
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated immunoisolation and protein analysis of circulating exosomes using microfluidic technology†‡
Mei
He§
a,
Jennifer
Crow
a,
Marc
Roth
a,
Yong
Zeng
*b and
Andrew K.
Godwin
*ac
aDepartment of Pathology and Laboratory Medicine, University of Kansas Medical Center, Kansas City, KS 66160, USA. E-mail: agodwin@kumc.edu; Fax: +1 (913) 945 6373; Tel: +1 (913) 945 6327
bDepartment of Chemistry, Ralph N Adams Institute for Bioanalytical Chemistry, and Bioengineering Graduate Program, University of Kansas, Lawrence, KS 66045, USA. E-mail: yongz@ku.edu; Fax: +1 (785) 864 5396; Tel: +1 (785) 864 8105
cUniversity of Kansas Cancer Center, Kansas City, KS 66160, USA
First published on 18th July 2014
Abstract
Developing blood-based tests is appealing for non-invasive disease diagnosis, especially when biopsy is difficult, costly, and sometimes not even an option. Tumor-derived exosomes have attracted increasing interest in non-invasive cancer diagnosis and monitoring of treatment response. However, the biology and clinical value of exosomes remains largely unknown due in part to current technical challenges in rapid isolation, molecular classification and comprehensive analysis of exosomes. Here we developed a new microfluidic approach to streamline and expedite the exosome analysis pipeline by integrating specific immunoisolation and targeted protein analysis of circulating exosomes. Compared to the conventional methods, our approach enables selective subpopulation isolation and quantitative detection of surface and intravesicular biomarkers directly from a minimally invasive amount of plasma samples (30 μL) within ~100 min with markedly improved detection sensitivity. Using this device, we demonstrated phenotyping of exosome subpopulations by targeting a panel of common exosomal and tumor-specific markers and multiparameter analyses of intravesicular biomarkers in the selected subpopulation. We were able to assess the total expression and phosphorylation levels of IGF-1R in non-small-cell lung cancer patients by probing plasma exosomes as a non-invasive alternative to conventional tissue biopsy. We foresee that the microfluidic exosome analysis platform will form the basis for critically needed infrastructures for advancing the biology and clinical utilization of exosomes.
Introduction
Developing non-invasive blood-based tests is extremely appealing for presymptomatic screening and early detection of cancers where obtaining tissue biopsy is highly invasive and costly. This is particularly true for many primary tumors and most metastatic diseases. Probing circulating exosomes becomes an emerging paradigm for cancer detection and monitoring response to treatment. Most eukaryotic cells release exosomes that are membrane vesicles derived from the endolysosomal pathway with a size range of ~30–150 nm.1 Exosomes play important biological roles via transfer of cargo consisting of proteins, RNAs,2,3 and mitochondrial DNA.4 They have been found to be abundant in plasma and malignant effusions derived from cancer patients.5–7 The constitutive release of exosomes with selectively enriched biomolecules presents distinctive opportunities for cancer diagnosis.8,9 However, exosome research has been severely constrained by the technical difficulties in isolation and molecular analysis of such nano-scale and molecularly diverse vesicles.1 Standard exosome isolation protocols heavily rely on multiple-step ultracentrifugation which is tedious, time consuming (>10 h) and inefficient.10 Moreover, ultracentrifugation co-purifies various vesicle subtypes secreted via different intracellular mechanisms, which may mask disease-related biosignatures.11 Size-exclusion methods normally do not concentrate exosomes and are prone to pressure-caused damage of vesicles12 and contaminations.13 Standard techniques widely used for exosome analysis, such as western blot, ELISA and mass spectrometry, require lengthy processes and large sample volumes, thus limiting clinical investigation. To date, there are no well-defined protocols for isolation and molecular characterization of exosomes.1,13Microfluidics has shown unique advantages for bioassays, such as high throughput,14,15 single-molecule and single-cell sensitivity,16–19 functional integration18,20–22 and automation.23,24 Although recent advancements in microfluidic technology have made an enormous impact on biological and medical sciences, much less efforts have been invested in applying microfluidic technology to accelerate exosome research. Recently, two flow-through microchips with surface-immobilized antibodies have been reported for solid-phase immunocapture and surface characterization of exosomes.20,25 A microfiltration system was developed for size isolation of microvesicles by integrating a porous polymer membrane.26 On-chip surface phenotyping of microvesicles has also been demonstrated by using miniaturized nuclear magnetic resonance27 and nano-plasmonic sensors.28 While these systems markedly improved the performance for exosome isolation and detection, they still rely on conventional analysis techniques to probe intravesicular constituents, limiting the ability for comprehensive characterization of exosomes.
Here we report for the first time an integrated microfluidic approach that enables on-chip immunoisolation and in situ protein analysis of exosomes directly from patient plasma. Specifically, a cascading microfluidic circuit was designed to streamline and expedite the pipeline for proteomic characterization of circulating exosomes, including exosome isolation and enrichment, on-line chemical lysis, protein immunoprecipitation, and sandwich immunoassays assisted by chemifluorescence detection. Compared to the conventional methods, our technology remarkably increases the sensitivity while reducing the assay time and sample requirement by two orders of magnitude. The integrative exosome analysis and the ability to probe intravesicular contents distinguish our platform from the existing microfluidic devices.
We applied the technology to analyze clinical plasma specimens, mainly from non-small-cell lung cancer (NSCLC) patients. Lung cancer is the leading cause of cancer-related deaths worldwide29 and NSCLC accounts for approximately 85% of lung cancer cases with an overall 5 year survival rate of only 15% (stage IIIA).30 Since the majority of NSCLC patients present with unresectable advanced disease, obtaining adequate tissue for diagnosis can be challenging. Furthermore, it is extremely difficult to obtain tissue biopsies prior to each therapy, which substantially limits the histologic and molecular information.31 Herein we demonstrated selective isolation of exosomes from NSCLC plasma and quantitative analysis of total expression and phosphorylation levels of type 1 insulin growth factor receptor (IGF-1R), a promising biomarker and therapeutic target for NSCLC.32 In contrast, current clinical assessment of IGF-1R expression primarily relies on immunohistochemical (IHC) tests of tumor tissues which are highly invasive.33 Because of the advantages of high sensitivity, fast speed, and small sample demand, the microfluidic exosome analysis technology developed here might open a new avenue for cancer diagnosis in a non-invasive manner, i.e., liquid biopsy.
Results and discussion
Integrated microfluidic exosome analysis platform
Our microfluidic technology uses a magnetic bead-based strategy to integrate and streamline the multi-step analysis of exosomes directly from human plasma (Fig. 1A). Compared to the surface-based exosome microchips,20,25 the immunomagnetic method allows for enrichment of captured exosomes and convenient sample preparation for transmission electron microscopy (TEM) characterization in addition to higher capture efficiency and analysis sensitivity due to the larger surface area.34 The PDMS chip that we have devised uses a cascading microchannel circuit to sequentially conduct exosome isolation and enrichment (1st-stage capture), chemical lysis and immunoprecipitation of intravesicular targets (2nd-stage capture), and chemifluorescence-assisted sandwich immunoassay (Fig. 1A and ESI,‡ Table S2). Briefly, the plasma sample pre-mixed with antibody-labeled magnetic beads was introduced through inlet #1 into the first chamber where the magnetic beads were retained and washed with PBS buffer (Fig. 1B(1), Movie S1‡). A lysis buffer was then introduced through inlet #2 to fill the chamber and then the flow was stopped to incubate the captured exosomes. The lysate was flowed into a serpentine channel and the antibody-labeled magnetic beads were injected from two side channels to capture the released intravesicular proteins (Fig. 1B(2)). The protein capture beads were magnetically retained in the 2nd chamber where detection antibodies and chemifluorescence reagents were sequentially introduced for sandwich immunodetection of protein markers of interest (Fig. 1B(3)). The buffers for binding and washing have been optimized to minimize bead aggregation and non-specific adsorption while maintaining the integrity of captured exosomes (see the ESI‡). The on-chip assay can be completed in less than 1.5 h and uses plasma sample volumes as low as 30 μL.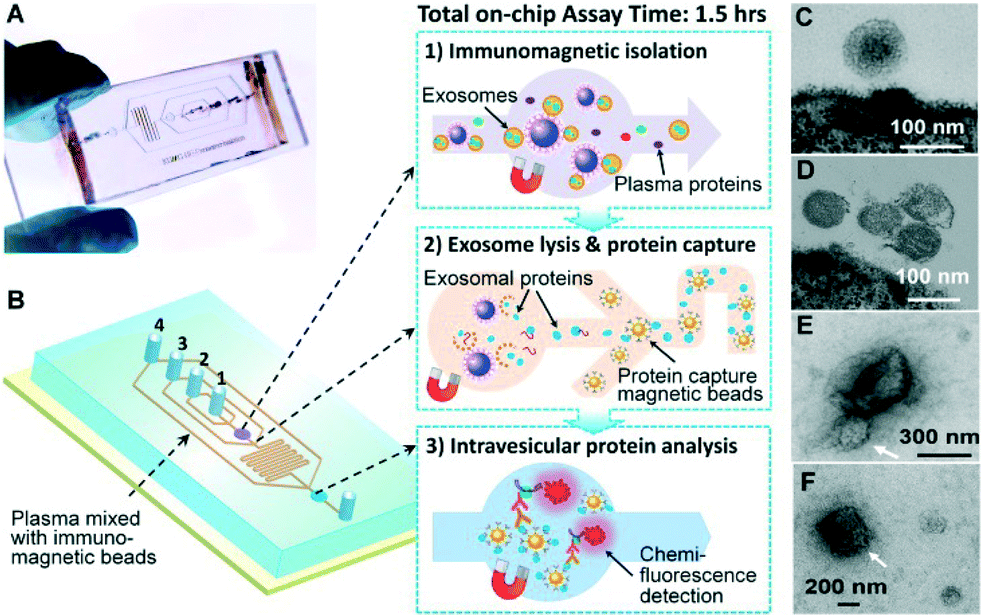 | ||
| Fig. 1 Integrated microfluidic exosome analysis directly from human plasma. (A) Image of the prototype PDMS chip containing a cascading microchannel network for multi-stage exosome analysis. (B) Streamlined workflow for on-chip immunomagnetic isolation, chemical lysis, and intravesicular protein analysis of circulating exosomes. #1–4 indicates the inlet for exosome capture beads, washing/lysis buffer, protein capture beads, and ELISA reagents, respectively. (C, D) Typical TEM images of exosomes from NSCLC (C) and ovarian cancer plasma (D) isolated by the microfluidic immunomagnetic method. The magnetic beads were conjugated with anti-EpCAM and anti-CA125 antibodies for NSCLC and ovarian cancer, respectively. (E, F) TEM images showing large aggregates (E) and other membranous particles (F) observed in the ultracentrifugation-purified vesicles, as indicated by the white arrows. | ||
Fig. 1C & D show the representative TEM images of on-chip isolated exosomes from NSCLC and ovarian cancer (OVCA), respectively. We observed a typical round, homogeneous morphology of exosomes which were carefully prepared by embedding and sectioning for TEM imaging. The cup shape of exosomes was often observed by electron microscopy, which likely resulted from drying-caused collapse of vesicles.11 A major size distribution of 40–150 nm was determined, which is consistent with the reported size range.35 For comparison, we purified the exosomes by the gold standard method, ultracentrifugation, and often observed a heterogeneous population of vesicles containing relatively large aggregates and other membranous particles (Fig. 1E & F). It is worth mentioning that at the early stage of technical development, we also explored the immunocapture of ultracentrifugation-purified exosomes for parallel evaluation of the one-step microfluidic isolation. However, the capture efficiency was found to be considerably low and variable, which may be attributed to the fact that the recovery rate of ultracentrifugation is low (5–25%)36 and further reduced by the additional immunocapture steps. In addition, we observed much more irregular vesicles bound to the beads by TEM, which appear to be collapsed or damaged (Fig. S1‡).
On-chip immunomagnetic isolation of circulating exosomes
We first investigated the magnetic capture of beads as it dictates the overall performance of exosome isolation and analysis. The beads suspended in a buffer solution were retained by a magnet placed underneath the capture chamber (Fig. 2A, inset), forming an aggregate (Fig. S2‡) induced by the dipolar interactions between the beads. It was reported that the amount of magnetically captured beads in a microchannel can be represented by the size of the bead aggregate, which increases linearly with time at a constant flow rate.37 We adopted this approach to conveniently assess the bead capture as a function of flow conditions (Fig. 2A). It was found that the aggregate size was linearly dependent on the total sample infusion volume regardless of the flow rates applied to reach certain infusion volumes (1–10 μL min−1, Fig. 2A). The independence on flow rate indicates the high bead capture efficiency and capacity of our system, which ensures quantitative measurement of exosomes over a wide range of flow conditions and sample volumes. We chose low flow rates for affinity capture of exosomes and protein targets released by chemical lysis (2 μL min−1 and 1 μL min−1, respectively), at which the bead recovery efficiency was determined to be >99.9% by counting the residual beads in the eluent.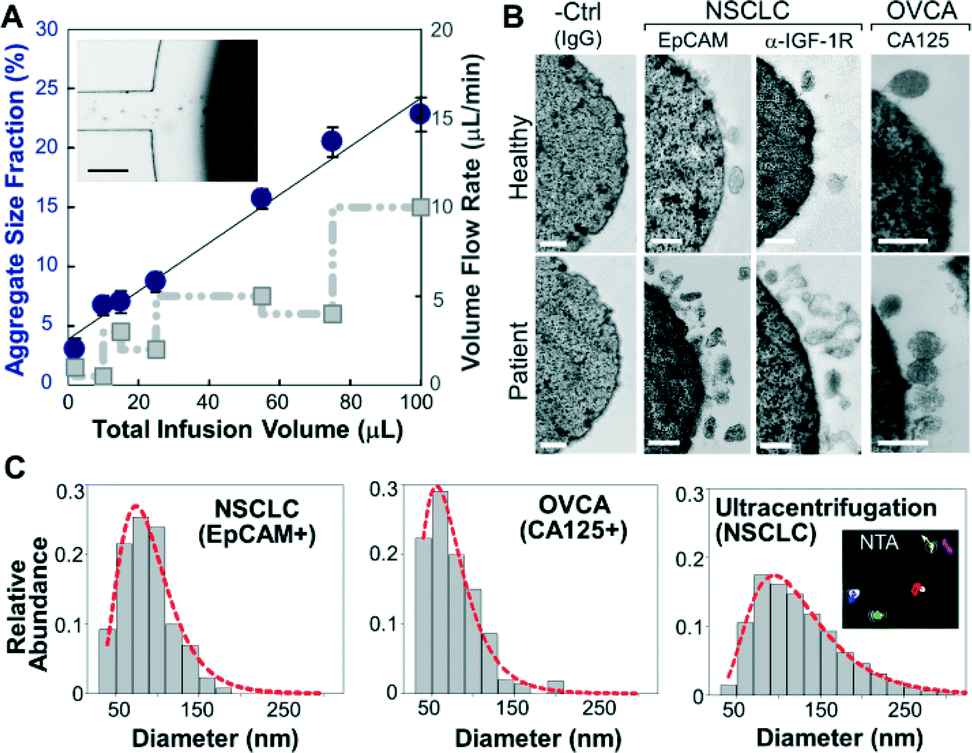 | ||
| Fig. 2 Microfluidic immunomagnetic capture of circulating exosomes. (A) Plot of the amount of beads captured in the chamber represented by the aggregate area fraction as a function of total infusion volume (circle) and flow rate (square). The error bars are standard deviations (n = 3). Inset: a bright-field image of the magnetic capture of beads in the 1st capture chamber. The scale bar is 100 μm. (B) Representative TEM images showing enriched exosomes on the surface of antibody-conjugated beads from NSCLC and ovarian cancer (OVCA) samples, while significantly fewer vesicles from healthy plasma and almost no vesicles on the negative control beads without specific antibodies were observed. (C) Representative size histograms of on-chip isolated exosomes from NSCLC (EpCAM+, n = 130) and OVCA (CA125+, n = 130) compared to that of ultracentrifugation-purified NSCLC vesicles measured by NTA using NanoSight (insets). Sizes were obtained by averaging five measurements. Red dot plots are log-normal fitting (R2 > 0.98). Scale bars: 100 nm. | ||
To verify the capture specificity and generalizability of our method, we compared the on-chip purification of exosomes from NSCLC, OVCA, and healthy plasma using beads labeled with monoclonal antibodies specific for epithelial cell adhesion molecules (EpCAM), IGF-1R α units (α-IGF-1R) or CA125. TEM examination shows that the antibody beads were densely coated with vesicles from the patient sample, while significantly fewer vesicles from healthy plasma and almost no vesicles on the negative control beads without specific antibodies were observed. These results confirm the specific binding of exosomes and effective washing to minimize non-specific binding. Moreover, we examined by TEM numerous exosomes isolated by targeting various surface markers. The majority of these exosomes remained intact, in contrast to the much more damaged vesicles observed for the immunocapture of vesicles pre-purified by ultracentrifugation (Fig. S1‡). This indicates the advantage of the direct one-step microfluidic immunoisolation to preserve vesicle integrity over the conventional ultracentrifugation-based protocols.10,11,20
The on-chip capture performance was further characterized by the size distribution of individual exosome subpopulations isolated by targeting both tumor-associated markers (EpCAM, α-IGF-1R, and CA125) and common exosomal markers (CD9, CD81, and CD63).38 Size is the most acceptable criterion for exosome identification39 and differentiation from other extracellular vesicle types.40 The current consensus is that exosomes originated from multivesicular endosome fusion are typically smaller than 150 nm while the majority of microvesicles derived from plasma membranes are relatively larger (150–2000 nm).1 Compared to nanoparticle tracking analysis (NTA) using NanoSight which requires ~1 mL of concentrated vesicles (~109 mL−1) for accurate size determination, TEM provides a robust means of sizing and counting exosomes in small volumes collected from microfluidic isolation without significant dilution (~30 μL). Most immunocaptured exosomes were found to be smaller than 150 nm with a notably smaller size range (e.g., 97% of EpCAM+ and CA125+ vesicles <150 nm) than those obtained by ultracentrifugation (72.1%) (Fig. 2C and S3‡). Current “gold standard” approaches based on ultracentrifugation yield a mixed population of various extracellular vesicle types with a wide size distribution.41 Indeed, our NTA analysis of ultracentrifugation-purified vesicles yielded a broader size variation and no distinct profiles between healthy and NSCLC cases (Fig. 2C and S4‡). These findings suggest that our microfluidic immunocapture method provides a more specific means of purifying exosomes than ultracentrifugation.
Profiling of exosome subpopulations defined by surface protein phenotypes
The surface protein composition of exosomes plays an important role in exosome-mediated effects42 and may provide tumor fingerprints.27,28 To demonstrate the ability to detect exosomal expression patterns associated with cancer, we conducted relative quantification of five exosome subpopulations defined by individual surface markers using TEM. Fig. 3A shows the results for two of the NSCLC samples that we have tested. Distinct subpopulation landscapes were observed as compared to the healthy controls with a 3- to 5-fold increase in abundance for the surface markers except CD63. We further demonstrated the adaptability of our method to other cancers by testing OVCA with the tumor markers (EpCAM and CA125) and exosomal markers (CD9, CD81, and CD63). The OVCA samples also provided a positive control for the NSCLC studies, as CD63 was found to be highly expressed in OVCA exosomes.43 As expected, high CD63 expression was observed, which validates our method and supports the observation of low CD63 expression in NSCLC cases. The abilities to discriminate disease from healthy subjects and to detect differential expression of markers (e.g., CD63) in cancers indicate the high immunocapture specificity of our microfluidic method.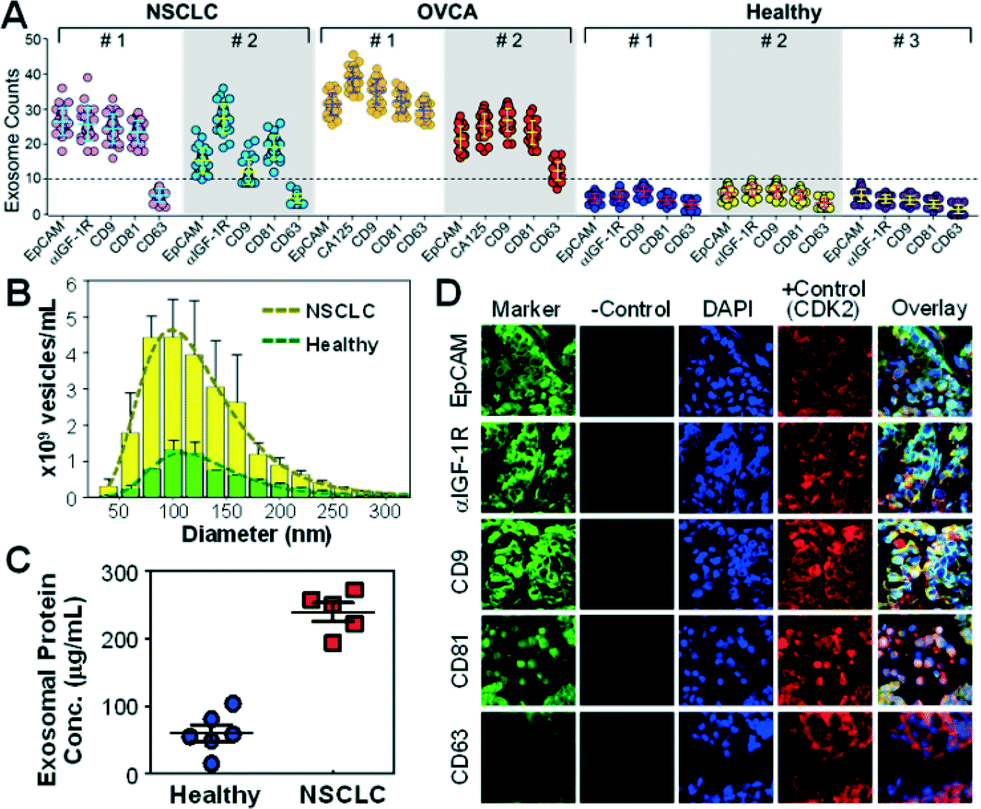 | ||
| Fig. 3 Microfluidic isolation and surface phenotyping of circulating exosomes in cancer. (A) The scattered dot plot of the abundance of bead-bound exosomes from NSCLC, OVCA and healthy plasma obtained by TEM (n = 25). A panel of surface markers (EpCAM, α-IGF-1R, CA125, CD9, CD81 and CD63) were used for exosome isolation. The dashed line indicates the highest exosome counts observed for healthy controls. (B) NTA analysis of the size distribution and abundance of vesicles purified from NSCLC and healthy controls by ultracentrifugation. The error bars are the standard deviations. The dashed lines are log-normal fitting (R2 > 0.98). (C) Bradford assay of total proteins in ultracentrifugation-purified exosomes from NSCLC patients (stage II) and healthy subjects (p = 0.0007). (D) Representative IFH images of the matched tumor tissue from NSCLC patient #1 in (A) showing high expression of the biomarkers except for CD63. | ||
To verify the microfluidic results, we performed parallel analyses of the NSCLC samples using standard ultracentrifugation and analytical methods. Exosome abundance in the patient plasma measured by NTA showed a ~4-fold increase on average compared to the healthy controls (p = 0.0001, Fig. 3B and S5‡), in line with the total protein levels determined by the Bradford assay (a 3.9-fold increase on average, p = 0.0007, Fig. 3C). Western blotting analysis showed increased exosomal expression of CD9, CD81, and IGF-1R markers but indiscernible or low CD63 levels in NSCLC patients of various stages (Fig. S6‡). Thus, the exosomes collected from a cell line (ovarian cancer C30) were included as a positive control for CD63 detection. Collectively, these standard studies verify the microfluidic analysis of the surface phenotypes of circulating exosomes. Both microfluidic and standard methods detected significant elevation of exosome abundance and exosomal markers in NSCLC and OVCA, suggesting the potential clinical value of circulating exosomes for cancer research and diagnostics. Our microfluidic technology provides not only a general approach for one-step isolation of exosomes directly from plasma but also the ability to purify molecularly defined subpopulations that are inaccessible to other physical methods such as ultracentrifugation,41 nanofiltration26 and size exclusion.13 Such capability would be beneficial for deconvoluting the complexity of extracellular vesicles to facilitate molecular classification and characterization of exosomes. The unique one-step isolation system also contrasts with the conventional bulk immunomagnetic methods in that it eliminates multiple, lengthy preparation steps of washing and manual buffer exchange which can cause damage and loss of exosomes.20,41,44
To determine if the plasma-derived exosomes in NSCLC show similar protein profiles to the tumor origin, we conducted a three-color immunofluorescence histological (IFH) study of patient-matched lung tumor tissues. High expression of EpCAM, α-IGF-1R, CD9 and CD81 and low expression of CD63 were detected in the tumor tissues (Fig. 3D and S7‡), in agreement with the subpopulation profiles of plasma exosomes obtained by the microfluidic technology and the standard analyses (Fig. 3 and S6‡). The matched molecular profiles between circulating exosomes and tumor tissues support the potential use of exosomes for non-invasive molecular profiling of solid tumor tissue. Our studies also provide experimental evidence to support recently arising questions on the use of CD63 as a general surface marker for exosome isolation.43 Decreased CD63 expression has been found in relation to tumour growth and invasiveness in lung and other cancers.45,46
Integrated exosome analysis for non-invasive detection of cancer biomarkers
Recent profiling results of microRNAs contained inside circulating exosomes have demonstrated the potential of exosomes as surrogate markers for tumor biopsy.44,47 There is increasing interest in proteomic characterization of exosomes. Our ultimate goal is to develop a microfluidic technology capable of measuring both surface and intravesicular proteins of circulating exosomes. In this proof-of-concept study, we demonstrated the integrated analysis of two targets in NSCLC: total IGF-1R and phosphorylated IGF-1R (p-IGF-1R). The IGF-1R pathway provides a potent proliferative signaling system implicated in tumorigenesis and metastasis. Phosphorylation of IGF-1R initiated by binding of ligands, such as IGF-1, is required for activation of MAPK, PI3K, AKT and other signaling pathways involved in cell proliferation and survival.48 Thus there has been an intense interest in the studies of IGF-1R and p-IGF-1R as diagnostic markers and therapeutic targets.48–50 However, currently immunohistochemical tests of tumor tissues predominate in the clinical assessment of IGF-1R expression which are invasive and problematic for regular monitoring of disease progression and response to treatment.51 To our best knowledge, no studies of exosomal IGF-1R and p-IGF-1R have been reported. As illustrated in Fig. 4A, IGF-1R is a transmembrane protein composed of two surface α subunits and two intravesicular β subunits containing a tyrosine kinase domain which can be phosphorylated. Thus total IGF-1R and p-IGF-1R provide good model targets for demonstrating microfluidic surface phenotyping and intravesicular protein analysis of exosomes. To avoid the interference from free proteins in plasma, we used a monoclonal EpCAM antibody for exosome capture and two antibodies that specifically recognize α-IGF-1R and p-IGF-1R.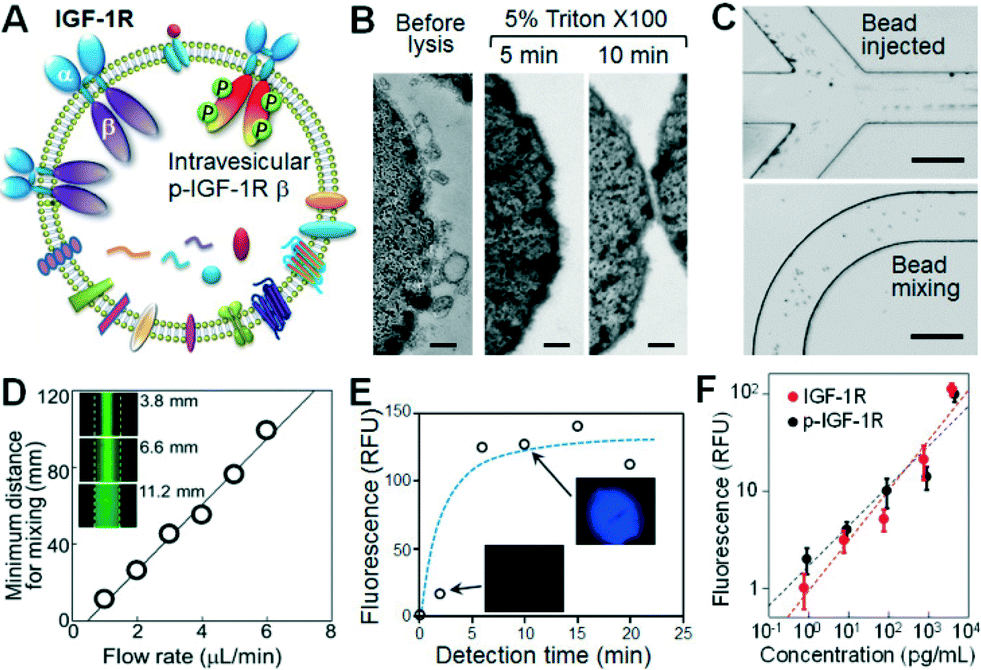 | ||
| Fig. 4 Integrated microfluidic exosome analysis. (A) Schematic of transmembrane IGF-1R in exosomes. We targeted the extravesicular IGF-1R α unit and phosphorylated β domain inside the vesicle for surface phenotyping and intravesicular protein analysis of exosomes. (B) Chemical lysis of exosomes by using Triton X-100 as a surfactant as observed by TEM. The scale bar is 100 nm. (C) Bright-field images of the injection (top) and mixing (bottom) of the protein capture beads in the serpentine channel. The scale bar is 200 μm. (D) The plot of the minimum flow distance required for uniform mixing as a function of flow rate ranging from 0.5 to 6 μL min−1. Inset: fluorescence images taken at various distances along the channel after mixing a stream of 0.1 μM of FITC-BSA solution with the bead suspension co-flowing at the same flow rate. (E) The effect of incubation time on chemifluorescence detection using alkaline phosphatase and the substrate DiFMU. (F) Calibration of on-chip capture and detection of IGF-1R and p-IGF-1R. | ||
The unique cascading microfluidic immunocapture strategy established here enables integration of exosome isolation with downstream processing and analysis, i.e., chemical lysis, flow mixing and protein capture, and chemifluorescence-based sandwich immunoassays. To chemically lyse the captured exosomes, a mild non-ionic detergent, Triton X-100, was used to lyse cells and yet retain the activity of proteins. The lysis conditions, including Triton X-100 concentrations and incubation times, were studied and 5 min of incubation with 5% Triton X-100 was found to be sufficient to completely lyse the exosomes (Fig. 4B). The exosome lysate was flushed into a serpentine microchannel to mix with magnetic beads conjugated with specific antibodies to capture the released protein targets. To enhance fluidic mixing, the suspension of protein capture beads was injected from two side channels to flank the lysate stream (Fig. 4C, top), facilitating the mass transfer across the channel.52 Uniform bead distribution across the 200 μm channel can be achieved within a travel distance of ~10 mm at 1 μL min−1 (Fig. 4C, bottom). Fluorescence imaging was also employed to study the mixing behaviour, revealing a linear response of the minimum distance for complete mixing to flow rate in the range of 0.5 to 6 μL min−1 (Fig. 4D). This result provides guidance to optimize the chip design and flow rate. In our system with a 25 cm mixing channel and a 4 mm microchamber, a flow rate of 1 μL min−1 was used to yield a long incubation time of ~3.8 min which allows efficient solid-phase affinity capture of proteins.53,54
We then optimized the on-chip bead-based immunoassay and chemifluorescence readout using a matched pair of capture/detection antibodies, an alkaline phosphatase (AP)-conjugated secondary antibody, and the DiFMUP substrate. The incubation time is an important factor for the small-scale enzymatic chemifluorescence detection. It was found that the fluorescence signal saturates after 6 min of incubation in the microchamber (Fig. 4E), allowing for fast on-chip protein detection. We then calibrated the on-chip human IGF-1R and p-IGF-1R assays by running protein standards through the entire process except the lysis step. As plotted in Fig. 4F, the microfluidic assay achieved quantitative detection of IGF-1R and p-IGF-1R over a dynamic range of 4 logs with a detection limit of 0.281 pg mL−1 and 0.383 pg mL−1, respectively (S/N = 3). Such sensitivity is at least 100-fold higher than that achieved by the commercial ELISA kits (Fig. S9‡),55 which indicates efficient immunoprecipitation of exosomal proteins in our microfluidic system.
With all the individual functions optimized, we finally implemented the integrated microfluidic analysis to examine the membrane protein IGF-1R and intravesicular p-IGF-1R directly in the plasma of early-stage NSCLC patients (stage II). The microfluidic results were compared to parallel ELISA analysis of ultracentrifugation-purified vesicles from the same patients (2 mL of plasma). To avoid interference from plasma IGF-1R, we used an EpCAM antibody to capture tumor-derived exosome subpopulations in 30 μL of patient plasma. A potential problem may arise from the cross-reactivity of the antibodies with insulin receptors (IRs) which share 80% homology with IGF-1R.56 To investigate this effect, we tested IGF-1R and p-IGF-1R antibodies from a number of vendors using a commercial IR ELISA kit (Table S1‡). No cross-reaction with IRs was detected for all the IGF-1R antibodies (Fig. S8‡). Fig. 5A shows that the NSCLC patients overexpress circulating exosomes with an EpCAM+/IGF-1R+ phenotype and can be well discriminated from the control group (p < 0.0001). The detected IGF-1R concentration was found to correlate linearly with the total abundance of plasma vesicles determined by NTA, while our method measured a fraction of the circulating vesicles (Fig. S10‡). In addition, the quantitative detection was achieved for vesicle concentrations much lower than the healthy levels. These results validate the method for sensitive and quantitative characterization of circulating exosomes in clinical samples. IGF-1R overexpression was also evident in the ELISA results presented in Fig. 5B (p < 0.01), consistent with the previous observations reported for NSCLC cell lines and tumor tissues.57–59 It is important to note that ELISA detects the total IGF-1R level in all vesicle types co-purified by ultracentrifugation, while our method enables characterization of molecularly defined subpopulations.
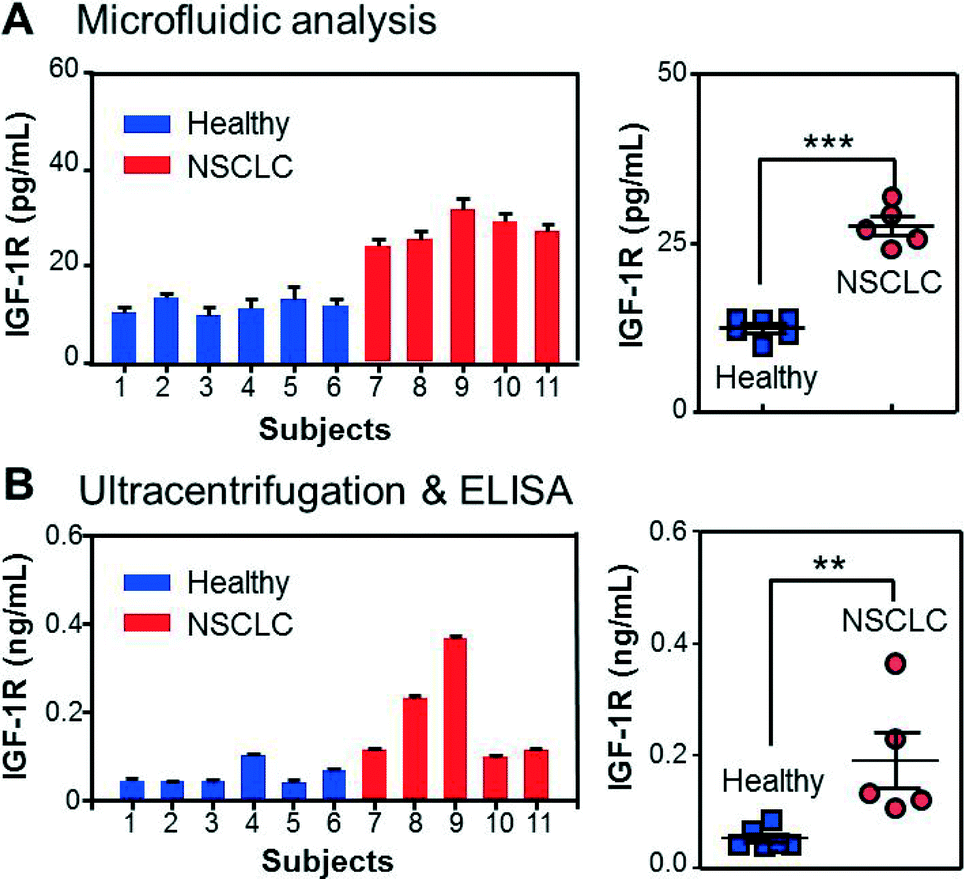 | ||
| Fig. 5 Quantitative detection of total IGF-1R in circulating exosomes directly from clinical plasma samples. (A) The results of the integrated microfluidic analysis presented in the bar (left) and scattered dot (right) plots show significant overexpression of IGF-1R in EpCAM+ exosomes of NSCLC patients compared to healthy controls (p = 0.0001, CV = 11.2%). (B) Parallel ELISA analysis confirmed the overexpression of IGF-1R in total exosomes purified from the same subjects by ultracentrifugation (p = 0.0097, CV = 56.4%). The error bars are standard deviations (n = 3) in all cases. | ||
The ability to probe the intravesicular contents of selected subpopulations is critical for the comprehensive characterization of exosomes and elucidation of their biological and pathological implications. To this end, we measured the intravesicular level of phosphorylated IGF-1R using the same samples as above. Although considerable cross-talking was observed between p-IGF-1R antibodies and IRs (Fig. S8‡), our method is able to specifically detect p-IGF-1R without IR interference because exosomal p-IGF-1R is captured by the monoclonal anti-IGF-1R beads while other exosomal species are removed by washing. As seen in Fig. S11A,‡ the p-IGF-1R profile showed no correlation with that of IGF-1R and the disease state, which was further confirmed by the ELISA analysis (Fig. S11B‡). This result confirms the specificity of the antibodies for detection of p-IGF-1R without discernible cross-reaction with IGF-1R. Previous studies have also reported the lack of correlation between p-IGF-1R and total IGF-1R levels in NSCLC.33,49,57,60 Understanding this phenomenon would require mechanistic studies of the IGF-1R signaling pathways, which are beyond the scope of this work. Overall, we have demonstrated microfluidic isolation and targeted proteomic analysis of exosomes directly from clinical plasma samples, all integrated in one rapid workflow with high sensitivity and specificity. Since many specific antibodies for cancer biomarkers are commercially available, our method can be readily extended to multiplexed proteomic analysis of circulating exosomes in various cancer types. The plasma volume required here was only ~1/100 of that for the conventional protocols, indicating highly efficient exosome immunocapture and sensitive protein analysis of our method. This advantage immediately addresses the challenges in exosome purification, a key setback in the clinical development of exosomal biomarkers.36,61
Conclusions
We have developed a microfluidic exosome analysis platform that integrates immunoaffinity isolation and protein analysis of tumor exosomes directly from human plasma. Relevant to future biomedical applications, we demonstrated profiling of surface phenotypes associated with cancer and quantitative analysis of surface and intravesicular biomarkers in a selected exosome subpopulation directly from minimally invasive plasma samples. Compared to the conventional methods, our technology remarkably increases the sensitivity while reducing the assay time and sample requirement. Owing to its simplicity and general applicability, the exosome analysis microchip can be readily scaled up for high-throughput screening of cancer as well as non-cancerous diseases. Therefore, we envision that this methodology holds the potential to facilitate the elucidation of biological functions and clinical implications of circulating exosomes.Acknowledgements
We would like to acknowledge the KU Cancer Center's Biospecimen Repository Core Facility staff for human specimens, the KU Microfabrication and Microfluidics Core facility for device fabrication, and Dr. Barbara Fegley from the KU Medical Center Electron Microscopy Research Laboratory for TEM imaging analysis. We thank Dr. Safinur Atay for her valuable suggestions. This study was supported in part by grants from the Mary Kay Foundation, the National Cancer Institute, R01 CA106588 and R01 CA140323, and the Kansas Bioscience Authority Eminent Scholar Program to A.K.G., the new faculty start-up funds to Y.Z. and the KUMC Auxiliary funds to M.H. The authors would also like to acknowledge support from the KU Cancer Center (P30 CA168524), and the Chancellors Distinguished Chair in Biomedical Sciences endowed Professorship to A.K.G. The funders did not have any involvement in the experimental design, data collection, analysis, or interpretation of the data, the writing of the article, or the decision to submit the article for publication.Notes and references
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- J. Skog, T. Wurdinger, S. van Rijn, D. H. Meijer, L. Gainche, M. Sena-Esteves, W. T. Curry Jr., B. S. Carter, A. M. Krichevsky and X. O. Breakefield, Nat. Cell Biol., 2008, 10, 1470–1476 CrossRef CAS PubMed.
- M. Guescini, S. Genedani, V. Stocchi and L. F. Agnati, J. Neural. Transm., 2010, 117, 1–4 CrossRef CAS PubMed.
- A. Hendrix and A. N. Hume, Int. J. Dev. Biol., 2011, 55, 879–887 CrossRef PubMed.
- R. H. Staals and G. J. Pruijn, Adv. Exp. Med. Biol., 2011, 702, 132–142 CrossRef.
- D. Schaeffer, A. Clark, A. A. Klauer, B. Tsanova and A. van Hoof, Adv. Exp. Med. Biol., 2011, 702, 79–90 CrossRef.
- R. B. Rountree, S. J. Mandl, J. M. Nachtwey, K. Dalpozzo, L. Do, J. R. Lombardo, P. L. Schoonmaker, K. Brinkmann, U. Dirmeier, R. Laus and A. Delcayre, Cancer Res., 2011, 71, 5235–5244 CrossRef CAS PubMed.
- S. Keller, J. Ridinger, A. K. Rupp, J. W. Janssen and P. Altevogt, J. Transl. Med., 2011, 9, 86 CrossRef CAS PubMed.
- D. D. Taylor, W. Zacharias and C. Gercel-Taylor, Methods Mol. Biol., 2011, 728, 235–246 CAS.
- A. Bobrie, M. Colombo, S. Krumeich, G. Raposo and C. Théry, J. Extracell. Vesicles, 2012, 1, 18397 CrossRef CAS PubMed.
- B. Gyorgy, K. Modos, E. Pallinger, K. Paloczi, M. Pasztoi, P. Misjak, M. A. Deli, A. Sipos, A. Szalai, I. Voszka, A. Polgar, K. Toth, M. Csete, G. Nagy, S. Gay, A. Falus, A. Kittel and E. I. Buzas, Blood, 2011, 117, e39–48 CrossRef CAS PubMed.
- K. W. Witwer, E. I. Buzas, L. T. Bemis, A. Bora, C. Lasser, J. Lotvall, E. N. Nolte-'t Hoen, M. G. Piper, S. Sivaraman, J. Skog, C. Thery, M. H. Wauben and F. Hochberg, J. Extracell. Vesicles, 2013, 2 Search PubMed.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146–2155 RSC.
- S. Cho, D. K. Kang, S. Sim, F. Geier, J. Y. Kim, X. Niu, J. B. Edel, S. I. Chang, R. C. Wootton, K. S. Elvira and A. J. deMello, Anal. Chem., 2013, 85, 8866–8872 CrossRef CAS PubMed.
- D. Witters, K. Knez, F. Ceyssens, R. Puers and J. Lammertyn, Lab Chip, 2013, 13, 2047–2054 RSC.
- Y. Zeng, R. Novak, J. Shuga, M. T. Smith and R. A. Mathies, Anal. Chem., 2010, 82, 3183–3190 CrossRef CAS PubMed.
- J. Shuga, Y. Zeng, R. Novak, Q. Lan, X. Tang, N. Rothman, R. Vermeulen, L. Li, A. Hubbard, L. Zhang, R. A. Mathies and M. T. Smith, Nucleic Acids Res., 2013, 41, e159 CrossRef PubMed.
- R. Novak, Y. Zeng, J. Shuga, G. Venugopalan, D. A. Fletcher, M. T. Smith and R. A. Mathies, Angew. Chem., Int. Ed., 2011, 50, 390–395 CrossRef CAS PubMed.
- C. Chen, J. Skog, C. H. Hsu, R. T. Lessard, L. Balaj, T. Wurdinger, B. S. Carter, X. O. Breakefield, M. Toner and D. Irimia, Lab Chip, 2010, 10, 505–511 RSC.
- Y. Sameenoi, K. Koehler, J. Shapiro, K. Boonsong, Y. Sun, J. Collett Jr., J. Volckens and C. S. Henry, J. Am. Chem. Soc., 2012, 134, 10562–10568 CrossRef CAS PubMed.
- U. Dharmasiri, S. K. Njoroge, M. A. Witek, M. G. Adebiyi, J. W. Kamande, M. L. Hupert, F. Barany and S. A. Soper, Anal. Chem., 2011, 83, 2301–2309 CrossRef CAS PubMed.
- M. He, J. Novak, B. A. Julian and A. E. Herr, J. Am. Chem. Soc., 2011, 133, 19610–19613 CrossRef CAS PubMed.
- S. M. Madren, M. D. Hoffman, P. J. Brown, D. T. Kysela, Y. V. Brun and S. C. Jacobson, Anal. Chem., 2012, 84, 8571–8578 CrossRef CAS PubMed.
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Lab Chip, 2014, 14, 1891–1900 RSC.
- R. T. Davies, J. Kim, S. C. Jang, E. J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed.
- H. Im, H. Shao, Y. I. Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- R. Siegel, D. Naishadham and A. Jemal, CA-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed.
- V. Hirsh, Curr. Oncol., 2012, 19, S86 CAS.
- L. M. Ofiara, A. Navasakulpong, N. Ezer and A. V. Gonzalez, Curr. Oncol., 2012, 19, S16–23 CAS.
- G. V. Scagliotti and S. Novello, Cancer Treat. Rev., 2012, 38, 292–302 CrossRef CAS PubMed.
- M. Nakagawa, H. Uramoto, S. Oka, Y. Chikaishi, T. Iwanami, H. Shimokawa, T. So, T. Hanagiri and F. Tanaka, Clin. Lung Cancer, 2012, 13, 136–142 CAS.
- A. H. C. Ng, U. Uddayasankar and A. R. Wheeler, Anal. Bioanal. Chem., 2010, 397, 991–1007 CrossRef CAS PubMed.
- V. Sokolova, A. K. Ludwig, S. Hornung, O. Rotan, P. A. Horn, M. Epple and B. Giebel, Colloids Surf., B, 2011, 87, 146–150 CrossRef CAS PubMed.
- H. G. Lamparski, A. Metha-Damani, J. Y. Yao, S. Patel, D. H. Hsu, C. Ruegg and J. B. Le Pecq, J. Immunol. Methods, 2002, 270, 211–226 CrossRef CAS.
- A. Sinha, R. Ganguly and I. K. Puri, J. Magn. Magn. Mater., 2009, 321, 2251–2256 CrossRef CAS PubMed.
- C. Thery, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CAS.
- H. Peinado, M. Aleckovic, S. Lavotshkin, I. Matei, B. Costa-Silva, G. Moreno-Bueno, M. Hergueta-Redondo, C. Williams, G. Garcia-Santos, C. Ghajar, A. Nitadori-Hoshino, C. Hoffman, K. Badal, B. A. Garcia, M. K. Callahan, J. Yuan, V. R. Martins, J. Skog, R. N. Kaplan, M. S. Brady, J. D. Wolchok, P. B. Chapman, Y. Kang, J. Bromberg and D. Lyden, Nat. Med., 2012, 18, 883–891 CrossRef CAS PubMed.
- J. C. Akers, D. Gonda, R. Kim, B. S. Carter and C. C. Chen, J. Neuro-Oncol., 2013, 113, 1–11 CrossRef PubMed.
- B. J. Tauro, D. W. Greening, R. A. Mathias, H. Ji, S. Mathivanan, A. M. Scott and R. J. Simpson, Methods, 2012, 56, 293–304 CrossRef CAS PubMed.
- S. Atay, S. Banskota, J. Crow, G. Sethi, L. Rink and A. K. Godwin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 711–716 CrossRef CAS PubMed.
- M. Jørgensen, R. Bæk, S. Pedersen, E. K. L. Søndergaard, S. R. Kristensen and K. Varming, J. Extracell. Vesicles, 2013, 2, 20920 Search PubMed.
- D. D. Taylor and C. Gercel-Taylor, Gynecol. Oncol., 2008, 110, 13–21 CrossRef CAS PubMed.
- M. S. Pols and J. Klumperman, Exp. Cell Res., 2009, 315, 1584–1592 CrossRef CAS PubMed.
- M. S. Kwon, S. H. Shin, S. H. Yim, K. Y. Lee, H. M. Kang, T. M. Kim and Y. J. Chung, Lung Cancer, 2007, 57, 46–53 CrossRef PubMed.
- G. Rabinowits, C. Gercel-Taylor, J. M. Day, D. D. Taylor and G. H. Kloecker, Clin. Lung Cancer, 2009, 10, 42–46 CAS.
- M. Pollak, Nat. Rev. Cancer, 2012, 12, 159–169 CAS.
- N. Peled, M. W. Wynes, N. Ikeda, T. Ohira, K. Yoshida, J. Qian, M. Ilouze, R. Brenner, Y. Kato, C. Mascaux and F. R. Hirsch, Cell. Oncol., 2013, 36, 277–288 CrossRef CAS PubMed.
- M. J. Fidler, D. D. Shersher, J. A. Borgia and P. Bonomi, Ther. Adv. Med. Oncol., 2012, 4, 51–60 CrossRef CAS PubMed.
- O. Larsson, A. Girnita and L. Girnita, Br. J. Cancer, 2005, 92, 2097–2101 CrossRef CAS PubMed.
- P. Sethu, M. Anahtar, L. L. Moldawer, R. G. Tompkins and M. Toner, Anal. Chem., 2004, 76, 6247–6253 CrossRef CAS PubMed.
- G. Proczek, A. L. Gassner, J. M. Busnel and H. H. Girault, Anal. Bioanal. Chem., 2012, 402, 2645–2653 CrossRef CAS PubMed.
- M. Herrmann, T. Veres and M. Tabrizian, Lab Chip, 2006, 6, 555–560 RSC.
- T. Wang, M. Zhang, D. D. Dreher and Y. Zeng, Lab Chip, 2013, 13, 4190–4197 RSC.
- A. Ullrich, A. Gray, A. W. Tam, T. Yang-Feng, M. Tsubokawa, C. Collins, W. Henzel, T. Le Bon, S. Kathuria and E. Chen, et al. , EMBO J., 1986, 5, 2503–2512 CAS.
- Y. Gong, E. Yao, R. Shen, A. Goel, M. Arcila, J. Teruya-Feldstein, M. F. Zakowski, S. Frankel, M. Peifer, R. K. Thomas, M. Ladanyi and W. Pao, PLoS One, 2009, 4, e7273 Search PubMed.
- K. A. Janeway, M. J. Zhu, J. Barretina, A. Perez-Atayde, G. D. Demetri and J. A. Fletcher, Int. J. Cancer, 2010, 127, 2718–2722 CrossRef CAS PubMed.
- C. Tarn, L. Rink, E. Merkel, D. Flieder, H. Pathak, D. Koumbi, J. R. Testa, B. Eisenberg, M. von Mehren and A. K. Godwin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8387–8392 CrossRef CAS PubMed.
- F. Cappuzzo, L. Toschi, G. Tallini, G. L. Ceresoli, I. Domenichini, S. Bartolini, G. Finocchiaro, E. Magrini, G. Metro, A. Cancellieri, R. Trisolini, L. Crino, P. A. Bunn Jr., A. Santoro, W. A. Franklin, M. Varella-Garcia and F. R. Hirsch, Ann. Oncol., 2006, 17, 1120–1127 CrossRef CAS PubMed.
- J. M. Street, P. E. Barran, C. L. Mackay, S. Weidt, C. Balmforth, T. S. Walsh, R. T. Chalmers, D. J. Webb and J. W. Dear, J. Transl. Med., 2012, 10, 5 CrossRef CAS PubMed.
Footnotes |
| † Author contributions: M. H., Y. Z. and A. K. G. conceived research; M. H. and Y. Z. designed and fabricated the devices; M. H., Y. Z., J. C., and M. R. performed the research; M. H., Y. Z., and A. K. G. analyzed the data; M. H., Y. Z., and A. K. G. wrote the manuscript. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00662c |
| § Current address: Department of Biological and Agricultural Engineering, Kansas State University, Olathe, KS 66061. |
| This journal is © The Royal Society of Chemistry 2014 |