaccharides) or with Ca2+ (UFH-Ca2+; 3.6 disaccharides/tetrasaccharides). (B) Rate of formation of unsaturated products by heparinase I action on UFH-Na+ and UFH-Ca2+. | |
Effect of Ca2+ on heparinase I activity acting on hep–Na+
The rate of enzymatic degradation of heparin in its sodium form (hep–Na+) was also investigated as a function of Ca2+ ions added to the digestion medium. The addition of Ca2+ up to 2 mM activated heparinase I two-fold (Fig. 3A) while addition of EDTA to this system abolishes stoichiometrically the enzyme activity, such that 1 mM EDTA completely inactivates heparinase I in the presence of 2 mM Ca2+ (Fig. 3B). Thus, hep–Na+ is not a substrate for heparinase I, but an inhibitor. The form of the curve of the initial velocity as a function of EDTA concentration is sigmoidal (Fig. 3B) resulting from a combination of two effects. As Ca2+ concentration decreases through increasing EDTA concentration, the concentration of heparin–Ca2+ formed also decreases, and, simultaneously, the concentration of the inhibitor, heparin–Na+, increases (Fig. 3B–D). The Hill plot (Fig. 3B, inset) revealed the presence of two Ca2+ binding sites affecting heparinase I activity. The complete removal of Ca2+ from the solution, achieved by increasing the EDTA concentration, reduced the initial velocity to zero [Fig. 3C]. Moreover, inhibition of heparinase I by saturating EDTA is reversible; the reintroduction of calcium ions into the system completely restored the enzymatic activity of heparinase I (data not shown). Furthermore, the titration of Ca2+ concentrations by fluorescent fura-2 dye showed that nominally zero calcium buffers contained about 30 μM Ca2+ (data not shown). These observations explain the small activation of heparinase I by the addition of Ca2+ and the powerful inhibition promoted by EDTA. Ca2+ is therefore an essential activator of heparinase I (Fig. 3C and D). Data shown in Fig. 3D (Lineweaver–Burk plot) were fitted by non-linear regression to eqn (S1) (ESI‡) yielding KSA = 1.4 ± 0.1 μM. Grant et al.14 suggested that when Ca2+ is added to a heparin–Na+ solution, heparin–Ca2+ complexes are formed with an initial stoichiometry of 1 Ca2+ ion per heparin tetrasaccharide unit, and further Ca2+ addition results in Ca2+ binding to form a complex with a stoichiometry of 1 Ca2+ ion per disaccharide unit. This stoichiometry, and the assumption that the optimum substrate for heparinase I is the heparin–Ca2+ complex, suggests that the proportion of products released by heparinase I (i.e. the ratio of disaccharides to tetrasaccharide products) is determined by the calcium ions interacting with heparin. The results shown in Fig. 2A, in which heparinase I was incubated with Na+–heparin in the presence of 2 mM Ca2+ and the enzyme released products in the ratio of 1.8 disaccharides/tetrasaccharides, and in Fig. 2B, in which heparin–Ca2+ was incubated with heparinase I and released products in the proportion 3.6 disaccharides/tetrasaccharides support this hypothesis. Moreover, the rate of formation of unsaturated products by heparinase I is twice as high when incubated with heparin–Ca2+ than when incubated with heparin–Na+ in the presence of 2 mM Ca2+ (Fig. 2B).
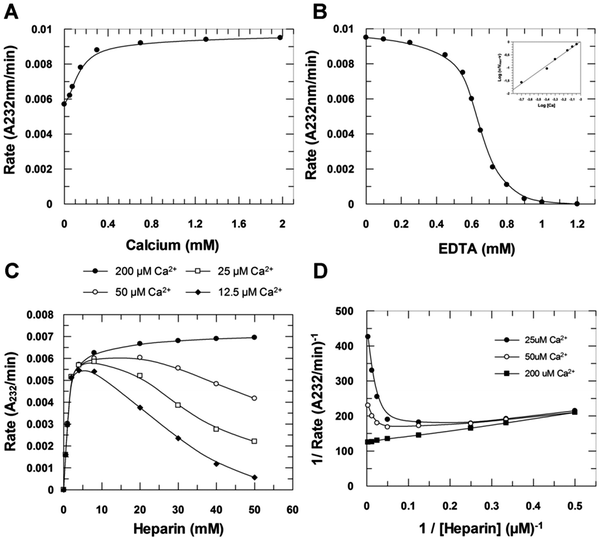 |
| | Fig. 3 Effect of Ca2+ on heparinase I activity. Heparinase I was incubated with 10 μM heparin in 50 mM tris-acetate buffer pH 7.0 at 26 °C, in different concentrations of Ca2+ (0–2 mM) (A) and in the absence of Ca2+ through addition of EDTA (B). (C) The influence of Ca2+ on heparinase I activity with increasing heparin concentrations. The same data are also shown as a Lineweaver–Burk plot (D). | |
Substrate specificity
Reduction of the carboxylate group of heparin (HepCarb-Red) abolished its enzymatic depolymerization, leading to the formation of a powerful competitive inhibitor (KI = 0.08 ± 0.01 μM) (Table 1). Thus, elimination of negative charges from the carboxyl group of the uronate residue of heparin does not disrupt its binding to the heparinase I active site, but does affect the catalytic activity of the enzyme. Table 1 also shows that specific desulfation (of the α-L-iduronic acid, 2-O-sulfate ring (hepIdoA,2-des)) or the desulfation of the acid α-L-iduronic, 2-O-sulfate ring, followed by epimerization of its iduronic to galacturonic acid (hepGalA) abolished its degradation by heparinase I, as well as leading to the formation of competitive inhibitors.
Table 1 Heparinase I dissociation constants for heparin derivatives
| Compound |
Dissociation constant (μM) |
| Heparin–Ca2+ |
K
SA = 1.4 ± 0.1 |
| HepCarb-Red |
K
I = 0.08 ± 0.01 |
| HepIdoA,2-de-S |
K
I = 2.8 ± 0.3 |
| HepGalA |
K
I = 2.3 ± 0.2 |
| HepNAc |
No inhibition |
The competitive inhibition dissociation constant observed for hepIdoA,2-de-S was KI = 2.8 ± 0.3 μM, and for hepGalA was KI = 2.3 ± 0.2 μM. These data demonstrate the importance of the 2-O-sulfate group of α-L-iduronic acid for heparinase I catalysis and this group is known to have an important influence on the conformation of the uronate ring.23N-Acetylation of the glucosamine moiety totally abolished its binding to the heparinase I active site, which may be a consequence of its distinct conformation, as proposed by Desai et al.3 and van Boeckel et al.,15N-acetylated heparin differs from intact heparin by the absence of a hydrogen bond involving the hydroxyl group at position-3 of the iduronate.24
The thermodynamic values associated with the formation of the complex enzyme–substrate (heparin–Ca2+) or the enzyme–inhibitor (heparin–Na+, Table 2) were determined by measuring the effect of temperature on their respective dissociation constant using the integrated Van't Hoff relationship. The effect of heparin binding is largely independent of the calcium concentration; the presence of 2 mM Ca2+ did not affect significantly the thermodynamic parameters of the heparin–heparinase I interaction (Table 2).
Table 2 Thermodynamic values for the binding of heparin by heparinase I
| |
ΔGbind (kcal mol−1) |
ΔHbind (kcal mol−1) |
ΔSbind (cal mol−1 K−1) |
| Heparin–Ca2+ |
8.0 ± 0.4 |
1.5 ± 0.1 |
−21.7 ± 0.9 |
| Heparin–Na+ |
7.8 ± 0.4 |
1.5 ± 0.2 |
−17.5 ± 0.7 |
The effect of temperature on heparinase I activity
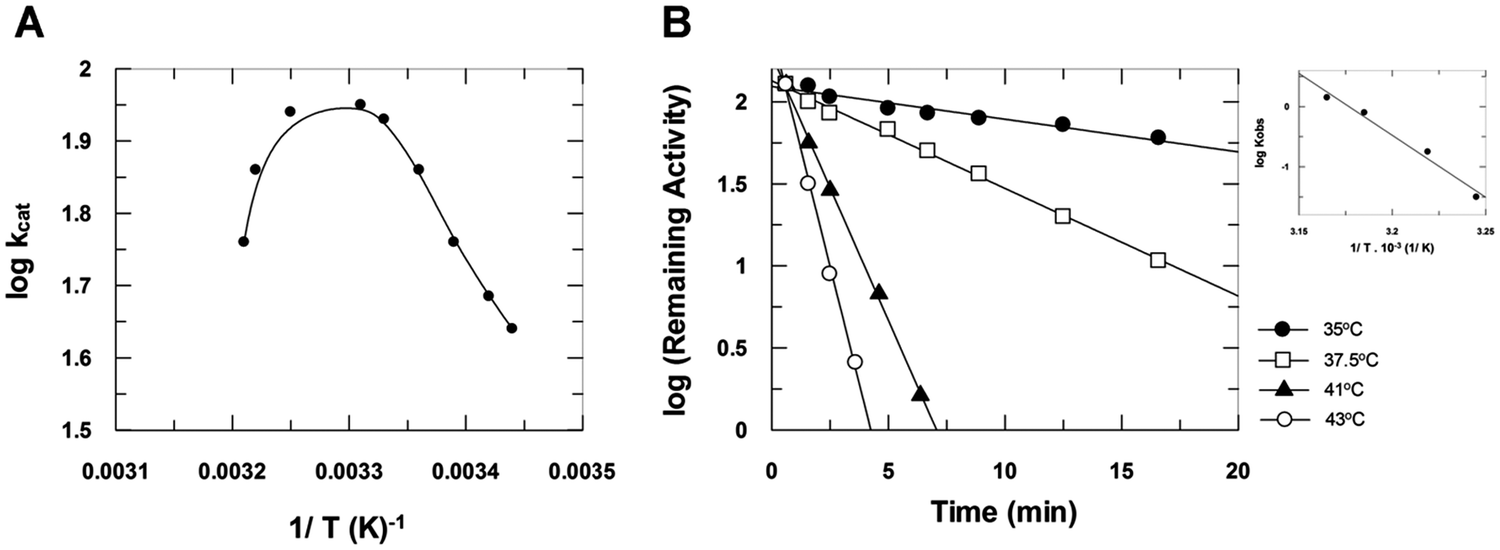
The plot of log kcatversus 1/T (Fig. 4A) shows the effect of temperature on heparinase I catalysis. The calculated activation energy of the catalytic step was Eacat = 14 ± 1 kcal mol−1. This plot also shows a sudden drop, indicating enzyme inactivation at temperatures above 35 °C, at which point heparinase I suffers thermal denaturation as a function of time (Fig. 4B). The activation energy observed during denaturation was Eadenat = 88 ± 5 kcal mol−1. Moreover, the data show that the binding of substrates to heparinase I protects the enzyme activity against thermal inactivation.
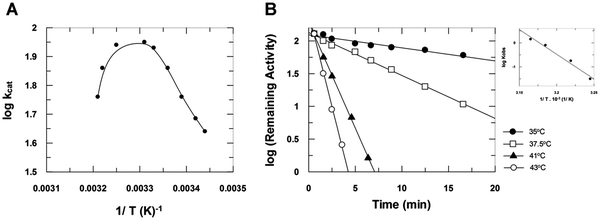 |
| | Fig. 4 The effect of temperature on heparinase I catalysis. (A) Heparinase I was incubated with 10 μM heparin and 10 mM CaCl2 in 50 mM tris-acetate buffer pH 7.0, at several temperatures. (B). Inactivation of heparinase I by temperature as a function of incubation time. Heparinase I was pre-incubated at denaturant temperatures in 50 mM of tris acetate buffer (pH 7.0) in the presence of 10 mM CaCl2. Heparinase I residual activity was measured at 25 °C and inactivation rates (Kobs) of heparinase I by temperature were obtained. (C) The Arrhenius plot of the process of heparinase I thermal denaturation. | |
The effect of temperature on the intrinsic tryptophan fluorescence of heparinase I
The conformational change in heparinase I induced by heparin binding in the absence or presence of 2 mM Ca2+ was assessed by monitoring changes in intrinsic tryptophan fluorescence as a function of time at several temperatures (Fig. 5). Fig. 5A shows that, at 30 °C, the heparinase I fluorescence emission spectra decay after 20 minutes, in both the absence and presence of 2 mM Ca2+. Fig. 5B shows the fluorescence intensity as a function of time at different temperatures. Increasing the temperature resulted in the progressive decrease of fluorescence intensity in the emission spectrum of the enzyme, suggesting that heparinase I has different conformational states that vary with temperature. The Arrhenius (Fig. 5C) and Eyring plots (Fig. 5D) of the heparinase I conformational rearrangement show a first-order rate dependence in the temperature range 15 to 45 °C. Heparin binding promoted a 2.3-fold increase in the first-order rate of conformational rearrangement of heparinase I; in the absence of heparin, the rate was 3.1 ± 0.2 ms−1, and in the presence of heparin, 7.1 ± 05 ms−1 (Table 3). As expected, this heparin binding effect is also independent of the calcium concentration (Fig. 5E). 2 mM Ca2+ promotes a small thermal stabilization of the enzyme; in the absence of Ca2+, the activation energy (Ea) of this process was 3.7 ± 0.2 kcal mol−1, and, in its presence, 5.6 ± 0.5 kcal mol−1 (Table 3). These data confirm that heparin–Na+ can bind to heparinase I with the same dissociation constant as its optimal substrate (Fig. 3 and Table 1), but cannot be processed by the enzyme. Heparin–Na+ is a competitive inhibitor of heparinase I with a dissociation constant of 1.9 ± 0.2 μM. Heparinase I, therefore, recognizes the heparin–Ca2+ complex as its substrate, with a dissociation constant of 1.4 ± 0.1 μM, forming a productive ternary complex heparinase I–Ca2+–heparin.
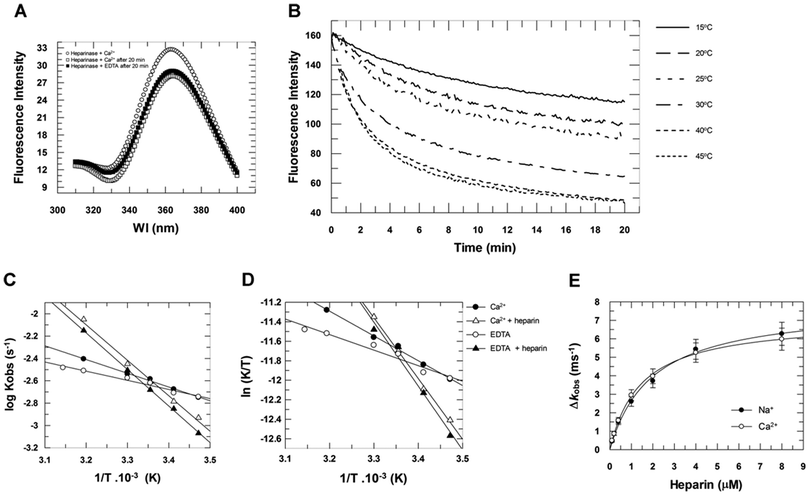 |
| | Fig. 5 The effect of temperature on the intrinsic tryptophan fluorescence of heparinase I. (A) Fluorescence spectrum of heparinase I in the presence or absence of Ca2+. (B) Fluorescence intensity as a function of the incubation time at different temperatures. Heparinase I was incubated at different temperatures in 50 mM Hepes buffer (pH 7.0) in the presence of 100 mM NaCl and 2 mM CaCl2. The Arrhenius plot (C) and the Eyring plot (D) of the process of heparinase I intrinsic tryptophan fluorescence decay in the presence or absence of Ca2+ and heparin. (E) Michaelis–Menten plot of the fluorescence intensity decay rates (kobs) of heparinase I, at 30 °C, in the presence or absence of Ca2+. | |
Table 3 Thermodynamic values for the intrinsic fluorescence decay of heparinase I
| Enzyme condition |
k
obs (ms−1) (40 °C) |
E
a (kcal mol−1) |
ΔH (kcal mol−1) |
ΔS (cal K−1 mol−1) |
| Heparinase I + Ca2+ |
3.93 ± 0.05 |
5.6 ± 0.5 |
5.1 ± 0.5 |
−54 ± 5 |
| Heparinase I + Ca2+ + heparin |
8.91 ± 0.06 |
15 ± 1 |
12.4 ± 0.4 |
−29 ± 2 |
| Heparinase I + EDTA |
3.09 ± 0.07 |
3.7 ± 0.2 |
3.2 ± 0.3 |
−60 ± 6 |
| Heparinase I + EDTA + heparin |
7.08 ± 0.04 |
14 ± 1.0 |
13.0 ± 1 |
−27 ± 5 |
The effect of pH on the heparinase I activity
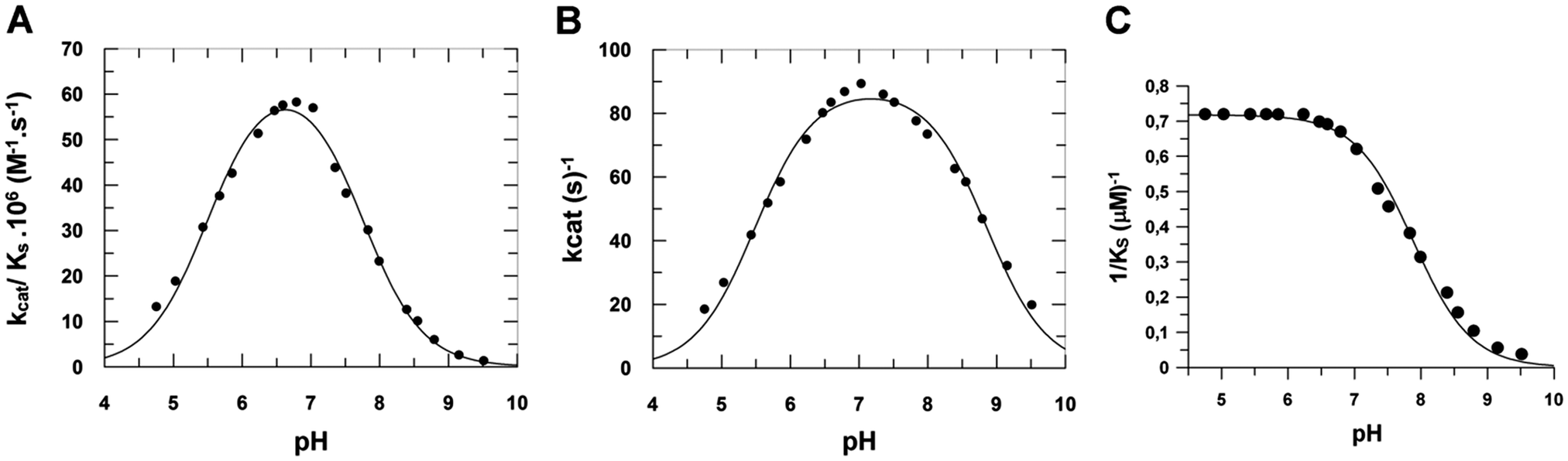
The amino acid identities of the prototropic groups present in the heparinase I active site were determined from the Vmaxapp/KSapp profile (Fig. 6A). The pKa values were obtained by fitting pH data to eqn (S2) (ESI‡) by non-linear regression analysis revealing two prototropic groups (pKE1 and pKE2) ionizing in the heparinase I active site at 25 °C. The first shows a pKE1 = 5.48 ± 0.05 and the second, a pKE2 = 7.85 ± 0.05. The pH optimum observed for heparinase I was 6.67 ± 0.05. In order to study the influence of substrate upon pKa values, hence the influence of pH on the heparinase I, the rate was measured (Fig. 6B).
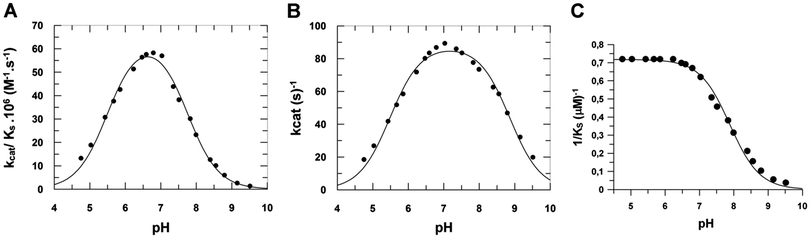 |
| | Fig. 6 The effect of pH on heparinase I activity. (A) The prototropic groups present in the heparinase I active site (pKE1 and pKE2) were determined from the Vmaxapp/KSaap profile. (B) The effect of pH on association constant (1/KSapp). (C) The effect of pH on heparinase I activity measured in a 50 mM tris-acetate and sodium acetate buffer system, pH range 4.50–9.50, in the presence of 10 mM CaCl2 at 25 °C. | |
The data show that the binding of heparin to heparinase I did not change significantly the pKE1 value (pKE1 = pKES1), however, the presence of heparin did induce an increase of pKE2 from 7.85 to 8.82 (Table 4). The plot of 1/KSappversus pH (Fig. 6C) shows that KSA values are constant at pH < 7.0. In accord with eqn (S3) (ESI‡), as [H+] increases to infinity, KSapp approaches a limit of KSA. KES1/KE1 = αKSA, giving a true KSA = 1.4 ± 0.1 μM, α = 1.0. When [H+] decreases to zero, the affinity of the enzyme for heparin is decreased. Since the ionization of heparin does not change above pH 7.0, this change of affinity can be attributed to the deprotonation of basic amino acids relevant to substrate binding. It is apparent from the pH profile that the activity of heparinase I can be described by a diprotic model, in which all heparinase I forms can bind heparin, but only the enzyme at neutral pH (En) is catalytically active; the forms (En+1) and (En−1) are not productive.
Table 4 pKa and thermodynamic values of prototropic groups from the heparinase I active site
| |
pKa (25 °C) |
ΔHion (kcal mol−1) |
ΔSion (cal mol−1 K−1) |
| pKE1 |
5.48 ± 0.05 |
+7.2 ± 0.2 |
−0.8 ± 0.3 |
| pKES1 |
5.45 ± 0.05 |
+7.3 ± 0.2 |
−0.7 ± 0.3 |
| pKE2 |
7.85 ± 0.05 |
+6.8 ± 0.4 |
−13 ± 2 |
| pKES2 |
8.82 ± 0.03 |
+6.8 ± 0.3 |
−18 ± 1 |
The temperature effect on the pKa values
The thermodynamic values from prototropic groups present at the heparinase I active site, summarized in Table 4, reveal the influence of temperature on pKE and pKES.
In line with its thermodynamic properties, the ionizable group responsible for the pKE1 value (5.48 ± 0.05) is a cationic group, probably a catalytically active histidine with ΔHion = 7.3 ± 0.2 kcal mol−1 and ΔSion = −0.7 ± 0.3 cal mol−1 K−1 (ref. 31) and heparin binding does not affect the thermodynamic properties of this group. The low pKa value observed for this histidine probably reflects its positively charged environment as described by other authors. In contrast, the thermodynamic values obtained for pKE2 did not permit immediate identification of the second prototropic group in the active site. The values of pKE2 = 7.85, ΔHion = 6.8 ± 0.4 kcal mol−1, ΔSion = −13 ± 1 cal mol−1 K−1 suggest the presence of either a tyrosine or a histidine residue. However, heparin binding to heparinase I induced a large increase of pKE2 from 7.85 to 8.82 and the entropy of ionization from −13 to −18 cal mol−1 K−1, without changing the enthalpy of ionization. The high negative value for the ionization of pKE2 indicates a neutral acid, which is compatible with the phenol group of a tyrosine residue.
Identification of amino acids involved in catalysis: solvent perturbation of acid pKa values
Previous findings have established that the presence of mixtures of aqueous organic solvents increases the pKa values of undissociated acids (sulfhydryl and phenolic groups) while it decreases, or does not alter, the pKa values of cationic acids (amine and histidine residues) and so offers a potential method for their differentiation. The size of the perturbation observed was dependent on the nature of the organic solvent and on the polarity of the neutral form of the acid.34 The solvent perturbation method was first proposed by Findlay et al.25 to distinguish between the ionizations of neutral acid residues and cationic acid residues on the basis of the difference in their behavior in mixed organic–aqueous solvent systems. The directions and magnitudes of the pKE1 and pKE2 perturbations by ethanol and acetonitrile are shown in Table 5. The presence of dioxane (7.5%) ethanol (20%) or acetonitrile (20%) did not significantly change the values of pKE1 and pKES1; ethanol altered the pKE1 from 5.48 ± 0.05 to 5.46 ± 0.05 (ΔpK = −0.02 ± 0.05), acetonitrile shifted it to 5.45 ± 0.05 (ΔpK = −0.03 ± 0.05) and dioxane (7.5%) did not alter the pKE1 values at all. No influence of heparin binding by solvent perturbation upon pKE1 values was noted. These data are in agreement with the perturbation of a cationic acid, corroborating the thermodynamic values measured for pKE1, which are appropriate for a histidine imidazole residue involved in heparinase I catalysis.
Table 5 Perturbations of pKa values in the active site of heparinase I by organic solvents
| Ionizable groups |
pKa values (25 °C) |
| Water |
Dioxane (7.5%) |
Ethanol (20%) |
Acetonitrile (20%) |
| pKE1 |
5.48 ± 0.05 |
5.48 ± 0.05 |
5.46 ± 0.05 |
5.45 ± 0.05 |
| pKES1 |
5.45 ± 0.05 |
5.44 ± 0.05 |
5.46 ± 0.05 |
5.45 ± 0.05 |
| pKE2 |
7.85 ± 0.05 |
>8.85 |
8.18 ± 0.05 |
8.20 ± 0.05 |
| pKES2 |
8.82 ± 0.03 |
>9.82 |
8.85 ± 0.03 |
9.15 ± 0.04 |
In contrast to their effects on the value of pKE1, dioxane, ethanol and acetonitrile all induced a significant increase in the observed values of pKE2; ethanol shifting the pKE2 from 7.85 ± 0.05 to 8.18 ± 0.05 (ΔpK = +0.33 ± 0.05), acetonitrile to 8.20 ± 0.05 (ΔpK = +0.35 ± 0.05) and dioxane 7.5% inducing a large increase in the pKE2 value, although it was not possible to measure its magnitude precisely due to the large shift promoted by dioxane; ΔpK > +1 and heparinase I was not stable at high pH (>9.6) values. It is important to note that the presence of substrates in the active site of heparinase I decreases the perturbation of the pKES2 value, the ΔpK value of ethanol was decreased from 0.33 to 0.03 and the ΔpK value of acetonitrile from 0.35 to 0.16, indicating that the behavior of the ionization observed in pKE2 by organic solvents is typical of a neutral acid, compatible with the ionization of a phenol group of a tyrosine residue. These data indicate the involvement of histidine (pKE1) and tyrosine (pKE2) residues, surrounded by basic amino acids in heparinase I catalysis.
Investigating proton transfer in the transition state: the isotope effect on heparinase I catalysis
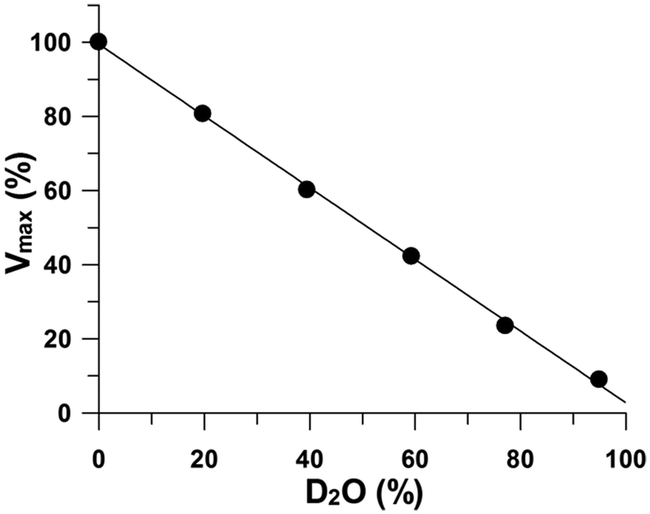
β-Elimination evolves proton donation to the GlnS,6S leaving group, after cleavage of the oxygen–aglycon bond.3,12 The magnitude of the immediate perturbation by deuterated water on reaction rates provides clues to the structure of the proton exchange steps in the transition state and can be useful to identify the rate-limiting reaction step.26,27 Solvent deuterium kinetic isotope effects were unity (Fig. 7), indicating that no kinetically significant proton transfer step is involved in the transition state. It can be concluded that the phenolic group (Table 5) of the Tyr residue assumes the role of a general acid, donating a proton to the O-leaving group of a glucosamine and reconstituting the hydroxyl group at the reducing end.
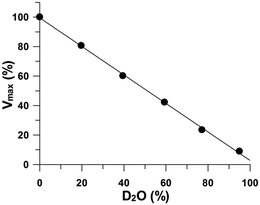 |
| | Fig. 7 The isotope effect on heparinase I catalysis conducted at 30 °C in 1085 μM heparin, 10 mM CaCl2, and 50 mM HEPES buffer at pH 7.0 prepared with increasing proportions of deuterium. | |
Calcium activation of heparinase I
Ca2+ is an essential activator of heparinase I, and the optimum substrate for heparinase I is the Ca2+–heparin complex, while heparin–Na+ is a competitive inhibitor of heparinase I. Ca2+ also controls the specificity of heparinase I for heparin, the proportion of Na+ and Ca2+ counter ions present in heparin determining the proportion of products released by heparinase I and the ratio of disaccharide and tetrasaccharide products (Fig. 2). Furthermore, it is known from NMR spectroscopy that Ca2+ specifically binds to the heparin chain affecting its structure, stabilizing the 1C4 conformation28 (ESI,‡ Fig. S3). The IdoA2S 1C4 form favors the anti β-eliminative reaction since it generates a less sterically hindered environment in which H5 is in the axial position away from the carboxylate, which will be attacked and eventually extracted from the iduronate ring by the catalytic amino acids, His and Tyr. The binding of Ca2+ to the heparin chain may help rigidify the polysaccharide chain in the bound state and/or assist in the formation of a bent conformation to allow easier access of the catalytic amino acids. Ultimately, saccharide cleavage to release two product fragments could then reasonably be expected to result in a favorable entropic change (ΔS positive).
Heparin binding induces conformational changes in heparinase I (Fig. 5). The effect of heparin on the first-order rate of heparinase I conformational rearrangement is characterized by a large enthalpy change of ΔHheparin − ΔHcontrol = 9 ± 1 kcal mol−1, which is compensated by a significant entropy gain ΔSheparin − ΔScontrol = 29 ± 4 cal K−1 mol−1. In the presence of heparin, a large value of Ea (15 ± 1 kcal mol−1) indicates substantial structural strain linked to the formation of the enzyme–substrate complex, either in the absence, or presence, of 2 mM Ca2+. During the formation of the heparinase I–heparin complex there is also a gain of entropy (ΔSheparin − ΔScontrol = 29 ± 4 cal K−1 mol−1) indicating that heparinase I can assume a more open conformation in the presence of heparin (Table 3). The activation energy of the catalytic step of heparinase I, Eacat = 14 ± 1 kcal mol−1, is very similar to the activation energy of the formation of the enzyme–substrate complex (15 ± 1 kcal mol−1, Table 3), indicating that the processes are linked.
Molecular dynamics and docking simulations suggest involvement of His203 and Tyr357 residues in the active site and the involvement of two Ca2+ ions. Molecular dynamics and docking simulations also suggest that the torsion angles about the heparin glycosidic linkage between the sugar residues in subsites +1 and −2 exhibit a non-standard geometry. At the heparinase I active site, the heparin chain makes a tight kink centered at GlcNS,6S4 in the −1 subsite. The sugar ring of the Ido2S6 residue in the −2 subsite assumes a higher energy twist-boat conformation. Carbohydrate hydrolases29 and lyases22 are known to force substrate distortion to bind with an energetically less favorable non-chair conformation. It has been shown that imposing strain on the bound substrate lowers the energy barrier of the transition state.22 Catalysis by the family of polysaccharide lyases has been proposed by Han22 as involving a stepwise mechanism in which (i) initial concerted α-proton abstraction and protonation of the carboxylic acid to generate an enol intermediate is followed by (ii) elimination of the β-leaving group from the enol. The present data show that Ca2+ is directly involved in the catalysis by neutralization of the carboxylate group of IdoA at the +1 subsite. Heparinase I in the absence of heparin binds a single calcium ion, and an additional calcium ion could bind between enzyme and substrate carboxylates occupying the +1 subsite. In this model, the neutralization of the negative charge on the carboxylate (C6 position) of the IdoA,2S6 in the subsite +1 by Ca2+ causes resonance stabilization of the carboxylate, thereby lowering the pKa value of the C5-bound hydrogen.22,30,31 The Nε2 atom of His203 from the heparinise I active site is 1.5 Å distant from the proton at C5 of the IdoA,2S in the subsite +1. The deprotonated imidazole group (Table 5) of His203 catalyzes the basic abstraction of the proton at C5 from the IdoA,2S in the subsite +1 resulting in β-elimination of the 4-O-glycosidic bond by an anti-β-eliminative process of the α(1 → 4) glycosidic linkage with the concomitant formation of a C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 double bond in the IdoA residue at the non-reducing end. Finally, the phenolic group (Table 5) of Tyr357 donates a proton to the glucosamine leaving group, whence the glycosidic linkage is broken and the double bond is formed in the product. The phenolic hydroxyl group of the Tyr357 is 0.8 Å distant from the 1-O-glycosidic bond of the glucosamine leaving group. In heparinase I, proton transfer to neutralize the O− leaving group of a glucosamine is not part of transition state formation, consistent with an enzyme-stabilized anionic 1-O-glucosamine as the leaving group.
C5 double bond in the IdoA residue at the non-reducing end. Finally, the phenolic group (Table 5) of Tyr357 donates a proton to the glucosamine leaving group, whence the glycosidic linkage is broken and the double bond is formed in the product. The phenolic hydroxyl group of the Tyr357 is 0.8 Å distant from the 1-O-glycosidic bond of the glucosamine leaving group. In heparinase I, proton transfer to neutralize the O− leaving group of a glucosamine is not part of transition state formation, consistent with an enzyme-stabilized anionic 1-O-glucosamine as the leaving group.
The activation of heparinase I by Ca2+ ions complexed to the heparin substrate is a further example of the influence of cations on glycosaminoglycans and their properties in association with proteins, which have previously been noted as resulting from structural changes in both conformation of heparin derivatives following cation binding,32 and in cell-signalling through fibroblast growth factors (FGFs) complexed with heparin and their subsequent modification of protein secondary structure.33,34 The present findings highlight an additional, important influence of cations on the degradation of the heparin substrate by a bacterial lyase. The details of the proposed mechanism for heparinase I will also be of wider interest to those studying this family of enzymes, whether from biochemical, biotechnological or industrial motivation.
Molecular dynamics simulations
In order to test the structural stability of the heparin substrate in the absence or presence of calcium ions in the active site of heparinase I, the distance between the side chain atoms of the catalytic Tyr357 and His203 residues (ESI,‡ Fig. S1) and the substrate glycosidic linkage was measured. The structural stability of the heparinase I–heparin complex (ES) was dramatically affected by the nature of the bound ion (ESI,‡ Fig. S1). The presence of Na+ ions in the complex heparinase I–heparin affected the optimum distance of the substrate to the catalytic Tyr357 and His203 residues, which was much more flexible than in the presence of calcium ions (ESI,‡ Fig. S2). Na+ in heparin led to higher Tyr357-distance values (1.8 ± 0.6 Å on average) than Ca2+ (0.8 ± 0.1 Å on average). Also, the presence of Na+ in heparin led to higher His203-distance values (3.9 ± 1.4 Å on average) than Ca2+ (1.5 ± 0.2 Å on average) in the ES complex.
Conclusion
Previous work in this area has proposed the involvement of a number of amino acid residues in the catalytic mechanism, including His203,18,19,22 with which the present findings concur but Cys13517 and Tyr35722 have been implicated. Thus, the mechanism of action of this enzyme, an exemplar of the important polysaccharide lyase class, has remained uncertain. We report that the cationic form of the heparin substrate available for enzyme activity determines both the efficiency of heparinase I digestion of heparin and the identity of the products of digestion. Furthermore, we show, through investigations into the properties of the active site, that alongside the known involvement of His203 in catalysis, a Tyr residue is implicated. Studies using molecular dynamics permit the active site to be proposed as involving heparin binding with two calcium ions, and the catalytic amino acids identified as His203 and Tyr357.
Among the implications of these findings are that heparin lysis by heparinase I is dependent on the cationic form of the substrate; a fact that is important for quality control during the production of pharmaceutical heparin and that the efficiency of removal (i.e. degradation) of heparin during surgical procedures employing heparinase I derivatised filters is also cation dependent. These findings will also be of interest to those employing heparinase I for research purposes.
Furthermore, we have proposed a mechanism involving His and Tyr residues in the catalytic step, which will have implications for the detailed understanding of the other members of the polysaccharide lyase family of enzymes.
Materials and methods
Materials
Unfractionated Heparin (UFH) from porcine intestinal mucosa (16 kDa, 190 UI mg−1 – USP) was obtained from Bioiberica (Barcelona, Spain). N-Ethylmaleimide (NEM), dithiotreitol (DTT), 1-ethyl-3-(3′-dimethylaminopropyl) carbodiimide (EDC), ethylenodiaminetetraacetic acid (EDTA) and bovine serum albumin (BSA) and 1,2-diaminoethane were purchased from Sigma-Aldrich. Heparinase I was prepared from F. heparinum (American Type Culture Collection – ATCC 13125, Rockville, ML, EUA) and purified until homogeneity as previously described. Furthermore, heparinase I was cloned and expressed as described (ESI,‡ Page S1). Heparin conversion to the Na+ and Ca2+ forms was achieved by exchange with cation exchange resin [Dowex 50W X8-400, which had been converted previously from the acid form following exhaustive washing with the relevant chloride salt (1 M), followed by deionized water].
Chemical modification of heparin
Heparin was N-desulfated by solvolysis in methanol and dimethyl sulfoxide,24 yielding the N-unsubstituted polysaccharide, HepNH3. N-Acetylation of HepNH3 was carried out using acetic anhydride35 yielding the fully N-acetylated polymer, HepNAc. Carboxy reduction of heparin (HepCarb-Red) employed a soluble carbodiimide (EDC) in the presence of sodium borohydride as previously described.36 Specific α-L-iduronic acid 2-O-desulfation of heparin (HepIdoA,2-des) and epimerization of heparin α-L-iduronic acid to α-L-galacturonic acid (HepGalA) were performed as described by Rej et al.37 Modified heparins were characterized by 1H and 13C NMR and were in agreement with published assignments.24
Chemical analysis of heparin
All chemical modifications of heparin were monitored by the following methods: hexosamine was measured after heparin acid hydrolysis; 4 M HCl, 100 °C, 6 hours. Labile and total sulfate were determined as previously described.38 Quantitation of uronic acid in heparin was performed by paper electrophoresis according to Kosakai et al.39 and using the carbazole reaction.40
Enzymatic assays
The enzymatic reactions were conducted on a U.V. spectrophotometer (U-2000 Spectrophotometer, Hitachi) equipped with a thermostatic cell. The reactions were performed in quartz cuvettes containing heparin (2–2171 μM), heparinase I and CaCl2 (0.05–10 mM) in tris-acetate, sodium acetate or HEPES buffer systems, in a final volume of 1000 μL. Heparin lyase activity was directly measured from the change of absorbance at 232 nm per unit time (EM = 5500 M−1 cm−1). The initial velocities (ΔA232 min−1) were calculated using only 5% or less of the initial heparin concentration (moles of uronic acid). Heparinase I kinetic parameters were determined using the non-linear regression software system (GraFit V.3.0, Erithacus Software). The degradation products were analyzed by paper chromatography and SAX-HPLC chromatography.41 The data from Ca2+ activation were analyzed by non-linear regression based on eqn (S1) (ESI,‡ Page S5).
Determinations of pKa, ΔHion and Ea
Precise identification of the catalytic residue is not a simple matter, since pKa values of amino acids vary strongly with their environment. However, analysis of the temperature effect on the ionization constant can be used as a tool by which investigation of the nature of the ionizable groups at the active site of heparinase I can be made. The pKa values of the prototropic groups of the heparinase I active site were determined by measuring the pH-dependence upon kcat/KSA, kcat and KSA. The data from pH-dependence of kcat/KSA and kcat were analyzed according to eqn (S2) (ESI,‡ Page S5), whereas the data concerning the influence of pH upon 1/KSA were analyzed based on eqn (S3) (ESI,‡ Page S5). The temperature effect on the pKa value of any dissociable group is the consequence of the enthalpy of ionization42 and the effect of changing temperature on pKa is given by the integrated Van't Hoff relationship (eqn (S4), ESI,‡ Page S5). The energy of activation of the heparinase I catalytic step was determined by evaluation of the temperature effect on the kcat (Eacat) value and the energy of activation of the enzyme denaturation step (Edenat) was measured by evaluation of the temperature effect on the first-order rate of enzyme denaturation (kdenat) (eqn (S5), ESI,‡ Page S6). Effects of pH and temperature on heparinase I activity were measured in a tris-acetate and sodium acetate buffer system, μ = 0.05 M, pH range 4.50–9.50 in the presence of 10 mM CaCl2. Since the pKa values of the buffers also change with the temperature, the pH values of each buffer were measured at each temperature. Heparinase I showed pH stability (in the range 4.50–9.50), for at least 15 minutes during incubation. It was assumed that the heparin ionization did not change appreciably in this pH range.30,31
Effect of organic solvents on heparinase I
The influence of organic solvents on pKa values of the heparinase I prototropic groups was determined using dioxane (7.5% v/v), ethanol (20% v/v) and acetonitrile (20% v/v) in a tris-acetate and sodium acetate buffer system in the presence of 10 mM CaCl2. The pKa values of buffers also change in the presence of organic solvents, so the pH values of each solution were measured at each solvent concentration in both cationic and neutral acid buffers. A pH meter (Corning 125) equipped with a glass combination electrode (Corning) was employed for all pH measurements. Previous studies showed that the glass electrode responds to proton activity in aqueous dioxane, ethanol and acetonitrile.43
Effect of deuterium isotopes on heparinase I catalysis
In order to ascertain whether protons were involved in the catalytic mechanism of heparinase I, the enzyme reaction was carried out in different ratios of deuterated/non-deuterated water.27 The activity of heparinase I on heparin was determined in the presence of 1085 μM heparin and 10 mM CaCl2, in 50 mM HEPES buffer at pH 7.0, with increasing ratios of deuterated/non-deuterated water. All reactions were carried out at 30 °C. Maximum velocity was calculated according to eqn (S6) (ESI,‡ Page S6).
Fluorescence spectroscopy
All fluorescence spectroscopy measurements were performed in buffer containing 50 mM HEPES, pH 7.0 and 100 mM NaCl. Steady-state fluorescence spectra were recorded using a Shimadzu RFPC 5301 spectrofluorometer and Stopped-flow fluorescence in a SX20 Stopped-Flow Spectrometer (Applied Photophysics). The excitation wavelength was 295 nm and the emission was measured from 310 to 400 nm. The excitation and emission slit widths were 3 and 5 nm, respectively, and the assays were carried out in 5 degree steps between 15 °C and 45 °C. Fluorescence spectroscopy measurements as a function of time were also performed in the same buffer and at the same temperatures. The excitation and emission wavelengths were 295 nm and 350 nm and the slit widths were 3 nm for excitation and 10 nm for emission. The thermodynamic parameters (Kobs, Ea, ΔH and ΔS) of intrinsic tryptophan fluorescence decay of heparinase I were determined.
Nuclear magnetic resonance (NMR)
1D and 2D NMR measurements were measured on a 600 MHz Bruker AVANCE III NMR spectrometer equipped with a triple resonance 5 mm probe. Chemical shifts are reported relative to TSP as a reference. All measurements were made at 25 °C with presaturation of the HOD signal at ∼4.7 ppm. NOESY experiments were recorded using standard Bruker sequences (noesyphpr) using a relaxation delay of 2 s. A spectral width of 2600 Hz was used in F1 and F2, and 1 K data points were collected in F2 at 128 t1 increments. A total of 32 transients were collected for each t1 increment.
Software and nomenclature
The disaccharide topology was generated using the PRODRG server44 and manipulation of the structures was performed using MOLDEN,45,46 VMD47 and PyMOL.48 Molecular dynamics (MD), including both calculations and analyses, was performed using the GROMACS simulation suite, version 4.5.1, and the GROMOS96 43a1 force field.49 The relative orientation of a pair of contiguous carbohydrate residues is described by two or three torsional angles at the glycosidic linkage. For the (1 → 4) linkages, the Φ and Ψ angles were defined as shown in eqn (S7) and (S8) (ESI,‡ Page S6). Octasaccharide Topology Construction and Energy Contour Plot Calculation, Docking Procedures and MD Simulations (ESI,‡ Pages S3 and S4) are further described in ESI.‡
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Carolina R. Córdula, Marcelo A. Lima, Helena B. Nader and Ivarne L. S. Tersariol contributed equally.
Abbreviations
| HepCarb-Red | Heparin carboxy-reduced |
| HepNAc | Heparin α-D-glucosamine N-acetylated |
| HepNH3 | Heparin α-D-glucosamine N-desulfated |
| HepGalA | Heparin α-L-galacturonate |
| HepIdoA | Heparin α-L-iduronate, 2-O-desulfated |
| UFH | Unfractionated heparin |
Acknowledgements
This manuscript was support by grants CNPq (Conselho Nacional de Desenvolvimento e Tecnológico), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo). EAY acknowledges CAPES for their support through a visiting professorship (172/2012). NMR data were collected both at UNICAMP and at the Advanced Magnetic Resonance Imaging and Spectroscopy (AMRIS) facility in the McKnight Brain Institute of the University of Florida and supported through FAPESP Multi-User Equipment Program and the National High Magnetic Field Laboratory, respectively.
References
- M. L. Garron and M. Cygler, Glycobiology, 2010, 20, 1547–1573 CAS.
- V. Lombard, T. Bernard, C. Rancurel, H. Brumer, P. M. Coutinho and B. Henrissat, Biochem. J., 2010, 432, 437–444 CrossRef CAS PubMed.
- U. R. Desai, H. M. Wang and R. J. Linhardt, Arch. Biochem. Biophys., 1993, 306, 461–468 CrossRef CAS PubMed.
- R. J. Linhardt, G. L. Fitzgerald, C. L. Cooney and R. Langer, Biochim. Biophys. Acta, 1982, 702, 197–203 CAS.
- H. B. Nader, M. A. Porcionatto, I. L. Tersariol, M. A. Pinhal, F. W. Oliveira, C. T. Moraes and C. P. Dietrich, J. Biol. Chem., 1990, 265, 16807–16813 CAS.
- A. S. Perlin, D. M. Mackie and C. P. Dietrich, Carbohydr. Res., 1971, 18, 185–194 CrossRef CAS.
- C. P. Dietrich, Biochem. J., 1968, 108, 647–654 CAS.
- R. Langer, R. J. Linhardt, S. Hoffberg, A. K. Larsen, C. L. Cooney, D. Tapper and M. Klein, Science, 1982, 217, 261–263 CAS.
- R. J. Linhardt, J. E. Turnbull, H. M. Wang, D. Loganathan and J. T. Gallagher, Biochemistry, 1990, 29, 2611–2617 CrossRef CAS.
- A. N. Payza and E. D. Korn, Nature, 1956, 177, 88–89 CrossRef CAS.
- C. P. Dietrich, M. E. Silva and Y. M. Michelacci, J. Biol. Chem., 1973, 248, 6408–6415 CAS.
- R. J. Linhardt, P. M. Galliher and C. L. Cooney, Appl. Biochem. Biotechnol., 1986, 12, 135–176 CrossRef CAS.
- M. E. Silva and C. P. Dietrich, J. Biol. Chem., 1975, 250, 6841–6846 CAS.
- D. Grant, W. F. Long, C. F. Moffat and F. B. Williamson, Biochem. J., 1992, 282(Pt2), 601–604 CAS.
- C. A. A. van Boeckel, S. F. van Aelst, G. N. Wagenaars, J. R. Mellema, H. Paulsen, T. Peters, A. Pollex and V. Sinnwell, Recl. Trav. Chim. Pays-Bas, 1987, 106, 19–29 CrossRef CAS.
- D. M. Whitfield and B. Sarkar, J. Inorg. Biochem., 1991, 41, 157–170 CrossRef CAS.
- R. Sasisekharan, D. Leckband, R. Godavarti, G. Venkataraman, C. L. Cooney and R. Langer, Biochemistry, 1995, 34, 14441–14448 CrossRef CAS.
- R. Godavarti, C. L. Cooney, R. Langer and R. Sasisekharan, Biochemistry, 1996, 35, 6846–6852 CrossRef CAS PubMed.
- R. Sasisekharan, G. Venkataraman, R. Godavarti, S. Ernst, C. L. Cooney and R. Langer, J. Biol. Chem., 1996, 271, 3124–3131 CrossRef CAS PubMed.
- A. D. Cardin and H. J. Weintraub, Arteriosclerosis, 1989, 9, 21–32 CAS.
- D. Liu, Z. Shriver, R. Godavarti, G. Venkataraman and R. Sasisekharan, J. Biol. Chem., 1999, 274, 4089–4095 CrossRef CAS PubMed.
- Y. H. Han, M. L. Garron, H. Y. Kim, W. S. Kim, Z. Zhang, K. S. Ryu, D. Shaya, Z. Xiao, C. Cheong, Y. S. Kim, R. J. Linhardt, Y. H. Jeon and M. Cygler, J. Biol. Chem., 2009, 284, 34019–34027 CrossRef CAS PubMed.
- D. R. Ferro, A. Provasoli, M. Ragazzi, G. Torri, B. Casu, G. Gatti, J. C. Jacquinet, P. Sinay, M. Petitou and J. Choay, J. Am. Chem. Soc., 1986, 108, 6773–6778 CrossRef CAS.
- E. A. Yates, F. Santini, M. Guerrini, A. Naggi, G. Torri and B. Casu, Carbohydr. Res., 1996, 294, 15–27 CAS.
- D. Findlay, A. P. Mathias and B. R. Rabin, Biochem. J., 1962, 85, 134–139 CAS.
- E. J. Enyedy and I. M. Kovach, J. Am. Chem. Soc., 2004, 126, 6017–6024 CrossRef CAS PubMed.
- D. B. Northrop, Annu. Rev. Biochem., 1981, 50, 103–131 CrossRef CAS PubMed.
- D. L. Rabenstein, J. M. Robert and J. Peng, Carbohydr. Res., 1995, 278, 239–256 CrossRef CAS.
- R. Kuroki, L. H. Weaver and B. W. Matthews, Science, 1993, 262, 2030–2033 CAS.
- L. A. Fransson, T. N. Huckerby and I. A. Nieduszynski, Biochem. J., 1978, 175, 299–309 CAS.
- D. Mikhailov, K. H. Mayo, A. Pervin and R. J. Linhardt, Biochem. J., 1996, 315(Pt2), 447–454 CAS.
- T. R. Rudd, S. E. Guimond, M. A. Skidmore, L. Duchesne, M. Guerrini, G. Torri, C. Cosentino, A. Brown, D. T. Clarke, J. E. Turnbull, D. G. Fernig and E. A. Yates, Glycobiology, 2007, 17, 983–993 CrossRef CAS PubMed.
- S. E. Guimond, T. R. Rudd, M. A. Skidmore, A. Ori, D. Gaudesi, C. Cosentino, M. Guerrini, R. Edge, D. Collison, E. McInnes, G. Torri, J. E. Turnbull, D. G. Fernig and E. A. Yates, Biochemistry, 2009, 48, 4772–4779 CrossRef CAS PubMed.
- T. R. Rudd, K. A. Uniewicz, A. Ori, S. E. Guimond, M. A. Skidmore, D. Gaudesi, R. Xu, J. E. Turnbull, M. Guerrini, G. Torri, G. Siligardi, M. C. Wilkinson, D. G. Fernig and E. A. Yates, Org. Biomol. Chem., 2010, 8, 5390–5397 CAS.
- T. Irimura, M. Nakajima and G. L. Nicolson, Biochemistry, 1986, 25, 5322–5328 CrossRef CAS.
- E. J. Paredes-Gamero, V. P. Medeiros, M. A. Lima, C. M. Accardo, E. H. Farias, G. I. Sassaki, P. T. Campana, A. Miranda, A. T. Ferreira, I. L. Tersariol and H. B. Nader, J. Cell. Biochem., 2012, 113, 1359–1367 CrossRef CAS PubMed.
- R. N. Rej, K. R. Holme and A. S. Perlin, Carbohydr. Res., 1990, 207, 143–152 CrossRef CAS.
- H. B. Nader and C. P. Dietrich, Anal. Biochem., 1977, 78, 112–118 CrossRef CAS.
- M. Kosakai and Z. Yosizawa, Anal. Biochem., 1975, 69, 415–419 CrossRef CAS.
- Z. Dische, J. Biol. Chem., 1947, 167, 189–198 CAS.
- M. A. Lima, A. J. Hughes, N. Veraldi, T. R. Rudd, R. Hussain, A. S. Brito, S. F. Chavante, I. I. Tersariol, G. Siligardi and H. B. Nader, Med. Chem. Commun., 2013, 4, 870–873 RSC.
- P. Maurel and P. Douzou, J. Biol. Chem., 1975, 250, 2678–2680 CAS.
- S. Grace and D. Dunaway-Mariano, Biochemistry, 1983, 22, 4238–4247 CrossRef CAS.
- A. W. Schuttelkopf and D. M. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 Search PubMed.
- N. J. Schaftenaar G, J. Comput.–Aided Mol. Des., 2000, 14, 123–134 CrossRef.
- G. Schaftenaar and J. H. Noordik, J. Comput.–Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS , 27–38.
- The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- L. Pol-Fachin, C. L. Fernandes and H. Verli, Carbohydr. Res., 2009, 344, 491–500 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Professor Carl P. Dietrich, a pioneer in the studies of heparin degrading enzymes. |
| ‡ Electronic supplementary information (ESI) available: Text and figures giving methodology, equations and molecular modeling data mentioned in the text. See DOI: 10.1039/c3mb70370c |
| § Joint first authors. |
| ¶ Present address: Cincinnati Childrens Hospital and Research Foundation, 240 Albert Sabin Way, S3.419, Cincinnati OH 45229-3039, USA. |
| || Present address: Department of Ophthalmology, University of Cincinnati, CARE/Crawley Bldg. RM 5860, 3230 Eden Avenue, Cincinnati, OH 45267-0838, USA. |
| ** Joint senior authors. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.