
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of fast protein phosphorylation kinetics in single cells on a microfluidic chip†
Matthias
Blazek
ab,
Tomas Silva
Santisteban
ab,
Roland
Zengerle
bc and
Matthias
Meier
*ab
aMicrofluidic and Biological Engineering, Department of Microsystems Engineering - IMTEK, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: matthias.meier@imtek.de
bCentre for Biological Signalling Studies - BIOSS, University of Freiburg, Germany
cLaboratory for MEMS Applications, Department of Microsystems Engineering - IMTEK, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany
First published on 14th November 2014
Abstract
In the present study, we developed a microfluidic large-scale integration (mLSI) platform for the temporal and chemical control of cell cultures to study fast kinetics of protein phosphorylation. For in situ protein analysis the mLSI chip integrates the Proximity Ligation Assay (PLA). To investigate cell-signaling events with a time resolution of a few seconds we first engineered and optimized the fluidic layout of the chip with 128 individual addressable cell culture chambers. The functionality of the cell culture operations and PLA is demonstrated by the determination of the minimum cell sample size for obtaining robust quantitative PLA signals at the single-cell level. We show that at least 350 cells per assay condition are required to statistically evaluate single cell PLA data. In the following we used the PLA chip with over 500 hundred cells per condition to record sequential phosphorylation reactions of the canonical protein kinase within the Akt pathway, which is activated in various human cancer types. This was achieved by stimulating mouse fibroblast cell cultures with either the platelet-derived growth factor (PDGF) or insulin-like growth factor (IGF-1). Fluidic cell stimulation pulses of 5 seconds were followed by precisely time shifted cell fixation pulses to obtain a temporal resolution of 10 seconds. PLA was then performed on all fixed arrays of cell cultures to extract the characteristic phosphorylation times at the single cell level for either the PDGF, or IGF-1 receptor and the Akt and GSK3β kinases. Characteristic phosphorylation times for the receptors were between 13 and 35 seconds, whereas for downstream kinases between 25 and 200 seconds. Thus we could reveal a molecular order of the phosphorylation reactions during the signal transduction through the Akt pathway. In dependence of the stimulus we found a temporal difference for the characteristic phosphorylation time of 20 and 150 seconds for the Ser-473 and Thr-308 residues on the Akt kinase, respectively. Temporal alteration of sequential phosphorylation reactions on Akt has been proposed as molecular mechanism to differentiate between stimuli and biophysically determined in the present study.
Introduction
Cells are information-processing devices. A predominant molecular mechanism for transferring signals from the extracellular microenvironment to the inner compartments of cells governs the sequential transfer and removal of phosphorus groups from proteins.1 Kinases and phosphatases are proteins that catalyze the phosphorylation and reverse de-phosphorylation reaction, respectively. Approaches for screening large-scale protein phosphorylation reactions,2,3 in combination with individual protein characterization studies, have led to the construction of complex physical maps of the phosphorylation network,4 involving the connections between kinases, phosphatases, and their substrates. Despite the high availability of phosphorylation data, our current understanding about the signal transduction remains static. One reason for this is the lack of quantitative detection technologies for probing protein phosphorylation in single cells with high sensitivity, specificity, and throughput.Recent advances of standard protein phosphorylation assay technologies, for example, western blotting analysis, mass spectrometry, and fluorescence-activated cell sorting (FACS) technique, start to fill this technical gap. An example of this progress is the miniaturization of the classical western blot assay onto a microfluidic chip,5 which leads to an increase in sensitivity and thus widens the scope of applications for analysis of single cells.6 Despite these promising advances, to obtain quantitative protein phosphorylation data still remains a challenge. More importantly, none of the existing techniques offer the temporal resolution required for characterizing the fast kinetics of protein phosphorylation reactions. This is due to an interrupted technical workflow between the sample preparation and protein analytical assay. Phosphorylation reactions in response to a cell stimulus occur in the second-to-minute time regime.7 Manually performed in situ cell experiments with standard flasks have reliable time resolutions of only one minute. Furthermore, the low number of repeats gives rise to large variances in the in situ experiments.
An alternative assay technology for quantifying proteins, their interactions, and posttranslational modifications is proximity ligation assay (PLA).8,9 PLA is an immuno-based assay, in which two oligonucleotide-labeled antibodies (Ab's) are primed to the same target at different epitopes. The proximity of the two Ab's is tested by a hybridization reaction of the oligonucleotide labels. A circular DNA template between the antibody labels and two additional oligonucleotide strands is formed. The circular DNA template is ligated and amplified by isothermal rolling circle amplification. As a result, a DNA polymer with a diameter of about 0.5 μm is generated at the cellular location of the protein event. The DNA polymer is stained with a fluorescent probe molecule. Although the in situ technology offers high sensitivity and specificity paired with cellular localization information, its throughput is severely limited by the complexity of the biochemical steps and end-point detection mode.
To overcome these drawbacks and to enable the PLA technology to quantify the protein phosphorylation dynamics, we integrated the PLA approach onto a chip in the previous study.10 The microfluidic large-scale integration11 (mLSI) technique combined the PLA platform with an array of independent cell culture chambers. The microchambers containing the cell cultures were arranged in a matrix format and allowed the screening of two independent parameters such as stimulation time and protein targets in PLA, in one in situ cell signaling experiment. Although the first-generation PLA platform could reveal the dynamics of protein phosphorylation in response to a cell stimulus, its quantification performance was hampered by low cell count statistics and a low time resolution of a few minutes.
Herein, we designed and engineered an mLSI platform consisting of individually addressable cell culture chambers in a serial configuration by using polydimethylsiloxane (PDMS). Different from the first PLA chip, the new mLSI chip was designed and optimized for tracing the response kinetics of protein phosphorylation events in the order of seconds. To demonstrate the advances in automation, miniaturization, and parallelization of the chip, we resolved the phosphorylation kinetics of four phosphorylation sites within the Akt signaling pathway upon the cell stimulation with different growth factors. The observed protein phosphorylation targets and their positions in the signaling cascade are summarized in Fig. 1. Of particular interest in this study is the double phosphorylation of the Akt kinase, which is an eponymous element of the pathway. The activation of the Akt kinase is a hallmark for several types of human cancer.12,13 It has been shown that the full activation of Akt kinase requires the phosphorylation of its residues Ser-473 and Thr-308.14 The temporal order of Akt phosphorylation remains a long-standing research question.15–17 The time traces of the Akt kinase phosphorylation recorded by the PLA chip in this study settled this question and enabled the application of this strategy for high-content cell-signaling studies.
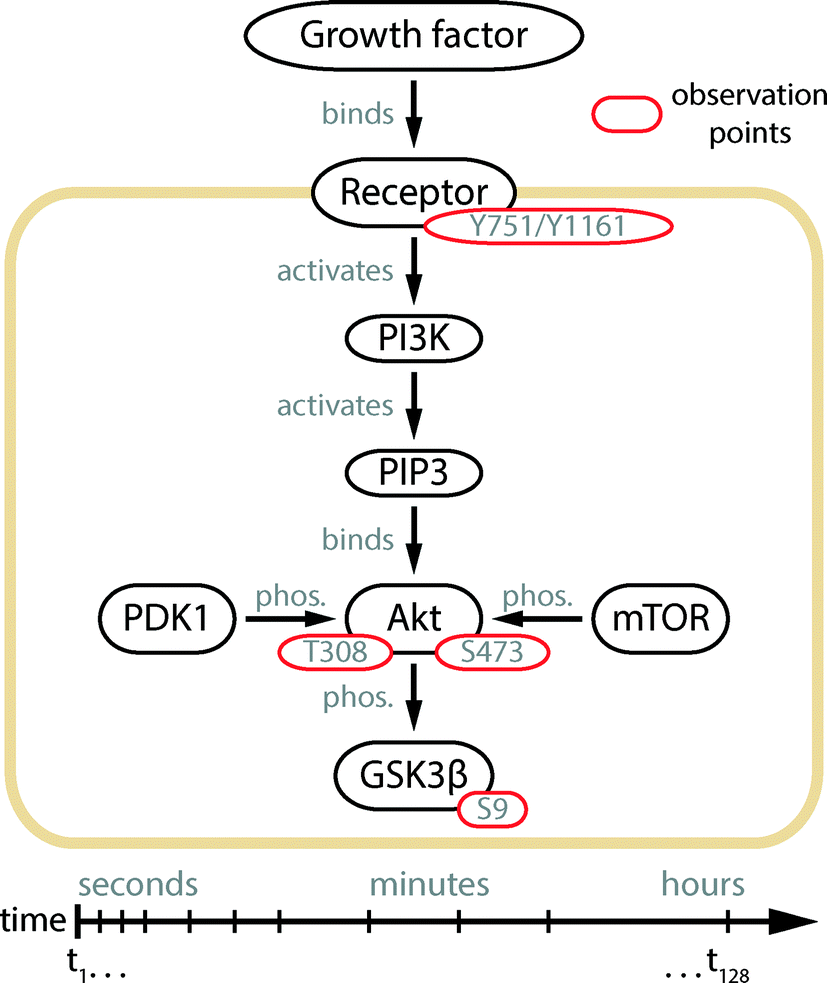 | ||
| Fig. 1 Illustration of the Akt signal transduction pathway.14 Extracellular signals are transmitted to the interior of the cell by a protein phosphorylation cascade starting at the cell membrane receptor. The substrate binding leads to the auto-phosphorylation of the receptor protein and activation of its kinase function. In the case of PDGF and IGF-1 receptors, the residues Tyr-751 and Tyr-1161 are auto-phosphorylated, respectively. The phosphorylation signal is passed to the phosphatidylinositol 3 kinase (PI3K), which produces phosphatidylinositol (3,4,5)-triphosphate (PIP3) at the inner surface of the cell membrane. In the following steps, Akt is recruited to the membrane and phosphorylated at its residues Thr-308 and Ser-473, by the kinases PDK118 and mTOR.16 Upon double phosphorylation, the kinase domain of Akt gains full activity and distributes the signal downstream to over a hundred substrates,14 where GSK3β is one of them.19 Yellow, black, and red enclosures indicate the cell membrane, proteins, and their phosphorylation sites, respectively. | ||
Experimental
Chip fabrication
PDMS chips were manufactured using the standard soft lithography methods20 for two-layer devices.11 Briefly, SU-8 3050 (MicroChem) and AZ 40 XT photoresists (MicroChem) were used for fabricating the molds of the cell culture chambers and connecting channel network in the flow layer, respectively. The mold for the control channels was fabricated by using SU-8 3025 (MicroChem). All features on the molds were 25 μm in height, except the cell culture chambers, which were 50 μm high. Chips were replicated from the molds by using Sylgard 184 PDMS (Dow Corning). The flow and control layers were bonded together using an off-ratio procedure. Pneumatic membrane valves of the chip were constructed in a pushdown configuration. In a final step the assembled PDMS chip was bonded to a glass carrier (Brain Laboratories) through oxygen plasma activation.Chip operation
The PDMS chips were mounted inside an on-stage microscopic cell incubator. Cell culturing media and buffers were stored in glass containers and supplied to the chip through Tygon tubings (Saint-Gobain). Assay reagents or water for the control channels were stored in Tygon tubings, which were directly inserted into the chip. All control and flow lines were separately pressurized via a solenoid valve at 200 and 40 kPa, respectively. A graphical user interface (GUI) within the Matlab programming environment (Mathworks) was used for automating the opening and closing cycles of the solenoid valves and for activating either the on-chip pneumatic membrane valves or the flow of the assay reagents.Cell culture
NIH3T3 mouse fibroblast cells (DMSZ) were cultured off-chip in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% PenStrep (all from Life Technologies) by following the standard procedures (ATCC). Cells were harvested and re-suspended at a concentration of 5 × 106 cells ml−1. Before seeding the cell suspension, all cell culture chambers in the PDMS chip were coated with a fibronectin (Sigma Aldrich) solution of 0.05%. Cells were cultured on-chip with full medium in a 5% CO2 atmosphere at 37 °C for 6 h and then starved under low-FBS conditions, i.e., 0.1% FBS, for 12 h. Medium exchange at every 90 min was automated by the protocols through the Matlab GUI. Cells were stimulated with platelet-derived growth factor PDGF-BB (Prospec Technogene) or insulin-like growth factor IGF-1 (Sigma Aldrich) at a concentration of 100 ng ml−1 or 200 ng ml−1, respectively. Both growth factors were diluted with cell starvation medium. After stimulation, the cells were fixed with 4% formaldehyde (ThermoFisher Scientific) for 16 min at room temperature and permeabilized with 0.05% Tween 20 (Sigma Aldrich) for 3 min.On-chip proximity ligation assay
Fixed cells were blocked (Olink) on-chip for 1 h. Next, monoclonal primary antibodies (Cell Signaling Technology) were diluted with antibody diluent (Olink) and incubated with the cell samples for 12 h with refresh cycles repeated every 2 h. The dilution ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 for Akt (Ser-473) (Cell Signaling, 4060), Akt (Thr-308) (Cell Signaling, 2965), and GSK-3β (Ser-9) (Cell Signaling, 9323), 1:25 for the PDGF β receptor (Tyr-751) (Cell Signaling, 3161), and 1:250 for the IGF-1 receptor (Tyr-1161) (Santa Cruz Biotechnology). Anti-rabbit PLA probes (anti-rabbit Plus and Minus, Olink) were diluted with Duolink antibody diluent at a ratio of 1:5 and incubated at room temperature for 2 h. DNA connectors (Olink) with the T4 ligase (Fermentas) were incubated at 32 °C for 1 h. Amplification reagents (Olink) were diluted according to the vendor's instructions and incubated at 32 °C for 2 h. Cell nuclei and cytoskeletons were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma Aldrich) and Phalloidin-Atto 488 (Sigma Aldrich), respectively.
:50 for Akt (Ser-473) (Cell Signaling, 4060), Akt (Thr-308) (Cell Signaling, 2965), and GSK-3β (Ser-9) (Cell Signaling, 9323), 1:25 for the PDGF β receptor (Tyr-751) (Cell Signaling, 3161), and 1:250 for the IGF-1 receptor (Tyr-1161) (Santa Cruz Biotechnology). Anti-rabbit PLA probes (anti-rabbit Plus and Minus, Olink) were diluted with Duolink antibody diluent at a ratio of 1:5 and incubated at room temperature for 2 h. DNA connectors (Olink) with the T4 ligase (Fermentas) were incubated at 32 °C for 1 h. Amplification reagents (Olink) were diluted according to the vendor's instructions and incubated at 32 °C for 2 h. Cell nuclei and cytoskeletons were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma Aldrich) and Phalloidin-Atto 488 (Sigma Aldrich), respectively.
Automation of the chip workflow
Two consecutive and automated protocols were developed for performing the cell stimulation experiments coupled with the PLA process. The on-chip valve configuration and flow pressure in each step was activated with the aid of a multichannel pressure-control system21 equipped with solenoid valves and a programmable interface. The flow pressure was fixed to 40 kPa in all following stimulation experiments. The first protocol included the steps: (1) coating of the glass channel surface with fibronectin, (2) cell seeding and culturing, (3) serum-starvation of the cells, (4) growth factor stimulation, and (5) cell fixation. The protocol consisted of 3200 automated flush steps completed over a period of 18 h. After the fixation of cells, the second protocol, including all PLA steps, was executed as follows: (6) cell permeabilization, (7) antigen blocking, (8) target detection with DNA-labeled antibodies, (9) ligation of the circular DNA template, (10) rolling circle amplification, and (11) fluorescence staining. The second protocol contained 3600 flush steps executed over a time period of 18 h. The detailed flush cycles, times, and temperatures are summarized in Table S1,† whereas the reagent compositions are given above.Image acquisition and analysis
Fluorescent images were acquired using the Axio Observer inverted microscope equipped with a 20× Plan Apochromat objective, AxioCam MRm camera, and Zen microscope software (all from Zeiss). Images were taken at eight positions in each cell culture chamber; and the filter sets of 43HE, 38HE, and 49 were used for the fluorescently stained PLA dots, cytoskeletons, and nuclei, respectively. The integration times were constant over the entire imaging procedure. The image analysis process, including cell segmentation and PLA dot identification, was performed by using Matlab image processing toolbox (Mathworks).Results
Microfluidic large-scale integration platform
The design of the mLSI chip with PLA functionality for recording in situ protein phosphorylation reactions is illustrated in Fig. 2a. The two-layer PDMS chip was manufactured by a rapid prototyping procedure.20 In the lower PDMS layer, a fluidic channel network addresses the 128 parallel-arranged cell culture chambers (blue). Each microchamber occupies an area of 0.5 mm2 and volume of 25 nl. The microchambers are organized into 16 logic blocks, and each consists of 8 chambers (Fig. 2a inset). Fluids were routed from the multiplexed inlet ports to individual cell culture chambers by 1622 pneumatic membranes valves. The corresponding control channel network was implemented into the top PDMS layer. The assembled PDMS chips were bonded to a glass carrier, which concomitantly provided the functional substrate for cell cultures.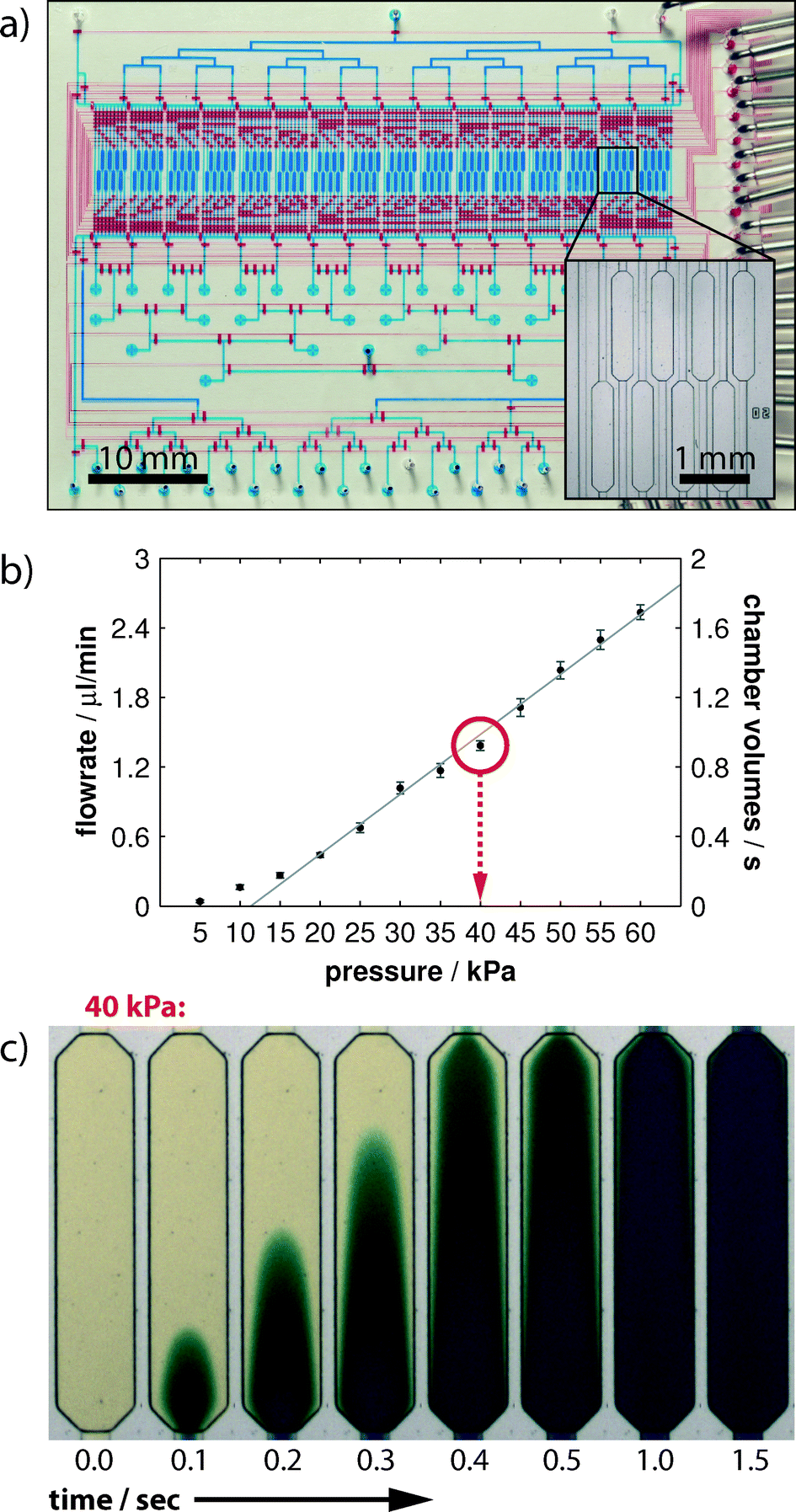 | ||
| Fig. 2 Microfluidic PLA-Chip design and fluid characterization. a) Layout of the fluidic chip. For the purpose of illustration, the microchannels of the control and flow layers are filled with red and blue aqueous solutions, respectively. The entire device covers an area of 65 × 48 mm2, within which the 128-cell culture chambers constitute the central elements. Scale bar denotes 10 mm. Inset: zoomed-in view of the microchambers for cell cultures. Scale bar denotes 1 mm. b) Plot of the volume flow rate from one inlet port through a cell culture chamber to the outlet against the applied flow pressure. A flow pressure of 40 kPa ensures the exchange of about one cell chamber volume (25 nl) within 1 s. c) Time-lapsed image series of colored aqueous solution streaming through one cell culture chamber at an applied flow pressure of 40 kPa. | ||
The theoretical time resolution for an on-chip cell stimulation experiment is given by the volume exchange time of one cell culture chamber. Therefore, we measured the rate of volume flow through the PLA chip in dependence of the applied flow pressure. During the measurements, the fluid was directed from the inlet ports through the channel network and one cell culture chamber to the outlet port. The expected linear dependence of volume flow rates (left axis) and applied flow pressures is shown in Fig. 2b. Error bars indicate one standard deviation from six independent experiments performed in different cell culture chambers. With the given volume of one cell culture chamber (25 nl), we calculated the volume exchange rate of one cell culture chamber and exchange time (in seconds) at different applied flow pressures (right axis). The exchange time of one chamber volume was one second at a flow pressure of 40 kPa. We further confirmed this value by performing time-lapse imaging of a cell culture chamber by using colored aqueous solutions. As shown in Fig. 2c, the fluid stream flowing through the chamber exhibited the characteristic Poiseuille flow profile from the time points of 0.1 s to 1.0 s. After 1.5 s, the fluid near the chamber side walls was completely stained and showed no difference in color intensity from the center region. The fluidic resistance from the inlet ports through each cell culture chamber to the outlet port was designed to be equal. Because the cell stimulation experiment with the subsequent cell fixation process requires the operation of two fluid routes, the highest achievable time resolution is therefore twice that of the volume exchange rate.
Statistics for robust single-cell PLA analysis
One of the advantages of the PLA technology is its ability to produce inherent single cell information. Single-cell information gives insight into the biological cell-to-cell variability, which is especially useful for the characterization of heterogeneities and robustness of signaling transduction.22,23 Quantitative protein information from single cells can be obtained through image analysis process, including cell segmentation, PLA dot identification, and dot-assignment to cell areas. However, the number of cells that are sampled with PLA dots for obtaining reproducible and statistically reliable single-cell PLA results remains unclear. From the engineering point of view, this number also concomitantly delimits the minimum size for the cell culture chamber.We addressed this question by systematically evaluating a large sample of mouse fibroblast cells (NIH-3T3) cultured in all 128 microchambers. Each chamber was seeded with about 400 cells, which, on average, covered 40% of the microchamber surface. Before fixation, cells were stimulated with 200 ng ml−1 IGF-1 for 10 min on-chip. IGF-1 triggered the Akt phosphorylation cascade. During the following PLA analysis, we targeted the Thr-308 phosphorylation site of Akt in all cells. A total number of 1024 microscopic images were acquired and analyzed for extracting the single-cell PLA dot distributions. Fig. 3b shows the distribution of the PLA dot counts of the phosphorylated Akt measured with more than 40000 cells. The mean PLA dot count per cell was 172 ± 76.
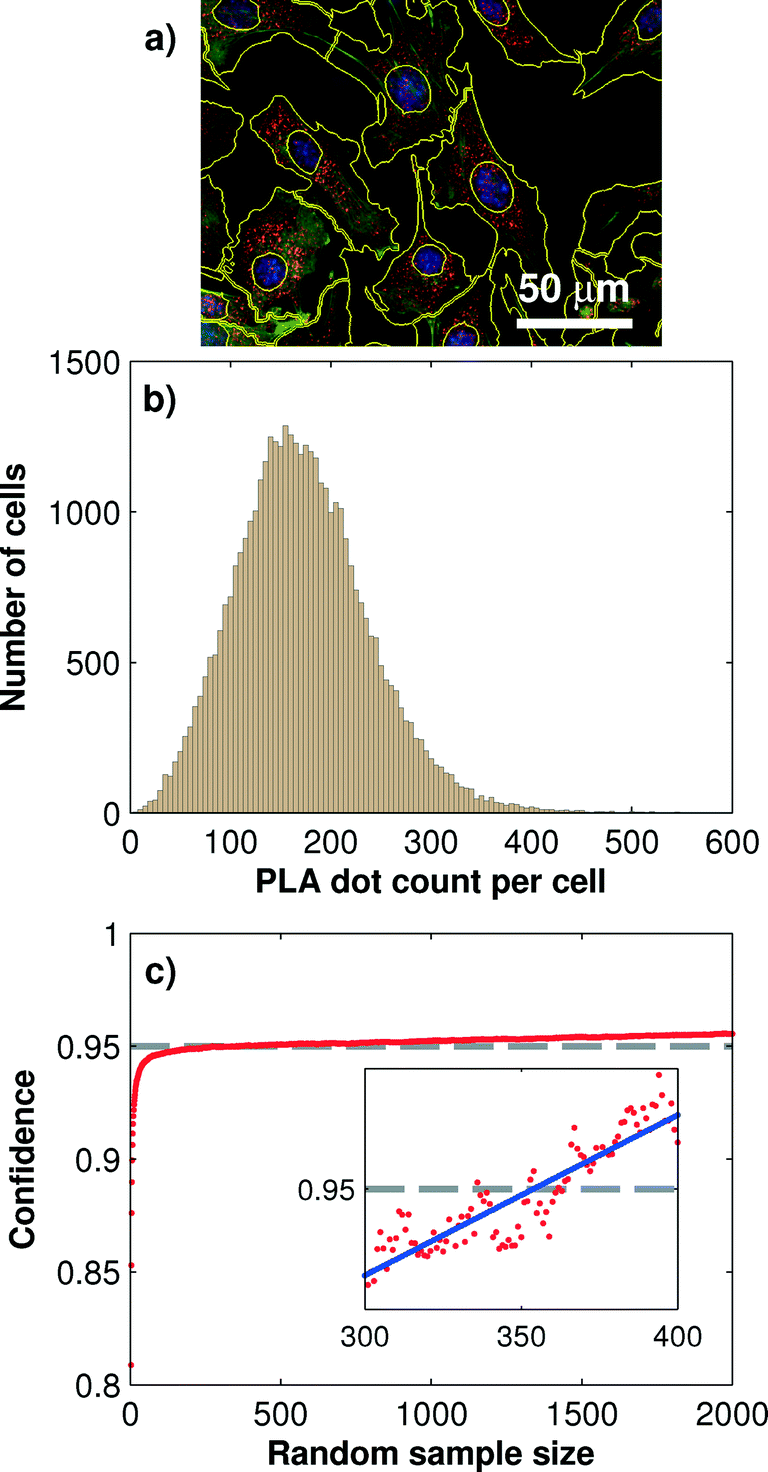 | ||
| Fig. 3 Statistical evaluation of the minimum cell sample size required for robust single-cell PLA results. a) Exemplary image of PLA results and single cell segmentation. b) PLA dot count distribution for the phosphorylated Akt at the Thr-308 residue obtained from 40000 single cells cultured under the same conditions. c) Monte Carlo simulation of the 95% confidence interval for different sample sizes. The current statistical setup indicates a recommended sample size of at least 350 cells per assay. | ||
To determine the minimum cell sample size for obtaining a robust on-chip PLA result, we performed a Monte Carlo simulation, which generated random sub-samples of the full data set. For each sub-sample, the arithmetic mean value, standard deviation, and 95% confidence interval, I95C, of the PLA result can be calculated by using the following equation:
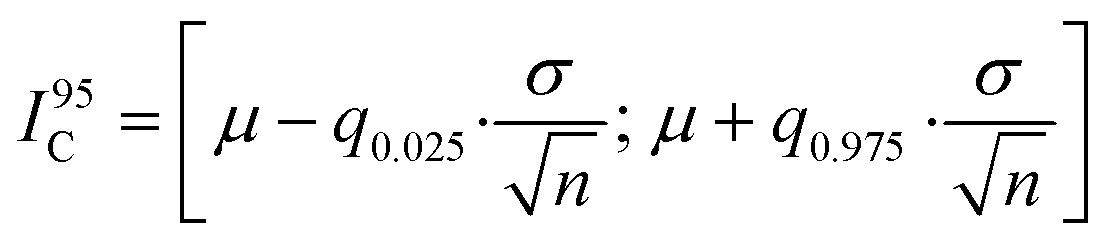
The minimum cell sample size depends on the absolute PLA dot count numbers per cell, which in turn is a function of the antibody concentration, affinity, incubation time, and/or fixation method used in the PLA experiment. To obtain a more general result for the minimal cell sample size we repeated the above analysis for three different protein targets in its phosphorylation state. The results are summarized in Table 1. It can be observed that with decreasing mean PLA dot count per cell the minimum cell sample size is increasing. At the lowest tested mean PLA dot count of 33 the minimum cell sample size was 470. At a cell confluency of 80%, about 800 fibroblast cells can be cultured in each of the 128 microchambers, and this number exceeds about twice the amount required for achieving the 95% confidence interval for single-cell PLA experiments.
| Antibody target | Cell sample size | PLA dots/cell | 95% confidence level |
|---|---|---|---|
| Akt Thr-308 | 40000 |
172 ± 76 | 350 |
| Akt Ser-473 | 24000 |
113 ± 58 | 320 |
| GSK3β Ser-9 | 18000 |
140 ± 66 | 290 |
| PDGFR Tyr-751 | 30000 |
33 ± 17 | 470 |
Fast phosphorylation kinetics of the Akt signaling pathway
In the next step, we recorded the phosphorylation kinetics of the Akt pathway at four observation points (see Fig. 1) upon cell stimulation with the growth factors PDGF and IGF-1. The phosphorylation rates of the receptors depend on the amount of growth factors in the microenvironment.24,25 Half-maximum activation concentrations (EC50) for the phosphorylation reaction of both PDGF and IGF-1 receptors are reported to be between 1 ng ml−1 and 10 ng ml−1.24–27 The downstream phosphorylation reactions of Akt are reported to be saturated at PDGF and IGF-1 concentrations of about 30 ng ml−1 and 75 ng ml−1, respectively.28,29 For comparing the phosphorylation kinetics triggered by the two growth factors, cells were stimulated with a growth factor three times higher than the signal saturation concentration of the hormones.Next, we exploited the cell culture chambers in the PLA chip to generate a time trajectory for one protein phosphorylation reaction. To achieve this, the cultured cells were starved with a serum-free media (0.1% FBS) for 12 h and then stimulated with a fluid pulse containing the growth factor. The fixation of the cells at different time points after stimulation maintained the molecular signaling response within the cells. We recorded the response with a time resolution of 10 s during the first 8 min after stimulation, from which onwards the time gap was increased to cover a time-frame between 30 min and 60 min depending on the protein target. The array of 128 fixed cell cultures was then subjected to the PLA assay. The automated imaging process and image analysis resulted in the time trajectories of single-cell PLA dot count distributions, as shown in Fig. 4.
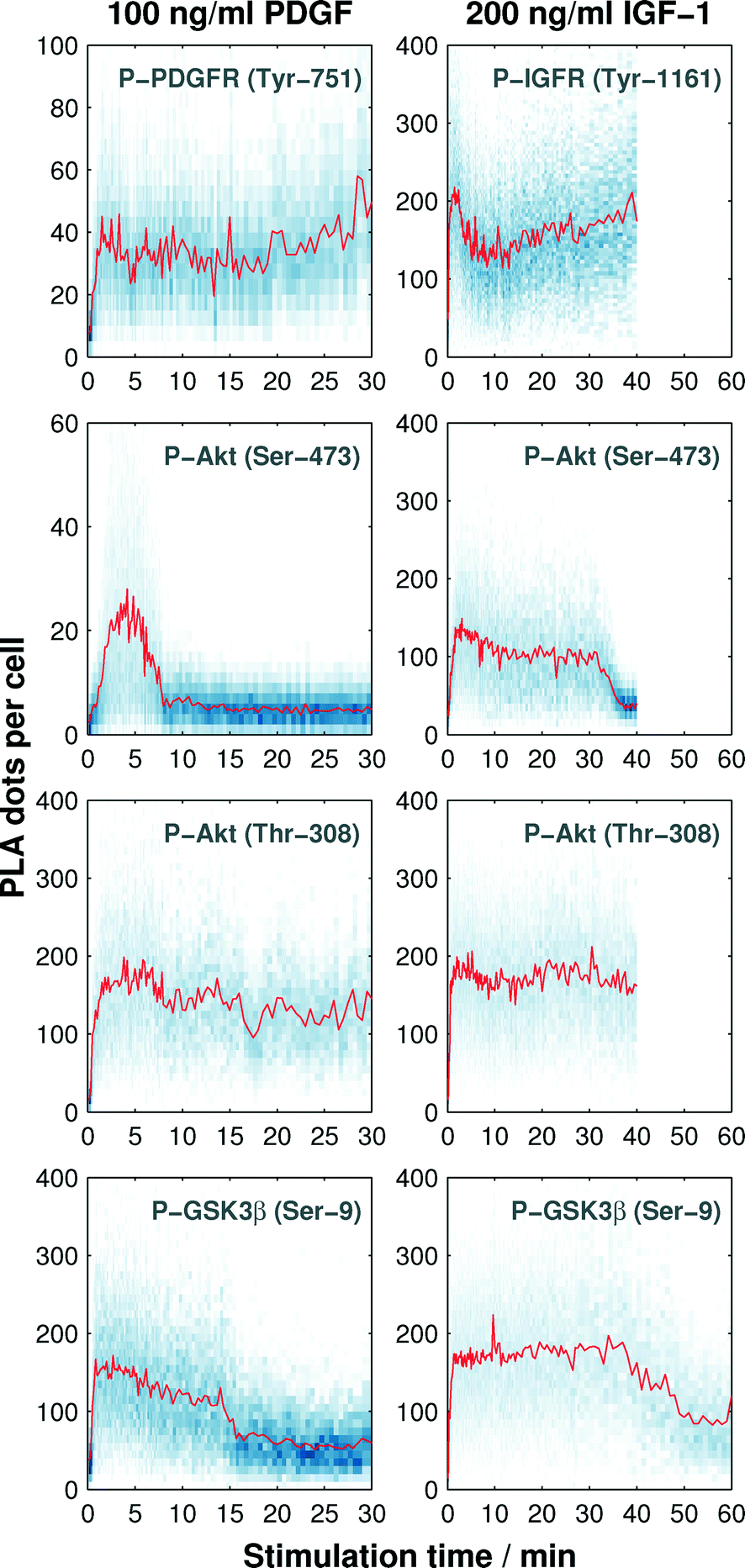 | ||
| Fig. 4 Time trajectories of single-cell PLA dot count distributions for the evaluation of protein phosphorylation kinetics. Left columns show the phosphorylation kinetics of the PDGF receptor and three kinases within the Akt pathway upon PDGF stimulation, and right columns represent the phosphorylation kinetics of the IGF-1 receptor and three kinases within the Akt pathway upon IGF-1 stimulation. Each time point shown in the graphs represents the normalized PLA dot count distributions (blue, in y-direction) evaluated from more than 400 single cells. Mean values are indicated in red. | ||
We observed an increase in the mean value (red line in Fig. 4) of the single-cell PLA dot count (blue dots along the vertical direction shown in Fig. 4) of all phosphorylation targets in response to the stimuli. The duration of the PLA dot count increase varied among the targets. PLA signal for both receptors and the Thr-308 residue of Akt did not return to the basal levels within the observation period. A response difference between the two growth factors was observed during the increase of the PLA signal of Akt at Ser-473 and GSK3β at Ser-9. As expected, the PLA signal of GSK3β, which is a substrate of Akt, decreases after the PLA signal of Akt in both cases.
To quantify the phosphorylation rates of the proteins, all single-cell PLA dot count distributions, up to the first 240 s, were fitted into a first order rate equation: 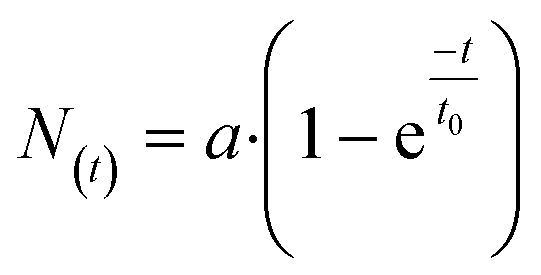 , with N(t), a, t, and t0 denoting the PLA dot count distribution, a constant, the time, and characteristic phosphorylation time, respectively. The temporal PLA dot count distributions and fitting results are shown in Fig. 5. As expected, the receptor kinases exhibit the fastest characteristic phosphorylation time among all pathway components, and the characteristic phosphorylation time of IGF-1 (t0 = 13.6 ± 1 s) is three times larger than that of PDGF (t0 = 35.6 ± 2.2 s). In addition, the phosphorylation reactions of the two Akt residues in response to the IGF-1 stimulation are faster than that in response to the PDGF stimulation. The comparison of the characteristic phosphorylation times revealed a difference between the two monitored residues of Akt upon stimulation with IGF-1 and PDGF. In both cases, the characteristic phosphorylation time of the Thr-308 residue was faster than that of the Ser-473 residue. Upon PDGF stimulation the characteristic phosphorylation time of the Akt Ser-473 residue (t0 = 200 ± 26.4 s) was four times slower than the characteristic phosphorylation time of the Akt Thr-308 residue (t0 = 43.7 ± 2.5 s). Upon IGF-1 stimulation the characteristic phosphorylation time of the Ser-473 (t0 = 53 ± 3.2 s) was still slower than the for the Thr-308 residue (t0 = 31.3 ± 1.6 s) but the difference between the two sites was only about 22 ± 3 s.
, with N(t), a, t, and t0 denoting the PLA dot count distribution, a constant, the time, and characteristic phosphorylation time, respectively. The temporal PLA dot count distributions and fitting results are shown in Fig. 5. As expected, the receptor kinases exhibit the fastest characteristic phosphorylation time among all pathway components, and the characteristic phosphorylation time of IGF-1 (t0 = 13.6 ± 1 s) is three times larger than that of PDGF (t0 = 35.6 ± 2.2 s). In addition, the phosphorylation reactions of the two Akt residues in response to the IGF-1 stimulation are faster than that in response to the PDGF stimulation. The comparison of the characteristic phosphorylation times revealed a difference between the two monitored residues of Akt upon stimulation with IGF-1 and PDGF. In both cases, the characteristic phosphorylation time of the Thr-308 residue was faster than that of the Ser-473 residue. Upon PDGF stimulation the characteristic phosphorylation time of the Akt Ser-473 residue (t0 = 200 ± 26.4 s) was four times slower than the characteristic phosphorylation time of the Akt Thr-308 residue (t0 = 43.7 ± 2.5 s). Upon IGF-1 stimulation the characteristic phosphorylation time of the Ser-473 (t0 = 53 ± 3.2 s) was still slower than the for the Thr-308 residue (t0 = 31.3 ± 1.6 s) but the difference between the two sites was only about 22 ± 3 s.
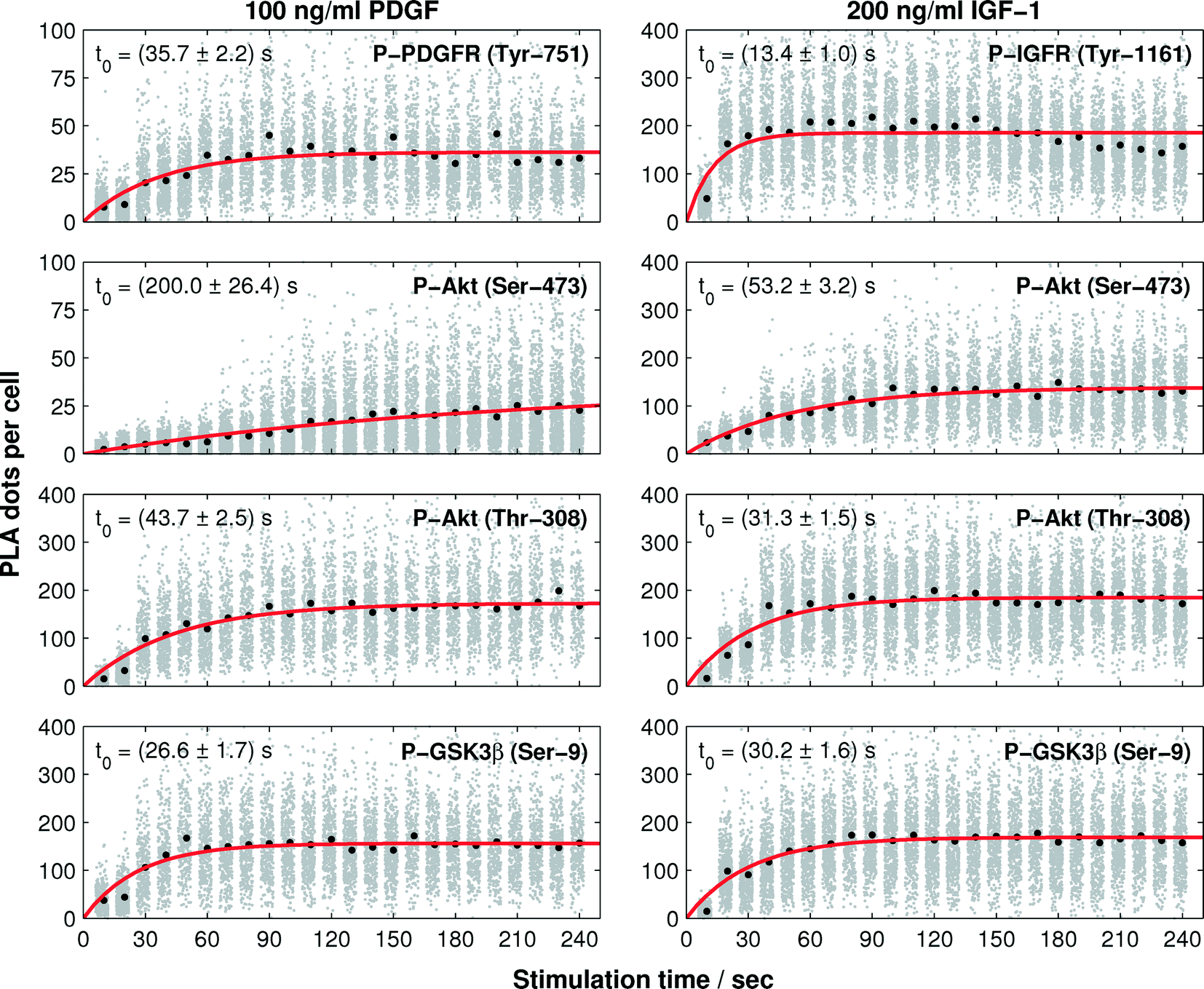 | ||
| Fig. 5 Phosphorylation rates evaluated from the time trajectories of single-cell PLA dot count distributions. The left and right columns represent the first 240 seconds of the protein phosphorylation time trajectories upon PDGF and IGF-1 stimulations, respectively. PLA dot counts of single cells are shown in gray. The mean values of the PLA dot counts are indicated by black dots. The simulated phosphorylation dynamics are shown in red. | ||
While the characteristic phosphorylation time of the receptors and downstream Akt kinase followed the expectation of a canonical signaling pathway, the characteristic phosphorylation time of the GSK3β Ser-9 residue was not in accordance with the signal processing image in Fig. 1. GSK3β is known to be a substrate of Akt. However, the present study revealed a faster characteristic phosphorylation time of the Ser-9 residue of GSK3β than that of Akt upon PDGF and IGF-1 stimulations. This result suggests either a strong catalytic activity of Akt towards GSK3β or a second kinase, which is activated faster than Akt, cross-activates the phosphorylation of GSK3β upon PDGF and IGF-1 stimulations.
Discussion
Akt pathway plays an important role in central cellular signaling transduction for the control of cell growth, apoptosis, and various metabolic processes. High-resolution kinetics of the Akt signal transduction next to quantitative data are missing but required for our understanding and prediction of cellular signal processing. In the present study, we developed a microfluidic tool with PLA functionality and demonstrated its capability for providing biophysical parameters for investigating the cell signaling dynamics.The minimum cell sample size to obtain reproducible phosphorylation signals with the on-chip PLA technology is in the order of 400 cells. This value provides an initial reference to all PLA studies, whereas, it could be modified according to the protein target, antibody, and number of dot count observed from the cells. Several hundreds to one thousand cell repeats have been previously used for obtaining statistically robust results from fluorescence in situ hybridization (FISH) experiments,30–32 and these should be in principle similar to the PLA experiments, which are also based on a fluorescent dot-based detection method.
In general, kinase phosphorylation within signal transduction cascades leads to various molecular causalities, e.g., change of kinase activity, sub-cellular location, and/or protein interaction partners. These molecular state changes of kinases are used for directing, distributing, and controlling the duration of signals within cells.33 The complexity of molecular events in response to a phosphorylation action led to controversial discussions regarding the order and causalities of the molecular changes.
Phosphorylation kinetics with resolutions of few seconds can reveal mechanistic details of the signal transduction process. The resolutions of the phosphorylation kinetics of PDGF and IGF-1 receptors in NIH 3T3 cells are reported to be in the order of 1–2 min. With the aid of on-chip PLA technology, we quantitatively revealed the phosphorylation kinetics of both hormone receptors upon the stimulation of NIH 3T3 cells. The improved time resolution of 10 seconds in the on-chip PLA experiments resulted in a precise characteristic phosphorylation time (t0 = 35 ± 2 s) of the PDGF receptor, whereas the accuracy of the phosphorylation rate of the IGF receptor suffered from a low resolution. Nevertheless, the time trajectories of PLA signals for the IGF-1 receptor were sufficient for predicting a fast characteristic phosphorylation time (t0 = 13 ± 1 s) and resolving the difference between the characteristic phosphorylation times of the receptors in response to their hormones. The direct comparison of the signal transduction pathway based on the phosphorylation kinetics of the two receptors oversimplifies their functions, since both are formed upon the binding of the substrate to higher-ordered protein complexes. Thus, the characteristic phosphorylation time of the receptor is the only reliable indicator of its activity. Activation rates of the downstream kinases are dependent on the fraction of the activated receptors and therewith on the concentration of growth factors in the cellular surrounding. Within the saturation regime of the receptors with growth hormones the phosphorylation rates of the downstream kinases become independent of the growth factor concentration. The calculated characteristic phosphorylation times of all downstream kinases within the IGF-1 pathway were slower than those of the receptor kinases. In case of the PDGF pathway, however the characteristic time constant for the GSK3β phosphorylation is faster than for the receptor, which argues that a simple linear phosphorylation reaction model can not describe the experimental data.
In general, to differentiate extracellular stimuli, kinases can form specific protein complexes and phosphorylation patterns and/or change the cellular position. Two hypotheses for the molecular events of Akt in response to cell stimulation with growth factors have been proposed based on experimental results: (i) the synthesis of PIP3 leads to the recruitment of Akt into the inner leaflet of the membrane, and this facilitates the association with PDK1 and the phosphorylation of the Thr-308 residue.18 Separately, the mTOR kinase complexed with the adaptor protein Rictor is activated and phosphorylates Akt at the Ser-473 residue16 at an undefined cellular location. (ii) Alternatively, mTOR activated in response to the growth factor stimulation phosphorylates Akt at the Ser-473 residue, followed by the binding of Akt to PIP3 and phosphorylation of the Thr-308 residue through PDK1.15 It has been suggested that neither order of the process is mutually exclusive; the prevalence of one or the other may be context-dependent.17 However, in vitro experiments have shown that Ser-473-phosphorylated Akt led to an increase in the affinity for PDK1; thus, the sequential phosphorylation of Akt was thought to start at Ser-473, followed by Thr-308.15
In this study, the recorded in situ phosphorylation rates of the two Akt residues in response to PDGF stimulation indicate that the sequential phosphorylation of Akt starts at the Thr-308 residue, followed by Ser-473. In the case of IGF-1 stimulation, a difference between the two phosphorylation rates of Akt was observed. Although the Thr-308 residue showed a larger rate constant than that of Ser-473, their phosphorylation reactions were not temporally separated. A further indication is that the signaling through the Akt kinase pathway is regulated at the molecular level depending on the stimulus, can be deduced from the duration of the Akt phosphorylation. The Ser-473 residue is in response to IGF-1 three times longer phosphorylated than upon PDGF stimulation.
As with GSK3β, which was expected to be the receiving kinase in the observed signaling cascade, the phosphorylation rate was larger than that of the proceeding kinase Akt. Apart from the Akt kinase, at least three additional kinases are reported to target the phosphorylation site Ser-9 of GSK3β.34 One of them is MAPK, which is known to be activated in parallel to the Akt kinase upon growth factor stimulation.35 A faster signal-processing through the MAPK kinase pathway than that through the Akt kinase pathway can explain the faster phosphorylation rate of GSK3β than that of Akt. It is worth noting that the rate constant does not exclude the possibility of phosphorylation of GSK3β by Akt. In fact the correlated duration of Ser-9 of GSK3β and Ser-473 of Akt phosphorylation upon IGF-1 and PDGF stimulations strongly indicates this and supports previous literature reports.19
In conclusion, the integration of highly parallel cell culture and processing performance with PLA functionality onto a microfluidic chip was successfully demonstrated by the measurement of the phosphorylation kinetics of the kinases within the Akt pathway. The variances of the PLA dot count distributions of all kinetic time traces were used for determining the kinetic rate constants. It is expected that the time trajectories of PLA dot count distributions at the single cell level can be useful for the detailed modeling of cell signaling cascades and deciphering the heterogeneities between multiple cell populations in the future.
Acknowledgements
This study was supported by the Excellence Initiative of the German Federal and State Governments (EXC-294) and by the German Research Foundation (Emmy-Noether grant ME3823/1-1). We thank Prof. M. Reth and G. Roth for helpful discussion.References
- G. Burnett and E. P. Kennedy, J. Biol. Chem., 1954, 211, 969–980 CAS.
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255–261 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- P. V. Hornbeck, J. M. Kornhauser, S. Tkachev, B. Zhang, E. Skrzypek, B. Murray, V. Latham and M. Sullivan, Nucleic Acids Res., 2011, 40, D261–D270 CrossRef PubMed.
- A. J. Hughes and A. E. Herr, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21450–21455 CrossRef CAS PubMed.
- A. J. Hughes, D. P. Spelke, Z. Xu, C.-C. Kang, D. V. Schaffer and A. E. Herr, Nat. Methods, 2014, 11, 749–755 CrossRef CAS PubMed.
- C. Salazar and T. Höfer, FEBS J., 2009, 276, 3177–3198 CrossRef CAS PubMed.
- S. Fredriksson, M. Gullberg, J. Jarvius, C. Olsson, K. Pietras, S. M. Gústafsdóttir, A. Ostman and U. Landegren, Nat. Biotechnol., 2002, 20, 473–477 CrossRef CAS PubMed.
- O. Söderberg, K.-J. Leuchowius, M. Gullberg, M. Jarvius, I. Weibrecht, L.-G. Larsson and U. Landegren, Methods, 2008, 45, 227–232 CrossRef PubMed.
- M. Blazek, C. Betz, M. N. Hall, M. Reth, R. Zengerle and M. Meier, Mol. Cell. Proteomics, 2013, 12, 3898–3907 CAS.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489–501 CrossRef CAS PubMed.
- K. M. Nicholson and N. G. Anderson, Cell. Signalling, 2002, 14, 381–395 CrossRef CAS.
- B. D. Manning and L. C. Cantley, Cell, 2007, 129, 1261–1274 CrossRef CAS PubMed.
- M. P. Scheid, P. A. Marignani and J. R. Woodgett, Mol. Cell. Biol., 2002, 22, 6247–6260 CrossRef CAS.
- D. D. Sarbassov, Science, 2005, 307, 1098–1101 CrossRef CAS PubMed.
- J. R. Hart and P. K. Vogt, Oncotarget, 2011, 2, 467–476 Search PubMed.
- K. S. Walker, M. Deak, A. Paterson, K. Hudson, P. Cohen and D. R. Alessi, Biochem. J., 1998, 331(Pt 1), 299–308 CAS.
- D. A. Cross, D. R. Alessi, P. Cohen, M. Andjelkovich and B. A. Hemmings, Nature, 1995, 378, 785–789 CrossRef CAS PubMed.
- J. McDonald and G. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- R. Gómez-Sjöberg, A. Leyrat, D. Pirone, C. Chen and S. Quake, Anal. Chem., 2007, 79, 8557 CrossRef PubMed.
- Q. Shi, L. Qin, W. Wei, F. Geng, R. Fan, Y. S. Shin, D. Guo, L. Hood, P. S. Mischel and J. R. Heath, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 419–424 CrossRef CAS PubMed.
- F. S. O. Fritzsch, C. Dusny, O. Frick and A. Schmid, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 129–155 CrossRef CAS PubMed.
- C. S. Park, J. Biol. Chem., 2003, 278, 37064–37072 CrossRef CAS PubMed.
- S. Favelyukis, J. H. Till, S. R. Hubbard and W. T. Miller, Nat. Struct. Biol., 2001, 8, 1058–1063 CrossRef CAS PubMed.
- S. Rankin and E. Rozengurt, J. Biol. Chem., 1994, 269, 704–710 CAS.
- H. Kato, T. N. Faria, B. Stannard, C. T. Roberts and D. LeRoith, J. Biol. Chem., 1993, 268, 2655–2661 CAS.
- M. Jarvius, J. Paulsson, I. Weibrecht, K.-J. Leuchowius, A.-C. Andersson, C. Wählby, M. Gullberg, J. Botling, T. Sjöblom, B. Markova, A. Ostman, U. Landegren and O. Söderberg, Mol. Cell. Proteomics, 2007, 6, 1500–1509 CAS.
- C. Pierre-Eugene, P. Pagesy, T. T. Nguyen, M. Neuillé, G. Tschank, N. Tennagels, C. Hampe and T. Issad, PLoS One, 2012, 7, e41992 CAS.
- Y. Buganim, D. A. Faddah, A. W. Cheng, E. Itskovich, S. Markoulaki, K. Ganz, S. L. Klemm, A. van Oudenaarden and R. Jaenisch, Cell, 2012, 150, 1209–1222 CrossRef CAS PubMed.
- T. Trcek, D. R. Larson, A. Moldón, C. C. Query and R. H. Singer, Cell, 2011, 147, 1484–1497 CrossRef CAS PubMed.
- N. Battich, T. Stoeger and L. Pelkmans, Nat. Methods, 2013, 10, 1127–1133 CrossRef CAS PubMed.
- F. Marks, U. Klingmüller and K. Müller-Decker, Cellular signal processing: an introduction to the molecular mechanisms of signal transduction, Garland Science, New York, NY, USA, 2009 Search PubMed.
- S. Frame and P. Cohen, Biochem. J., 2001, 359, 1–16 CrossRef CAS.
- Y. Saito, J. R. Vandenheede and P. Cohen, Biochem. J., 1994, 303, 27–31 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00797b |
| This journal is © The Royal Society of Chemistry 2015 |