
Use of vacuum bagging for fabricating thermoplastic microfluidic devices†
Christopher L.
Cassano
a,
Andrew J.
Simon
a,
Wei
Liu
ab,
Carl
Fredrickson
a and
Z.
Hugh Fan
*acd
aInterdisciplinary Microsystems Group, Dept. of Mech. & Aerospace Eng., Univ. of Florida, P.O. Box 116250, Gainesville, FL 32611, USA. E-mail: hfan@ufl.edu
bSchool of Chem. and Chem. Eng., Shaanxi Normal Univ., Xi'an 710062, PR China
cJ. Crayton Pruitt Family Dept. of Biomed. Eng., Univ. of Florida, P.O. Box 116131, Gainesville, FL 32611, USA
dDept. of Chem., Univ. of Florida, P.O. Box 117200, Gainesville, FL 32611, USA
First published on 15th October 2014
Abstract
In this work we present a novel thermal bonding method for thermoplastic microfluidic devices. This simple method employs a modified vacuum bagging technique, a concept borrowed from the aerospace industry, to produce conventional thick substrate microfluidic devices, as well as multi-layer film devices. The bonds produced using this method are superior to those obtained using conventional thermal bonding methods, including thermal lamination, and are capable of sustaining burst pressures in excess of 550 kPa. To illustrate the utility of this method, thick substrate devices were produced, as well as a six-layer film device that incorporated several complex features.
Introduction
The vast majority of fabrication processes employed in the creation of polymer microfluidic substrates result in open channels, which must be bonded with another layer to create closed channels. The bond strength between the substrate and cover is of critical importance, since many of the analytical methods used in microfluidics require an imposed pressure gradient in order to function. Additionally, the type of bonding method employed can have a direct impact on the life span of a device (either in storage or in service), solvent compatibility, surface chemistry, and a host of other factors that may limit the robustness or usefulness of a particular device.Efforts have been made in developing various methods for bonding polymer devices, each with widely varying bond strengths and levels of difficulty in implementation.1 The bonding methods used for polymer devices fall into three broad categories: adhesive bonding,2,3 solvent bonding,4,5 and thermal bonding.6,7 A comparison among different bonding methods has been reviewed in the literature.1
Thermal bonding is a particularly attractive option for producing thermoplastic microfluidic devices for two reasons. First, the entanglement of polymer chains makes it possible to bond polymer substrates and obtain finished devices with bond strengths on par with the ultimate stress of the bulk material. Second, the absence of intermediate layers of adhesives allows devices to be constructed with homogeneous channel surfaces.
Vacuum bagging is a well-developed process,8,9 used across the aerospace and automotive industries, to manufacture high-performance laminated composite materials.10 The process of vacuum bagging employs a pressure differential acting across two sides of a flexible membrane to create a uniformly distributed load. In traditional composites work, this “clamping” force is used to evenly disperse and remove excess adhesive resin. This also forces conformal contact between the composite and the surface of a mold.
Since bonding of sheets requires a high level of surface contact between mating surfaces, the uniform clamping force produced by vacuum bagging lends itself to adaptation to produce strong bonds. Here, we report a simple and universal method to quickly produce strong polymer devices by adapting vacuum bagging for use with thermal bonding. We demonstrated this method for cyclic olefin copolymers (COC) using both “normal” two-layer thick substrate devices produced by hot embossing,11 as well as multi-layer film devices produced using a rapid prototyping approach. We also examined the effect of temperature on bond strength of traditional two layer devices, and illustrated the function of a complex multi-layer device – created with this bonding method – using a chemiluminescent flow injection assay.
Vacuum bagging process
The process used in vacuum bagging is straightforward (Fig. 1 and Fig. S1†), and it only requires a few minutes to set up and a handful of raw materials, resulting in a strong, optically transparent bond. While solvents or surface treatments could be incorporated into this method without significant alteration to the protocol, they are not required. This is especially important since the volatility of common solvents for bonding COC and other polymers place significant limitations on the useful time before alignment must be completed. As a result, vacuum bagging is considerably more forgiving than solvent-based approaches, which is one of the great benefits.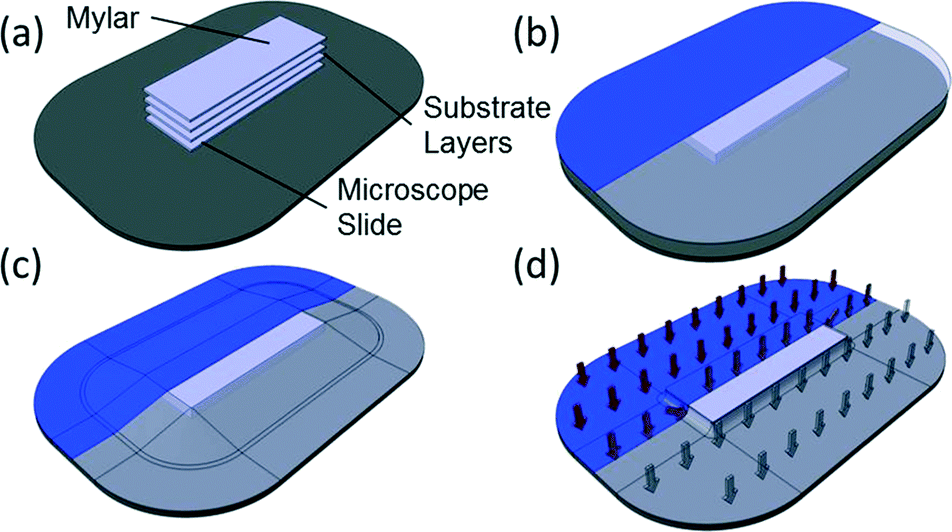 | ||
| Fig. 1 Assembly of a vacuum bag for device bonding. (a) Cleaned substrate layers are oriented and aligned. The aligned substrate layers are placed on top of a glass slide on the backing plate and covered with a layer of Mylar film (as a sacrificial layer). (b) The layers are covered by a sheet of flexible bagging film. (c) The bagging film is secured to the backing plate using vacuum sealant tape. (d) Vacuum is applied to the completed bag through the vacuum port, creating a distributed load across the entire surface of the vacuum bag. The whole assembly is then baked under vacuum. | ||
Despite being simple and forgiving there are several factors that must be considered in order to obtain a consistent bond and a high quality product. First, the cleanliness of the polymer layers is critical to obtaining a good bond. Interfacial polymer chain diffusion is central to the thermal bonding process, hence surface contaminants including solvents, ink, skin oil, or dirt/dust will interfere with the diffusion process and limit the strength of the bond, and in some cases completely inhibit it.
Second, a smooth contact surface between the vacuum bagging film and the devices is very important. As the internal pressure of the vacuum bag decreases the bagging film will begin to deform around any solid object within the bag. This is necessary, as it allows the distributed load to transfer evenly to the devices. However, as air is evacuated from the bag, the surface area of the bag decreases significantly (analogous to the surface area of a pyramid versus the area of its base). As the film presses down, wrinkles form where excess material gathers. Wrinkles that form above the devices will transfer into the device surface leaving marks in the surface layers. More importantly, they can also act as local stress concentrations, which can, in turn, result in channel deformations in that region. The best solution to this problem is to guide the wrinkles away from critical device areas. A Mylar film placed over the top of the devices as a sacrificial layer during bonding can also help improve the surface finish.
Third, for reasons that will be discussed later, vacuum intensity plays a significant role in the resulting strength of bonded devices. Improperly sealed vacuum bags and unstable vacuum contribute to batch-to-batch variability, which can be seen in the clustered failure pressures shown in Fig. S2b† for devices produced at 85 °C. In this case a small vacuum leak resulted in a change in burst pressure resulting from a decrease in wetting.
An equally significant problem arises from spatially varying vacuum within the vacuum bag, the results of which can be seen in Fig. S2b† for devices produced at 90 and 95 °C. As the distance from the vacuum source to the device increases the amount of vacuum decreases. This can be combatted, to some extent, by proper sealing of the vacuum bag and the use of permeable breather material, however, the best solution is to try to maintain uniform spacing between the devices and the vacuum source.
Finally, the combination of temperature (approaching the glass transition temperature, Tg), extended thermal soak time, and the distributed load can lead to permanent deformation or creeping12–14 of the polymer materials. Deformation is the root cause of channel collapse or channel shape distortion in thermally bonded microfluidic devices, and should be avoided. Unfortunately, deformation is in direct competition with bond strength, since both increase with increasing time, temperature, and applied pressure. For example, diffusion bond strength of polymers tends to increase as the fourth root of time,15 so very long thermal soaks lead to stronger bonds, however, in addition to being very inconvenient, it leads to significant deformation. As a result, deformation tends to be the limiting factor for bonding, and parameters must be chosen to minimize deformation while maximizing bond strength. This phenomenon is especially evident in wide channels, where the large unsupported cover layer is more susceptible to deform and deflect.
Practical starting points
Typical processes for COC and acrylic device fabrication can be found in the ESI,† but for other thermoplastic materials there are several key starting points that can be used to begin optimization. In practice, holding the bonding temperature 3–5 °C below the glass transition temperature yields a relatively strong bond without significant distortion. This is true for COC and acrylics, and would be a good starting point for optimization for other types of thermoplastics. If the glass transition temperature cannot be found, the Vicat softening point may be used instead, as it is typically several degrees lower than the glass transition temperature.A two-to-three hour thermal soak is a good starting point for optimization, and is generally sufficient to produce a high quality bond without causing significant deformation. A heat soak shorter than 2 hours usually results in a very weak bond resembling static cling. Heat soaks exceeding 6 hours tend to result in extensive deformation. For applications where deformation is a serious issue, another approach would be to use a staged bonding protocol consisting of a shorter duration heat soak under vacuum to allow for initial bonding and wetting, followed by re-pressurization, then a higher temperature and longer duration of heat soak to strengthen the bond.
Finally, higher vacuum strengths lead to stronger bonds, but it is not necessary to use an expensive ultra-high vacuum pump. It is our experience that strong, usable bonds are readily achieved with 80 to 90 kPa of vacuum, however, vacuum strengths less than 60 to 70 kPa result in poor bonding and should be avoided.
Tips and tricks
There are several tips that should make this process easier and more successful to employ. The most important thing to consider is that substrates must be completely clean and free from contaminants like dust, and especially skin oil. If the surfaces are not completely clean it will not be possible to use the low temperature and short heat soak that are necessary to avoid deformation.Devices must also be completely dry before bonding. Trapped moisture will expand during the bonding process and can lead to voids between the layers or a rough exterior surface (Fig. S3a†). Placing unbonded layers in a warm oven to drive off the excess moisture works very well for this purpose.
Corners and edges that are in direct contact with the vacuum bagging film deflect more than the center of a device (Fig. S3b†). This is usually a minor issue, and can be solved by using a caul plate to uniformly press against the surface and prevent the bagging material from deforming the edges, or substrates can be left sufficiently large that the edge deformation does not impact the flow channels.
If devices are not bonding properly, it can be tempting to increase the thermal soak time beyond four hours. However, strength improvements from the additional heat soak time are outpaced by deformation and usually yield very poor end results. Instead, if devices are not bonding properly, it is better to examine the process, including the vacuum seal or more carefully cleaning the substrates.
Finally, very wide channels with large unsupported spans tend to deflect significantly, even without the application of vacuum. These kinds of problems can be addressed by using a thicker cover sheet, which will naturally resist the load.
Bond strength
As mentioned before, vacuum bagging is capable of creating devices with bond strengths exceeding those of other thermal bonding methods. The large variety of COC and cyclic olefin polymer (COP) available, as well as the number of strength testing protocols used, make it difficult to make a direct comparison of relative bond strengths based solely upon the literature. Therefore, for the purposes of comparison, specimens were made using vacuum bonding and thermal lamination, followed by burst pressure tests. It can be seen in Fig. S2b,† that the resulting strength of vacuum bonded bi-material COC/COP devices is significantly stronger than similar laminated devices. It is also clear that the resulting devices are significantly stronger as the temperature approaches the Tg of the Zeonor® 1020R substrate. It is important to note that maintaining the heat soak temperature below the Tg of the thick substrate not only allows for a strong bond, but it also prevents deformation of the microchannels.There are three possible mechanisms by which the bond strength is increased compared to thermal lamination. First, a pressure-dependent depression in Tg might result in a shift from thermal diffusion bonding to thermal welding (melting) with an accompanying increase in bond strength. Second, the increased duration of the thermal treatment might increase the strength by allowing for increased diffusion. Third, improved surface contact and wetting between the layers might enhance the diffusion of polymer chains.
Among three possible reasons above, we did the following analysis. While the Tg of polymers can be pressure dependent,16,17 the slope of dTg/dP is typically small and positive.18 As a result, it is likely that applied pressure would result in an insignificant increase in Tg rather than a decrease, thus the applied pressure of the vacuum bag is not likely to cause the materials to fuse rather than diffuse. Consequently, a slow thermal diffusion bonding, as opposed to a rapid thermal weld, is more likely. It is also known that diffusion bonding is a temperature- and time-dependent process, hence elevated temperatures used in conjunction with increased soak times will result in a stronger bond. However, we found that vacuum bag failure occurring early in the process resulted in negligible bond strength, even at elevated temperatures. Therefore, it is believed that the most likely reason for the enhanced bond strength is the superior surface contact between polymer layers.
Thick substrate devices
Thermal bonding is particularly useful in the field of microfluidics, and is commonly used to produce devices with thick substrate channel layers.1 Unfortunately, the high temperatures and pressures used to bond devices result in a significant risk of channel deformation during bonding. At the same time, the low thermal conductivity of most plastics (three to four orders of magnitude less than metals) means that it takes a relatively long time for heat to diffuse to the bonding interface, which can be especially problematic if mechanical substrates of differing thicknesses are used.On the other hand, the relatively long duration and low-temperature of the heat soak, and small, uniform clamping force provided by the vacuum bag make it significantly easier to create thick substrate devices without channel deformation. In fact, in our experience, the quality of the bond, and absence of channel deformation, is relatively consistent across a wide range of vacuum strength and thermal soak durations, which makes this bonding method particularly useful.
To demonstrate the effectiveness of this method, a thick substrate device – used to conduct two-dimensional (2D) protein separation19 – was produced using two 1.5 mm-thick Zeonor® 1020R substrates. The significant thickness of the resulting device (Fig. 2a) would make it difficult to produce using other thermal bonding techniques without significant channel deformation. However, the channels of this device remain open and uniform. As can be seen in Fig. 2b, there are no leaks, voids, or collapses in the channels.
 | ||
| Fig. 2 A thick substrate device used for 2D electrophoresis. (a) Both the channel layer and the cover have been made out of 1.5 mm thick COC and bonded together using a vacuum bag. (b) Fluorescent micrograph of a part of the device after introducing 5 μM fluorescein. | ||
Multi-layer film devices
In addition to the ability to create thick devices, vacuum bagging completely eliminates time constraints resulting from using solvents, adhesives, or surface treatments, making it possible to align a large number of layers. These features make vacuum bonding amenable to creating multi-layer film devices, effectively enabling quasi-additive manufacturing. To illustrate this unique capability of vacuum bag bonding, a six-layer microfluidic device (Fig. 3) was created. | ||
| Fig. 3 Layout of a 6-layer vacuum-bonded film device used for chemiluminescent detection. (a) Individual film layers are cut using a digital craft cutter. This production technique allows the integration of both 3D features, such as the transverse groove mixers (layer 5), as well as a 3D flow path, demonstrated by the two overlaid serpentine channel layers which serve as both mixers and detection zones (layers 2 and 4). (b) Order and orientation of the film layers. (c) Solid model of an assembled device. (d) A completed device with threaded interfaces attached. | ||
To demonstrate the tremendous flexibility of the quasi-additive manufacturing process, several useful, complex features were incorporated into the design of the device. These include transverse groove mixers and a three-dimensional flow path and detection zone (Fig. 3c and Fig. S4a†). This device has been used to conduct a flow injection assay, the results of which can be found in the ESI.†
Conclusions
A novel method for bonding thermoplastic devices has been presented, which is based on the vacuum bagging procedure used by the aerospace industry. This method produces strong bonds – without oxygen plasma or solvents – and can be used to make thick substrate or multi-layer film devices. We demonstrated the method for bonding either COC or poly(methyl methacrylate) (PMMA), but it should be applicable to other thermoplastic materials. This method is, in many cases, superior to other thermal bonding approaches, while being significantly simpler, and more forgiving to accomplish than solvent or adhesive bonding methods. Although this method is not scalable to roll-to-roll lamination, it will offer stronger bonding, ability to laminate at a lower temperature, and single-step assembly of multiple layers.Acknowledgements
This work is supported in part by National Institute of Health (R21GM103535 and K25CA149080), National Science Foundation (OISE-0968313), and the University of Florida. We would also like to thank Mr. Harvey Freitag for his help in producing laminated devices.Notes and references
- C.-W. Tsao and D. DeVoe, Microfluid. Nanofluid., 2009, 6, 1–16 CrossRef CAS.
- J. Do and C. H. Ahn, Lab Chip, 2008, 8, 542–549 RSC.
- B. R. Flachsbart, K. Wong, J. M. Iannacone, E. N. Abante, R. L. Vlach, P. A. Rauchfuss, P. W. Bohn, J. V. Sweedler and M. A. Shannon, Lab Chip, 2006, 6, 667–674 RSC.
- K. W. Ro, J. Liu and D. R. Knapp, J. Chromatogr. A, 2006, 1111, 40–47 CrossRef CAS PubMed.
- G. Chen, F. Svec and D. R. Knapp, Lab Chip, 2008, 8, 1198–1204 RSC.
- M. F. Bedair and R. D. Oleschuk, Anal. Chem., 2006, 78, 1130–1138 CrossRef CAS PubMed.
- C. K. Fredrickson, Z. Xia, C. Das, R. Ferguson, F. T. Tavares and Z. H. Fan, J. Microelectromech. Syst., 2006, 15, 1060–1068 CrossRef CAS.
- J. Summerscales and T. J. Searle, Proc. Inst. Mech. Eng., Part L, 2005, 219, 45–58 Search PubMed.
- C. Williams, J. Summerscales and S. Grove, Composites, Part A, 1996, 27, 517–524 CrossRef.
- L. G. Stringer, Composites, 1989, 20, 441–452 CrossRef CAS.
- K. Liu, P. Gu, K. Hamaker and Z. H. Fan, J. Colloid Interface Sci., 2012, 365, 289–295 CrossRef CAS PubMed.
- P. Gu, T. Nishida and Z. H. Fan, Electrophoresis, 2014, 35, 289–297 CrossRef CAS PubMed.
- Y. Luo, M. Xu, X. D. Wang and C. Liu, J. Phys.: Conf. Ser., 2006, 48, 1102 CrossRef.
- M. Heckele and W. K. Schomburg, J. Micromech. Microeng., 2004, 14, R1 CrossRef CAS.
- Y. M. Boiko and R. E. Prud'Homme, Macromolecules, 1997, 30, 3708–3710 CrossRef CAS.
- U. Bianchi, A. Turturro and G. Basile, J. Phys. Chem., 1967, 71, 3555–3558 CrossRef CAS.
- H. A. Schneider, B. Rudolf, K. Karlou and H. J. Cantow, Polym. Bull., 1994, 32, 645–652 CrossRef CAS.
- A. Quach and R. Simha, J. Appl. Phys., 1971, 42, 4592–4606 CrossRef CAS PubMed.
- C. Das, J. Zhang, N. D. Denslow and Z. H. Fan, Lab Chip, 2007, 7, 1806–1812 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: materials and reagents, chemiluminescent assay procedure, device fabrication procedure, and burst pressure testing. See DOI: 10.1039/c4lc00927d |
| This journal is © The Royal Society of Chemistry 2015 |