 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhaseguide assisted liquid lamination for magnetic particle-based assays†
Chayakom
Phurimsak
a,
Ender
Yildirim
bc,
Mark D.
Tarn
a,
Sebastiaan J.
Trietsch
bd,
Thomas
Hankemeier
b,
Nicole
Pamme‡
a and
Paul
Vulto§
bd
aDepartment of Chemistry, The University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: n.pamme@hull.ac.uk; Fax: +44 (0)1482 466410; Tel: +44 (0)1482 465027
bLeiden Academic Centre for Drug Discovery, University of Leiden, 2333 CC Leiden, The Netherlands. E-mail: p.vulto@lacdr.leidenuniv.nl; Tel: +31 (0)71 527 4509
cMechanical Engineering Department, Cankaya University, Ankara, Turkey
dMimetas B.V., P.O. Box 11002, 2301 EA Leiden, The Netherlands
First published on 25th April 2014
Abstract
We have developed a magnetic particle-based assay platform in which functionalised magnetic particles are transferred sequentially through laminated volumes of reagents and washing buffers. Lamination of aqueous liquids is achieved via the use of phaseguide technology; microstructures that control the advancing air–liquid interface of solutions as they enter a microfluidic chamber. This allows manual filling of the device, eliminating the need for external pumping systems, and preparation of the system requires only a few minutes. Here, we apply the platform to two on-chip strategies: (i) a one-step streptavidin–biotin binding assay, and (ii) a two-step C-reactive protein immunoassay. With these, we demonstrate how condensing multiple reaction and washing processes into a single step significantly reduces procedural times, with both assay procedures requiring less than 8 seconds.
Introduction
Magnetic microparticles have been used extensively in clinical procedures for a number of years now, and for good reason.1–4 Typically consisting of a core of iron oxide nanoparticles in a polymer or silica matrix, these microparticles exhibit superparamagnetic properties, meaning that they are only magnetic in the presence of a magnetic field; when the field is removed they lose their magnetic properties and are able to redisperse in a solution. Their small size yields high surface-to-volume ratios, and they are available commercially with a range of surface functionalities that make them applicable to a number of applications. Such applications can include isolation and separation of target analytes (cells, proteins, DNA), DNA analysis, cell assays, and immunoassays. Conventional magnetic particle-based assays, involving the sequential transfer of reagents and washing buffers to/from sample vials, offer high capture efficiencies and minimal particle loss. However, they are batch processes, and suffer from having multiple manual steps that are both laborious and time-consuming.The incorporation of magnetic particle manipulation into microfluidic immunoassay procedures5–7 over the last decade or so has allowed the benefits of the former to be combined with the typical advantages associated with miniaturisation. These include a reduction in sample/reagent volumes and diffusion distances, and a small footprint that makes portability and point-of-care feasible. Many microfluidic-based magnetic particle assays are performed using a “trap-and-release” methodology, in which a plug of functionalised particles is trapped within a microchannel and has solutions of reagents and washing solutions washed over it consecutively.8–11
In recent years there has been a shift towards performing continuous flow assays in order to reduce these multi-step procedures into a single step, decreasing processing times and introducing a degree of automation. Such methods are derived from continuous flow separation techniques in which magnetic particles in a sample stream are pulled into an adjacent buffer stream to extract a target analyte that has become bound to the particles.12–16 However, by generating multilaminar flow streams in a microfluidic chamber, consisting of alternating streams of reagents and washing solutions, particles can be deflected consecutively through each stream, thus performing sequential reactions/binding events.17–24
This concept can also be flipped, in that rather than deflecting magnetic particles through continuously flowing streams of solutions via a fixed magnet, they are instead pulled by a moving magnet through static solutions. These principles can be observed in some droplet-based systems, in which aqueous droplets containing samples, reagents, and buffers are immersed in an immiscible phase such as oil25–29 or air,30–33 and a magnet used to move particles between the droplets. A recent development utilising a similar principle involves the generation of aqueous regions of samples, reagents and washing buffers, separated by an immiscible phase that forms barriers between the aqueous solutions. This prevents samples and reagents from mixing together, yet allows magnetic particles to cross the immiscible phase when pulled by an external magnet, thus transferring the particles between the reaction and labelling regions.
A prime example of the transfer of particles across immiscible barriers is “immiscible filtration assisted by surface tension” (IFAST), in which microfluidic chambers, linked by small “gate” regions, are alternately filled with aqueous and oil-based solutions.34 IFAST, and variants such as VerIFAST35,36 and SNARE (selective nucleic acid removal via exclusion),37 has mostly seen applications in DNA purification34,37 and cell isolation,35,36,38 but has also been demonstrated for immunoassays.39 Several other devices have been developed that utilise a variety of immiscible phases, including liquid wax,40 paraffin,41 and air.42–45
However, while the movement of magnetic particles across static regions of assay solutions has developed as a fast and robust technique, it is not particularly necessary to use immiscible solutions. Becker et al.46,47 have shown a “stationary fluidics” device in which magnetic particles are pulled through a chain of reaction compartments to perform DNA washing and amplification, using only aqueous solutions. The geometry of the compartments was designed to minimise crossover of reagents between chambers. A variant of this system from Gottheil et al.48 was also recently demonstrated in which phaseguides were applied to the bubble-free filling of the device, and the chambers were connected by a passive valve that formed a small bridge between the compartments.
Here, we present a new method of filling a microfluidic device with stationary fluids for magnetic particle-based assays, which harnesses phaseguide technology for the generation of adjacent regions of aqueous liquids. This essentially allows the device to “mimic” platforms that use immiscible solutions, while actually consisting entirely of miscible solutions.
Phaseguides are lines of microstructures that are incorporated onto the floor of a microfluidic channel, and are around one-quarter the height of the microchannel.49,50 Starting with a dry, empty device, the phaseguides direct the advancing air–liquid interface as a solution is introduced into the channel, with the meniscus being pinned on the phaseguide. Crucially, the advancing liquid will be unable to cross the phaseguide until the section of chip bordered by the phaseguide is full, at which point the pressure build-up across the meniscus is such that the phaseguide is breached and liquid will advance into the next section of the chip. The liquid overflows the phaseguide at the point of least resistance, hence by incorporating this fact into the design of the phaseguides the route by which the liquid takes during filling of the chip can be precisely controlled. This powerful technique is therefore capable of allowing complex channel geometries, in particular those with sharp corners, to be filled without having air bubbles become trapped within the device, since all of the air is forced out by the advancing liquid. Microfluidic platforms that have taken advantage of phaseguide technology have included those for the separation and purification of analytes,51 the recovery of dielectrophoretically separated cells and particles,49 the extraction of low molecular weight RNA,52 the enrichment of bacteria by free-flow electrophoresis,53,54 isothermal amplification of DNA,55 lysis of bacteria and purification of the released RNA,56 and stratified 3D cell culture.57
We report the application of phaseguide technology for the lamination of “lanes” of aqueous solution through which functionalised magnetic particles are manoeuvred (Fig. 1). Immunoassays are performed by generating alternating lanes of reagent and washing buffer solutions, and as the particles move sequentially through each lane the target analyte becomes bound to the particles and is subsequently labelled. Two proof-of-principle assay procedures are described here to highlight the platform's potential suitability for clinical diagnostics: (i) a one-step streptavidin–biotin binding assay (Fig. 1a), and (ii) a two-step C-reactive protein sandwich immunoassay (Fig. 1b). This phaseguide-assisted platform has the benefits of allowing the filling of the device without the need for external pumping mechanisms (e.g. syringe pumps), while the assay itself is controlled by manual movement of the magnet across the top of the chip. These aspects yield a simple yet rapid process in terms of both chip preparation and of performing the magnetic particle-based assay.
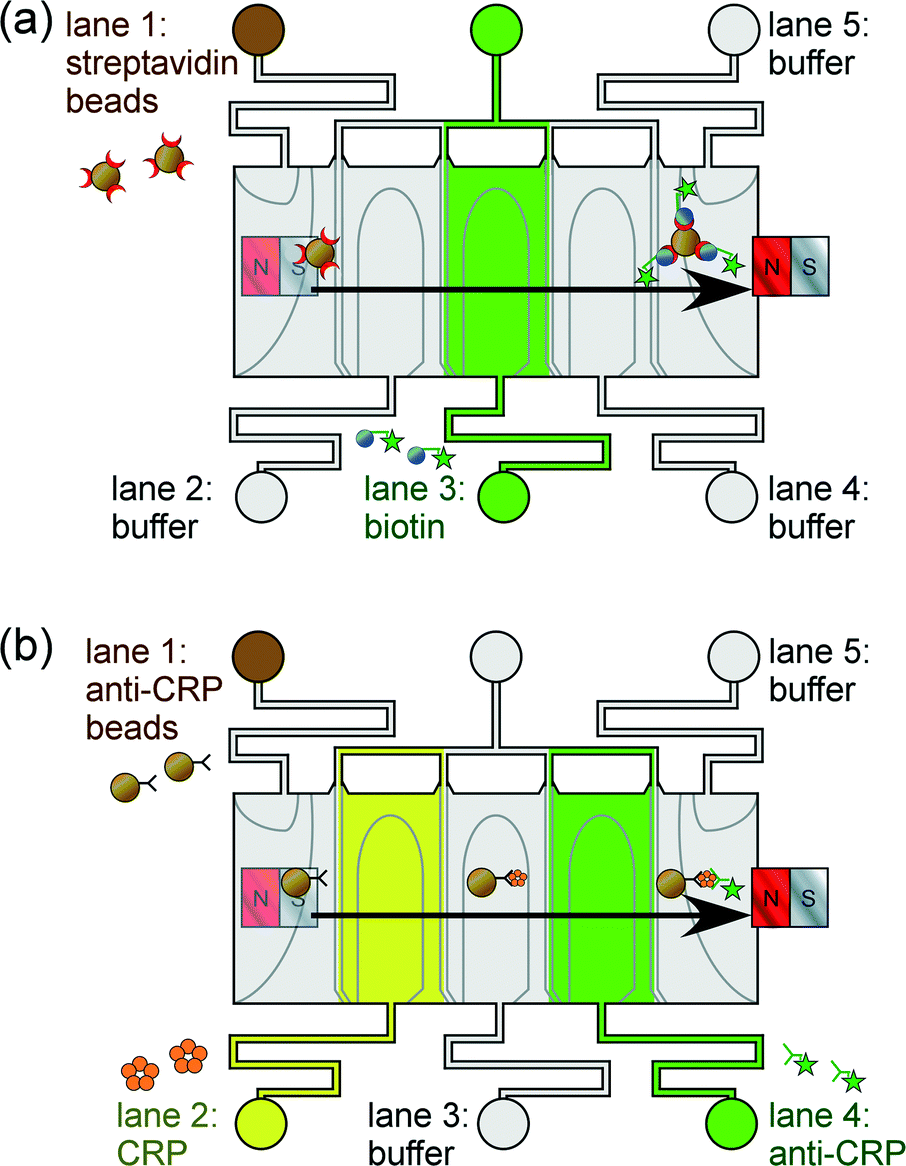 | ||
| Fig. 1 Principles of magnetic particle-based assays employing lamination of reagents and buffers for (a) a one-step streptavidin–biotin assay, and (b) a two-step CRP sandwich immunoassay. Functionalised magnetic particles are pulled through lanes of reagents and washing buffers formed via the use of phaseguides (shown as grey lines in the microfluidic chamber). | ||
Experimental
Chip design and fabrication
The design of the microfluidic device is shown in Fig. 2a. The layout featured a large rectangular chamber (4 mm long × 10 mm wide) that was split into five “lanes”, each having a width of 2 mm and a length of 4 mm. Each lane was fed by a single inlet channel (200 μm width), and air vent channels (200 μm width) were situated at the top of the chamber, between the adjacent lanes, to allow air to be expelled during the filling of the system. Each lane featured phaseguides that were designed to direct the advancing air–liquid in such a manner as to fill only one lane from each inlet hole, whilst expelling all of the air from that lane. Each lane was separated by two parallel lines of phaseguides that “kinked” near the lower wall of the chamber (shown in Fig. 2a with angles α1 and α2), with these kinks determining where the air–liquid interface overflowed into the adjacent lane.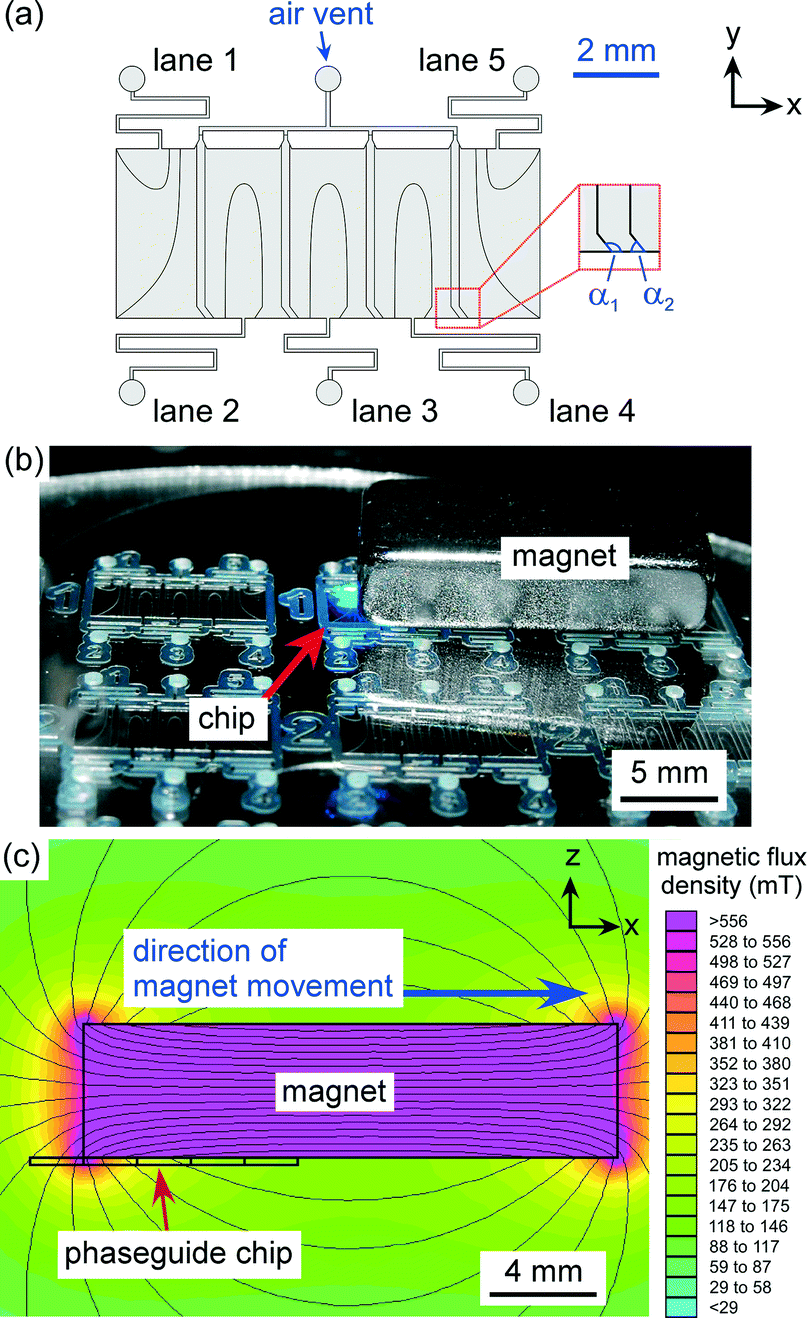 | ||
| Fig. 2 (a) Design of the microfluidic device, illustrating the phaseguide pattern that generates five individual lanes. (b) Photograph of the microfluidic device and the 20 × 10 × 5 mm3 NdFeB magnet used to manipulate the magnetic particles. (c) Simulation of the magnetic field produced by the magnet in relation to the phaseguide chip (side view). | ||
The device was fabricated on a glass substrate using a dry film resist, Ordyl SY-330 (Elga Europe, Italy), as the structural material.58,59 The Ordyl was laminated onto a soda lime glass wafer (Berlin Glass, Germany) and exposed with 240 mJ cm−2 using a mask aligner to pattern the phaseguide layout onto the film resist. An additional three layers of Ordyl were laminated and exposed in the same manner to build up the chamber/channel geometry, before being baked at 85 °C for 1 min. The resist was developed in three baths of BMR developer (Elga Europe) with increasing purity, followed by rinsing with isopropanol and water, and finally being spun dry. The process yielded phaseguides that were approximately 30 μm in height and 50 μm in width, and a channel structure that was 120 μm in height.
Access holes were ultrasonically drilled into a second soda lime glass wafer. After cleaning the drilled wafer, it was bonded to the patterned wafer using a heated press at a temperature of 95 °C and a pressure of approximately 60 N cm−2 for 30 min.
Preparation of solutions
All solutions were prepared using high purity water (18.2 MΩ cm at 25 °C) that had been double-filtered (0.05 μm, ELGA Option 4 feeding into an ELGA UHG PS system, ELGA Process Water, Marlow, UK). Phosphate buffered saline solution (PBS, pH 7.45) was prepared by dissolving a 5 g tablet (Invitrogen, Life Technologies, Paisley, UK) in 500 mL water, with 0.05% w/v Tween20 (Sigma-Aldrich, Dorset, UK) added to prevent particles sticking to the microchannel surfaces and to each other. A solution of fluorescently labelled biotin was prepared by dissolving biotin-4-fluorescein (λex = 494 nm, λem = 521 nm, Invitrogen) in PBS solution.Lyophilised recombinant C-reactive protein (CRP, R&D Systems, Abington, UK) was reconstituted in 20 mM tris buffer (pH 8, Sigma-Aldrich), with 0.1% bovine serum albumin (BSA, Sigma-Aldrich) added, to a stock concentration of 200 μg mL−1 as per the manufacturer's instructions. Biotinylated primary CRP antibody (1° Ab, monoclonal mouse anti-human C-reactive protein, R&D Systems) was purchased as a lyophilised powder and reconstituted in PBS to a stock concentration of 50 μg mL−1, as per the manufacturer's instructions. Fluorescently labelled secondary CRP antibody (2° Ab-FITC, polyclonal goat anti-human C-reactive protein conjugated to fluorescein isothiocyanate, λex = 495 nm, λem = 521 nm) was purchased as an aqueous solution at a concentration of 10 mg mL−1 (Abcam plc., Cambridge, UK).
Preparation of particle suspensions
Stock magnetic particle suspensions were prepared by dispersing streptavidin-functionalised 2.8 μm diameter particles (Dynabeads M-270 Streptavidin, Invitrogen) in PBS solution. 10 μL of the particle suspension provided (6.5 × 108 particles mL−1) was added to a 1.5 mL Eppendorf tube (VWR, Leicester, UK), before being washed three times with PBS. 1000 μL of PBS was added to the tube, after which the tube was vortexed and the magnetic particles pulled to the side of the tube via an external magnet. This allowed the supernatant to be removed via a pipette while the particles remained in the tube, enabling a further 1000 μL of PBS to be added in order to repeat the washing process. From here, the particles would be treated in one of two manners depending on whether they were to be used for the one-step streptavidin–biotin binding assay or the two-step CRP sandwich immunoassay.In order to perform the one-step biotin–streptavidin binding assay, the particles were resuspended in 50 μL of PBS to a concentration of 1.3 × 108 particles mL−1, and were stored at 4 °C until ready for use.
The magnetic particles required further treatment when performing the two-step CRP assay. Following the final PBS washing step, the supernatant was removed and 200 μL of 1° Ab solution (10 μg mL−1) was added to the Eppendorf tube, then allowed to incubate for 15 min with agitation. This allowed the biotinylated primary antibody to bind to the streptavidin groups on the particles. The particles were washed three times with PBS and finally suspended in 50 μL of PBS to a concentration of 1.3 × 108 particles mL−1, and stored at 4 °C until ready for use.
Chip setup and operation
The microfluidic device was filled with liquids by consecutively introducing aqueous solutions (1.5 μL) into the inlet holes (1–5) via a pipette (Eppendorf). Each pipetting step filled up one lane of the assay. The chip was then placed onto the sample stage of an inverted fluorescence microscope (TE-2000, Nikon, Surrey, UK) for visualisation of the magnetic particles. Images were captured via a CCD camera (Retiga-EXL, QImaging, Media Cybernetics, Buckinghamshire, UK) and Image-Pro Plus 6.2 software (Media Cybernetics, UK). Fluorescence measurements of the particles were performed using ImageJ software (http://rsb.info.nih.gov/ij/).A large, rectangular neodymium–iron–boron (NdFeB) magnet (20 × 10 × 5 mm3, Magnet Sales, Swindon, UK) was placed on top of the chip, above the chamber in such a position that the left edge of the magnet was located over the interface between lanes 1 and 2 in the chip (Fig. 2b). Fig. 2c shows a simulation of the magnetic field generated across the microfluidic chamber (from a side view) at this starting position, which was generated using FEMM 4.2 software (http://femm.foster-miller.net). It can clearly be seen that the bottom-left corner of the magnet provides a very large magnetic field and gradient, thus the particles in the chip will be attracted towards this edge and follow it as the magnet is moved to the right.
During experiments, magnetic particles were introduced into lane 1 of the microfluidic device, while lanes 2–4 contained different combinations of reagents and washing buffer solutions (depending on the experiment) and lane 5 consisted of PBS washing buffer. To perform an assay, the magnet was moved by hand from left-to-right across the chip. This caused the magnet to move from its starting location to a position at which the left edge of the magnet was situated over the right-hand side of lane 5. As the magnet was moved across the chamber, the magnetic particles followed the magnet, crossing each lane from lane 1 to lane 5 where they came to a stop in the PBS wash buffer. Images of the particles were then captured for fluorescence analysis, and compared to images taken before the magnet was moved. This technique was used to perform two types of assay: (i) a proof-of-principle one-step streptavidin–biotin binding assay, and (ii) a two-step CRP sandwich immunoassay.
Streptavidin–biotin binding assay
In order to first test the system for performing magnetic particle-based assays, a one-step streptavidin–biotin binding assay was attempted. Streptavidin functionalised magnetic particles (1.3 × 108 particles mL−1, yielding 2 × 105 particles in the chamber) were introduced into lane 1 of the microfluidic device, while fluorescently labelled biotin was introduced into lane 3, and PBS washing buffer into lanes 2, 4 and 5 (Fig. 1a). Biotin concentrations of 1 to 100 μg mL−1 were tested.C-reactive protein immunoassay
A sandwich immunoassay was set up by introducing 1.5 μL of primary antibody (1° Ab) coated magnetic particles (1.3 × 108 particles mL−1, yielding 2 × 105 particles in the chamber) into lane 1 of the microfluidic device, CRP into lane 2, fluorescently labelled secondary antibody (2° Ab-FITC, 100 μg mL−1) into lane 4, and PBS washing buffer into lanes 3 and 5 (Fig. 1b). CRP concentrations of 0.1 to 1 μg mL−1 were investigated.Results
Liquid lamination
Fig. 3a–c shows the chip filling process in ordered lanes using orange and blue dyes as an example. Firstly, liquid is introduced into lane 1 to the point where the lane is full but has not overflowed past the phaseguide separating it from lane 2 (Fig. 3a). Liquid is then injected into lane 2 until the lane is full but there has been no overflow across the separation phaseguides. Finally, a small burst of extra pressure from the pipette forces the liquid in lane 2 to cross the phaseguides separating it from lane 1 (but not lane 3) at the kinked sections of the phaseguides (Fig. 3b). This bridges the liquids in lanes 1 and 2 and forces the air out of the vent at the top of the chamber, thereby allowing lamination of the two liquids in a static form. This process of adding liquids to adjacent channels is repeated in turn for lanes 3 to 5 until the entire microfluidic chamber has been filled that consists of five lanes of laminated solutions (Fig. 3c). By changing the solutions in each lane it was possible to prepare the chip in such a manner as to perform one-step and two-step assays. | ||
| Fig. 3 (a) Filling of lanes 1 and 2 with ink, with the blue ink in lane two crossing into the space between the lanes at position α2. (b) Applying a little extra pressure bridges the gap between the lanes, forcing the air out of the vent and laminating the two liquids. (c) Filled device featuring five laminated lanes of different aqueous solutions. | ||
Streptavidin–biotin binding assay
Streptavidin functionalised magnetic particles were introduced into lane 1 of the microfluidic chamber, with fluorescently labelled biotin in lane 3 and PBS in lanes 2, 4 and 5 (Fig. 4a). The filling of each lane with 1.5 μL solution required ~30 s by pipette, and so the total filling of the chamber required ~150 s (2.5 min). Placing the magnet in its starting location required only a few seconds as it was not required that the position be particularly accurate, as long as the left edge of the magnet was somewhere over lane 1. When the magnet was moved from left-to-right across the top of the chip the magnetic particles followed its movement, passing through the lane of biotin and into the lanes of PBS washing buffer. The buffer solution provided a “washing step” as the particles moved through the solution, removing unbound material from the surfaces of the particles. Importantly, the time taken to move the magnet over the chamber, and thus the time needed to move the particles through the lanes, was only 8 s, thereby making the assay incredibly fast.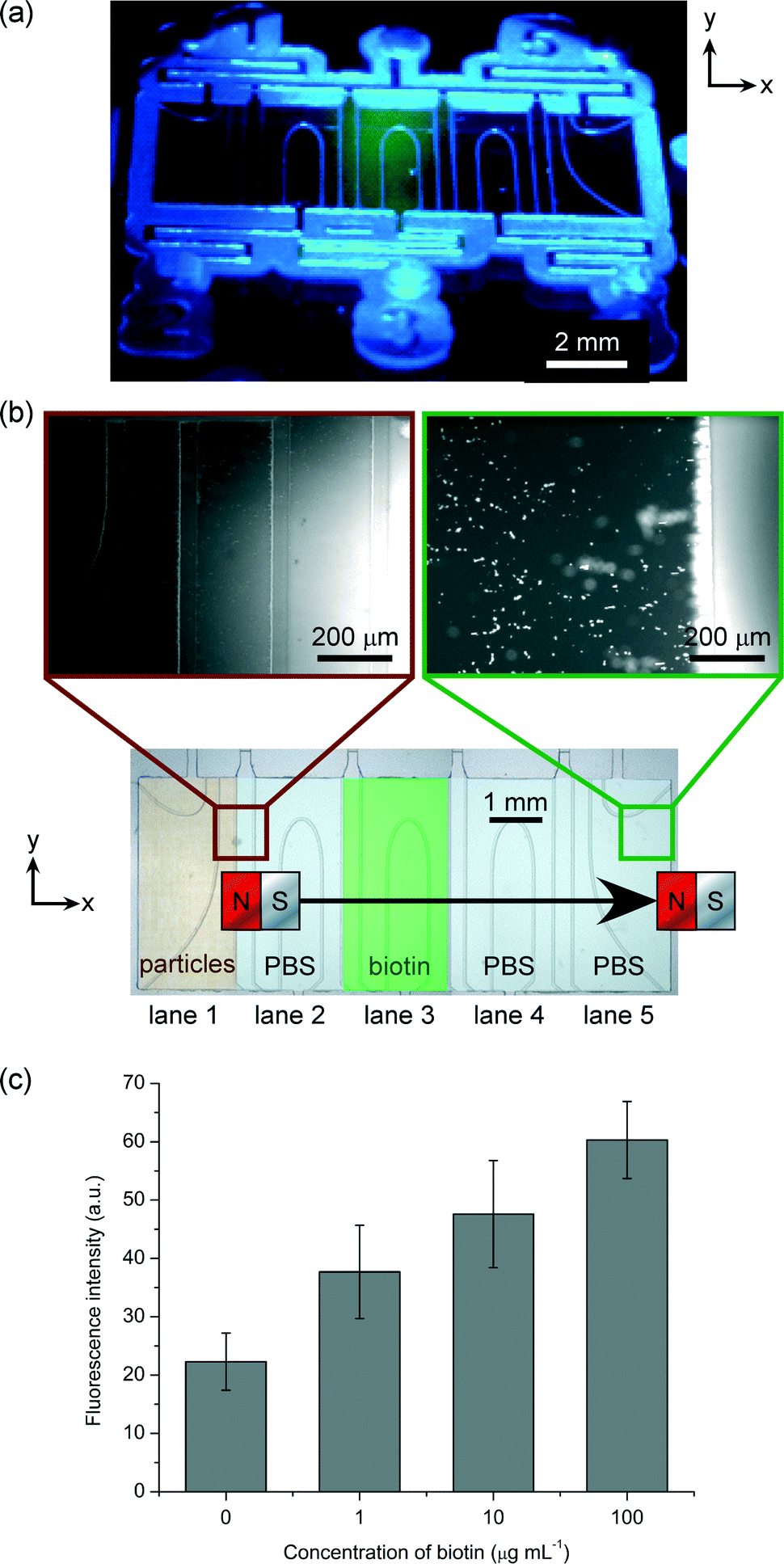 | ||
| Fig. 4 (a) Photograph of the phaseguide chip containing a central lane of fluorescent biotin. (b) Photographs of particles before and after passing through the biotin lane. The overview of the entire chip is a false-coloured photograph to highlight where the lanes would be. (c) Fluorescence intensities of the particles after passing through different concentrations of biotin. | ||
Fig. 4b shows photographs of the magnetic particles before and after passing through the lane of fluorescently labelled biotin. The images clearly demonstrate the increase in fluorescence intensity once they have migrated through the reagent, indicating successful binding of the biotin to the streptavidin functionalised particles. The assay was repeated with biotin concentrations of 1, 10 and 100 μg mL−1, and the fluorescence intensities of the particles were measured when the particles were in lane 5 (having traversed the biotin lane). The fluorescence intensities of the particles in lane 1 before beginning the assay were also measured, since the polystyrene matrix of the particles exhibits auto-fluorescence. Twenty particles were measured in each instance, and the results are shown in Fig. 4c. Here, it was found that increasing the biotin concentration yielded an increase in fluorescence intensity of the particles, and while optimisation of the platform is still required the results nonetheless demonstrate its potential for quantitative analysis.
An important factor to consider when dealing with laminated aqueous solutions is the extent of diffusion between the reagent and the buffer solutions. Over time, the two will slowly diffuse into each other, affecting the concentration of biotin through which the particles pass and therefore limiting the ability to perform quantitative assays. Observing the extent of diffusion of biotin over time showed that 5 min after filling the chip, some diffusion had occurred but the biotin was largely contained as a well-defined column in the chamber (Fig. 5a). However, 10 min after filling the chip the effect of diffusion was far more noticeable, with the biotin dispersing across a large region of the chip and having different concentrations of biotin in different areas (Fig. 5b). Bearing in mind, however, that once the chip was filled it required several seconds to position the magnet and then 8 s to perform the assay, the amount of diffusion would largely be negligible.
 | ||
| Fig. 5 Photographs showing diffusion of a lane of fluorescent biotin after (a) 5 min, and (b) 10 min after complete filling of the chip with solutions. | ||
Combined with the fact that the chip requires only a few minutes to set up and several seconds to perform the assay, this would make it a powerful tool for performing rapid, quantitative assays without the need for a pumping mechanism. In order to investigate the potential of the platform for more clinically relevant immunoassays, the inflammatory biomarker, C-reactive protein, was chosen for initial tests.
C-reactive protein immunoassay
C-reactive protein (CRP) is an acute phase protein found in blood whose concentration increases in response to inflammation or infection.60,61 As such, it is routinely measured in clinical diagnostics. Normal levels of CRP in serum are typically 1–10 μg mL−1, while levels of 10–40 μg mL−1 indicate mild inflammation or viral infection, and levels of 40–200 μg mL−1 suggest active inflammation or bacterial infection. Furthermore, chronic, minor elevations in CRP may also be indicative of cardiovascular disease.62,63To perform a sandwich CRP assay, lane 1 of the microfluidic device was filled with a suspension of primary antibody (1° Ab) functionalised magnetic particles, lane 2 with a solution of CRP, lane 4 with fluorescently labelled secondary antibody (2° Ab-FITC), and lanes 3 and 5 with PBS washing buffer. When the magnet was moved across the top of the chip, the particles were pulled through the lane containing CRP, whereupon the protein became bound to the antibodies on the particles, before entering the lane of wash solution in order to remove unbound material from the particle surfaces. The particles then passed through the lane of 2° Ab-FITC, which became bound to the CRP now present on the particles, thereby fluorescently labelling them. Finally, the particles passed into the final lane of wash solution and to the edge of the chamber, allowing unbound material to be washed from the particles and for images to be captured for fluorescence analysis. As in the one-step streptavidin–biotin reaction, the entire sandwich immunoassay required only 8 s: the time taken for the magnet to be moved across the chip.
Fig. 6a shows photographs of the magnetic particles before and after passing through the chamber, and clearly demonstrates the increase in fluorescence intensity of the particles after they had passed through each of the reagent and washing lanes. As before, the particles exhibited a slight fluorescence signal due to auto-fluorescence of the polystyrene matrix. The experiment was repeated with varying concentrations of CRP: 0.1, 1 and 10 μg mL−1. Results showed that the fluorescence intensity of the particles increased with increasing concentration (Fig. 6b, twenty particles were analysed in each case). However, it can be observed that the signal is approaching saturation as the concentration of CRP reaches 10 μg mL−1. This is likely due to the CRP approaching, or even reaching, the binding capacity of the magnetic particles at these higher concentrations, which would saturate the fluorescence signal and prevent further increases even if higher CRP concentrations were used. Despite the fluorescence approaching saturation between 1 and 10 μg mL−1, a one-tailed Welch's T-test showed a statistically significant difference at a confidence level of 95%, although at higher confidence levels (≥97.5%) the difference between these two signals was no longer statistically significant. The differences between the signals at the other CRP concentrations (i.e. between 0 and 1 μg mL−1) remained significant even at 99.5% confidence levels. Thus, the assay shows promise for the quantitative detection of CRP, although optimisation will be required to ensure statistical significance between all CRP concentrations tested, and to investigate the linear range and limit of detection.
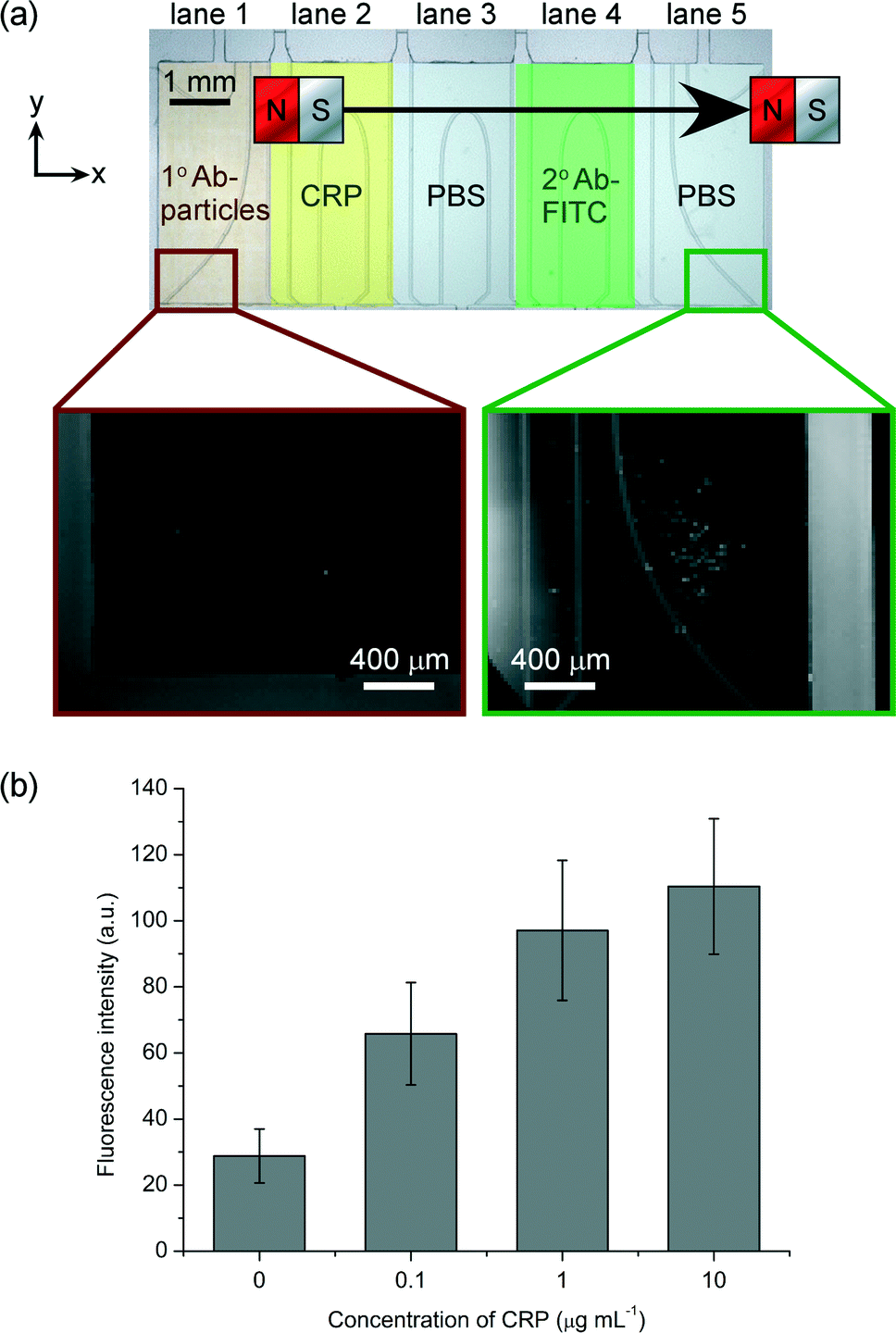 | ||
| Fig. 6 (a) Photographs of magnetic particles before and after passing through the reagent and washing lanes during the two-step CRP sandwich immunoassay. An increase in fluorescence intensity of the particles represents the binding and labelling of CRP. The overview photograph of the chip is false-coloured to allow visualisation of the position of the lanes. (b) Fluorescence intensities of the magnetic particles at different CRP concentrations. | ||
Negative control tests were also performed in which streptavidin functionalised particles were pulled through each of the lanes, with the results (not shown) illustrating that no increase in fluorescence could be obtained without using 1° Ab functionalised particles. When only streptavidin functional groups were present on the particles surfaces, the CRP would not be able to bind to the particles, and so there would be nothing for the 2° Ab-FITC to bind to as the particles passed through lane 4. This demonstrates that non-specific binding is not a problem, as any biomolecules that have absorbed onto the particles will be washed off as they are pulled through the washing lanes. Furthermore, no fluorescence increase was observed of 1° Ab particles that had been pulled through the CRP lane until they then passed through the 2° Ab-FITC lane. Off-chip tests were also performed in Eppendorf tubes to ensure that the on-chip processes followed the same progression as the off-chip procedures.
This proof-of-principle immunoassay demonstrates the potential of the platform for quantitative analysis of clinically relevant biomarkers. The system, which utilises liquid lamination via the incorporation of phaseguide structures into the microfluidic device, offers a simple yet controllable means of filling the device with miscible solutions, offering fast chip setup times (a few minutes) and rapid immunoassays (8 s). The platform shows great promise for point-of-care analysis, particularly as it does not require the use of a pumping mechanism, instead needing only manual pipetting of the reagents, making it promising as a portable system. However, before this can be realised there are several factors that could be optimised or incorporated into the platform, which will be discussed in the next section.
Discussion and outlook
This paper reports for the first time a stratification of miscible liquids in orderly lanes. Phaseguides have been used for stratification in earlier publications; however this required gelation steps to maintain the stratified profile. Stratification of miscible liquids is not possible in microfluidic chambers without adding an additional engineering complexity. Without phaseguides, liquid would spread radially and multiple liquids spread as one once they are brought in contact. The trick in the work presented here is that fluids are (1) manipulated in a rectangular shape while (2) keeping them separated until the filling process is near completion, followed by (3) filling of the small air gap that exists between the two liquids by (4) forcing phaseguide overflow at a position distal from a venting channel.The phaseguide-assisted liquid lamination platform has been shown here to be capable of performing magnetic particle-based assays in both one-step (streptavidin–biotin) and two-step (CRP) formats. Given that these were proof-of-principle assays designed to showcase the core principles of the system, the next steps in terms of assays would be investigations into other clinically relevant biomarkers (e.g. for cardiac disease, sexually transmitted infections, and various diseases), as well as the transition to looking at real clinical samples rather than standard solutions. In addition to immunoassays, it should be relatively easy to extend the technique to solid-phase extraction and nucleic acid assays, enabling DNA and RNA analysis from a sample matrix.
Clearly, as different biomarkers are investigated they may require slight changes to the way in which the procedure is carried out. For example, some assays may require longer incubation times due to slow reaction kinetics. This could be accounted for by increasing the width of the reagent lanes to increase the length of time the particles spend in each lane, moving the magnet more slowly across the chip, or simply holding the magnet in place over a particular reagent lane for a period of time to ensure the reaction occurs.
The current setup employs a conventional inverted fluorescence microscope; a somewhat limiting factor when attempting to transfer a technology to the portability and point-of-care stage. However, given the recent advances in miniaturised optical detection systems,64,65 we envisage the incorporation of such technology into a more complete setup at a later stage of development. Regarding the potential for mass production of the device itself, current chip fabrication is achieved by lamination methods that are already commonplace in the printed circuit board and biosensor industries. Still, it would also be possible to fabricate chips by injection moulding or hot embossing via the use of a precision milled master, and this has very recently been demonstrated for phaseguide chip production by Gottheil et al.48 Nonetheless, the design principles would remain the same for mass produced chips as for the prototype shown here.
Filling the lanes of the microfluidic chip with reagents and buffers is straightforward due to the incorporation of the phaseguide structures. The manual pipetting removes the need for an external syringe pump, yielding a static fluidic system in the chip, and requires only around 30 s of filling per lane. However, manual pipetting can sometimes succumb to human error during the filling of the system, particularly when “bridging” the gap between two lanes as the filling of the second lane nears completion. An accidental burst of pressure can force the liquid across the gap and into the adjacent lane, distorting the “shape” of the liquids within the lanes. When this occurs it seldom has a significant effect on the rest of the procedure, but in extreme circumstances it could affect the width of the reagent lanes and therefore the volume of reagent that the particles interact with. To prevent this from happening, steps could be taken to ensure a more reproducible filling procedure. In particular, blister packs66 containing the solutions could be placed over the inlet holes of the device, and actuated using a mechanical system that could be operated by hand (e.g. a “rolling pin” type assembly that rolls over the surface the device, bursting each blister pack in turn, or a series of pins that can be pressed onto the blister packs). The blister packs could be designed to contain just the right amount of volume to fill a lane, and a controlled mechanism for bursting them would ensure a reproducible filling.
When storing reagents in aqueous form is not viable (due to short lifetimes of the reagents) it may be possible to lyophilise the reagents67 as spots in the inlet channels of the microfluidic device, such that when a blister of buffer solution is burst it reconstitutes the reagent and fills the lane. Returning to the issue of reagent diffusion discussed earlier, the effects of diffusion could be greatly limited by the proposed mechanical setup, since always having the same chip preparation time followed immediately by the assay will ensure that the effect of diffusion is always the same, rather than running the risk of having diffusion affect one experiment more than another when performed manually.
As can be seen in Fig. 2b, multiple microfluidic devices can be produced on a single wafer, which could be exploited to perform calibration assays alongside an assay of a sample, thereby generating all the data necessary for a quantitative analysis on one device. The incorporation of phaseguide-based mixing structures55 into the device could also enable automated dilution of reagents for a calibration, with the phaseguides allowing differing volumes of buffer solution to mix with a reagent solution in order to generate a dilution series without needing to pre-dilute the reagents.
Currently, the magnet is moved by hand in order to pull the particles across the chamber. This has the major benefits of keeping the platform simple, easy to use and power-free. However, it can also introduce some human error since moving the magnet at different rates would affect the exposure time of the particles to the sample and reagents. Despite this, repeats of experiments have shown that there is little difference in results, and so long as the movement of the magnet is slow enough to allow the binding events to occur then the movement speed should not cause any great discrepancies. Clearly, moving the magnet too fast would also result in the particles being unable to follow, and so there will be a compromise between the desire for a fast procedure and the time required for the binding events to occur. A total movement time of 8 s was sufficient for the assays performed here, but other reactions, assays and extractions may require longer residence times in the sample or reagent lanes, as explained earlier. In principle, a lead screw and motor system could be constructed for automated, controllable and reproducible movement of the magnet, but this would add a degree of complexity that we are trying to avoid in this platform in terms of keeping the device simple and easy to use.
The various tools proposed above represent potential directions in which to take the development of the magnetic particle-based assay platform for future devices. However, the core elements of the system have been demonstrated here: (i) the formation of static, adjacent lanes of reagent and washing solutions via phaseguide-assisted filling of the device, thereby eliminating the need for external syringe pumps, (ii) rapid magnetic particle-based assays, in both one-step and two-step format, in which the chip setup takes only a few minutes and the assay itself requires only 8 seconds. Future work would involve optimisation of the device and its operation, which would allow studies into linear ranges and limits of detection for different biomarkers.
Conclusions
We have demonstrated the use of phaseguide structures to achieve the sequential lamination of five aqueous liquids in a microfluidic chamber. Filling of the chamber is achieved via manual pipetting, eliminating the need for external pumping systems, and requires only a few minutes. Magnetic particle-based assays have been performed in the system by drawing functionalised magnetic particles consecutively through each “lane” of reagent and washing buffer via an external magnet, requiring only 8 seconds to perform one-step and two-step reactions. Extension of the protocol to solid-phase extraction and nucleic acid assays should be straightforward, making the platform far more versatile in the future, and we can envisage a number of technological developments for optimisation of the system and procedure.Acknowledgements
C. P. thanks the Royal Thai Government (Thailand) for financial support. E. Y. acknowledges the Scientific and Technological Research Council of Turkey for financial support. S. J. T., T. H. and P. V would like to thank technology foundation STW and The Netherlands Metabolomics Centre for financial support. The authors gratefully acknowledge Koen Sweering and Athanasios Giannitsis for fabrication of chips.Notes and references
- M. A. M. Gijs, Microfluid. Nanofluid., 2004, 1, 22–40 CAS.
- N. Pamme, Lab Chip, 2006, 6, 24–38 RSC.
- M. A. M. Gijs, F. Lacharme and U. Lehmann, Chem. Rev., 2010, 110, 1518–1563 CrossRef CAS PubMed.
- N. Pamme, Curr. Opin. Chem. Biol., 2012, 16, 436–443 CrossRef CAS PubMed.
- A. H. C. Ng, U. Uddayasankar and A. R. Wheeler, Anal. Bioanal. Chem., 2010, 397, 991–1007 CrossRef CAS PubMed.
- M. D. Tarn and N. Pamme, Expert Rev. Mol. Diagn., 2011, 11, 711–720 CrossRef CAS PubMed.
- T. Verch and R. Bakhtiar, Bioanalysis, 2012, 4, 177–188 CrossRef CAS PubMed.
- J. W. Choi, K. W. Oh, J. H. Thomas, W. R. Heineman, H. B. Halsall, J. H. Nevin, A. J. Helmicki, H. T. Henderson and C. H. Ahn, Lab Chip, 2002, 2, 27–30 RSC.
- S. Bronzeau and N. Pamme, Anal. Chim. Acta, 2008, 609, 105–112 CrossRef CAS PubMed.
- Y. N. Yang, H. I. Lin, J. H. Wang, S. C. Shiesh and G. B. Lee, Biosens. Bioelectron., 2009, 24, 3091–3096 CrossRef CAS PubMed.
- R. Venu, B. Lim, X. H. Hu, I. Jeong, T. S. Ramulu and C. G. Kim, Microfluid. Nanofluid., 2013, 14, 277–285 CrossRef CAS.
- N. Pamme and A. Manz, Anal. Chem., 2004, 76, 7250–7256 CrossRef CAS PubMed.
- N. Pamme and C. Wilhelm, Lab Chip, 2006, 6, 974–980 RSC.
- P.-H. Shih, J.-Y. Shiu, P.-C. Lin, C.-C. Lin, T. Veres and P. Chen, J. Appl. Phys., 2008, 103, 07A316 Search PubMed.
- S. H. S. Lee, T. A. Hatton and S. A. Khan, Microfluid. Nanofluid., 2011, 11, 429–438 CrossRef CAS.
- B. Eickenberg, F. Wittbracht, P. Stohmann, J.-R. Schubert, C. Brill, A. Weddemann and A. Huetten, Lab Chip, 2013, 13, 920–927 RSC.
- M. D. Tarn, M. J. Lopez-Martinez and N. Pamme, Anal. Bioanal. Chem., 2014, 406, 139–161 CrossRef CAS PubMed.
- S. A. Peyman, A. Iles and N. Pamme, Chem. Commun., 2008, 1220–1222 RSC.
- S. A. Peyman, A. Iles and N. Pamme, Lab Chip, 2009, 9, 3110–3117 RSC.
- L. A. Sasso, A. Undar and J. D. Zahn, Microfluid. Nanofluid., 2010, 9, 253–265 CrossRef CAS PubMed.
- S. A. Peyman, H. Patel, N. Belli, A. Iles and N. Pamme, Magnetohydrodynamics, 2009, 45, 361–370 Search PubMed.
- L. A. Sasso, K. Aran, Y. Guan, A. Ündar and J. D. Zahn, Artif. Organs, 2013, 37, E9–E17 CrossRef PubMed.
- L. Sasso, I. Johnston, M. Zheng, R. Gupte, A. Ündar and J. Zahn, Microfluid. Nanofluid., 2012, 13, 603–612 CrossRef CAS.
- Y. Gao, A. W. Y. Lam and W. C. W. Chan, ACS Appl. Mater. Interfaces, 2013, 5, 2853–2860 CAS.
- U. Lehmann, C. Vandevyver, V. K. Parashar and M. A. M. Gijs, Angew. Chem., Int. Ed., 2006, 45, 3062–3067 CrossRef CAS PubMed.
- M. Shikida, K. Takayanagi, H. Honda, H. Ito and K. Sato, J. Micromech. Microeng., 2006, 16, 1875–1883 CrossRef.
- H. Tsuchiya, M. Okochi, N. Nagao, M. Shikida and H. Honda, Sens. Actuators, B, 2008, 130, 583–588 CrossRef CAS PubMed.
- R. S. Sista, A. E. Eckhardt, V. Srinivasan, M. G. Pollack, S. Palanki and V. K. Pamula, Lab Chip, 2008, 8, 2188–2196 RSC.
- C. H. Chiou, D. J. Shin, Y. Zhang and T. H. Wang, Biosens. Bioelectron., 2013, 50, 91–99 CrossRef CAS PubMed.
- J. Pipper, M. Inoue, L. F. P. Ng, P. Neuzil, Y. Zhang and L. Novak, Nat. Med., 2007, 13, 1259–1263 CrossRef CAS PubMed.
- J. Pipper, Y. Zhang, P. Neuzil and T.-M. Hsieh, Angew. Chem., Int. Ed., 2008, 47, 3900–3904 CrossRef CAS PubMed.
- A. H. C. Ng, K. Choi, R. P. Luoma, J. M. Robinson and A. R. Wheeler, Anal. Chem., 2012, 84, 8805–8812 CrossRef CAS PubMed.
- Y. Zhang, S. Park, K. Liu, J. Tsuan, S. Yang and T.-H. Wang, Lab Chip, 2011, 11, 398–406 RSC.
- S. M. Berry, E. T. Alarid and D. J. Beebe, Lab Chip, 2011, 11, 1747–1753 RSC.
- B. P. Casavant, D. J. Guckenberger, S. M. Berry, J. T. Tokar, J. M. Lang and D. J. Beebe, Lab Chip, 2013, 13, 391–396 RSC.
- B. P. Casavant, L. N. Strotman, J. J. Tokar, S. M. Thiede, A. M. Traynor, J. S. Ferguson, J. M. Lang and D. J. Beebe, Lab Chip, 2014, 14, 99–105 RSC.
- L. Strotman, R. O'Connell, B. P. Casavant, S. M. Berry, J. M. Sperger, J. M. Lang and D. J. Beebe, Anal. Chem., 2013, 85, 9764–9770 CrossRef CAS PubMed.
- S. M. Berry, L. N. Strotman, J. D. Kueck, E. T. Alarid and D. J. Beebe, Biomed. Microdevices, 2011, 13, 1033–1042 CrossRef PubMed.
- S. M. Berry, L. J. Maccoux and D. J. Beebe, Anal. Chem., 2012, 84, 5518–5523 CrossRef CAS PubMed.
- K. Sur, S. M. McFall, E. T. Yeh, S. R. Jangam, M. A. Hayden, S. D. Stroupe and D. M. Kelso, J. Mol. Diagn., 2010, 12, 620–628 CrossRef CAS PubMed.
- R. C. den Dulk, K. A. Schmidt, G. Sabatte, S. Liebana and M. W. J. Prins, Lab Chip, 2013, 13, 106–118 RSC.
- H. Bordelon, N. M. Adams, A. S. Klemm, P. K. Russ, J. V. Williams, H. K. Talbot, D. W. Wright and F. R. Haselton, ACS Appl. Mater. Interfaces, 2011, 3, 2161–2168 CAS.
- K. M. Davis, J. D. Swartz, F. R. Haselton and D. W. Wright, Anal. Chem., 2012, 84, 6136–6142 CrossRef CAS PubMed.
- N. M. Adams, A. E. Creecy, C. E. Majors, B. A. Wariso, P. A. Short, D. W. Wright and F. R. Haselton, Biomicrofluidics, 2013, 7, 014104 Search PubMed.
- O. Strohmeier, A. Emperle, G. Roth, D. Mark, R. Zengerle and F. von Stetten, Lab Chip, 2013, 13, 146–155 RSC.
- H. Becker, C. Carstens, D. Kuhlmeier, C. Zilch and C. Gaertner, Proceedings of the MicroTAS 2012 Conference, 2012, pp. 791–793 Search PubMed.
- H. Becker, C. Carstens, D. Kuhlmeier, N. Sandetskaya, N. Schroeter, C. Zilch and C. Gaertner, Stationary microfluidics: molecular diagnostic assays by moving magnetic beads through non-moving liquids, in Proceedings of SPIE: Microfluidics, BioMEMS, and Medical Microsystems XI, 2013, vol. 8615, p. 86150B Search PubMed.
- R. Gottheil, N. Baur, H. Becker, G. Link, D. Maier, N. Schneiderhan-Marra and M. Stelzle, Biomed. Microdevices, 2014, 16, 163–172 CrossRef CAS PubMed.
- P. Vulto, G. Medoro, L. Altomare, G. A. Urban, M. Tartagni, R. Guerrieri and N. Manaresi, J. Micromech. Microeng., 2006, 16, 1847–1853 CrossRef.
- P. Vulto, S. Podszun, P. Meyer, C. Hermann, A. Manz and G. A. Urban, Lab Chip, 2011, 11, 1596–1602 RSC.
- P. Vulto and G. Urban, Integrated microfluidic component for purifying analyte molecules and purification method, US 08431339, 2013 Search PubMed.
- P. Vulto, G. Dame, U. Maier, S. Makohliso, S. Podszun, P. Zahn and G. A. Urban, Lab Chip, 2010, 10, 610–616 RSC.
- D. Puchberger-Enengl, S. Podszun, H. Heinz, C. Hermann, P. Vulto and G. A. Urban, Biomicrofluidics, 2011, 5, 044111 Search PubMed.
- S. Podszun, P. Vulto, H. Heinz, S. Hakenberg, C. Hermann, T. Hankemeier and G. A. Urban, Lab Chip, 2012, 12, 451–457 RSC.
- S. Hakenberg, M. Huegle, M. Weidmann, F. Hufert, G. Dame and G. A. Urban, Lab Chip, 2012, 12, 4576–4580 RSC.
- P. Vulto, P. Kuhn and G. A. Urban, Lab Chip, 2013, 13, 2931–2936 RSC.
- S. J. Trietsch, G. D. Israels, J. Joore, T. Hankemeier and P. Vulto, Lab Chip, 2013, 13, 3548–3554 RSC.
- P. Vulto, N. Glade, L. Altomare, J. Bablet, L. D. Tin, G. Medoro, I. Chartier, N. Manaresi, M. Tartagni and R. Guerrieri, Lab Chip, 2005, 5, 158–162 RSC.
- P. Vulto, T. Huesgen, B. Albrecht and G. A. Urban, J. Micromech. Microeng., 2009, 19, 077001 CrossRef.
- B. Clyne and J. S. Olshaker, J. Emerg. Med., 1999, 17, 1019–1025 CrossRef CAS.
- S. Black, I. Kushner and D. Samols, J. Biol. Chem., 2004, 279, 48487–48490 CrossRef CAS PubMed.
- W. K. Lagrand, C. A. Visser, W. T. Hermens, H. W. M. Niessen, F. W. A. Verheugt, G. J. Wolbink and C. E. Hack, Circulation, 1999, 100, 96–102 CrossRef CAS.
- J. P. Casas, T. Shah, A. D. Hingorani, J. Danesh and M. B. Pepys, J. Intern. Med., 2008, 264, 295–314 CrossRef CAS PubMed.
- L. Novak, P. Neuzil, J. Pipper, Y. Zhang and S. Lee, Lab Chip, 2007, 7, 27–29 RSC.
- H. Zhu, S. O. Isikman, O. Mudanyali, A. Greenbaum and A. Ozcan, Lab Chip, 2013, 13, 51–67 RSC.
- A. Disch, C. Mueller and H. Reinecke, Low cost production of disposable microfluidics by blister packaging technology, in 2007 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 2012, vol. 1–16, pp. 6323–6326 Search PubMed.
- A. Ahlford, B. Kjeldsen, J. Reimers, A. Lundmark, M. Romani, A. Wolff, A.-C. Syvanen and M. Brivio, Analyst, 2010, 135, 2377–2385 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Video demonstrating the phaseguide-assisted filling of a device with aqueous solutions of orange and blue inks. See DOI: 10.1039/c4lc00139g |
| ‡ Corresponding author for magnetic particle-based assays. |
| § Corresponding author for phaseguide design. |
| This journal is © The Royal Society of Chemistry 2014 |