Networks of genes modulating the pleiotropic drug response in Saccharomyces cerevisiae†
Ploi
Yibmantasiri
,
Peter W.
Bircham
,
David R.
Maass
,
David S.
Bellows
and
Paul H.
Atkinson
*
School of Biological Sciences, Victoria University of Wellington, Room 321, MacDiarmid Building, Kelburn, Wellington, 6012, New Zealand. E-mail: Paul.Atkinson@vuw.ac.nz; Fax: +64 4 463 5331; Tel: +64 4 04 463 7444
First published on 8th November 2013
Abstract
The pleiotropic drug response (PDR) or multidrug resistance (MDR) are cellular defence mechanisms present in all species to deal with potential toxicity from environmental small molecule toxins or bioactives. The rapid induction of MDR by xenobiotics in mammalian cells and PDR in budding yeast (S. cerevisiae) has been well studied but how pathway specificity is achieved across different structural classes of xenobiotics is not well understood. As a novel approach to this problem we investigated the genome-wide network of genes modulating the yeast PDR. Fluorescently-tagged ABC pumps Pdr5p-GFP and Yor1p-GFP were used as real-time reporters for the Pdr1p/Pdr3p controlled response. Using the yeast non-essential gene deletion set fifty-four gene deletions that suppressed up-regulation of reporter fluorescence to the cell surface in the presence of atorvastatin were identified by high content confocal automated microscopy. Secondary validation using spot dilution assays to known PDR substrates and Western blot assays of Pdr5p expression confirmed 26 genes able to modulate the PDR phenotype. By analysis of network connectivity, an additional 10 genes that fell below the primary screen cut-off were predicted to be involved in PDR and confirmed as above. The PDR modulating genes taken together were enriched in signalling (Rho-GTPase, MAPK), Mediator complexes, and chromatin modification (subunits of ADA and SAGA complexes). Many of the gene deletions cause extra sensitivity in Δpdr1Δpdr3 strains strongly suggesting that there are alternative pathways to upregulate PDR, independently of Pdr1p/Pdr3p. We present here the first high-content microscopy screening for PDR modulators, and identify genes that are previously unsuspected regulators of PDR apparently contributing via network interactions.
Introduction
Multidrug resistance or pleiotropic drug resistance (PDR) is a phenotype achieved by interactions between transcriptional activators, Mediator complexes and efflux pumps forming a robust network.This phenomenon is conserved across all kingdoms of life and is a major cause of resistance to antifungal and chemotherapeutic treatments.1–4 The presence of xenobiotics rapidly induces PDR gene expression which in S. cerevisiae is under the control of two homologous transcription factors, Pdr1p and Pdr3p.2,5–7 Although the downstream efflux pumps and targets of Pdr1p, Pdr3p are well annotated, the modulating events between sensing of chemically different xenobiotics and transcriptional activation are less well understood. Whereas xenobiotics may bind directly to Pdr1p/Pdr3p activating them,8 there may be other processes involved. There appears to be a basal generic PDR response activating a common core of 7 genes dependent on Pdr1p/Pdr3p called the early PDR (ePDR). This set of genes is rapidly activated by chemically unrelated xenobiotics (fluphenazine, benomyl). The ePDR acts in combination with additional transcriptional responses leading to drug specific responses.9 In these comparative studies with genome wide microarrays, the authors concluded that even for ePDR there must be “upstream” (of Pdr1p binding) signals mediating the ePDR that are completely unknown. In this current study we investigated PDR-upstream events using functional assays based on the genome-wide yeast deletion set and C-terminus GFP-tagged plasma membrane protein reporters. The intracellular levels and distribution of Pdr5p and Yor1p were investigated using automated high-content confocal microscopy. Both proteins are important ABC transporters under the transcriptional control of Pdr1p/Pdr3p. This allows GFP fluorescence to serve as a sensitive real-time indicator of Pdr1p/Pdr3p transactivation. To show that our reporters respond to xenobiotic treatment cells were treated with atorvastatin, a known PDR substrate. These two transporters were selected as reporters owing to their differences in Pdr1p/Pdr3p dependency2 in which Pdr5p showed more dependency on Pdr1p/Pdr3p than Yor1p. Using proteins with differential Pdr1p/Pdr3p dependency may allow us to identify the major common modulators of the PDR pathway. The design of the assay with our rationale is seen in Fig. 1. We also investigated whether PDR could be affected through network connectivity.
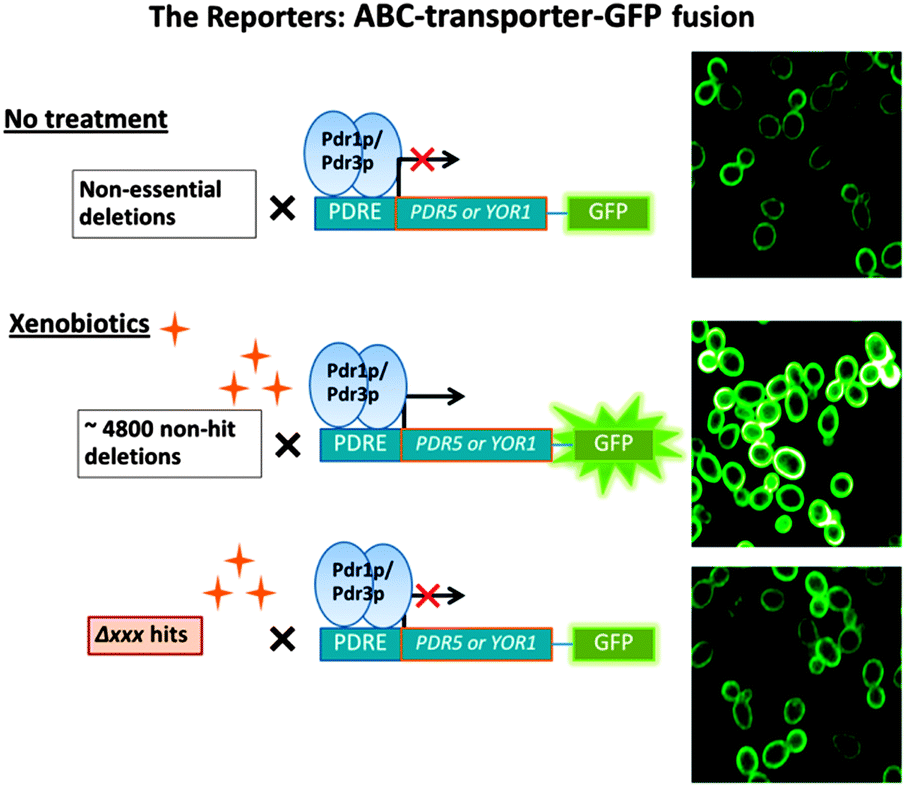 | ||
| Fig. 1 Schematic presentation of the experimental rationale for use of PDR efflux pumps GFP reporters. The top panel shows plasma membrane location of the reporters expressed on a deletion mutant array background in the absence of xenobiotic. The middle panel shows the amplification of reporters in gene deletions not involved in PDR in the presence of xenobiotic. The bottom panel shows attenuation of reporters by specific gene deletions in the presence of xenobiotic. | ||
Results
Fidelity of GFP-labelled efflux pump reporters to xenobiotic activation
To verify that C-terminal GFP tagging does not affect Pdr5p or Yor1p functionality, susceptibility to the PDR substrate cycloheximide (CHX) was tested. There was no significant difference in sensitivity between the reporter strains and wild-type (Fig. 2 top panel). Reporter activation was measured after exposure to the PDR substrate atorvastatin. There was an increase in GFP intensity of 2–2.5 fold for each reporter compared to untreated cells (Fig. 2 bottom panel). The red fluorescent protein (RFP mCherry) under a constitutive promoter was expressed in the cytoplasm and used as the internal control when comparing GFP intensity between control and atorvastatin treated groups.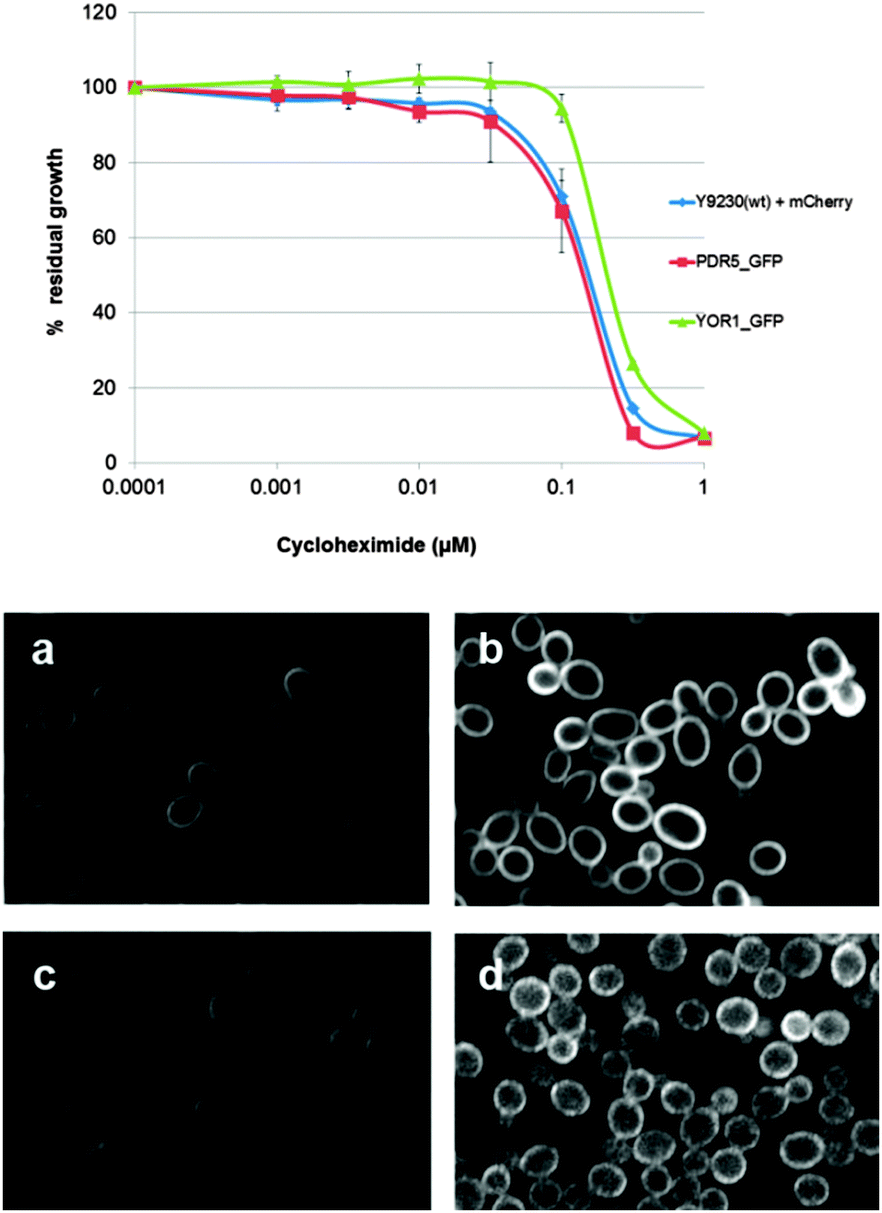 | ||
| Fig. 2 Properties of ABC-GFP reporters. Top panel: dose response of Pdr5p and Yor1p GFP reporters compared with wild-type (Y9320 + mCherry) at various concentrations of cycloheximide. Each reporter strain contained an internal control cytoplasmic mCherry under a constitutive promoter at the LYP1 locus (see strains used in Materials and methods). Bottom panel: ABC transporter GFP reporters. Left panels: DMSO controls. Right panels: 35 μM atorvastatin treatment for 4 hours before images were recorded by the OPERA confocal microscope and analysed by Acapella software. (a and b) PDR5-GFP (YCG285). (c and d) YOR1-GFP (YCG334). | ||
Enrichment of signalling and transcriptional related genes in a primary screen modulating the PDR response
Each reporter strain was mated with the yeast gene deletion set10 to screen for gene deletions which prevent the upregulation of the PDR reporters in the presence of xenobiotics. The PDR-reporter deletion libraries were grown on agar plates overnight before being transferred into microtitre plates. These were then incubated for four hours in the presence of atorvastatin to induce the PDR response. Following this, live cell images were captured using an Opera high throughput confocal microscope. Gene deletions that cause mis-localisation of a generic plasma membrane protein not involved in PDR were considered false positives and excluded from further studies. These genes were previously identified using a housekeeping plasma membrane protein Mrh1p-GFP as a reporter screen on the deletion set background. These gene deletions are involved in ER processing and the secretory pathway11 and are PDR-independent. A list of these genes is in Table S1 (ESI†). Examples of deletions that affect plasma membrane protein location are in Fig. S1 (ESI†). Gene deletions which caused change in RFP internal control were also excluded from further analysis.Five screens were carried out, three with the Pdr5p reporter and, due to time constraint, only two with the Yor1p reporter. We scored the common gene deletions affecting, the location of both reporters. A gene deletion was considered as a “hit” if it suppresses plasma membrane location in 2 out of the 3 Pdr5p reporter screens and 1 of the Yor1p reporter screens. The “expression” scores were measured using Acapella Texture analysis software measuring “Ridge”, “Bright”, and “Edge” parameters (Materials and methods). Two out of three of these parameters must show no significant changes between control and atorvastatin treated gene-deletion strains for the gene deletion to be considered as a hit (Materials and methods).
We initially identified 54 gene deletions (“hits”) that prevented the expected increase in Pdr5p and Yor1p expression upon treatment with atorvastatin (Fig. 3, Fig. S2, ESI†). Descriptions for these genes are available in Table S2 (ESI†). Hits appeared in signalling networks (Rho-GTPase, MAPK), RNA polymerase II Mediator complexes, chromatin modification (subunits of SAGA histone acetyltransferase complexes), and sphingophospholipid synthesis.
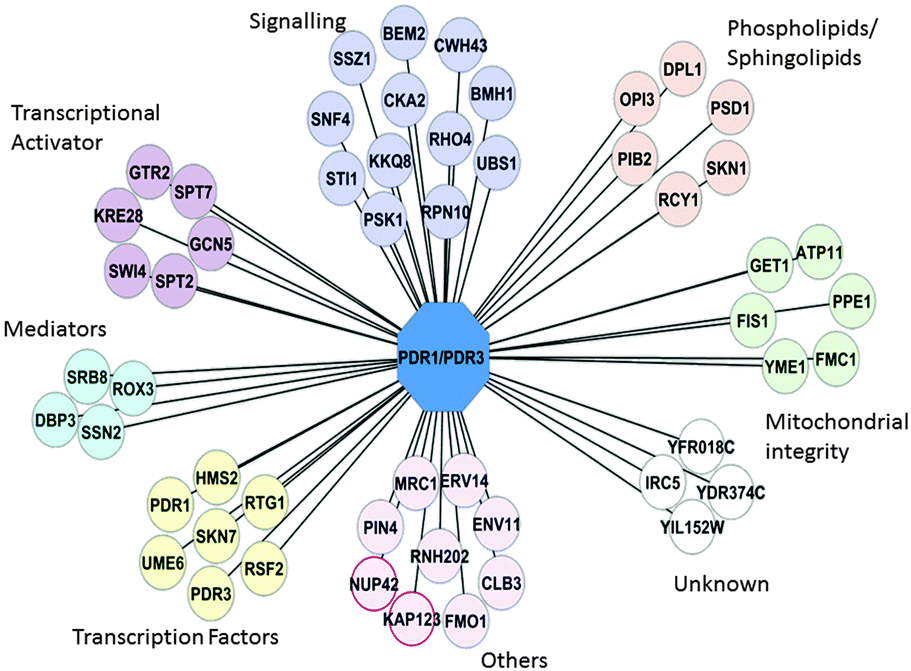 | ||
| Fig. 3 PDR Primary screen network. Gene deletions that prevented normal upregulation of Pdr5p-GFP and Yor1p-GFP upon atorvastatin treatment. PDR1/PDR3 are in the central node to represent the known master regulators of the PDR phenotype. Each line of connection represents a possible regulatory relationship between gene XXX and PDR1/PDR3. Groups with the * symbol are genes that shows significant GO enrichment with p-value < 0.05. | ||
Gene Ontology (GO) slim-analysis in the function category showed significant enrichment of 11 gene deletions (including PDR1) in transcription activation and co-activation (p-value 0.05 Table S3, ESI†). In addition there was enrichment of deletion mutants that are involved in kinase activity (PSK1, SNF4, KKQ8 and BMH1, (see “Signalling” Fig. 3)), sphingolipid and phospholipid biosynthesis (DPL1, SKN1, PIB2), and mitochondria integrity (PSD1, FMC1, PPE1, FIS1, ATP11, and YME1). Mediator and transcriptional activators such as Gal11p and Ngg1p have previously been shown to regulate or facilitate the PDR response.8,12 However, enrichment of the other transcription factors and activators seen in Fig. 3 in the regulation of PDR1/PDR3 has not previously been described. Such an observation suggests a program of gene transcription is switched on in response to xenobiotics. This is consistent with earlier observations of Fardeau et al. (2007), who suggested signalling pathways upstream of Pdr1p might play a role in the activation of the early PDR response (ePDR).9
To validate the primary screen results, spot dilution assays were carried out with six known PDR substrates (oligomycin, cycloheximide, fluconazole, ketoconazole, atorvastatin, benomyl), according to the description in Experimental methods. Examples of spot dilutions can be seen in Fig. S3 (ESI†). If these genes were important to the cell's survival because of a central role in the PDR response it would be expected that their deletion would show increase PDR-drug sensitivity in all cases. Sub-inhibitory concentrations (not lethal to wild type cells) of the drugs were used. A strain with double deletion of PDR1/PDR3 were used as a positive control, and showed sensitivity to all the compounds used. We found that 17 gene deletions showed an increase in sensitivity toward one or more compounds, whereas only 8 were only sensitive to atorvastatin. Interestingly deletion of PDR1 alone was sufficient to sensitise cells to atorvastatin but none of the other compounds (Table 1). These differential sensitivities further supported existence of an upstream and/or independent pathway of Pdr1p/Pdr3p present in yeast cells.
| Gene (ORF) | Sensitivity in Δxxx | Sensitivity in ΔxxxΔpdr1Δpdr3 | Decreased Pdr5p abundance detected by Western blot | Description |
|---|---|---|---|---|
| “—” means the deletion did not confer sensitivity in spot dilution assay or reduction of Pdr5p in Western blot. Sensitivity observed in ΔxxxΔpdr1Δpdr3 were compared to Δpdr1Δpdr3 control strain. | ||||
| BEM2 (YER155C) | Atorvastatin | Atorvastatin | — | Rho GTPase activating protein (RhoGAP) |
| Benomyl | Cycloheximide | |||
| Cycloheximide | Fluconazole | |||
| Ketoconazole | Ketoconazole | |||
| Oligomycin | ||||
| BMH1 (YER177W) | — | Fluconazole | Yes | 14-3-3 protein regulation of exocytosis, vesicle transport, Ras/MAPK signalling, signalling |
| Ketoconazole | ||||
| CWH43 (YCR017C) | — | Oligomycin | — | Putative sensor/transporter protein involved in cell wall biogenesis |
| DPL1 (YDR294C) | Cycloheximide | — | — | Dihydrosphingosine phosphate lyase, regulates intracellular levels of sphingolipid |
| FMC1 (YIL098C) | Cycloheximide | Atorvastatin | — | Required for assembly or stability at high temperature of the F1 sector of mitochondrial F1F0 ATP synthase |
| Oligomycin | Fluconazole | |||
| Ketoconazole | ||||
| Oligomycin | ||||
| GCN5 (YGR252W) | — | Atorvastatin | — | Catalytic subunit of the ADA and SAGA histone acetyltransferase complexes |
| Cycloheximide | ||||
| Fluconazole | ||||
| Ketoconazole | ||||
| Oligomycin | ||||
| GTR2 (YGR163W) | — | Fluconazole | Yes | Putative GTP binding protein that negatively regulates Ran/Tc4 GTPase cycle; activates transcription |
| Ketoconazole | ||||
| KAP123 (YER110C) | — | Negative genetic interaction | — | Karyopherin beta, mediates nuclear import of ribosomal proteins prior to assembly into ribosomes and import of histones H3 and H4 |
| MDM39 (GET1) (YGL020C) | — | Fluconazole | — | Subunit of the GET complex; involved in insertion of proteins into the ER membrane |
| Ketoconazole | ||||
| OPI3 (YJR073C) | — | Oligomycin | — | Phospholipid methyltransferase catalyzes the last two steps in phosphatidylcholine biosynthesis |
| PDR1 (YGL013C) | Atorvastatin | Negative genetic interaction | Yes | Master regulator involved in recruiting other zinc cluster proteins to pleiotropic drug response elements (PDREs) |
| PIB2 (YGL023C) | Cycloheximide | Fluconazole | Yes | Protein binding phosphatidylinositol 3-phosphate |
| Ketoconazole | Ketoconazole | |||
| PIN4 (YBL051C) | Atorvastatin | — | — | G2/M phase progression contains an RNA recognition motif, NLS |
| Benomyl | ||||
| Cycloheximide | ||||
| PSD1 (YNL169C) | Benomyl | — | — | Mitochondrial inner membrane, converts phosphatidylserine to phosphatidylethanolamine |
| Ketoconazole | ||||
| RCY1 (YJL204C) | Atorvastatin | Negative genetic interaction | — | F-box protein involved in recycling plasma membrane proteins |
| Ketoconazole | ||||
| RHO4 (YKR055W) | Benomyl | — | — | Small GTPase of the Rho/Rac subfamily of Ras-like proteins |
| Cycloheximide | ||||
| Fluconazole | ||||
| Ketoconazole | ||||
| Oligomycin | ||||
| ROX3 (YBL093C) | Atorvastatin | Negative genetic interaction | Yes | Subunit of the RNA polymerase II Mediator complex |
| Cycloheximide | ||||
| Fluconazole | ||||
| Ketoconazole | ||||
| Oligomycin | ||||
| RPN10 (YHR200W) | Cycloheximide | Cycloheximide | — | Non-ATPase base subunit of the 19S regulatory particle (RP) of the 26S proteasome |
| Ketoconazole | (Oligomycin?) | |||
| SNF4 (YGL155W) | Atorvastatin | Oligomycin | — | Activating gamma subunit of the AMP-activated Snf1p kinase complex |
| Oligomycin | ||||
| SPT7 (YBR081C) | Cycloheximide | Cycloheximide | Yes | Subunit of the SAGA transcriptional regulatory complex |
| Fluconazole | Fluconazole | |||
| Ketoconazole | Ketoconazole | |||
| Oligomycin | Oligomycin | |||
| SRB8 (YCR018W) | Atorvastatin | Atorvastatin | Yes | Subunit of the RNA polymerase II Mediator complex for Pol-II? |
| Cycloheximide | Cycloheximide | |||
| Fluconazole | ||||
| Oligomycin | ||||
| SSZ1 (YHR064C) | Cycloheximide | Cycloheximide | — | Hsp70 protein; also involved in pleiotropic drug resistance via activation of PDR1 and PDR5 |
| Oligomycin | Fluconazole | |||
| Ketoconazole | ||||
| Oligomycin | ||||
| SWI4 (YER111C) | Cycloheximide | Cycloheximide | — | DNA binding component of the SBF complex (Swi4p–Swi6p), a transcriptional activator |
| Fluconazole | ||||
| Ketoconazole | ||||
| UME6 | Atorvastatin | Atorvastatin | — | Key transcriptional regulator of early meiotic genes |
| (YDR207C) | Benomyl | Cycloheximide | ||
| Cycloheximide | Fluconazole | |||
| Ketoconazole | ||||
| Oligomycin | ||||
| YME1 (YPR024W) | — | Atorvastatin | — | Mitochondrial inner membrane i-AAA protease complex, responsible for degradation of unfolded or misfolded proteins |
| Cycloheximide | ||||
| Fluconazole | ||||
| Ketoconazole | ||||
| Oligomycin | ||||
Gene deletions increase sensitivity in a Δpdr1Δpdr3 double mutant background, suggesting alternative drug resistance pathways
To investigate if any of the genes identified in the primary screens are able to cause extra sensitivity in double knockouts of transcription factors PDR1 and PDR3, we mated the 54 identified gene deletion strains with a strain lacking both PDR1 and PDR3 (YCG326; see Material and methods). The rationale behind this was that gene deletions that conferred extra sensitivity in the already sensitive Δpdr1Δpdr3 background strain should, in theory, be the genes that are independent of Pdr1p/Pdr3p and control other compensatory PDR pathways.We found that most genes that caused sensitivity when deleted by themselves also caused extra sensitivity to one or more PDR-substrates when tested in the Δpdr1Δpdr3 background. However, we found that deletion of CWH43, YDR532C, BMH1, GET1, OPI3, GTR2, YME1, and GCN5 only caused increased sensitivity in the Δpdr1Δpdr3 background (Table 1, Fig. S3, ESI†). Deletion of RCY1, KAP123, and ROX3 showed strong negative genetic interaction (no growth) with Δpdr1Δpdr3, while single deletions of these genes individually were viable (Table 1). Interestingly, while single Δrcy1 and Δrox3 mutations increased sensitivity to PDR substrate compounds tested (Table 1), Δkap123 as a single mutation did not cause increased sensitivity in the spot dilution assays. The network of gene deletions that conferred sensitivity in spot dilution assays grouped by GO function is shown in Fig. 4.
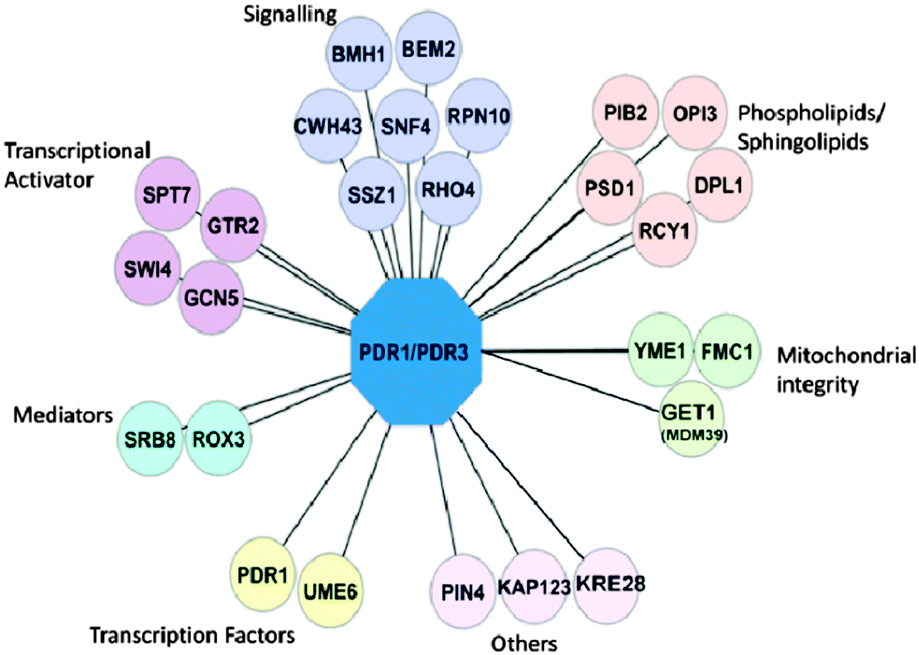 | ||
| Fig. 4 Network of genes from PDR secondary screen. Gene deletions from the primary screens were subjected to spot dilution assay as Δxxx, and Δxxx Δpdr1 Δpdr3. Deletions which confer sensitivity toward at least one the six test compounds tested or reduced abundance of Pdr5p on the genome-wide deletion background are shown here. PDR1/PDR3 are shown as the hub connecting known regulators of the PDR. Each connection line represents a possible regulatory relationship between gene XXX and PDR1/PDR3. | ||
The twenty-six gene deletions that increased sensitivity to xenobiotics in the Δpdr1/Δpdr3 background (Table 1) were further subjected to Western blot analysis for Pdr5p (Yor1p was not measured here because no antibody was available). This was to examine if the increase in xenobiotic sensitivity conferred by the deletion was due to decrease in Pdr5p abundance. We found that in addition to Δpdr1 and Δpdr3, several other gene deletions reduced Pdr5p abundance relative to the Pma1p control which showed no alteration in abundance on Western blots. These genes were Δrox3, Δspt7, Δgtr2, and Δbmh1 (Table 1). Since the expression of another plasma membrane protein, Pma1p, an internal control, did not change upon deletion of these genes (Fig. S3 and S4, ESI†), their effect was specific for the PDR response, and not just a global transcriptional activation effect.
Network analysis predicts gene deletions that contribute to PDR phenotype
From the results reported here, the network of genes that affect the PDR response in S. cerevisiae can be seen to include PDR1, PDR3, as well as gene deletions that showed GO enrichment such as Mediators, the SAGA complex, and the genes involved in signalling. To identify other possible PDR regulatory/modifying genes, we explored the network connectivity between these genes shown in Fig. 4. Using the published (http://thebiogrid.org/) physical and genetic interactions of the 26 genes that we found necessary for the PDR phenotype (Table 1), we identified the most highly connected hubs among these genes (Fig. S4, ESI†). Genes that connected between these hubs and/or interacted with PDR1 (Pdr1p) and PDR3 (Pdr3p) were then identified. This curated list of genes was then used to “screen” the genes that registered in our primary screen but were below the threshold used in this study for declaring them as “hits”. A further 18 gene deletions were identified that were subsequently tested to see if they showed sensitivity in spot dilution assays and/or reduction of Pdr5p abundance. We found that 10 deletions showed sensitivity toward at least one of the PDR substrate compounds tested (Table 2) or reduced Pdr5p in the Western blots, or both.| Gene | Sensitivity in single deletion | Sensitivity in Δpdr1Δpdr3 background | Decreased Pdr5p abundance detected by Western blot | Description |
|---|---|---|---|---|
| N/T = not tested. | ||||
| ADA2 (YDR448W) | Cycloheximide | Not conclusive | N/T | Transcription coactivator, component of the ADA and SAGA transcriptional adaptor/HAT (histone acetyltransferase) complexes |
| DEP1 (YAL013W) | Cycloheximide | Fluconazole | Yes | Transcriptional modulator involved in regulation of structural phospholipid biosynthesis |
| Oligomycin | ||||
| ERG3 (YLR056W) | Atorvastatin | No | Yes | C-5 sterol desaturase, catalyzes the introduction of a C-5(6) double bond into episterol, a precursor in ergosterol biosynthesis |
| Cycloheximide | ||||
| Oligomycin | ||||
| HEK2 (YBL032W) | No | Ketoconazole | N/T | RNA binding protein |
| Oligomycin | ||||
| HEX3 (SLX5) (YDL013W) | Atorvastatin | Fluconazole | N/T | Subunit of the Slx5–Slx8 SUMO-targeted ubiquitin ligase (STUbL) complex |
| Oligomycin? | Ketoconazole | |||
| HSP82 (YPL240C) | N/T | N/T | Yes | Hsp90 chaperone required for pheromone signalling |
| LGE1 (YPL055C) | No | No | Yes | Protein of unknown function |
| MNN10 (YDR245W) | Atorvastatin | Atorvastatin | N/T | Subunit of a Golgi mannosyltransferase complex |
| Benomyl | Cycloheximide | |||
| Fluconazole | ||||
| Ketoconazole | ||||
| NGG1 (YDR176W) | Atorvastatin | N/T | No | Transcriptional regulator; component of transcriptional adaptor and histone acetyltransferase complexes, the ADA complex, the SAGA complex, and the SLIK complex |
| Cycloheximide | ||||
| Ketoconazole | ||||
| Oligomycin | ||||
| SPT8 (YLR055C) | Atorvastatin | Fluconazole | Yes | Subunit of the SAGA transcriptional regulatory complex but not present in SAGA-like complex |
| Fluconazole | Ketoconazole | |||
| Ketoconazole | ||||
Additional genes identified using network interactions predictions were enriched in transcription factors, the SAGA/transcriptional activator and chromatin remodelling (DEP1, NGG1, SPT8, ADA2) as seen by GO analysis.
This result validated the overall screening methodology but also suggested that the PDR phenotypes can be affected, perhaps effected, through genetic networks.
Discussion
Development of new reporters for investigating possible PDR modulator networks
We used a novel reporter system to identify genes involved in sensing xenobiotics and signalling to Pdr1p/Pdr3p transcription factors. More than half of the gene deletions identified in this screen increased xenobiotic sensitivity in the absence of Pdr1p/Pdr3p i.e. the triple mutants ΔxxxΔpdr1Δpdr3. In addition some of these gene deletions reduced Pdr5p abundance, while others did not. These observations strongly suggest there are upstream and compensatory pathways to achieve a PDR response independent of Pdr1p/Pdr3p.It is possible is that xenobiotics bind directly to Pdr1p.8 Thakur et al. 2008 observed that ketoconazole has a binding KD of 39 μM. However, we found that in wild type cells 20 μM ketoconazole is toxic in YPD agar, i.e. cells are already inviable below the dissociation constant of this putative Pdr1p substrate binding site. It is not known if such direct binding is general for xenobiotics or if it is specific for ketoconazole as shown by Thakur et al. 2008. It is possible Xenobiotic binding alters the conformation of Pdr1p, which then might allow binding of PDR-stress response-specific modifying proteins to bind and activate transcription of PDR-specific genes.13 This view would fit with the interpretations of Fardeau et al. (2007) who proposed that Pdr1p is a stress response factor and a promoter regulator. It has been shown Pdr1p is involved in both basal expression (no xenobiotics), and the rapid upregulation of the PDR genes in the presence of xenobiotics.9
Our study has provided evidence that PDR phenotypes may also be effected through genetic and physical interactions networks. An illustrative example is LGE1, a gene that encodes a protein of unknown function. LGE1 fell below the threshold and was not identified as a “hit” in the primary screen. We hypothesised that deletion of LGE1 might affect the PDR phenotype because LGE1 is synthetic lethal with SWI4, and UME6. As shown in Table 2, deletion of LGE1 did not increase xenobiotic sensitivity in any deletion background. However, it did reduce Pdr5p abundance. This effect might be due to an LGE1 function being associated with the network hubs PDR3, SWI4 and UME6, engaged in transcriptional regulation. By assessing the network, in such a way we may be able to ascertain the pathways in which Lge1p might work.
Overall we observed enrichment of genes involved in signalling in particular those that are involved with Rho-GTPase, Ras, and MAPK. This finding complements our chemical screen in which the GFP labelled ABC-reporters were screened against the 1280 compound Library of Pharmacologically Active Compounds (LOPAC). Several of the compounds that strongly upregulated our ABC-reporters are known to be signalling and kinase inhibitors (Table S4, ESI†). Similar complementary results have been observed in C. albicans. LaFayette et al. (2010) showed that protein kinase inhibitors in a LOPAC screen that have little toxicity on their own, enhanced the toxicity of fluconazole in C. albicans, leading them to propose that kinase signalling regulates multidrug resistance in this pathogenic yeast species.14 These reports combined with the screening results reported here strongly suggest that kinase-signalling pathways are involved in controlling the PDR phenotype.
GO analysis performed on the gene deletions that conferred sensitivity to xenobiotics in the spot dilution assays also showed significant enrichment in genes involved in enzyme activation and protein binding. These genes are BEM2 (a Rho GTPase activating protein), BMH1, and SNF4 (p-value 0.03599). All three genes when deleted individually displayed a decrease in resistance towards many xenobiotics (www.yeastgenome.org). They are also linked with signalling and response to stress (Table 1), especially Bmh1p which controls proteome at post-transcriptional level and involved in many cellular processes from signalling to stress response. Bmh1p and Snf4p are found in plasma membrane, cytoplasm and nucleus and can potentially relay signal from plasma membrane to the nucleus. This adds to the suggestion that a signalling pathway, probably kinase proteins (RAS/MAPK), are required to relay xenobiotic information sensed at the plasma membrane to Pdr1p/Pdr3p in the nucleus. These genes also can also act independently of Pdr1p/Pdr3p (i.e. they all conferred extra sensitivity to strains deleted in Δpdr1Δpdr3), suggesting that they act upstream of Pdr1p/Pdr3p and may also switch on PDR compensatory pathway in the absence of Pdr1p/Pdr3p.
Our assays also revealed additional genes that work independently of PDR1 and PDR3 and contribute to the PDR phenotypes. GO function analysis showed significant enrichment of genes with the SAGA, SWI/SNF chromatin modification and transcriptional activity/co-activation namely UME6, SWI4, SKN7, RSF2, RTG1, ROX3, SRB8, SSN2, and GCN5. We suggest that these genes are likely to be PDR specific modulators and different mediators and chromatin modifications are used to differentiate different stress responses. Some might share the same targets. It is noteworthy that these gene deletions did not confer H2O2 sensitivity, an oxidative stress inducer. Oxidative stress has been linked to PDR target genes.15–19 This result further supports that these genes independently contribute to PDR.
Srb8p has been shown to be linked with the upregulation of PDR5 in cells that have lost the mitochondria genome (ρ0 cells),4 but not in wild-type cells. Our results that showed Srb8p plays a role in normal upregulation of Pdr5p and Yor1p in the presence of xenobiotics suggest that Srb8p might have more roles in the PDR phenotype than is currently known. The SAGA, SWI/SNF chromatin modification complex though often global in function may provide cells with a specific minor backup pathway for Pdr1p/Pdr3p. We propose that in the absence of the major regulator Pdr1p/Pdr3p, these minor regulator components become essential. The importance of minor transcriptional machinery has been proposed by Venter 2009.20 Both SPT8 and SWI4 show negative genetic interactions with GAL11 supporting our proposal that the SAGA and SWI complexes provide cells with an independent alternative pathway to achieve the PDR phenotype. This might be through bypassing the requirement of Gal11p or Pdr1p/Pdr3p altogether. This could explain why the GAL11 deletion does not appear as a hit in our primary screen or spot dilution assay as we anticipated it would. This hypothesis also explains why most of the strains containing deletion of genes in this category were sensitive to xenobiotics and become hypersensitive in Δpdr1Δpdr3 background.
Chromatin remodelling complexes and co-activators, such as RNA polymerase II and Mediator subunits, affect transcriptional activity of gene expression globally.21,22 However, these functions also occur for specific gene expression (“chromatin signalling pathway”).23,24 Chromatin modification/remodelling complexes have been associated with the drug resistant phenotype in S. cerevisiae, C. albicans, and mammalian breast cancer cell lines.25–27 These pathways may provide new targets for overcoming drug resistance, especially if the drug resistant phenotype is achieved from mechanisms other than hyperactivated Pdr1p/Pdr3p. Small molecule inhibitor library screening using such deletion backgrounds and looking for hypersensitivity should provide more insight of how these genes might work together to influence PDR phenotype.27,28
In summary we propose that there is a signalling network of genes upstream of Pdr1p/Pdr3p that conveys the presence of xenobiotics to Pdr1p/Pdr3p in the nucleus inducing a strong and rapid PDR response. These are likely to involve RAS/MAPK signalling pathway(s). We also propose that chromatin remodelling helps cells to achieve a maximum PDR response enhancing Pdr1p/Pdr3p transcriptional activity and/or provide cells with alternative pathways.
Future work will be to analyse the genes that we identified to be involved in PDR signalling. For example we might overexpress genes which show sensitivity in triple deletions with PDR1 PDR3, and see if the over expression rescues the xenobiotic sensitivity phenotype of Δpdr1Δpdr3 strains.
Materials and methods
Yeast cells were grown in yeast peptone dextrose (YPD) under non-selective conditions, or in synthetic complete (SC) media under selective conditions at 30 °C or at 30 °C with shaking for liquid culture.29Yeast dose response growth assay
Cells at a concentration of 5 × 105 cells per ml were treated with various concentrations of cycloheximide. DMSO was used as a negative control. After 18 hour incubation at 30 °C, cell density was determined by measuring absorbance at 590 nm using a spectrophotometer (Perkin-Elmer Wallac Envision 2012). Percent residual growth was calculated using the formula
Strains used
Yeast strains used in this study were derived from S288C and their genotypes are listed in Table 3.| Strain | Genotype | Description |
|---|---|---|
| BY4741 | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Wild-type (derived from S288C) |
| YCG255 | MAT α, can1Δ::STE2pr-Sp-URA3; lyp1Δ::mCherry-NAT; his3Δ1; leu2Δ0; ura3Δ0; LYS2+ | SGA starting strain |
| – mCherry-NAT is red fluorescent protein (RFP) linked with the NAT (nourseothricin) dominant selectable marker under a constitutive promoter. It has a cytoplasmic location | ||
| – RFPs act as internal control and as marker for automated cell recognition software | ||
| YCG285 | MAT α, can1Δ::STE2pr-URA3; lyp1Δ::mCherry-NAT; PDR5-GFP-HIS3MX6; leu2Δ0 his3Δ1 met15Δ0 ura3Δ0 | Pdr5p reporter strain with C-terminal GFP linked |
| YCG334 | MAT α can1Δ::STE2p-URA3; lyp1Δ::mCherry-NAT; YOR1-GFP-HIS3MX6; leu2Δ0 his3Δ1 met15Δ0 ura3Δ0 | Yor1p-GFP reporter strain |
| YCG326 | Mat α pdr1Δ::NAT pdr3Δ::URA3 can1Δ::STE2pr_Sphis5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 LYS2+ | Pdr1 Pdr3 knockout |
Strain construction
Yeast strains YCG285 and YCG344 were generated by tetrad dissection of spores from crossing YCG255 × Pdr5p-GFP or Yor1p-GFP respectively. All the GFP strains were commercial strains from Invitrogen with C-terminal labelled green fluorescent protein (GFP). The haploid parental strain genotype is MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0. The commercial GFP strains were mated against SGA starting strains so that the final progeny strains had the genotype suitable for SGA methodology.The method for tetrad dissection was as follows:
Fifty microliters of cells grown in liquid sporulation media for 7 days was spun down at 7000 rpm for 15 seconds, and supernatant was discarded. The cell pellet was re-suspended in 50 μl of zymolyase in sorbitol (0.5 mg ml−1 zymolyase in 1 M sorbitol), and incubated for 10 minutes at 30 °C. Three hundred microliters of sterile distilled water was added to the suspension and the tube was gently inverted to mix. Fifteen microliters of the suspension was spotted onto YPD plates to separate tetrads. Tetrads were dissected using Singer Dissection Microscope.
The dissected spores were grown for 2–3 days at 30 °C, then replica-plated onto selective media using standard procedures.29
Synthetic genetic array (SGA) and drug screening
Five screens were conducted; three with the Pdr5p-GFP reporter and two with the Yor1p-GFP reporter. Analysis of each screen was conducted as in Materials and methods. In the primary screens, gene deletions of interest are those showing no difference between the mean of the control condition and drug treated condition (identified with Z-scores below 5). These are the genes that when deleted result in an inability to upregulate PDR5 and YOR1 upon drug treatment, and therefore should be the common modulators of the PDR response.All analyses involving measurement of GFP intensity were normalized against the intensity of an internal RFP control.
SGA analysis was carried out according to the method as previously described30 using a Singer RoToR™ HDA bench-top replicator robot (Singer, Somerset, UK) in which the MATα query strain was crossed en masse with the MATa yeast deletion mutant array in 384 colony format. After 2 days incubation at 30 °C final selections plates were then pinned to SC plates in which plates were arrayed so that each gene deletion was duplicated side by side in 384 format. Plates were then incubated at 30 °C overnight, before cells were transferred from colonies to liquid in 384 PekinElmer Cell Carrier plates containing DMSO (control) and drug (atorvastatin 35 μM) in alternate columns. After 4 hours of incubation, cell images were taken using a PerkinElmer Opera® High content microscope. The set up was as follows: 60× Water Immersion lens NA = 1.2 utilising 488 nm and 561 nm lasers with power of 4110 μW and 731 μW respectively. Filter for camera 1 (GFP): 520/35 nm, camera 2 (RFP): 600/40 nm, Detect Dichro: 568, Primary Dichro: 405/488/561/640. Images were captured utilising “binning” of 2 and exposure time of 400 milliseconds. The images were analysed using Acapella™ software to automatically identify cells using RFP cytoplasmic mCherry as described previously.11 Acapella SER Texture feature analysis allowed the observation of subtle intra-cellular changes of biological relevance that could be missed by looking at fluorescence intensity or location alone. SER texture features “Ridge”, “Bright”, and “Edge” were selected due to the fact that they best represented the correct localization of GFP-reporter (Pdr5p, and Yor1p) and gave reproducible results across the different screens.
Identification of possible regulators of pleiotropic drug resistance
In each experiment in the primary screens, each parameter Z scores from Texture analysis for each deletion were calculated.where:
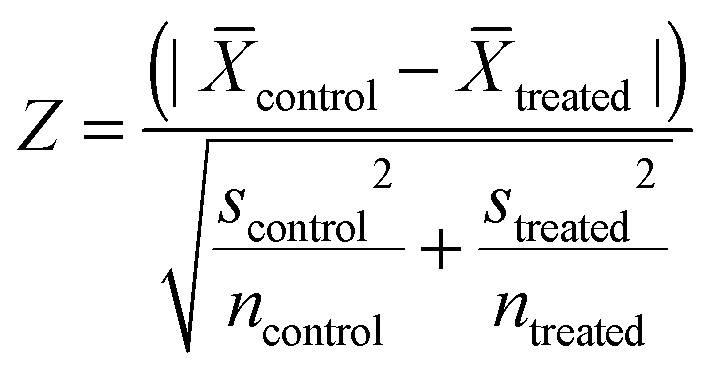
![[X with combining overline]](https://www.rsc.org/images/entities/i_char_0058_0305.gif) = the mean of texture properties (ridge, bright or edge) from texture analysis in Acapella software from each group (control or treated). s = standard deviation of the texture properties (ridge, bright or edge) from texture analysis in Acapella software from each group (control or treated). n = number of cells identified by Acapella software from each group (control or treated). Gene deletions with the Z score lower than 5‡ in 2 out of 3 texture analysis (“Ridge”, “Bright”, “Edge”) were counted as hits. Gene deletions that appeared as hits from 4 out of 5 screens were subjected to further validation.
= the mean of texture properties (ridge, bright or edge) from texture analysis in Acapella software from each group (control or treated). s = standard deviation of the texture properties (ridge, bright or edge) from texture analysis in Acapella software from each group (control or treated). n = number of cells identified by Acapella software from each group (control or treated). Gene deletions with the Z score lower than 5‡ in 2 out of 3 texture analysis (“Ridge”, “Bright”, “Edge”) were counted as hits. Gene deletions that appeared as hits from 4 out of 5 screens were subjected to further validation.
Spot dilution assay
All experiments were repeated 3 times.Yeast cells at a concentration of 5 × 108 cells per ml were serially diluted in water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10), and were 2 μl was spotted on YPD plates (unless stated differently) containing drugs of concentration stated in Table 4. Plates were incubated for 2 days at 30 °C and growth phenotype was observed for each strain.
:10), and were 2 μl was spotted on YPD plates (unless stated differently) containing drugs of concentration stated in Table 4. Plates were incubated for 2 days at 30 °C and growth phenotype was observed for each strain.
| Compound | Concentration used in Δxxx background | Concentration used in Δxxx Δpdr1Δpdr3 background |
|---|---|---|
| Cycloheximide (CHX) | 0.15 μg ml−1 | 40 nM |
| Ketoconazole | 1 μM | 0.5 μM |
| Fluconazole | 31 μM | 12 μM |
| Oligomycin (in YPGE plate) | 0.35, 0.5 μg ml−1 | 0.35 μg ml−1 |
| Benomyl (SC plate) | 60 μM with 25 mM HEPES | Was not used |
| Atorvastatin (SC plate) | 35 μM | 5 μM |
Yeast cell extract and Western blot
Cell lysate and protein extraction was conducted according to (Egner, Mahe et al., 1995).31 Cells from different strains were grown in YPD to the same OD600 of 2, and were then harvested by centrifuging at 4000 rpm for 2 minutes, then washed once with sterile distilled water. Cell extract was prepared by lysing the pellet with 150 μl of 1.85 M NaOH–7.5% 2-mercaptoethanol for 10 minutes on ice, followed by addition of 150 μl of 50% trichloroacetic acid and 10 minutes incubation on ice. The pellet was collected by centrifugation at 13000g for 3 minutes and then re-suspended in 10 μl of 1 M Tris base and 50 μl sample buffer (40 mM Tris-HCl pH 6.8, 8 M urea, 5% sodium dodecylsulfate (SDS), 0.1 M EDTA, 1% 2-mercaptoethanol, 0.01% bromophenol blue). Samples were then heated at 42 °C for 15 minutes, then 20 μl of each sample was loaded to a 7.5% polyacrylamide-SDS gel and electrophoresed for 2 hours at 200 V. The running buffer used was 3.3 g Tris-base, 14.4 g glycine, 1 g SDS, and distilled water added up to 1000 ml. Proteins were then transferred to an Immobilon™ transfer membrane (IPVH00010) pore size 0.45 μm which had been washed in methanol for 1–2 minutes, at 20 V for 17 hours. The transfer buffer used was 3.3 g Tris-base, 14.4 g glycine, 200 ml methanol, and distilled water added up to 1000 ml.
After 17 hours, the membrane was blocked with 5% bovine serum albumin (BSA) in 0.1% Tween in Tris-buffered saline (TBST) for 1 hour to reduce non-specific binding. Membrane was then washed with TBST for 10 minutes three times. The membrane was then incubated with a Pdr5p primary antibody (polyclonal Pdr5 (yN-18)) antibody raised in goats (Santa Cruz Co., California, USA stock #SC-27253) diluted 1:500 in 5% TBST for 3 hours at room temperature. The membrane was then washed with TBST for 10 minutes three times before being incubated with the secondary antibody, Donkey polyclonal antigoat antibody labelled with Cy5 obtained from abcam® ab6566 was diluted 1:4000 in 5% TBST for 1 hour in the dark.
The membrane was washed in Tris-buffered saline (TBS) for 10 minutes three times before scanning at 635 nm using Fujifilm FLA-5100 series fluorescent scanner. Pma1p/Pma2p, which are plasma membrane H+-ATPase were used as internal control. Primary antibody for Pma1p/Pma2p raised in rabbits was obtained from Santa Cruz, SC-33735 (diluted at 1:500). Pma1p/Pma2p primary antibody and Cy5 anti-rabbit antibody diluted at 1:4000 were incubated and detected after scanning for Pdr5p.
Conclusions
This is the first reporter screen that identifies upstream modulator networks around Pdr1p/Pdr3p. Our method can be adapted to many other cellular pathway systems to identify potential regulators, both activators and suppressors. The assay can be used to investigate subtle morphological changes caused by mutation or bioactive compounds in cells which might have been missed by less sensitive methods.By establishing genetic networks upstream of PDR1 PDR3, we can derive insights about pathways that modify or compensate the major pathway Pdr1p/Pdr3p-dependent PDR response. Typical of complex gene expression, manipulation of such modifying genes, in principle, will allow observation of specific phenotypes and hence a better understanding of complex drug resistance traits.
Acknowledgements
We would like to thank Dr Arun Kanakkanthara for help with Western blot assays. Prof. Scott Moye-Rowley, University of Iowa, for his helpful advice. The PDR deletion strains Δpdr1Δpdr3 were generated by Namal Coorey in this laboratory. We would like to thank the Maurice Wilkins Centre of the University of Auckland and the Foundation for Research Science and Technology for financial support.Notes and references
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- K. Gulshan and W. S. Moye-Rowley, Eukaryotic Cell, 2007, 6, 1933–1942 CrossRef CAS PubMed.
- Z. A. Kanafani and J. R. Perfect, Clin. Infect. Dis., 2008, 46, 120–128 CrossRef PubMed.
- P. Shahi, K. Gulshan, A. M. Näär and W. S. Moye-Rowley, Mol. Biol. Cell, 2010, 21, 2469–2482 CrossRef CAS PubMed.
- E. Balzi and A. Goffeau, J. Bioenerg. Biomembr., 1995, 27, 71–76 CrossRef CAS.
- Y. M. Mamnun, R. Pandjaitan, Y. Mahé, A. Delahodde and K. Kuchler, Mol. Microbiol., 2002, 46, 1429–1440 CrossRef CAS.
- S. Moye-Rowley, Prog. Nucleic Acid Res. Mol. Biol., 2003, 73, 251–279 CrossRef.
- J. K. Thakur, H. Arthanari, F. Yang, S.-J. Pan, X. Fan, J. Breger, D. P. Frueh, K. Gulshan, D. K. Li, E. Mylonakis, K. Struhl, W. S. Moye-Rowley, B. P. Cormack, G. Wagner and A. M. Naar, Nature, 2008, 452, 604–609 CrossRef CAS PubMed.
- V. Fardeau, G. l. Lelandais, A. Oldfield, H. l. n. Salin, S. Lemoine, M. Garcia, V. r. Tanty, S. p. Le Crom, C. Jacq and F. d. r. Devaux, J. Biol. Chem., 2007, 282, 5063–5074 CrossRef CAS PubMed.
- E. A. Winzeler, D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B. Andre, R. Bangham, R. Benito, J. D. Boeke, H. Bussey, A. M. Chu, C. Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury, S. H. Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M. Laub, H. Liao, N. Liebundguth, D. J. Lockhart, A. Lucau-Danila, M. Lussier, N. M'Rabet, P. Menard, M. Mittmann, C. Pai, C. Rebischung, J. L. Revuelta, L. Riles, C. J. Roberts, P. Ross-MacDonald, B. Scherens, M. Snyder, S. Sookhai-Mahadeo, R. K. Storms, S. Véronneau, M. Voet, G. Volckaert, T. R. Ward, R. Wysocki, G. S. Yen, K. Yu, K. Zimmermann, P. Philippsen, M. Johnston and R. W. Davis, Science, 1999, 285, 901–906 CrossRef CAS.
- P. W. Bircham, D. R. Maass, C. A. Roberts, P. Y. Kiew, Y. S. Low, M. Yegambaram, J. Matthews, C. A. Jack and P. H. Atkinson, Mol. BioSyst., 2011, 7, 2589–2598 RSC.
- J. A. Martens, J. Genereaux, A. Saleh and C. J. Brandl, J. Biol. Chem., 1996, 271, 15884–15890 CrossRef CAS PubMed.
- A. J. Prunuske, J. K. Waltner, P. Kuhn, B. Gu and E. A. Craig, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 472–477 CrossRef CAS PubMed.
- S. L. LaFayette, C. Collins, A. K. Zaas, W. A. Schell, M. Betancourt-Quiroz, A. A. L. Gunatilaka, J. R. Perfect and L. E. Cowen, PLoS Pathog., 2010, 6, e1001069 Search PubMed.
- A.-M. Alarco, I. Balan, D. Talibi, N. Mainville and M. Raymond, J. Biol. Chem., 1997, 272, 19304–19313 CrossRef CAS PubMed.
- A. Kolaczkowska and A. Goffeau, Drug Resist. Updates, 1999, 2, 403–414 CrossRef CAS PubMed.
- S. Tenreiro, A. R. Fernandes and I. Sá-Correia, Biochem. Biophys. Res. Commun., 2001, 280, 216–222 CrossRef CAS PubMed.
- F. Wendler, H. Bergler, K. Prutej, H. Jungwirth, G. Zisser, K. Kuchler and G. Högenauer, J. Biol. Chem., 1997, 272, 27091–27098 CrossRef CAS PubMed.
- A. Wu, J. A. Wemmie, N. P. Edgington, M. Goebl, J. L. Guevara and W. S. Moye-Rowley, J. Biol. Chem., 1993, 268, 18850–18858 CAS.
- B. J. Venters and B. F. Pugh, Genome Res., 2009, 19, 360–371 CrossRef CAS PubMed.
- S. Hahn and E. T. Young, Genetics, 2011, 189, 705–736 CrossRef CAS PubMed.
- G. Niederacher, E. Klopf and C. Schüller, Int. J. Mol. Sci., 2011, 12, 4758–4769 CrossRef CAS PubMed.
- D. Hnisz, M. Tscherner and K. Kuchler, Epigenomics, 2011, 3, 129–132 CrossRef CAS PubMed.
- E. Smith and A. Shilatifard, Mol. Cell, 2010, 40, 689–701 CrossRef CAS PubMed.
- C. Gao, L. Wang, E. Milgrom and W.-C. W. Shen, J. Biol. Chem., 2004, 279, 42677–42686 CrossRef CAS PubMed.
- J. Morschhäuser, K. S. Barker, T. T. Liu, J. Blaß-Warmuth, R. Homayouni and P. D. Rogers, PLoS Pathog., 2007, 3, e164 Search PubMed.
- M. Toth, I. M. Boros and E. Balint, Cancer Sci., 2012, 103, 659–669 CrossRef CAS PubMed.
- S. A. Wacker, B. R. Houghtaling, O. Elemento and T. M. Kapoor, Nat. Chem. Biol., 2012, 8, 235–237 CrossRef CAS PubMed.
- D. Amberg, D. Burke and J. Strathern, Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Cold Spring, 2005 edn, 2005 Search PubMed.
- A. H. Y. Tong, M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader, N. Pagé, M. Robinson, S. Raghibizadeh, C. W. V. Hogue, H. Bussey, B. Andrews, M. Tyers and C. Boone, Science, 2001, 294, 2364–2368 CrossRef CAS PubMed.
- R. Egner, Y. Mahé, R. Pandjaitan and K. Kuchler, Endocytosis and vacuolar degradation of the plasma membrane-localized Pdr5 ATP-binding cassette multidrug transporter in Saccharomyces cerevisiae, Mol. Cell. Biol., 1995, 15, 5879–5887 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mb70351g |
| ‡ 4.753 is the corrected Z value for multiple testing with equivalent significant level of 0.01. Correction for multiple testing was required because we compared control versus treated for 4500 genes = 4500 multiple tests. The new corrected p-value was 0.01/4500 = 0.000002. |
| This journal is © The Royal Society of Chemistry 2014 |