 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCircle-to-circle amplification on a digital microfluidic chip for amplified single molecule detection†
Malte
Kühnemund
a,
Daan
Witters
b,
Mats
Nilsson
*c and
Jeroen
Lammertyn
*b
aScience for Life Laboratory, Department of Immunology, Genetics and Pathology, Uppsala University, Box 815, 751 08 Uppsala, Sweden
bKU Leuven – University of Leuven, BIOSYST-MeBioS, Willem de Croylaan 42, Leuven, Belgium. E-mail: jeroen.lammertyn@biw.kuleuven.be; Tel: +32 16 32 14 59
cScience for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Box 1031, Se-171 21 Solna, Sweden. E-mail: mats.nilsson@scilifelab.se; Tel: +46 (0)762 756 161
First published on 5th June 2014
Abstract
We demonstrate a novel digital microfluidic nucleic acid amplification concept which is based on padlock probe mediated DNA detection and isothermal circle-to-circle amplification (C2CA). This assay platform combines two digital approaches. First, digital microfluidic manipulation of droplets which serve as micro-reaction chambers and shuttling magnetic particles between these droplets facilitates the integration of complex solid phase multistep assays. We demonstrate an optimized novel particle extraction and transfer protocol for superparamagnetic particles on a digital microfluidic chip that allows for nearly 100% extraction efficiencies securing high assay performance. Second, the compartmentalization required for digital single molecule detection is solved by simple molecular biological means, circumventing the need for complex microfabrication procedures necessary for most, if not all, other digital nucleic acid detection methods. For that purpose, padlock probes are circularized in a strictly target dependent ligation reaction and amplified through two rounds of rolling circle amplification, including an intermediate digestion step. The reaction results in hundreds of 500 nm sized individually countable DNA nanospheres per detected target molecule. We demonstrate that integrated miniaturized digital microfluidic C2CA results in equally high numbers of C2CA products μL−1 as off-chip tube control experiments indicating high assay performance without signal loss. As low as 1 aM synthetic Pseudomonas aeruginosa DNA was detected with a linear dynamic range over 4 orders of magnitude up to 10 fM proving excellent suitability for infectious disease diagnostics.
Introduction
DNA analysis plays an important role in molecular diagnostics. Traditional laboratory analysis methods, however, require time-consuming manual handling and sophisticated instrumentation. Recently, integrated microfluidic lab-on-chip devices emerged that can overcome these limitations by miniaturization and the automation of laboratory methods,1–4 making them attractive for research and point-of-care (POC) diagnostic applications. One elegant way of microfluidic liquid handling is the transportation of nano- to microliter sized droplets by electrowetting-on-dielectric (EWOD) on digital microfluidic (DMF) chips.5–7 On DMF chips, each droplet serves as a discrete reaction compartment that can be addressed individually and independently.8 This allows fluid droplets to be controlled with maximum flexibility and programmability, an important asset for bio-assays requiring many complex handling steps to be executed simultaneously on one single device. DMF chips have shown to be capable of manipulating magnetic particles with unprecedented precision, speed and flexibility.9–11 As such, magnetic particles can be separated, washed, mixed and incubated with new reagents in a highly automated and miniaturized way allowing for the performance of multistep solid-phase bio-assays.9,10,12 Efficient and rapid mixing is promoted by a droplet internal vortex that is created by the transportation of droplets by EWOD.6,7 Recently, a magnetic extraction protocol for 3 μm sized ferromagnetic particles on a digital microfluidic chip was described that allowed for the performance of a solid-phase immunoassay in presence of a spatially fixed permanent magnet.13 Here we report on 2 critical particle extraction parameters that have not been addressed before and illustrate a novel highly efficient extraction and transfer protocol for 1 μm sized superparamagnetic particles on a DMF chip using a spatially movable magnet allowing to shuttle particles between droplets with high simplicity and perform particle-based enzymatic reactions that require a homogeneous particle suspension.One of the critical issues for automated DNA detection and analysis has been the difficulty of integrating DNA amplification assays on microfluidic platforms. The gold standard for DNA amplification, the polymerase chain reaction (PCR), requires precise cycling of temperatures which is challenging to achieve in integrated micro-systems. Nevertheless, Chang et al. succeeded in integrating a PCR assay on a DMF chip making use of the capability to shuttle droplets between heating zones to achieve cycling.14 Besides PCR, several isothermal amplification methods have emerged that are promising alternatives to PCR. Amongst them are helicase dependent amplification (HDA),15 strand displacement amplification (SDA),16 loop mediated isothermal amplification (LAMP)17 and rolling circle amplification (RCA).18 LAMP assays have successfully been integrated into micro-total analysis systems with high speed and sensitivity.19 However, LAMP assays are poorly multiplexable and the target dependent design of primer sets rather complicated. Hyper branched RCA is a simple and rapid method20 but the branching primer pairs often lead to unspecific background amplification.
High specificity and sensitivity are major requirements for DNA detection in diagnostics. Specificity can be achieved by the requirement of two or more hybridization events and a DNA ligation event that generates amplifiable DNA reporter molecules.21 Padlock probes are linear oligonucleotides that are ligated in a strictly target sequence-dependent manner to form a circle that can subsequently be amplified.22 Circularization by ligation requires that both target complementary probe arms hybridize to the target sequence with perfect match. The high discrimination rate of thermophilic DNA ligases enables single nucleotide mismatch detection.23 Amplification of padlock probes by RCA is exclusive to circularized probes which adds another step of specificity.24,25 The RCA product, a concatemer of hundreds of padlock probe replicates, collapses into a distinct 500 nm sized spheric DNA coil that, when labeled with fluorescent oligonucleotides, can be digitally counted after deposition on a microscope slide26 or while flowed through a microfluidic channel.27 As these readout methods only detect a fraction of the generated RCA products an additional amplification step is needed when high sensitivity is required. Circle-to-circle amplification (C2CA) generates hundreds of RCA products from one initial RCA product by monomerization of the concatemer, re-circularization of monomers and amplification of the newly generated circles in a second RCA.28 C2CA is an isothermal amplification method with low amplification bias and variation coefficient facilitating high multiplexed analysis.28 The use of magnetic particles as a solid support significantly increases assay sensitivity and speed by separation of circles from unreacted padlock probes and sample remains.29,30 Furthermore, in contrast to most digital single molecule detection methods, RCA and C2CA do not require compartmentalization for multiplexed or digital analysis. Single molecules are amplified to individually countable objects which enables digital read out.26,27 However, besides high analytical performance and isothermal reaction conditions, C2CA requires multiple steps which make its application in diagnostics less favorable. Therefore the integration and automation of C2CA into microfluidic lab-on-chip devices is of utmost importance in order to bring its advantages to diagnostic application.
Only a few studies have been reported in which microfluidic chips are used for the detection of RCA amplified molecules.27,31,32 Sato et al. (2010) have described an on chip micro-bead based RCA assay achieving low femtomolar detection limits. The counting of RCA products, however, required tedious confocal microscopy in order to detect RCA products distributed around the micro-beads in the channel.32 In another study C2CA was performed in the sample-well of a microfluidic chip for gel-electrophoretic separation of the C2CA products.33 Despite the detection of C2CA products on microfluidic systems the assay has, until now, not been performed integrated on a microfluidic chip.
Here we report, for the first time, the performance of C2CA on a DMF chip with all assay steps integrated on-chip, except for heating. We demonstrate how DMF's reconfigurable and automated droplet actuation principle, combined with an improved protocol for shuttling magnetic particles between droplets that serve as discrete reaction microchambers, offers a unique and novel way of performing miniaturized particle-based C2CA. C2CA performed on a DMF chip proved as efficient as compared to reaction tube conditions, a major achievement rendered by DMF's excellent droplet mixing capabilities and the new particle manipulation protocol described in this paper. We applied DMF C2CA for the detection of ultra-low amounts of synthetic P. aeruginosa DNA illustrating the suitability for infectious disease diagnostics.
Results and discussion
Digital microfluidic C2CA assay layout
The integration of C2CA on the DMF chip is illustrated in Fig. 1. The droplets containing the DNA sample, ligation reaction mixture, magnetic particles and washing buffer were initially placed on the chip before the assay was started. The RCA, digestion and ligation & 2nd RCA droplets were consecutively introduced during the assay, as described in the experimental section.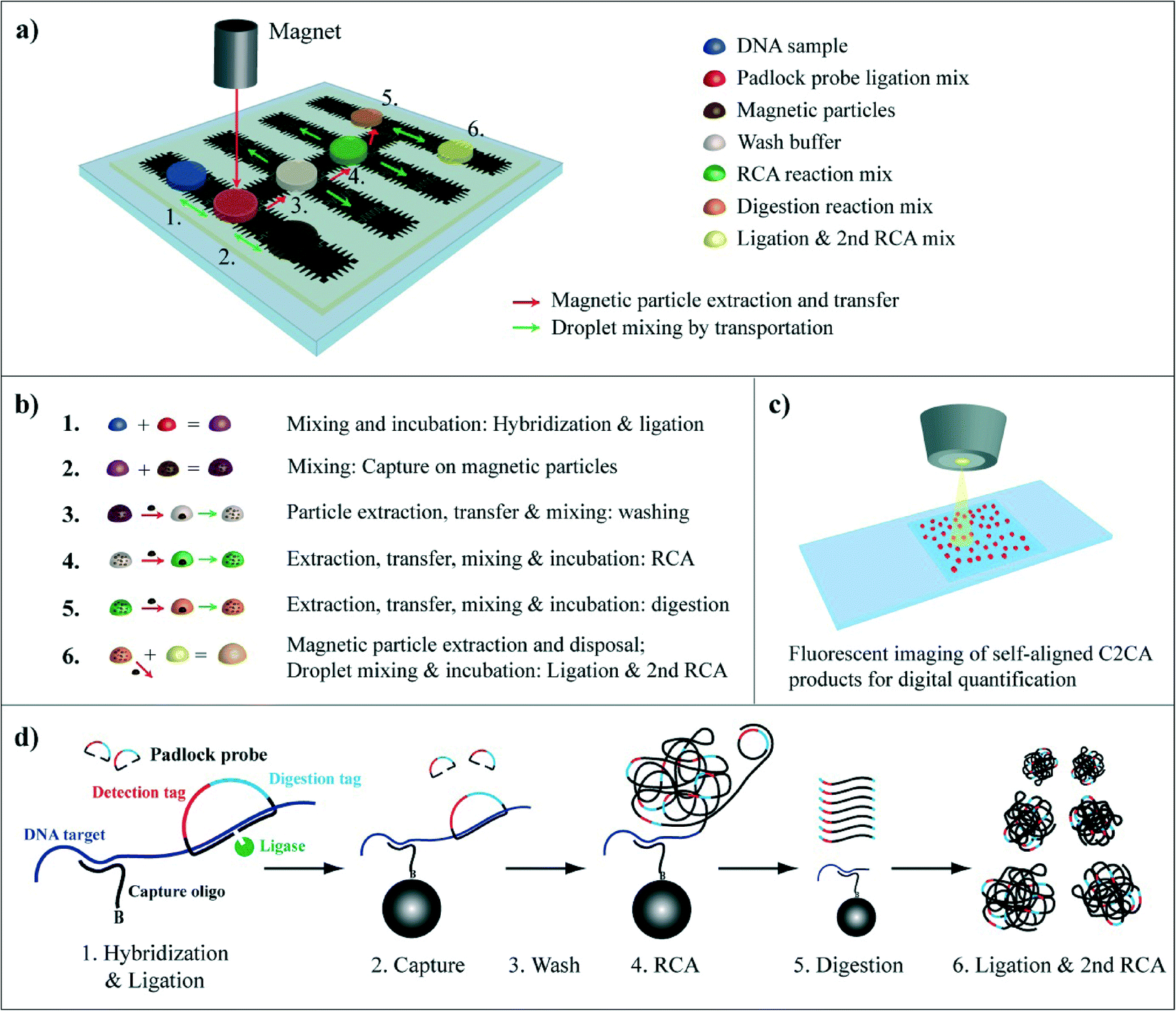 | ||
| Fig. 1 Circle-to-circle amplification protocol on a DMF chip. a) DMF chip and assay layout. Positions of droplets with reaction mixes are depicted. Mixing of two droplets is illustrated with green arrows with double arrowheads. Mixing of single droplets is illustrated by green arrows with single arrowhead. Magnetic particle transfer between droplets is indicated with red arrows. b) Flow chart illustrating magnetic particle extraction & transfer and droplet mixing; droplet color code as in 1a. c) Fluorescence imaging of individual RCA products that randomly align on a microscope slide promoting digital read-out. d) C2CA: (1) a ligation mix containing target specific padlock probes and biotinylated capture oligonucleotides is mixed with a DNA sample. (2) After hybridization and ligation, streptavidin coated magnetic particles are mixed with the ligation complex to bind the ligation complex to magnetic particles via the biotinylated capture oligonucleotide. (3) The particles are magnetically separated, transferred to the washing droplet and washed. (4) The particles are separated again and mixed into RCA reaction mix. (5) After RCA the particles with coupled RCA products are extracted and transferred into the digestion mix where the RCA products are monomerized. (6) The droplet with monomers is mixed with a droplet for ligation + the 2nd RCA (for more details see Dahl et al., 2004 ref. 28). | ||
In brief, a droplet containing ligase, target specific padlock probes and biotinylated capture oligonucleotides is merged with a DNA sample (Fig. 1a and b step 1). After 5 min hybridization and ligation the capture oligonucleotide is annealed and the padlock probe ligated on the DNA strand (Fig. 1d). Thereupon, streptavidin coated magnetic particles are merged with the sample and mixed by droplet transportation promoting the capture of the ligation complex by biotin–straptavidin bond (Fig. 1a, droplet movement is indicated by green arrows; Fig. 1b step 2). The particles are magnetically extracted and transferred to the washing droplet by moving the particles along with the magnet to the washing droplet (illustrated in Fig. 1a by red arrows and in Fig. S1†). Next, the particles are dispersed and, hence, washed by droplet transportation along the electrode row (Fig. 1a, green arrows; Fig. 1b step 3). The particles are then separated and transferred to and mixed in the RCA reaction mixture (Fig. 1b step 4). After 20 min RCA and 2 min heat inactivation the particles with coupled RCA products are transferred into the digestion mix (Fig. 1b step 5) and the RCA products are enzymatically monomerized during 2 min incubation and 2 min heat inactivation. After particle extraction and disposal the droplet with monomers is mixed with a second ligation and RCA mix (Fig. 1b step 6) where the monomers are re-circularized and amplified in a second RCA step for 25 min. The total assay time is ~60 min. The C2CA products are labeled with fluorescent oligonucleotides, imaged on a microscope slide and counted digitally (Fig. 1c).
Highly efficient extraction of 1 μm sized superparamagnetic particles on a DMF chip
Magnetic particles play an important role in the automation of C2CA by supplying a solid phase for molecule capture and for washing away sample remains and unreacted padlock probes. In order to achieve a high performance with the assay layout described above the first objective of this study was to develop an extraction and transfer protocol for 1 μm sized superparamagnetic particles on a DMF chip (Fig. S1†). Ng et al. (2012)10 have implemented a particle extraction and washing protocol with a spatially removable magnet on a DMF chip, however, no information about the particles that were used in the study was provided. Vergauwe et al. have developed an extraction protocol for ferromagnetic particles on a DMF chip with a spatially fixed permanent magnet.13 They investigated the effect of the capillary force of the moving droplet on the extraction efficiency for different ferromagnetic particle concentrations. The automation of the C2CA protocol, however, requires the use of superparamagnetic particles because the particles need to be homogenously dispersed and incubated in enzyme reaction mixtures in absence of a magnet. In contrast to ferromagnetic particles, which after magnetic attraction stay magnetized and which are hardly dispersible, superparamagnetic particles can easily be resuspended. Droplet transportation in DMF creates a recirculating flow inside the droplet which leads to very efficient mixing7 and superparamagnetic particles can, after extraction from a droplet, be rapidly resuspended in a new droplet.9,10In addition to the parameters tested by Ng et al. (2012)10 and Vergauwe et al.13 we investigated the influence of (i) the distance between the magnet and the particles on the particle extraction efficiency by placing the magnet on top plates with different thicknesses; and (ii) the position of the magnet edge above the electrodes.
The distance between the magnet and the particles has a strong effect on the particle extraction efficiency. When using a 1.1 mm thick top plate, the extraction of 1 × 106 particles μL−1 (the concentration usually used in C2CA) was insufficient as a high amount of particles remained in the droplet when the droplet was transported away from the magnet (Fig. 2c; movie S3†). The incomplete extraction of low particle concentrations is likely due to insufficient magnetic force on the particles as an increased distance between the magnet and the magnetic particles decreases the magnetic force on the particles.10,12 At the point of extraction the droplet meniscus starts necking and the particle pellet eventually pinches off when the neck closes behind the extracted pellet.34 However, if the magnetic force is insufficient the capillary force of the closing droplet breaks the particle pellet and pulls a fraction of the particles back into the moving droplet. The loss of such large amounts of particles would drastically decrease the assay efficiency.
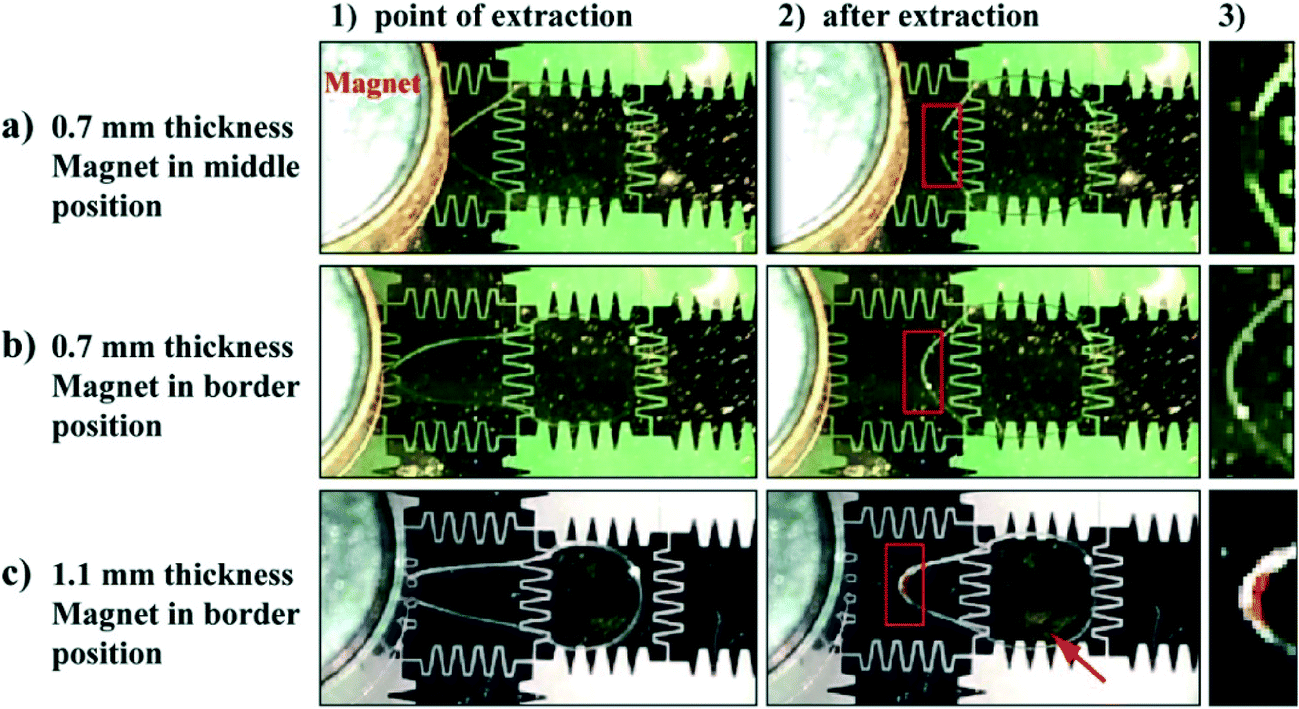 | ||
| Fig. 2 Video clip captures of magnetic particle extraction from a droplet moving on the DMF chip under a magnet placed on top of a 0.7 mm (a and b) or 1.1 mm thick top plate (c). The magnet is positioned in either the middle (a) or on the border between two electrodes (b and c). 1) Video clip capture at the point of extraction: magnetic particles are pelleted at the edge of the magnet as the droplets move from the left to the right. 2) The same droplets a few milliseconds after extraction. The red arrow in c-2) indicates remaining particles in the droplet. Red boxes in 2) indicate the meniscus of the droplet which is enlarged and illustrated in 3). The parameters were tested with 3 repeated extraction attempts. | ||
The position of the magnet above the electrode was found to play an important role in the particle extraction process, as well. The extraction of low particle concentrations (105–106 particles μL−1) was successful only when the magnet edge was placed on the border between two electrodes (Fig. 2b and movie S4†) but insufficient when positioned in the middle of the electrode. In the latter case a high amount of particles was left inside the droplet when the droplet moved away from the magnet (Fig. 2a; movie S5†). This effect can be explained in relation to the length of the droplet meniscus stretch during the particle extraction process. With the magnet edge positioned on the border between 2 electrodes, the magnetic particles are concentrated at the edge of the droplet. When the droplet moves to the neighboring electrode, the droplet creates a longer stretched meniscus to the particle pellet than when the magnet edge is positioned in the middle of the electrode (compare point of extraction (1) and after extraction (2) in Fig. 2a and b). The longer meniscus stretch forms a narrower neck and facilitates the pinching-off to a greater extent than a shorter stretch. Consequently, the particles get extracted.
With the magnet positioned on the border between two electrodes and a 0.7 mm thick top plate, 1 μm sized superparamagnetic particles with a concentration as low as 1.6 × 105 particles μL−1 were extracted successfully. Using these parameters, extraction efficiencies were also determined for 1 μm sized T1 dynabeads and 2.8 μm sized M280 beads. T1 dynabeads could be extracted in the same concentration (1.6 × 105 particles μL−1) as C1 dynabeads and M280 beads could be extracted in 3 × 104 particles μL−1 concentration (Fig. S2†).
Since the loss of particles during magnetic extraction and transfer between reagent droplets results in signal decrease we investigated the number of 1 μm sized particles that were lost during extraction using the optimized particle extraction parameters. For that purpose the total number of particles that remained in the enzyme reaction droplets after extraction was determined, as described in the experimental section. The amount of particles that were lost during extraction from the ligation (2183.25 ± 584), washing buffer (2611 ± 1248) and RCA droplets (1807.25 ± 1281) was very low (Fig. 3, black bars), proving that particles are extracted with very high efficiencies (~99.5% for all 3 extraction steps; illustrated by grey bars in Fig. 3, n = 4). With an optimized magnetic particle extraction and transfer protocol available, C2CA can be performed on the DMF chip.
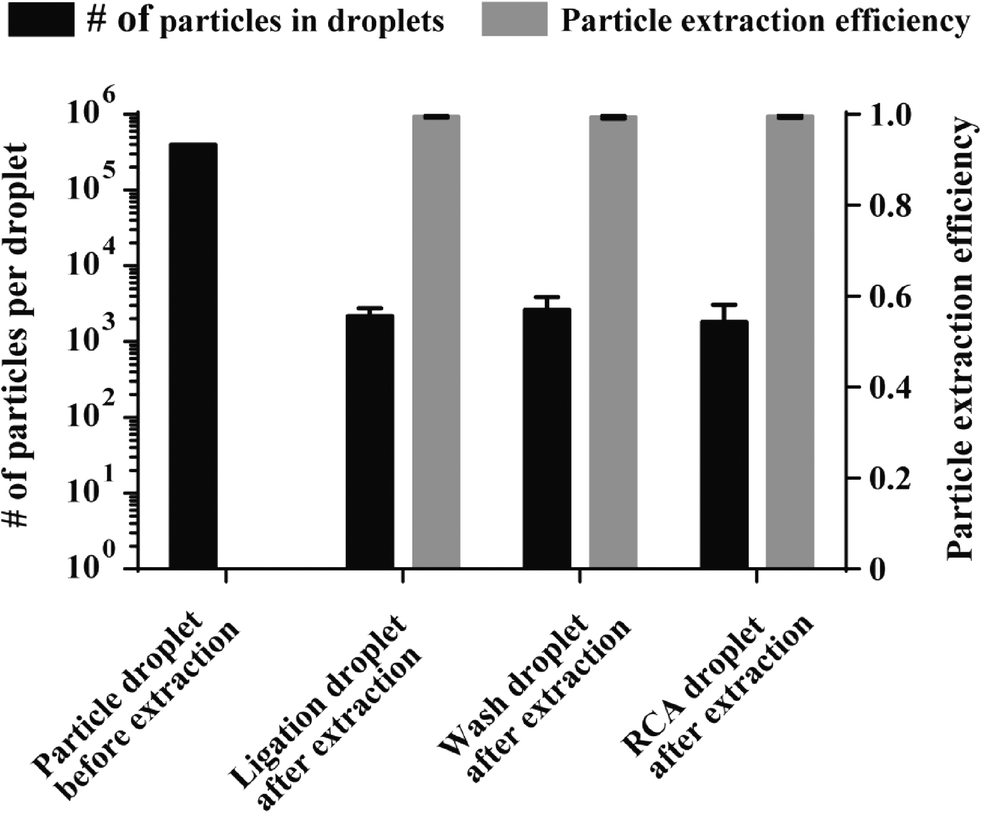 | ||
| Fig. 3 Magnetic particle extraction efficiency. Black bars indicate the average number of particles counted after extraction from the ligation, washing and RCA droplets and the number of particles per droplet before extraction (black bar on the left). Grey bars show the particle extraction efficiencies calculated as 1 – number of particles in droplets after extraction divided by the number of particles in droplets before extraction (n = 4 each, error bars represent 1 stdev. from the mean). | ||
Ligation, RCA and digestion reaction efficiencies on the DMF chip
C2CA consists of 6 subsequent steps that require optimal droplet transportation, mixing and magnetic particle extraction. The mobility of droplets carrying proteins on hydrophobic surfaces, as it is the case in the Teflon-AF covered surface of DMF chips, can be inhibited by bio-fouling effects.35 The reaction mixtures that are used in C2CA include BSA and enzymes which both can interact with hydrophobic surfaces. Absorption of enzymes does not only contribute to problems with on-chip droplet transportation but would also reduce the reaction efficiency. We added Pluronics to enzyme mixtures which was shown to reduce bio-fouling effects and to improve the transport of reagents on DMF chip surfaces.36 The presence of Pluronic F68 in ligation, RCA and digestion reaction mixtures significantly improved droplet mobility on the DMF chip and had no negative effect on the activity of the enzymes involved in C2CA, as investigated in off-chip experiments (data not shown).To test the on-chip enzymatic efficiencies the ligation, RCA and digestion reaction steps were performed on the DMF chip individually. For that purpose enzymatic reaction mixes were prepared in larger volumes and 2.5 μL was pipetted on the DMF chip. In this series of experiments all other reaction steps except the investigated one were performed off-chip in reaction tubes (Fig. 4, table above graph; Fig. 1b, step 1: hybridization & ligation, step 4: RCA and step 5: digestion). The droplets were transported over a few electrodes and then incubated without movement for 5 min (ligation), 20 min (RCA) and 2 min (digestion). After these incubations, the droplets were again briefly transported to disperse particles that had settled during the incubation and then the droplets were aspirated from the chip and subsequent steps were performed in reaction tubes. As a control, C2CA was performed in parallel under standard reaction conditions in reaction tubes in a 20 μL volume. All C2CA products were labeled with fluorescent detection oligonucleotides and imaged on a microscope slide. The products were counted with the particle analyzer function of ImageJ. The enzymatic reaction efficiencies are plotted as the ratio of the resulting counts to the counts of the tube control experiment (Fig. 4). All 3 enzymatic reactions worked with high efficiency as the ratios between on-chip and tube-control experiments for all 3 enzymatic steps were close to 1. No significant difference was found between the performances of enzymatic reactions on-chip compared with tube control experiments (2-tailed Student's t-test).
 | ||
| Fig. 4 Enzymatic reaction efficiencies on the DMF chip. The C2CA enzymatic reaction steps were individually tested on-chip (The respective step that was performed on the DMF chip is indicated with “✓” in the table above the graph). Prior and subsequent steps were performed off-chip in reaction tubes (as indicated with “—” in the table above the graph). A RCA control with 2.5 μL reaction volume was performed in tubes testing for the effect of reaction volume reduction. After remaining steps were performed in tubes the C2CA products were counted. The reaction efficiencies are plotted as the ratio to the counted C2CA product number of a 20 μL tube control experiment. Error bars represent 1 stdev. from the mean (n = 3 for ligation and RCA 2.5 μL, n = 2 for RCA and digestion; experiment repeated twice). | ||
To test whether the mere decrease in reaction volume has an influence on the efficiency a 2.5 μL RCA reaction was performed in reaction tubes. 2.5 μL of particles in RCA mixture were separated from a 20 μL volume and all subsequent steps were performed in 2.5 μL volume in tubes. No significant difference in reaction efficiency was observed (Fig. 4, right column). However, relatively high variations were obtained for the ligation step and the RCA in the 2.5 μL tube control, as indicated by the high standard deviations in Fig. 4 (n = 3 replicates, two experimental repetitions). The high variations very likely result from inaccurate manual pipetting and handling of particles in 2.5 μL volumes during the various reaction steps in tubes. Since the ligation is the first step of the assay, and all subsequent steps were performed in small reaction volumes off-chip, this experiment was most prone to pipetting errors.
Conclusively, ligation, RCA and digestion reactions can be performed on DMF chips without loss of efficiency. Hydrophobic surface interactions seem not to influence the enzymatic reactions as the reaction efficiencies on-chip were comparable with tube experiments. Hence, C2CA is suitable for the integration into DMF chips.
Circle-to-circle amplification on DMF for the detection of ultra-low DNA concentrations
After successfully testing the individual enzymatic reaction steps on the DMF chip, which showed comparable enzymatic reaction efficiencies on-chip and off-chip, the complete C2CA with all sequential steps was performed on-chip as illustrated in Fig. 1a and described in the experimental section.To determine the limit of detection (LOD) the target DNA was diluted in 10-log steps, and C2CA was performed on the DMF chip and in reaction tubes in parallel. In Fig. 5a, representative images of C2CA products imaged on microscope slides after on-chip and tube control amplifications are depicted. Each distinct C2CA product represents an amplified single molecule that can be digitally counted. One smaller region per image is enlarged and the zoomed image is located at the top right corner of each image for visualization of individual C2CA products. With increasing DNA concentration increasing numbers of C2CA products cover the 2 dimensional area on the microscope slide, until the slide is densely packed and the signal is saturated (images from 100 fM amplification in Fig. 5a show a saturated slide).
 | ||
| Fig. 5 Fluorescence microscope read out of amplified single molecule detection with all steps of the C2CA implemented on a DMF chip. a) Fluorescent images of self-aligned C2CA products on microscope slides after probing different amounts of target DNA and C2CA on DMF chip (1) and in reaction tubes (2). In total 6 areas were imaged per sample and the C2CA products counted with ImageJ. One representative image per sample is shown with an enlarged region within the image for the negative control, 100 aM and 100 fM concentrations. b) Logarithmic plot of the number of RCA products counted in the sum of 6 images as a function of DNA concentration. Error bars represent 1 stdev. from the mean (n = 2). | ||
As little as 100 ymol (~60 molecules) in 10 μL (10 aM concentration) were detected in reaction tubes which correlates well with results that have been achieved with this amplification method.27 The on-chip amplification resulted in comparable detection efficiency. Due to a very low number of C2CA products with very low standard deviation generated from the negative control on-chip (average 6 ± 1.4, n = 2), the number of C2CA products generated from 1 aM concentration (average 17.6, n = 2) was higher than three standard deviations of the negative control (Fig. 5b) indicating a limit of detection of 1 aM for DMF C2CA. Linear regression analysis shows linearity between 1 aM and 10 fM with an R2-value of 0.99 for tube reaction values and an R2 = 0.97 for DMF chip values for the same dynamic range. No statistically significant differences between the tube control and on-chip experiments were observed for any of the tested concentrations (2-tailed Student's t-test).
Conclusively, we demonstrated how C2CA can be performed on DMF platforms using the novel magnetic particle shuttling protocol to enable DNA detection with very high sensitivity, reproducibility and as high assay efficiency as when performed under optimal standard tube conditions, a result that has not been reached on other microfluidic platforms until now.
This study aimed to investigate key parameters necessary for full automation and optimize the assay performance on-chip. In this study the RCA, digestion and ligation & 2nd RCA reaction mixtures were manually pipetted onto the chip and transported to their respective position after heat inactivation of the previous enzymatic step. Currently, efforts are made for the integration of local microheater elements on-chip. The possibility to locally heat selected droplets while keeping the temperature on the rest of the chip constant is an opportunity for total assay automation. Additionally, isothermal approaches for enzyme inactivation, such as enzyme inactivating washing buffers, are being investigated.
In comparison to qPCR14 or LOOP19 DNA amplification methods on microfluidic chips, C2CA offers great multiplexing capability while maintaining high specificity,26,28,37 an important feature for clinical diagnostic applications. Moreover, the digital read-out format of counting individual C2CA products facilitates high sensitivity and a broad dynamic range.27,28 While quantitative PCR performed on a digital microfluidic chip requires intermediate fluorescent measuring steps,14 the read out of C2CA only requires a simple end point measurement reducing the complexity of the detector unit.
Experimental
Reagents
Phi29 polymerase and T4 DNA ligase, dNTP mix, ATP and phi29 10× reaction buffer were obtained from Thermo Scientific (Göteborg, Sweden). AluI restriction enzyme was acquired from New England Biolabs (Bionordika, Stockholm, Sweden). Amp ligase was bought from Epicentre (Nordic Biolabs AB, Täby, Sweden). Pluronic F68 was acquired from Sigma Aldrich (Bornem, Belgium). Vapor-Lock oil was bought from Qiagen (Hilden, Germany). Oligonucleotides were obtained from Integrated DNA Technologies (Heverlee, Belgium). Superparamagnetic MyOne dynabeads C1, T1 and M280 with streptavidin surface functionalization were acquired from Life Technologies (Oslo, Norway). All reagents for photolithography were purchased from Rohm and Haas (Marlborough, MN, USA). Parylene-C and Silane A174 were obtained from Plasma Parylene Coating services (Rosenheim, Germany), and Teflon-AF was obtained from Dupont (Wilmington, DE, USA).Chip fabrication and droplet manipulation parameters
DMF chips were fabricated as described previously.11 Briefly, bottom plates of the DMF chips with a total dimension of 32 × 32 mm2 were fabricated by first sputtering a thin layer of chromium (100 nm) on glass wafers. A pattern of chromium actuation electrodes with 2.8 × 2.8 mm2 electrode size was then created by using standard photolithography and wet etching. After stripping the remaining photoresist the substrates were primed with Silane A174 before being coated with a layer of Parylene-C (3.5 μm thickness) by means of chemical vapor deposition. Finally, a thin layer of Teflon-AF (120 nm) was spin-coated on top of the Parylene-C dielectric layer.Two different top plates were fabricated for DMF chip experiments for the optimization of magnetic particle extraction: (i) top plates of indium tin oxide (ITO) coated glass slides with 1.1 mm thickness (Delta Technologies, Stillwater, USA) were spin coated with Teflon-AF (120 nm) to render the surface hydrophobic. (ii) gold top plates were fabricated by sputtering a 12 nm thin layer of gold on top of 0.7 mm thick glass wafers. Here the gold served as a ground electrode. Subsequently, a thin layer of Teflon-AF (120 nm) was spin-coated on top.
Droplets of 2.5 μL were manually pipetted on the bottom substrate before being sandwiched between the bottom and the top plate using a 140 μm thick tape that was placed on the bottom plate, as a spacer. Alternatively, droplets were pipetted into the space between bottom and top plate and transported to their respective position by electrode actuation. Droplets were actuated with a 120 VAC actuation voltage by amplifying the signal of a function generator operating at 1 kHz. Droplet movement along the actuation electrodes with 2.5 s electrode activation time was controlled by using custom made Labview (National instruments, Austin, USA) and Matlab (Mathworks, Natick, USA) programs.
Oligonucleotides
Table 1 lists the oligonucleotide sequences that were used in this study written in 5′ → 3′ orientation. Target hybridization sites of the capture oligonucleotide (orange – italic) and the padlock probe (green – bold) and digestion (blue – underlined) and detection (red – underlined-italic) tag sequences are highlighted accordingly. Circularization of the padlock probe on the DNA target is illustrated in Fig. 1d, step 1.

|
Extraction and transport of superparamagnetic particles on the DMF chip and investigation of the extraction efficiency
For the optimization of superparamagnetic particle extraction on the DMF chip two parameters were investigated in relation to particle concentration: (i) the thickness of the top plate. The use of 0.7 mm thick top plates with a 12 nm conductive gold layer was compared to 1.1 mm thick ITO coated top plates for particle extraction efficiency; (ii) the effect of the magnet position on the extraction efficiency. The minimal particle concentration needed for magnetic particle extraction from a droplet was determined for 3 types of streptavidin functionalized superparamagnetic particles, C1 dynabeads, T1 dynabeads and M280 dynabeads. The particles were washed 3 times in PBS with 0.05% Pluronic F68 and diluted to different concentrations. The particle dilutions were tested for particle extraction on-chip and the minimal concentration required for successful extraction was determined.For the particle extraction and transfer between droplets a magnet (NdFeB, 6 mm diameter, 12.7 N, Supermagnete, Gottmadingen, Germany) was placed on top of the top plate in proximity to the particle containing droplet (see Fig. 1 and S1†). Extraction of the magnetic particles occurs when the droplet moves away from the magnet. The extracted magnetic particles are surrounded by a thin shell of liquid and oil. Next, the extracted particle pellet was moved to another droplet by moving the magnet. Finally the particles were released in a new droplet and resuspended again by droplet transportation (movie S1†). During the C2CA assay the particles were, after extraction, transported one row forward to the next droplet by moving the magnet, as illustrated by the red arrows in Fig. 1a.
The performance of the abovementioned extraction protocol was investigated by counting the total number of particles that remained in the droplets after extraction. For that purpose, the droplets were aspirated from the chip and applied to a nanoliter well microscope slide chip (Picovitro, Stockholm, Sweden). Each droplet was spread out into 4 separate 500 nl wells and the wells were sealed with a rubber gasket. Additionally, a positive control with a 1/10 dilution of the particle concentration (1 × 106 particles μL−1) used in the assay was applied. After 30 min of sedimentation to the bottom of the wells the particles were imaged with bright field microscopy. The particles were counted using the particle analyzer function of ImageJ analysis software (NiH, USA).
DNA detection with padlock probes and circle-to-circle amplification on the digital microfluidic chip
The assay layout for DMF C2CA is depicted in Fig. 1a. Moreover, a flow chart illustrating the magnetic particle extraction & transfer and droplet mixing scheme is provided in Fig. 1b. The chip was placed on the temperature controllable heating plate of a thermocycler (Biometra, Göttingen, Germany) to apply the required temperatures on-chip.Dynabeads T1 magnetic particles were washed 3 times in PBS with 0.05% Pluronic F68 and were diluted to 1 × 106 particles μL−1. 2.5 μL of particles and 2.5 μL washing buffer (PBS with 0.05% Pluronic F68) were initially positioned on-chip as illustrated in Fig. 1a. Then, 1.25 μL DNA sample and 1.25 μL padlock probe ligation mix, containing 50 nM biotinylated capture oligonucleotide, 50 nM padlock probe, 20 mM Tris–HCl (pH 8.3), 25 mM KCl, 10 mM MgCl2, 0.5 mM NAD, 0.2 μg μL−1 BSA, 0.01% Triton® X-100, 0.05% Pluronic F68 and 625 mU amp ligase, were positioned and mixed on-chip and incubated at 50 °C for 5 min. After incubation, the particles and the sample-ligation mix were merged and mixed by transporting the droplet over the neighboring electrodes on the first electrode row of the DMF chip for 5 min (see Fig. 1a, green arrows). The particles were extracted with the magnet and moved to the washing buffer droplet (see red arrows in Fig. 1a) and washed by 30 s droplet transportation between the 5 electrodes on the second electrode row (see grey droplet and green arrows indicating direction of droplet transportation in Fig. 1a and b). Afterwards, the RCA reaction droplet (phi29 buffer (33 mM Tris–acetate, 10 mM Mg–acetate, 66 mM K–acetate, 0.1% Tween 20, 1 mM DTT), 0.2 μg μL−1 BSA, 0.05% Pluronic F68, 125 μM dNTP mix and 500 mU phi29 polymerase) was pipetted into the space between the bottom and top plate, dragged under the top plate and transported to its position by electrode actuation (as already demonstrated by Ng et al. 201210 and herein illustrated in movie S2†). The magnetic particles were then extracted from the washing droplet and transferred to and mixed in the RCA droplet. After incubation at 37 °C for 20 min and 65 °C for 2 min, the droplet was briefly mixed to re-dissolve settled particles. The digestion droplet (phi29 buffer, 0.2 μg μL−1 BSA, 0.05% Pluronic F68, 120 nM digestion oligonucleotide and 300 mU AluI restriction digest enzyme) was then inserted like the RCA droplet and the particles were extracted and transferred from the RCA into the digestion droplet. The chip was incubated at 37 °C for 2 min and 65 °C for 2 min. The particles were extracted and the particle pellet left behind. The droplet for ligation and 2nd RCA (phi29 buffer, 0.2 μg μL−1 BSA, 0.05% Pluronic F68, 125 μM dNTP mix, 0.67 mM ATP, 35 mU T4 DNA ligase and 300 mU phi29 polymerase) was inserted and mixed with the digestion droplet and incubated at 37 °C for 25 min and at 65 °C for 2 min. This sequential insertion of RCA, digestion and ligation & 2nd RCA enzyme mixes by on-chip pipetting was required because the heat inactivation between the enzymatic steps did not allow for all droplets to be placed on-chip from the beginning.
Positive and negative controls were run in reaction tubes: 10 μL target DNA was mixed with 10 μL padlock probe ligation mix. 20 μL magnetic particles in 1 × 106 particles μL−1 concentration was added after ligation and incubated for 5 min with slow tilt rotation. The particles were then washed in 80 μL washing buffer and incubated in 20 μL RCA reaction mix. Subsequently, 5 μL digestion mix was added. After digestion, the monomers were transferred to new reaction tubes and mixed with 25 μL ligation & 2nd RCA reaction mix.
Digital quantification of C2CA products
After the 2nd RCA the 5 μL droplet was aspirated from the chip, mixed with 5 μL labeling mix (1 M NaCl, 20 mM EDTA, 20 mM Tris–HCl, 0.01% Tween-20 and 5 nM Cy3-labeled detection oligonucleotide) and incubated at 75 °C for 2 min and 55 °C for 10 min. 5 μL of the fluorescently labeled C2CA products were applied on Superfrost slides (VWR, Belgium) and covered with a 2.25 cm2 cover slip. The C2CA products are negatively charged which prevents them from interfering with each other and to bind to the positively charged microscope slides aligning themselves in a random pattern as illustrated in Fig. 1c. Six randomly chosen areas were imaged with a 20× objective and the pictures analyzed with ImageJ software. Representative images are shown in Fig. 5a. C2CA products were counted with the ImageJ particle analyzer function. For more information regarding C2CA product counting on the 2 dimensional slide surface see ESI.†Conclusions
Despite several advantages over other amplification methods, C2CA consists of many manual steps making the integration into microfluidic platforms a complicated task. Until now, all reported efforts of microfluidic C2CA have suffered high efficiency loss.33,38 Hence, the automation on a microfluidic chip that facilitates high assay efficiencies is of utmost importance in order to bring the advantages of C2CA to diagnostic application. We demonstrated in this paper a microfluidic platform that is highly suitable for performing this assay without efficiency loss.We showed the effect of 2 critical particle extraction parameters for 1 μm sized superparamagnetic particles that have not been addressed in previously reported particle extraction studies.10,12,13 We described a protocol for extracting and transferring these particles in concentrations as low as 1.6 × 105 particles μL−1 with nearly 100% extraction efficiency using a new simple tool for extraction efficiency quantification. The ultra-high particle recovery of this protocol, together with the strong droplet mixing capabilities of DMF, facilitate the performance of complex multistep assays, as herein demonstrated with C2CA, with near 100% assay efficiencies.
Moreover, we showed that enzymatic ligation, RCA and digestion reactions can be performed on a DMF chip with equally high efficiency as in reaction tubes, indicating no or little unspecific absorption of enzymes on the hydrophobic chip surface. We illustrated how the automated liquid handling of small reaction volumes together with the highly efficient particle extraction protocol on the DMF facilitates the miniaturization of the C2CA protocol with low variation as opposed to the manual handling of small reaction volumes which results in high variations.
With these prerequisites, the complete C2CA was successfully performed on the DMF chip with high reproducibility, broad dynamic range and outstanding sensitivity in an overall assay time of ~60 min. A 1 aM concentration of synthetic P. aeruginosa DNA was detected above background, with a linear dynamic range over 4 orders of magnitude up to 10 fM. In contrast to other microfluidic C2CA approaches, in which the on-chip assay efficiency is significantly lower than in reaction tubes,33 the assay efficiency on the DMF chip in this study was equally high as in tube controls proving the unique advantages of our herein presented DMF particle manipulation protocol for performing multistep magnetic particle-based assays.
The use of either an integrated local heating element or an isothermal enzyme inactivation procedure is required for total automation. However, the necessary parameters for total automation and the integration concept were presented in this paper. We believe that this report can have a major impact on the implementation of RCA and C2CA assays for laboratories and diagnostic settings, as the manual performance of these assays is otherwise cumbersome.
This study presents, to our knowledge, the first integrated digital single molecule nucleic acid amplification on a DMF chip. The need for compartmentalization for digital read-out is solved by employing an amplification method that results in individually countable amplification products rather than by using microfabrication procedures for compartmentalization. Exploiting the great multiplexing capability of padlock probes the DMF C2CA platform offers a great tool for infectious disease diagnostics. However, the range of other possible applications goes beyond diagnostics as this amplification method is unique in creating precise copies of single stranded DNA which is of importance in DNA aptamer synthesis or DNA origami technologies.
Acknowledgements
The research leading to the reported results has received funding from the European Commission's Seventh Framework Programme (FP7/2007-2013) under the grant agreement BIOMAX (project no. 264737) (MK), the Fund for Scientific Research Flanders (project G.0997.11 and G.0861.14), the Swedish Research Council, Science for Life Laboratory, and KU Leuven (OT 13/058 and IDO 10/012).References
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef CAS.
- A. Manz, E. Verpoorte, D. E. Raymond, C. S. Effenhauser, N. Burggraf and H. M. Widmer, Micro Total Analysis Systems, Mesa Monographs, 1995, pp. 5–27 Search PubMed.
- S. Shoji and M. Esashi, J. Micromech. Microeng., 1994, 4, 157–171 CrossRef CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- A. R. Wheeler, Science, 2008, 322, 539–540 CrossRef CAS PubMed.
- M. G. Pollack, R. B. Fair and A. D. Shenderov, Appl. Phys. Lett., 2000, 77, 1725–1726 CrossRef CAS PubMed.
- M. G. Pollack, A. D. Shenderov and R. B. Fair, Lab Chip, 2002, 2, 96–101 RSC.
- K. Choi, A. H. Ng, R. Fobel and A. R. Wheeler, Annu. Rev. Anal. Chem., 2012, 5, 413–440 CrossRef CAS PubMed.
- Y. Fouillet, D. Jary, C. Chabrol, P. Claustre and C. Peponnet, Microfluid. Nanofluid., 2008, 4, 159–165 CrossRef.
- A. H. Ng, K. Choi, R. P. Luoma, J. M. Robinson and A. R. Wheeler, Anal. Chem., 2012, 84, 8805–8812 CrossRef CAS PubMed.
- D. Witters, K. Knez, F. Ceyssens, R. Puers and J. Lammertyn, Lab Chip, 2013, 13, 2047–2054 RSC.
- N. Vergauwe, D. Witters, F. Ceyssens, S. Vermeir, B. Verbruggen, R. Puers and J. Lammertyn, J. Micromech. Microeng., 2011, 21, 054026 CrossRef.
- N. Vergauwe, S. Vermeir, J. B. Wacker, F. Ceyssens, M. Cornaglia, R. Puers, M. A. M. Gijs, J. Lammertyn and D. Witters, Sens. Actuators, B, 2014, 196, 282–291 CrossRef CAS PubMed.
- Y. H. Chang, G. B. Lee, F. C. Huang, Y. Y. Chen and J. L. Lin, Biomed. Microdevices, 2006, 8, 215–225 CrossRef CAS PubMed.
- M. Vincent, Y. Xu and H. Kong, EMBO Rep., 2004, 5, 795–800 CrossRef CAS PubMed.
- G. T. Walker, M. C. Little, J. G. Nadeau and D. D. Shank, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 392–396 CrossRef CAS.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- A. Fire and S. Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4641–4645 CrossRef CAS.
- Y. Hataoka, L. Zhang, Y. Mori, N. Tomita, T. Notomi and Y. Baba, Anal. Chem., 2004, 76, 3689–3693 CrossRef CAS PubMed.
- P. M. Lizardi, X. Huang, Z. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- T. Conze, A. Shetye, Y. Tanaka, J. Gu, C. Larsson, J. Goransson, G. Tavoosidana, O. Soderberg, M. Nilsson and U. Landegren, Annu. Rev. Anal. Chem., 2009, 2, 215–239 CrossRef CAS PubMed.
- M. Nilsson, H. Malmgren, M. Samiotaki, M. Kwiatkowski, B. P. Chowdhary and U. Landegren, Science, 1994, 265, 2085–2088 CAS.
- M. Nilsson, K. Krejci, J. Koch, M. Kwiatkowski, P. Gustavsson and U. Landegren, Nat. Genet., 1997, 16, 252–255 CrossRef CAS PubMed.
- J. Baner, M. Nilsson, M. Mendel-Hartvig and U. Landegren, Nucleic Acids Res., 1998, 26, 5073–5078 CrossRef CAS PubMed.
- M. Nilsson, Histochem. Cell Biol., 2006, 126, 159–164 CrossRef CAS PubMed.
- J. Goransson, C. Wahlby, M. Isaksson, W. M. Howell, J. Jarvius and M. Nilsson, Nucleic Acids Res., 2009, 37, e7 CrossRef PubMed.
- J. Jarvius, J. Melin, J. Goransson, J. Stenberg, S. Fredriksson, C. Gonzalez-Rey, S. Bertilsson and M. Nilsson, Nat. Methods, 2006, 3, 725–727 CrossRef CAS PubMed.
- F. Dahl, J. Baner, M. Gullberg, M. Mendel-Hartvig, U. Landegren and M. Nilsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4548–4553 CrossRef CAS PubMed.
- J. Goransson, R. Ke, R. Y. Nong, W. M. Howell, A. Karman, J. Grawe, J. Stenberg, M. Granberg, M. Elgh, D. Herthnek, P. Wikstrom, J. Jarvius and M. Nilsson, PLoS One, 2012, 7, e31068 Search PubMed.
- R. Q. Ke, A. Zorzet, J. Goransson, G. Lindegren, B. Sharifi-Mood, S. Chinikar, M. Mardani, A. Mirazimi and M. Nilsson, J. Clin. Microbiol., 2011, 49, 4279–4285 CrossRef CAS PubMed.
- J. Melin, H. Johansson, O. Soderberg, F. Nikolajeff, U. Landegren, M. Nilsson and J. Jarvius, Anal. Chem., 2005, 77, 7122–7130 CrossRef CAS PubMed.
- K. Sato, A. Tachihara, B. Renberg, K. Mawatari, Y. Tanaka, J. Jarvius, M. Nilsson and T. Kitamori, Lab Chip, 2010, 10, 1262–1266 RSC.
- L. Mahmoudian, N. Kaji, M. Tokeshi, M. Nilsson and Y. Baba, Anal. Chem., 2008, 80, 2483–2490 CrossRef CAS PubMed.
- R. C. den Dulk, K. A. Schmidt, G. Sabatte, S. Liebana and M. W. Prins, Lab Chip, 2013, 13, 106–118 RSC.
- V. N. Luk, G. Mo and A. R. Wheeler, Langmuir, 2008, 24, 6382–6389 CrossRef CAS PubMed.
- S. H. Au, P. Kumar and A. R. Wheeler, Langmuir, 2011, 27, 8586–8594 CrossRef CAS PubMed.
- O. Ericsson, J. Jarvius, E. Schallmeiner, M. Howell, R. Y. Nong, H. Reuter, M. Hahn, J. Stenberg, M. Nilsson and U. Landegren, Nucleic Acids Res., 2008, 36, e45 CrossRef PubMed.
- L. Mahmoudian, J. Melin, M. R. Mohamadi, K. Yamada, M. Ohta, N. Kaji, M. Tokeshi, M. Nilsson and Y. Baba, Anal. Sci., 2008, 24, 327–332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00348a |
| This journal is © The Royal Society of Chemistry 2014 |