 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiffusion-based microfluidic PCR for “one-pot” analysis of cells†
Sai
Ma
a,
Despina Nelie
Loufakis
b,
Zhenning
Cao
a,
Yiwen
Chang
b,
Luke E. K.
Achenie
b and
Chang
Lu
*b
aSchool of Biomedical Engineering and Sciences, Virginia Tech, Blacksburg, Virginia 24061, USA
bDepartment of Chemical Engineering, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: changlu@vt.edu
First published on 28th May 2014
Abstract
Genetic analysis starting with cell samples often requires multi-step processing including cell lysis, DNA isolation/purification, and polymerase chain reaction (PCR) based assays. When conducted on a microfluidic platform, the compatibility among various steps often demands a complicated procedure and a complex device structure. Here we present a microfluidic device that permits a “one-pot” strategy for multi-step PCR analysis starting from cells. Taking advantage of the diffusivity difference, we replace the smaller molecules in the reaction chamber by diffusion while retaining DNA molecules inside. This simple scheme effectively removes reagents from the previous step to avoid interference and thus permits multi-step processing in the same reaction chamber. Our approach shows high efficiency for PCR and potential for a wide range of genetic analysis including assays based on single cells.
Analysis of genes from target cells based on polymerase chain reaction (PCR) is routinely required for illustrating the fundamental molecular biology involved in cellular events and detecting abnormal pathways involved in disease development. Microfluidic devices offer potential for genetic analysis based on tiny amounts of cell samples with high sensitivity and a high degree of integration.1–8 Genetic analysis of cells often starts with cell lysis which releases genes from cells. The released genes typically require isolation and purification before they are analyzed by PCR-based amplification.9,10 The integration of these steps on a microfluidic platform warrants careful consideration and arrangement. The entire process involves various chemical and biological reagents; as such, some reagents may strongly interfere with the functions of others. Most notably, chemical reagents used for cell lysis (such as sodium dodecyl sulphate and Triton X-100) may inhibit PCR by reducing polymerase activity.11–13 Furthermore, the intracellular molecules such as proteins,14 polysaccharides,15 and ions16 (including Ca2+, Fe3+ and EDTA) in the cell lysate may also interact with a polymerase and affect the PCR result. So far there have been three strategies to remove or alleviate the impact of cell pretreatment on PCR: 1. Institute some type of isolation step to remove the lysis reagent and undesired intracellular molecules while preserving nucleic acids. This is typically done by adsorption of nucleic acids on solid surfaces (e.g. beads or matrices) while replacing the solution.2,3,17,18 The adsorption and desorption processes involved in these assays increase the complexity of the procedure. Additional chambers and structures may also be needed on the chip to accommodate the procedure. 2. Use alternative lysis methods that interfere with PCR to a lesser degree than surfactants, typically in combination with dilution. Freeze–thaw19 or heating20,21 has been applied to cell lysis in the context of genetic analysis. These methods are less efficient than surfactant-based lysis.22 3. Use a Direct PCR kit based on Phusion polymerase that is tolerant to surfactant-based lysis reagents.23 However, Phusion polymerase can be inhibited by SYBR green and other dyes24 and lacks 5' to 3' exonuclease activity.25 Thus commonly used fluorescence-based quantification is impossible with the Phusion polymerase system. To summarize, microfluidic strategies that permit simple operation and device design and are compatible with Taq polymerase are still in high demand. Such a strategy will be particularly beneficial for precise and demanding operations such as genetic analysis based on single cells.26–28
In this work, we demonstrate a simple scheme for conducting microfluidic PCR starting from cells, taking advantage of the difference in the diffusivity between DNA and various reagents/intracellular molecules. We use the same reaction chamber for both cell lysis and PCR and this is analogous to “one-pot synthesis” (i.e. using one reactor for successive reactions without separation and purification). The lysis buffer and the PCR mix were introduced into the chamber by concentration-gradient-driven diffusion. During such diffusion, the new solution replaces the solution and molecules from the previous step without removing the slow-diffusing large DNA molecules. We show that this scheme allows highly efficient genetic analysis of a low number of cells (including single cells). The single chamber (“one-pot”) design drastically minimizes the complexity of the microfluidic device. We envision that this may be a general approach for on-chip multi-step assays on sizable DNAs.
Our microfluidic device has a simple structure that includes a reaction chamber connected with two loading chambers on both sides (Fig. 1a). The connections between the reaction chamber and the two loading chambers (i.e. a number of channels) could be cut off by closing two-layer valves.29,30 A hydration line structure was also added in the control layer so that pressure and water inside the hydration line minimize the evaporation during PCR.2,31 The temperature required by PCR (up to 95 °C) accelerated water evaporation through the gas-permeable PDMS structure. The water in the hydration line (under 25 psi during PCR) moved through the PDMS membrane (~40 μm thick) by osmosis to compensate water loss due to evaporation. The hydration line also created high pressure in the reaction chamber during PCR which also decreased evaporation. With the volume of the loading chambers substantially larger than that of the reaction chamber (by a factor of 16), this simple microfluidic platform allowed the replacement of small molecules (either reagents or intracellular molecules) inside the reaction chamber by diffusion from/into the loading chambers under the concentration gradient, while preserving large DNAs inside throughout such process. The diffusion process is governed by Fick's first law:
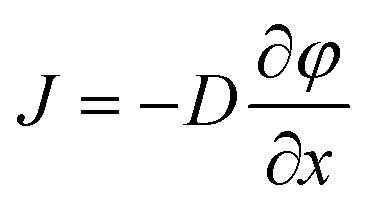 | (1) |
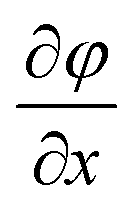 is the concentration gradient. There is a substantial difference in the diffusivity of involved species: at 25 °C, the diffusivity of genomic DNA fragments is estimated to be 2.0 × 10−13 m2 s−1 (by assuming ~50 kb as the average size and calculating using the empirical equation32), compared to 4.7 × 10−11 m2 s−1 for Taq polymerase,33 8.0 × 10−11 m2 s−1 for intracellular proteins34 (considering ~53 kDa as the average size), 1.0 × 10−10 m2 s−1 for primers (~20 bp ssDNA),35 3.7 × 10−10 m2 s−1 for dNTP,36 3.0 × 10−10 m2 s−1 for Triton X-10037 and 1.0 × 10−9 m2 s−1 for small ions,38 such as Mg2+, K+ and Cl−. Fig. 1b shows a COMSOL model of our microfluidic design. Only 1.4% of genomic DNA fragments diffuse out of the reaction chamber in 1 h. In comparison, 87.5% of the lysis reagent Triton X-100 and 44.3% of the intracellular proteins diffuse out of the reaction chamber (into the loading chambers) in the same period. 1 h loading of new reagents causes the concentrations of Taq polymerase and primers in the reaction chamber to reach 32% and 58%, respectively, of the concentrations in the loading chambers, compared to 90% for small ions. Thus the concentrations in the loading chambers may need to be high in order to supply the reaction chamber with desired concentrations for a given short loading period. It is worth emphasizing that the modeling was conducted by assuming the size of DNA to be ~50 kb which was the typical size of genomic DNA after lysis with shearing, as opposed to the size of a complete genomic DNA. A scaling law of D ~ L−v (v = 0.571 ± 0.014) applies for linear DNA molecules from 6 to 290 kb, where D is the diffusivity, L is the length of DNA and v is the scaling exponent.39 Larger DNA sizes which have smaller diffusivity will further facilitate our scheme.
is the concentration gradient. There is a substantial difference in the diffusivity of involved species: at 25 °C, the diffusivity of genomic DNA fragments is estimated to be 2.0 × 10−13 m2 s−1 (by assuming ~50 kb as the average size and calculating using the empirical equation32), compared to 4.7 × 10−11 m2 s−1 for Taq polymerase,33 8.0 × 10−11 m2 s−1 for intracellular proteins34 (considering ~53 kDa as the average size), 1.0 × 10−10 m2 s−1 for primers (~20 bp ssDNA),35 3.7 × 10−10 m2 s−1 for dNTP,36 3.0 × 10−10 m2 s−1 for Triton X-10037 and 1.0 × 10−9 m2 s−1 for small ions,38 such as Mg2+, K+ and Cl−. Fig. 1b shows a COMSOL model of our microfluidic design. Only 1.4% of genomic DNA fragments diffuse out of the reaction chamber in 1 h. In comparison, 87.5% of the lysis reagent Triton X-100 and 44.3% of the intracellular proteins diffuse out of the reaction chamber (into the loading chambers) in the same period. 1 h loading of new reagents causes the concentrations of Taq polymerase and primers in the reaction chamber to reach 32% and 58%, respectively, of the concentrations in the loading chambers, compared to 90% for small ions. Thus the concentrations in the loading chambers may need to be high in order to supply the reaction chamber with desired concentrations for a given short loading period. It is worth emphasizing that the modeling was conducted by assuming the size of DNA to be ~50 kb which was the typical size of genomic DNA after lysis with shearing, as opposed to the size of a complete genomic DNA. A scaling law of D ~ L−v (v = 0.571 ± 0.014) applies for linear DNA molecules from 6 to 290 kb, where D is the diffusivity, L is the length of DNA and v is the scaling exponent.39 Larger DNA sizes which have smaller diffusivity will further facilitate our scheme.
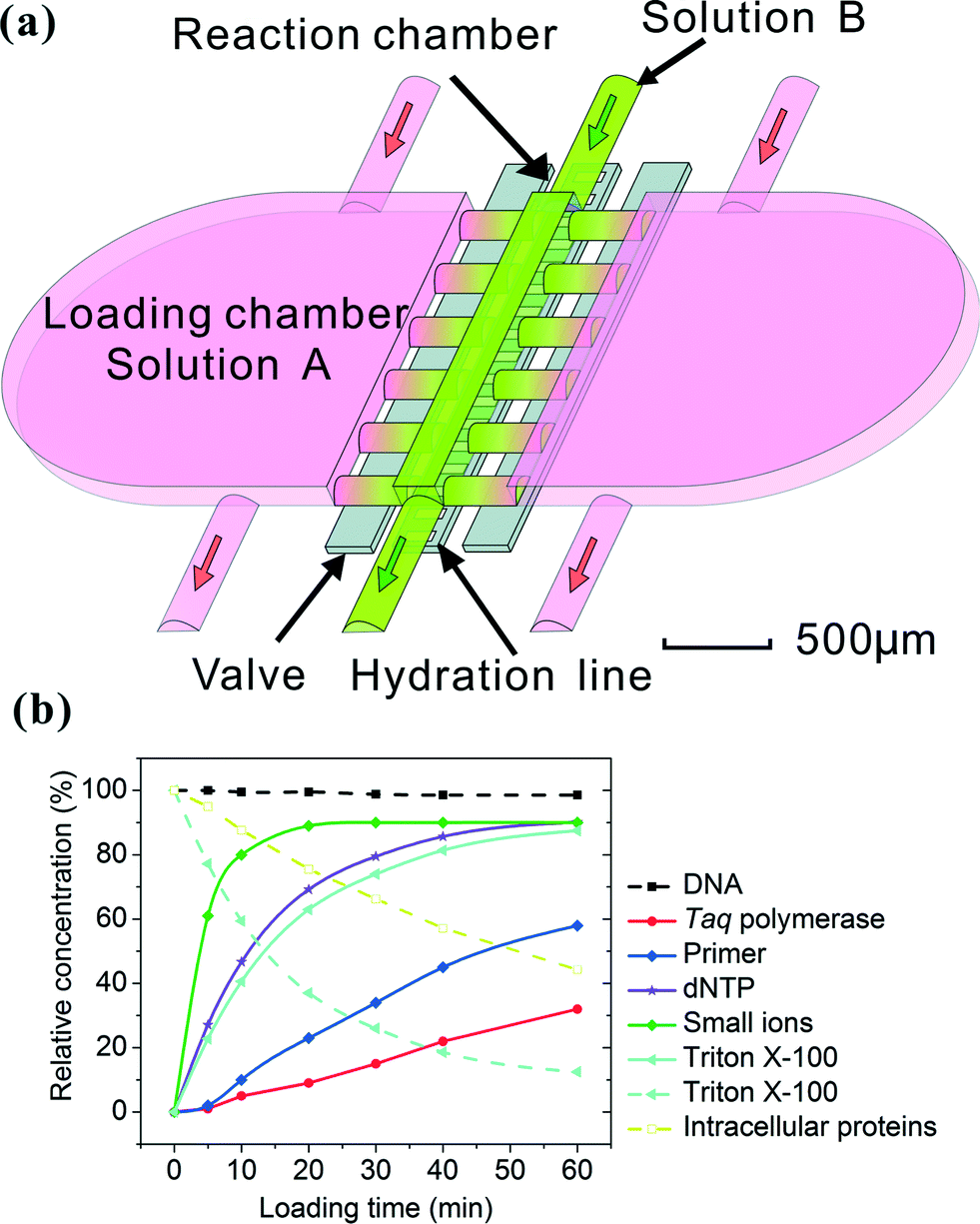 | ||
| Fig. 1 The principle of solution replacement based on diffusion. (a) The design of the microfluidic device (with a 24 nl reaction chamber). The device consists of two PDMS layers. Control and hydration are implemented in the same control layer by actuating the valves and supplying water under pressure (~25 Psi). The fluidic layer has a small reaction chamber connected with two large loading chambers. When the valves are open, molecules in solution A may enter the reaction chamber by diffusion, effectively replacing solution B. (b) The entry (solid lines) and release (broken lines) of various molecules into/out of the reaction chamber, modeled by COMSOL Multiphysics. The initial concentration in the loading chamber (in the cases of entry into the reaction chamber) or that in the reaction chamber (in the case of release out of the reaction chamber) was used as the reference (i.e. having a value of 100%). | ||
As an initial test, we used our platform for PCR of purified human genomic DNA produced from a lymphoblastoid cell line (GM12878). In the experiment, we first loaded genomic DNA in water into the reaction chamber (about 500 copies of DNA molecules in the 24 nl reaction chamber). We then filled the loading chambers with PCR mix having designed concentrations (250 U ml−1Taq polymerase, 1.2 μM for each primer, 0.4 mM for each of dNTPs, 6 mM MgCl2 and 100 mM KCl, 0.8% PEG 8000, 0.08% Tween-20) and allowed various periods for the loading times (0–30 min). It is worth noting that the concentrations of Taq polymerase and primers in the loading chamber were significantly higher than the desired concentrations (50 U ml−1 and 0.4 μM, respectively) in order to speed up the diffusion-based material transfer. During loading, the PCR mix (with primers targeting the GAPDH gene) slowly replaced the original solution in the reaction chamber by diffusion. The reaction chamber was then sealed off by the valves and the microfluidic chip was placed on a flat-plate thermal cycler for PCR amplification. After the PCR amplification, the PCR product was quantified using qPCR to obtain the copy number of the amplified gene. As shown in Fig. 2, we observed increasing amplification with longer loading times (the grey bars). To verify that the PCR result was dictated by the diffusion process, we used COMSOL to model the molecular transport during loading and generated values for Taq polymerase (0, 0.2, 2, 13 and 38 U ml−1) and primers (0, 3, 20, 120, 400 nM) in the reaction chamber under various loading times (0, 1, 3, 10, 30 min). We then conducted on-chip PCR in the reaction chamber containing PCR mixes with the above Taq polymerase/primer concentrations (striped bars). We found that the amount of the PCR product was very similar in these two cases. This confirms that the increase in the amplification under longer loading time was due to higher reagent concentrations in the reaction chamber after longer diffusion and the diffusion of Taq polymerase and primers was the limiting step.
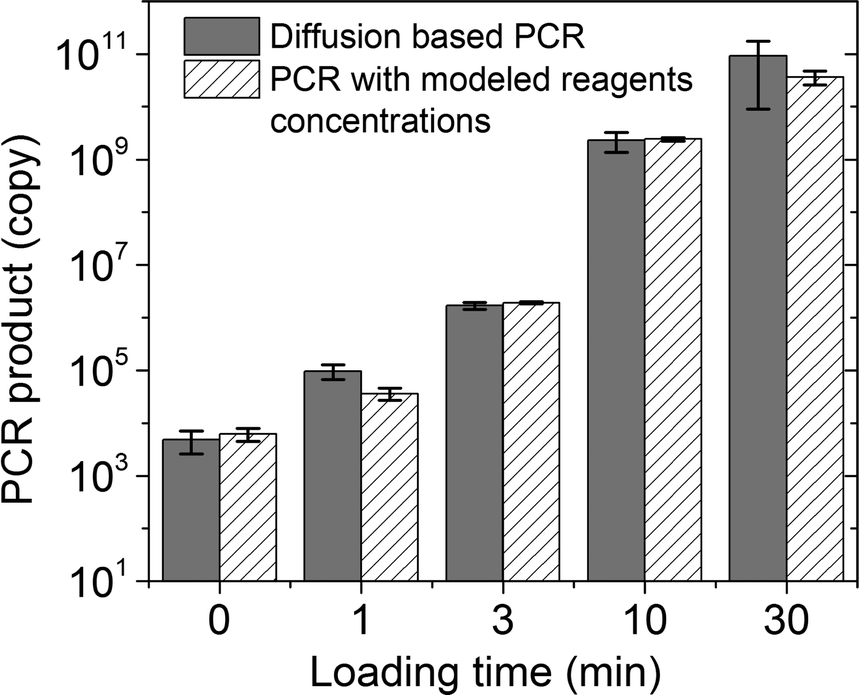 | ||
| Fig. 2 Diffusion-based PCR in a 24 nl reaction chamber starting with genomic DNA purified from GM12878 cells. The detection targeted the GAPDH gene and the amplification run for 30 cycles. The copy number of the PCR product was quantified using qPCR. The results with various loading times (grey bars, 0, 1, 3, 10 and 30 min) are compared with those of on-chip PCR with various Taq polymerase concentrations (0, 0.2, 2, 13 and 38 U ml−1) and primers concentrations (0, 3, 20, 120, 400 nM) (striped bars, these values were generated by COMSOL modelling of the diffusion process for the corresponding loading times) to confirm the impact of the diffusion on PCR results. | ||
Based on this understanding, we designed the multi-step PCR process starting from cells (Fig. 3a). Compared to the device described in Fig. 1 and 2, we reduced the volume of the reaction chamber to 3 nl (with those of the loading chambers also decreased in proportion), used it to process 1–100 cells and applied 45 thermal cycles for the amplification. GM12878 cells were first loaded into the reaction chamber. Triton X was flowed into the loading chambers (with the reaction chamber closed) and then diffused into the reaction chamber for lysing the cells with a loading time of 10 min (shown in Fig. 3b). The reaction chamber was then closed for 5 min for lysis to complete while the PCR mix (250 U ml−1Taq polymerase, 1.2 μM for each primer, 0.4 mM each of dNTPs, 6 mM MgCl2 and 100 mM KCl, 0.8% PEG 8000, 0.08% Tween-20) was flowed into the loading chambers. The PCR reagents were then diffused into the reaction chamber with a loading time of 30 min, while the lysis reagent Triton X and intracellular molecules diffused out. We run the thermal cycles (45 cycles) while the reaction chamber was closed. The PCR product was then flushed out of the reaction chamber for quantification by qPCR off-chip. Fig. 3c shows the results of the diffusion-based PCR for detecting samples ranging from single cells to 50 cells. Real-time PCR used in our experiment has the resolution to differentiate various numbers of cells in the range of 1–1000 (as shown in Fig. S1 in the ESI†). Fig. 3c shows that various numbers of cells (1, 5, 15 and 50) were easily differentiated based on the amount of the PCR product after amplification of 45 cycles. It is worth noting that the signal from the blank sample (0 cell) was due to the dimerization of primers as suggested by the melting curves (Fig. S2 in the ESI†). The parameters of the above process were carefully designed to maximize the efficiency of the on-chip PCR. Our model shows that the residual Triton X in the reaction chamber during PCR was less than 0.1% which has a minimal effect on PCR performance based on our test results (Fig. S3 in the ESI†). As a further confirmation, the diffusion-based PCR starting with cells yielded similar results compared to those starting with purified DNA for the same number of cells (Fig. S4 in the ESI†), indicating that there was no inhibition from the lysis reagent or intracellular molecules.
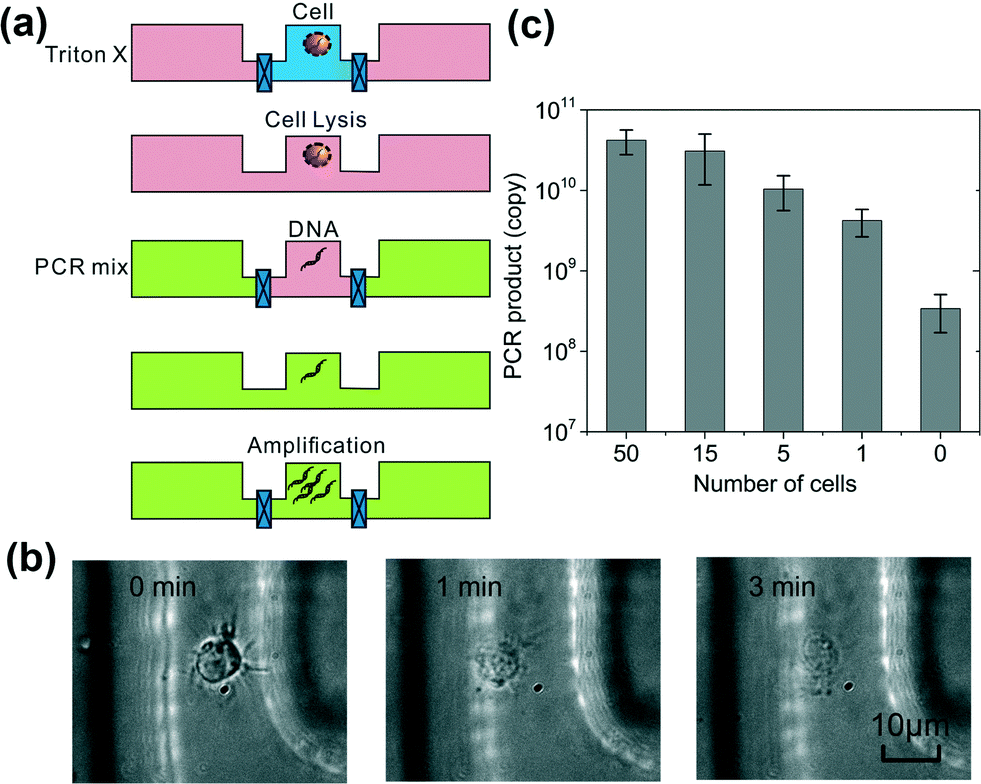 | ||
| Fig. 3 Combined lysis and PCR starting with GM12878 cells in a 3 nl reaction chamber. (a) A schematic of the procedure. Triton X lysis buffer replaces the original cell buffer by diffusion. Then the Triton X lysis reagents are replaced by PCR mix by diffusion. Finally PCR amplification occurs for 45 cycles. Triton X lysis buffer and PCR mix have loading times of 10 and 30 min, respectively. (b) The cell lysis observed under a differential interference contrast (DIC) microscope. Cells are completely lysed after the loading of Triton X lysis buffer for 3 min. (c) The combined lysis and PCR procedure on cells of various numbers (0–50). The PCR signal resulted from the control sample (0 cells) is from primer dimerization as shown by the melting curves (Fig. S2 in the ESI†). | ||
The use of diffusion for replacing small-molecule reagents in a microfluidic reactor is universally applicable to assays involving large DNA molecules isolated from cells. Our approach efficiently eliminates DNA isolation and purification steps and drastically simplifies the design of the microfluidic device. These advantages will be critical for assays starting from a low number of cells or processes involving large-scale parallel operations (e.g. analysis of single cell arrays). The major drawback of the approach is the added time required for diffusion. This additional assay time can be shortened by elevation of the temperature and increase in the concentration in the loading chamber. When complex cell samples (e.g. clinical samples involving multiple cell types) are used, the isolation of the target cell type from the mixture before the use of our protocol may be helpful for thorough removal of the inhibition of PCR by intracellular molecules.
Acknowledgements
We acknowledge financial support from Virginia Tech ICTAS NanoBio Trust, NSF CBET grants 1016547 and 0967069, NIH NCI R21CA174577.Notes and references
- E. A. Ottesen, J. W. Hong, S. R. Quake and J. R. Leadbetter, Science, 2006, 314, 1464–1467 CrossRef CAS PubMed.
- J. S. Marcus, W. F. Anderson and S. R. Quake, Anal. Chem., 2006, 78, 3084–3089 CrossRef CAS PubMed.
- C. J. Easley, J. M. Karlinsey, J. M. Bienvenue, L. A. Legendre, M. G. Roper, S. H. Feldman, M. A. Hughes, E. L. Hewlett, T. J. Merkel, J. P. Ferrance and J. P. Landers, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19272–19277 CrossRef CAS PubMed.
- R. Muddu, Y. A. Hassan and V. M. Ugaz, Angew. Chem., Int. Ed., 2011, 50, 3048–3052 CrossRef CAS PubMed.
- N. Agrawal, Y. A. Hassan and V. M. Ugaz, Angew. Chem., Int. Ed., 2007, 46, 4316–4319 CrossRef CAS PubMed.
- R. C. Bailey, G. A. Kwong, C. G. Radu, O. N. Witte and J. R. Heath, J. Am. Chem. Soc., 2007, 129, 1959–1967 CrossRef CAS PubMed.
- A. J. Qavi and R. C. Bailey, Angew. Chem., Int. Ed., 2010, 49, 4608–4611 CrossRef CAS PubMed.
- M. R. Hartman, D. Yang, T. N. N. Tran, K. Lee, J. S. Kahn, P. Kiatwuthinon, K. G. Yancey, O. Trotsenko, S. Minko and D. Luo, Angew. Chem., Int. Ed., 2013, 52, 8699–8702 CrossRef CAS PubMed.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 RSC.
- L. Chen, A. Manz and P. J. R. Day, Lab Chip, 2007, 7, 1413–1423 RSC.
- I. G. Wilson, Appl. Environ. Microbiol., 1997, 63, 3741–3751 CAS.
- P. Rådström, R. Knutsson, P. Wolffs, M. Lövenklev and C. Löfström, Mol. Biotechnol., 2004, 26, 133–146 CrossRef PubMed.
- H. A. Erlich, PCR technology. Principles and applications for DNA amplification, Stockton press, 1989 Search PubMed.
- A. Trombley Hall, A. McKay Zovanyi, D. R. Christensen, J. W. Koehler and T. Devins Minogue, PLoS One, 2013, 8, e73845 Search PubMed.
- L. Monteiro, D. Bonnemaison, A. Vekris, K. G. Petry, J. Bonnet, R. Vidal, J. Cabrita and F. Mégraud, J. Clin. Microbiol., 1997, 35, 995–998 CAS.
- W. A. Al-Soud and P. Rådström, J. Clin. Microbiol., 2001, 39, 485–493 CrossRef CAS PubMed.
- L. A. Legendre, J. M. Bienvenue, M. G. Roper, J. P. Ferrance and J. P. Landers, Anal. Chem., 2006, 78, 1444–1451 CrossRef CAS PubMed.
- J. W. Hong, V. Studer, G. Hang, W. F. Anderson and S. R. Quake, Nat. Biotechnol., 2004, 22, 435–439 CrossRef CAS PubMed.
- N. M. Toriello, E. S. Douglas, N. Thaitrong, S. C. Hsiao, M. B. Francis, C. R. Bertozzi and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20173–20178 CrossRef CAS PubMed.
- A. K. White, M. VanInsberghe, I. Petriv, M. Hamidi, D. Sikorski, M. A. Marra, J. Piret, S. Aparicio and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13999–14004 CrossRef CAS PubMed.
- K.-Y. Lien, W.-C. Lee, H.-Y. Lei and G.-B. Lee, Biosens. Bioelectron., 2007, 22, 1739–1748 CrossRef CAS PubMed.
- T. W. Gilbert, T. L. Sellaro and S. F. Badylak, Biomaterials, 2006, 27, 3675–3683 CAS.
- D. P. Manage, Y. C. Morrissey, A. J. Stickel, J. Lauzon, A. Atrazhev, J. P. Acker and L. M. Pilarski, Microfluid. Nanofluid., 2011, 10, 697–702 CrossRef CAS.
- F. Mao, W.-Y. Leung and X. Xin, BMC Biotechnol., 2007, 7, 76 CrossRef PubMed.
- Y. Wang, D. E. Prosen, L. Mei, J. C. Sullivan, M. Finney and P. B. Vander Horn, Nucleic Acids Res., 2004, 32, 1197–1207 CrossRef CAS PubMed.
- R. Novak, Y. Zeng, J. Shuga, G. Venugopalan, D. A. Fletcher, M. T. Smith and R. A. Mathies, Angew. Chem., Int. Ed., 2011, 50, 390–395 CrossRef CAS PubMed.
- P. Liu, X. Li, S. A. Greenspoon, J. R. Scherer and R. A. Mathies, Lab Chip, 2011, 11, 1041–1048 RSC.
- K. Leung, H. Zahn, T. Leaver, K. M. Konwar, N. W. Hanson, A. P. Pagé, C.-C. Lo, P. S. Chain, S. J. Hallam and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7665–7670 CrossRef CAS PubMed.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- M. A. Unger, H.-P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- K. A. Heyries, C. Tropini, M. VanInsberghe, C. Doolin, O. I. Petriv, A. Singhal, K. Leung, C. B. Hughesman and C. L. Hansen, Nat. Methods, 2011, 8, 649–651 CrossRef CAS PubMed.
- G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman and A. Verkman, J. Biol. Chem., 2000, 275, 1625–1629 CrossRef CAS PubMed.
- Z. Bu, R. Biehl, M. Monkenbusch, D. Richter and D. J. Callaway, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17646–17651 CrossRef CAS PubMed.
- A. Nenninger, G. Mastroianni and C. W. Mullineaux, J. Bacteriol., 2010, 192, 4535–4540 CrossRef CAS PubMed.
- T. Mamedov, E. Pienaar, S. E. Whitney, J. R. TerMaat, G. Carvill, R. Goliath, A. Subramanian and H. J. Viljoen, Comput. Biol. Chem., 2008, 32, 452–457 CrossRef CAS PubMed.
- M. J. Hubley, B. R. Locke and T. S. Moerland, Biochim. Biophys. Acta, Gen. Subj., 1996, 1291, 115–121 CrossRef.
- D. G. Leaist, J. Solution Chem., 1991, 20, 187–197 CrossRef CAS.
- Y.-H. Li and S. Gregory, Geochim. Cosmochim. Acta, 1974, 38, 703–714 CrossRef CAS.
- R. M. Robertson, S. Laib and D. E. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7310–7314 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00498a |
| This journal is © The Royal Society of Chemistry 2014 |