 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefined topologically-complex protein matrices to manipulate cell shape via three-dimensional fiber-like patterns†
Christopher
Moraes
ab,
Byoung Choul
Kim
abc,
Xiaoyue
Zhu‡
a,
Kristen L.
Mills§
d,
Angela R.
Dixon
a,
M. D.
Thouless
*de and
Shuichi
Takayama
*abcf
aDepartment of Biomedical Engineering, College of Engineering, University of Michigan, 2200 Bonisteel Blvd, Ann Arbor, MI 48109, USA. E-mail: takayama@umich.edu
bBiointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA
cMacromolecular Science and Engineering Center, College of Engineering, University of Michigan, 2300 Hayward St., Ann Arbor, MI 48109, USA
dDepartment of Mechanical Engineering, College of Engineering, University of Michigan, 2350 Hayward St., Ann Arbor, MI 48109, USA
eDepartment of Materials Science & Engineering, College of Engineering, University of Michigan, 2300 Hayward St., Ann Arbor, MI 48109, USA. E-mail: thouless@umich.edu
fDivision of Nano-Bio and Chemical Engineering WCU Project, UNIST, Ulsan, Republic of Korea
First published on 26th February 2014
Abstract
Culturing cells in three-dimensional (3D) environments has been shown to significantly influence cell function, and may provide a more physiologically relevant environment within which to study the behavior of specific cell types. 3D tissues typically present a topologically complex fibrous adhesive environment, which is technically challenging to replicate in a controlled manner. Micropatterning technologies have provided significant insights into cell-biomaterial interactions, and can be used to create fiber-like adhesive structures, but are typically limited to flat culture systems; the methods are difficult to apply to topologically-complex surfaces. In this work, we utilize crack formation in multilayered microfabricated materials under applied strain to rapidly generate well-controlled and topologically complex ‘fiber-like’ adhesive protein patterns, capable of supporting cell culture and controlling cell shape on three-dimensional patterns. We first demonstrate that the features of the generated adhesive environments such as width, spacing and topology can be controlled, and that these factors influence cell morphology. The patterning technique is then applied to examine the influence of fiber structure on the nuclear morphology and actin cytoskeletal structure of cells cultured in a nanofibrous biomaterial matrix.
Introduction
The use of micropatterning technologies has generated significant insights into the physical mechanisms underlying cell viability,1 differentiation,2 morphogenesis,3 migration,4 and into the fundamental relationships between cells and their environment.5–7 In micropatterning studies, cells attach and spread on spatially defined, microfabricated adhesive patterns, designed to mimic specific aspects of the native cellular microenvironment in terms of geometry, architecture, composition, mechanics and dynamics.8 Though extremely useful, micropatterning technologies are typically applied only to two-dimensional substrates. This is a significant drawback, as three-dimensional (3D) culture systems provide physiologically relevant environments for certain cell types, and are known to significantly influence cell function,9–11 morphology,12 viability,13 proliferation,14 motility15 and differentiation.16 Furthermore, it has been shown that in vitro 3D environments can better replicate realistic in vivo responses to therapies,17,18 enhancing the translatability of in vitro discoveries towards clinical applications. Hence, it is desirable to conduct experiments in 3D environments, and the ability to micropattern well-defined 3D adhesive patterns for cell culture should yield considerable insight into 3D cell-environment interactions.The architecture of 3-D biomaterial cell-culture systems is significantly more complex than that of their 2-D counterparts. In 3-D, both artificial and natural culture environments can often be considered as a mesh of adhesive fibers to which cells can attach (Fig. 1A). Cells spread through this mesh, taking on 3D-specific morphologies.19 Given the demonstrated importance of morphology on cell function,7 the adhesive mesh presented by the fibers probably plays a central role in directing 3D functionality. This hypothesis is strongly supported by recent studies demonstrating that cells cultured on fiber-like ‘1-D’ linear adhesive patterns exhibit morphologies and migration behaviors that more closely mimic cells in 3-D cultures, as compared to cells cultured on conventional 2-D surfaces.15,20 However, adhesive fiber patterns differ greatly between and within biomaterial systems: the dimensions, alignment and topology of fibers are challenging to control simultaneously, with each parameter depending upon intrinsic biomaterial properties and the material processing techniques being used. Currently, there exists no simple method to prescribe these features a priori, making it challenging to identify the precise role that each one plays in driving cell function. In this work, we develop a technique to fabricate topologically-complex “fiber-like” adhesive patterns in a controlled fashion. We do this by decorating topologically complex microfabricated surfaces (microgrooves in this initial demonstration) with adhesive ‘fiber-like’ patterns.
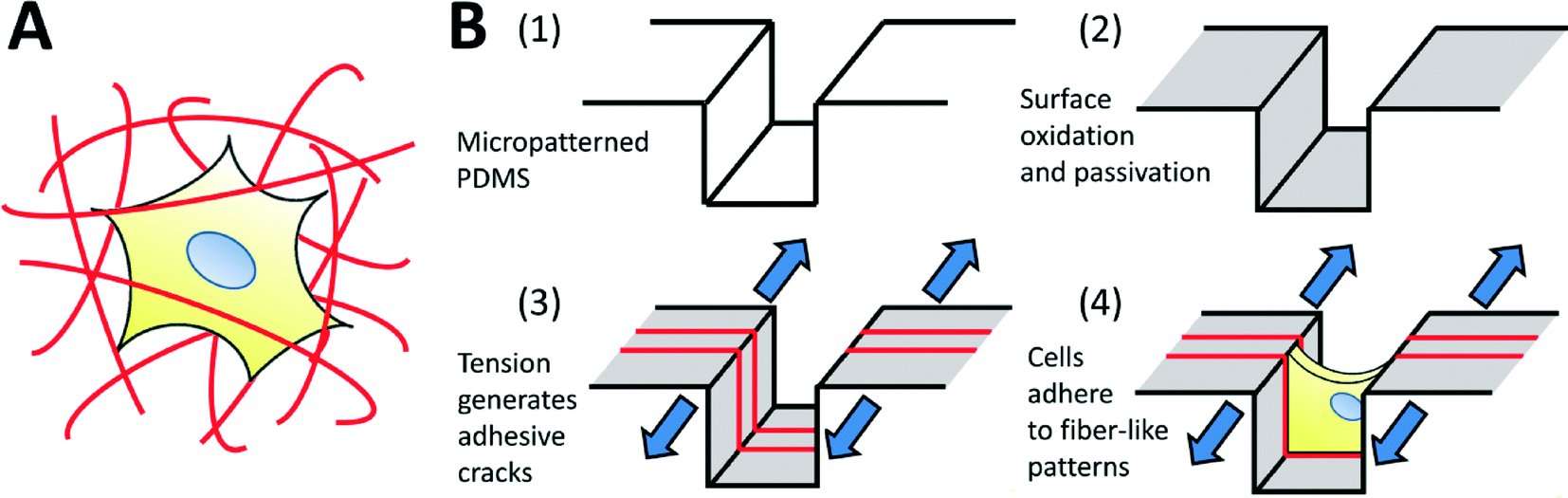 | ||
| Fig. 1 (A) Schematic of a biological cell encapsulated within a three-dimensional fibrous matrix. Cell morphology is dictated by the arrangement of surrounding topologically complex adhesive fibers. (B) Fabrication process to generate topologically complex adhesive sites to control cell shape in three dimensions. (1, 2) Micropatterned PDMS substrates are plasma oxidized to generate a thin, brittle silica-like layer, and surface modified to prevent cell adhesion. (3) The mismatch in toughness and modulus between the bulk PDMS and the silica-like oxidized layer drives the formation of a stable array of cracks in the surface when the system is placed under applied tension. Adhesive proteins selectively adsorb to these crack structures, forming precisely defined topologically complex adhesive ‘fibers’ for (4) cell attachment and control of morphology. | ||
While it is possible to carefully engineer a fibrous scaffold to culture cells in a defined 3D environment,21,22 such techniques are typically limited in terms of throughput and spatial resolution. The spatial resolutions needed to create micron- and sub-micron scale adhesive fibers are challenging to achieve even using micropatterning technologies such as microcontact printing (μCP).23 μCP involves coating matrix proteins onto the raised features of an elastomeric mold, and then transferring the proteins by a stamping process onto a candidate cell culture surface. Although small features can be fabricated into the stamp, they are likely to collapse or deform during stamping; this limits the resolution of the protein patterns that can be obtained.24 Furthermore, μCP is unable to transfer patterns onto topologically complex substrates. Alternative techniques such as projection photolithographic patterning,25 direct laser writing,26,27 and dip-pen nanolithography28 address these issues of spatial resolution and topographic patterning, but require highly specialized equipment and expertise, unavailable in most wet labs. Furthermore, these sequential techniques are not easily amenable to increasing throughput, which is generally necessary for biological studies.
In order to address these issues, we explore the use of fracture-based micropatterning technologies29–33 to generate fiber-like patterns with three-dimensional topologies. Thin brittle films supported on an elastomeric membrane form a stable array of cracks under applied mechanical tension, and we have leveraged this phenomenon to create dynamic and controllable adhesive cracks at the nano- to micron-scale for patterned cell culture.20,34 For the purposes of this initial demonstration, we adapt this approach to generate fiber-like patterns that transverse the topography of a micro-grooved slab of polydimethylsiloxane (PDMS; Fig. 1B). We do this by using non-directional plasma oxidation to generate a conformal oxidized layer on a microstructured substrate. The brittle oxidized layer is functionalized to be non-adhesive to cells, and then cracked under applied tension. These cracks expose the underlying adhesive PDMS which is then coated with extracellular matrix proteins. This process creates an array of fiber-like adhesive patterns on all surfaces of the microgrooves. Cells cultured on these well-defined protein matrices adopt distinct morphologies depending on the presentation of the adhesive patterns. We further demonstrate that protein patterns can be used to dissect the effects of microenvironmental structure on nuclear shape and actin cytoskeletal structure in nanofibrous biomaterials.
Methods
Microstructure fabrication
Negative relief SU-8 (Microchem) masters were fabricated following protocols from the manufacturer. SU-8 2025 was spin-coated onto cleaned silicon wafers to a thickness of ~30 μm, and patterned by conventional photolithography to yield an array of 2 cm long microgrooves ranging in width from 20 to 80 μm. Regions of the mold were intentionally designed without microgrooves to serve as flat surfaces for control experiments. The resulting structures were hard-baked at 120 degrees overnight, and silanized by exposure to (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane while under vacuum for 30 minutes. Polydimethylsiloxane (PDMS; Sylgard 184, Dow Corning, MI) base and curing agents were thoroughly mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (w/w), degassed under vacuum, cast against the SU-8 master and cured at 60 °C for at least 4 hours. The positive replica PDMS slab was then carefully peeled from the master. A well structure, designed to hold sufficient medium for cell culture, was made by gluing the patterned face of the microstructured PDMS to the bottom of a square PDMS gasket. The gasket was fabricated by manually cutting a 1 cm × 1 cm hole through a 4 cm × 4 cm × 3 mm PDMS cuboid. With the ridge-and-groove microstructures facing up, the relief substrate was glued to the bottom of the gasket using uncured PDMS. The assembly was cured at 150 °C for at least 12 hours, to ensure complete curing of the material. To facilitate rapid production of these devices, the positive relief structures were oxidized, silanized and replicated in epoxy (EPOXY Technology, Inc), using protocols supplied by the manufacturer. The resulting epoxy masters were then used to produce monolithic microstructured samples with gaskets in a single cast.
:1 ratio (w/w), degassed under vacuum, cast against the SU-8 master and cured at 60 °C for at least 4 hours. The positive replica PDMS slab was then carefully peeled from the master. A well structure, designed to hold sufficient medium for cell culture, was made by gluing the patterned face of the microstructured PDMS to the bottom of a square PDMS gasket. The gasket was fabricated by manually cutting a 1 cm × 1 cm hole through a 4 cm × 4 cm × 3 mm PDMS cuboid. With the ridge-and-groove microstructures facing up, the relief substrate was glued to the bottom of the gasket using uncured PDMS. The assembly was cured at 150 °C for at least 12 hours, to ensure complete curing of the material. To facilitate rapid production of these devices, the positive relief structures were oxidized, silanized and replicated in epoxy (EPOXY Technology, Inc), using protocols supplied by the manufacturer. The resulting epoxy masters were then used to produce monolithic microstructured samples with gaskets in a single cast.
Generation of adhesive fiber-like patterns
The procedure to generate adhesive crack structures on the microgroove-patterned and flat PDMS surfaces is outlined in Fig. 1B. The PDMS devices were oxidized to produce a thin brittle silica-like layer on the device surface, using a plasma oxidation system (Covance-MP; Femtoscience). To allow uniform oxidation of all groove surfaces, the flat metal grounding electrode that generates highly directional plasma fields was removed from the system, and the PDMS devices were oxidized under vacuum at 200 W for between 7.5 and 15 minutes. The surfaces of the plasma-oxidized devices were then modified to render them non-adhesive to cells, following previously published protocols.34 The surfaces were first silanized by placing them in a dessicator with 100 μL of a 1:1 mixture of mineral oil and (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane, under vacuum for 30 minutes. To confirm successful silianization, which renders the surface hydrophobic, a droplet of water was put onto a test surface to verify that any wetting was minimal. The devices were incubated for one hour with 0.1% Pluronic F-108 (BASF, in water), which binds to the hydrophobic silane, creating a cell repellent surface. The final step of the preparation was to rinse the surfaces with copious amounts of double distilled water to remove any unbound Pluronic.
The devices were mounted onto Microvice holders (S.T. Japan USA; FL, USA) using clamps provided by the manufacturer. The Microvice holders were used to subject the devices to linear strains of 2.5, 5, 10 or 15%, and generate cracks in the silica-like oxidized PDMS layer. The freshly exposed cracked surfaces were not resistant to protein binding, and incubation with solubilized candidate extracellular matrix (ECM) proteins generated patterned ECM protein arrays. For all cell culture experiments described in this work, incubation with 40 μg mL−1 of fibronectin in phosphate-buffered saline (PBS; Sigma) for 30 minutes was used to generate the adhesive structures. For visualization purposes, TRITC-labelled bovine serum albumin (TRITC-BSA) was used as the matrix protein. After the protein patterns had been created, the applied strain was released to close the cracks, and the devices were washed thoroughly in PBS to remove any non-adsorbed ECM. In all cases, the strain was applied along the direction of the microstructures, generating cracks perpendicular to the direction of the channels. In this way, ‘fiber-like’ protein patterns are deposited along the walls of the PDMS microgrooves. Cracks formed ‘1-D’ adhesive linear patterns on the flat surfaces, and were used for control experiments. The crack spacings and widths were characterized using a 3D scanning laser interferometer (LEXT OLS4000; Olympus).
Cell culture
The patterned protein matrices were sterilized under germicidal UV light for 30 minutes and stretched to the desired strain for each experiment. The PDMS devices were loaded with 150 μL of serum-free culture media (Advanced Dulbecco's Modified Eagle Media (Advanced DMEM; Gibco); supplemented with 4 mM L-glutamine), and degassed under low vacuum for 30 minutes to ensure that air bubbles in the crack features are removed. NIH 3T3 fibroblast and C2C12 myoblast cells were cultured on tissue-culture plastic in fully-supplemented growth media (DMEM supplemented with 10% fetal bovine serum and 1% antimycotics–antibiotics; 5% CO2, 37 °C) on tissue-culture plastic until 60–70% confluent. The cells were then trypsinized with 0.25% trypsin for 5 minutes, pelleted by centrifugation, and re-suspended in Advanced DMEM at the desired concentration. The cells were then seeded on the PDMS devices at 10000 cells cm−2, and allowed to spread on the protein patterns overnight. They were then fixed in 4% paraformaldehyde for 15 minutes at room temperature, and stored at 4 °C until ready for staining and analysis. The strain applied to the devices was released at this point, as the cells retained their morphology and structure after fixation.
Fluorescent labeling, imaging and analysis
Fixed samples were washed three times in PBS, and permeabilized using 0.1% Triton-X in PBS for 20 minutes at room temperature. Fibronectin (FN) matrix proteins were fluorescently labeled using standard indirect immunostaining protocols. The samples were blocked with 3% bovine serum albumen (BSA) for 30 minutes at 37 °C, and incubated overnight at 4 °C in a humidified chamber with a 1:200 dilution of anti-FN primary antibodies in 0.1% BSA solution. The samples were then washed three times in PBS, blocked with 10% goat serum for 30 minutes at room temperature, and incubated with a secondary Alexafluor 488 antibody (Invitrogen) for 1 hour at room temperature. Direct labeling for actin fibers was conducted by incubating samples with 0.1 μM FITC-labeled phalloidin (Sigma) in 0.1% BSA for 30 minutes at room temperature. Nuclei were labeled with Hoechst 33258 (10 μg mL−1 in 0.1% BSA) for 15 minutes at room temperature. In all cases, the samples were washed in PBS thoroughly, and mounted on a glass coverslip using Fluoromount G (Southern Biotech).
The samples were imaged using either epifluorescent (TE300, Nikon) or confocal (Leica SP5) microscopy. Image reconstruction and analysis were conducted in ImageJ (NIH). Nuclear dimensions were calculated by fitting ellipses to nuclear images using native ImageJ functions, and extracting dimensions of long and short axes.
Statistical analysis
Data are reported as a mean ± a standard deviation, and statistical comparisons were based on one-way analysis of variance (ANOVA). Post-hoc comparisons were performed with the Tukey test for post-ANOVA pairwise comparisons in a one-way ANOVA using SigmaStat 3.5 (San Jose, CA).Results & discussion
Multiscale surface features that span the nano- to micro- regimes have previously been demonstrated to be critically important in regulating cell function in a variety of cell-culture model systems.6 For example, micro- and nanotopography significantly influence differentiation of stem cells35,36 and the organization and function of engineered cardiac tissue.37 Fabricating controlled multiscale features is particularly challenging when mimicking three-dimensional cell culture environments, which present cells with topographically complex adhesive structures that span both the nano- and micro-scale regimes, and typically take the form of an encapsulating fibrous mesh (Fig. 1A). Topographical and adhesive features of the fibrous mesh likely play a significant role in directing cell function, and are hence particularly important to manipulate at both the micro- and nano-scales. In order to replicate the fibrous nature of 3D cell-culture environments in a controlled manner, we generated an array of fiber-like adhesive patterns along the surfaces of a microstructured substrate. In this work, linear adhesive patterns were generated by fracture along the surfaces of microgrooves with rectangular cross-sections (Fig. 1B) to produce a cellular environment consisting of adhesive ‘fiber-like’ patterns that surrounded the cell on three sides. The dimensions and characteristics of adhesive patterns could be tuned, enabling control over the properties of the fibrous adhesive structure presented to cultured cells.Microgroove structures were successfully fabricated with dimensions appropriate for cell culture by creating PDMS replicas from SU-8 molds. The surfaces were passivated to block cell adhesion, using a protocol demonstrated to reduce adhesion by two orders of magnitude (ESI† Fig. S1), and decorated with fiber-like adhesive patterns using crack patterning. Control over the resulting adhesive structures was then studied and characterized for this system. Finally, the system was used to study the effect of adhesive fiber-like architecture on the nuclear and cytoskeletal morphology of fibroblasts, and the results compared to studies of cells cultured in electrospun nanofibrous scaffolds with similar adhesive properties.
Fabrication of 3D fiber-like adhesive protein patterns
A weakly-directional oxygen plasma was applied to the sample in order to coat the microgroove surfaces with a conformal brittle silica-like oxidized layer. The directionality of the plasma was minimized by removing the grounding plate in a Covance Femtoscience plasma oxidation system, forcing the cylindrical metal chamber to serve as the ground electrode. Although the resulting plasma was certainly not isotropic, the strong planar directionality imparted by the flat metal grounding plate was reduced, generating a conformal oxidized film on the microgroove structures. Contiguous cracks were demonstrated to form on the top, bottom and side walls of the structures under applied tension (Fig. 2A) and confocal fluorescent microscopy revealed that these adhesive fractures were continuous along all the surfaces of the microgrooves (Fig. 2B, C, ESI† movies 1A, B). In contrast, a strongly directional plasma generated cracks that were not continuous along intersecting surfaces.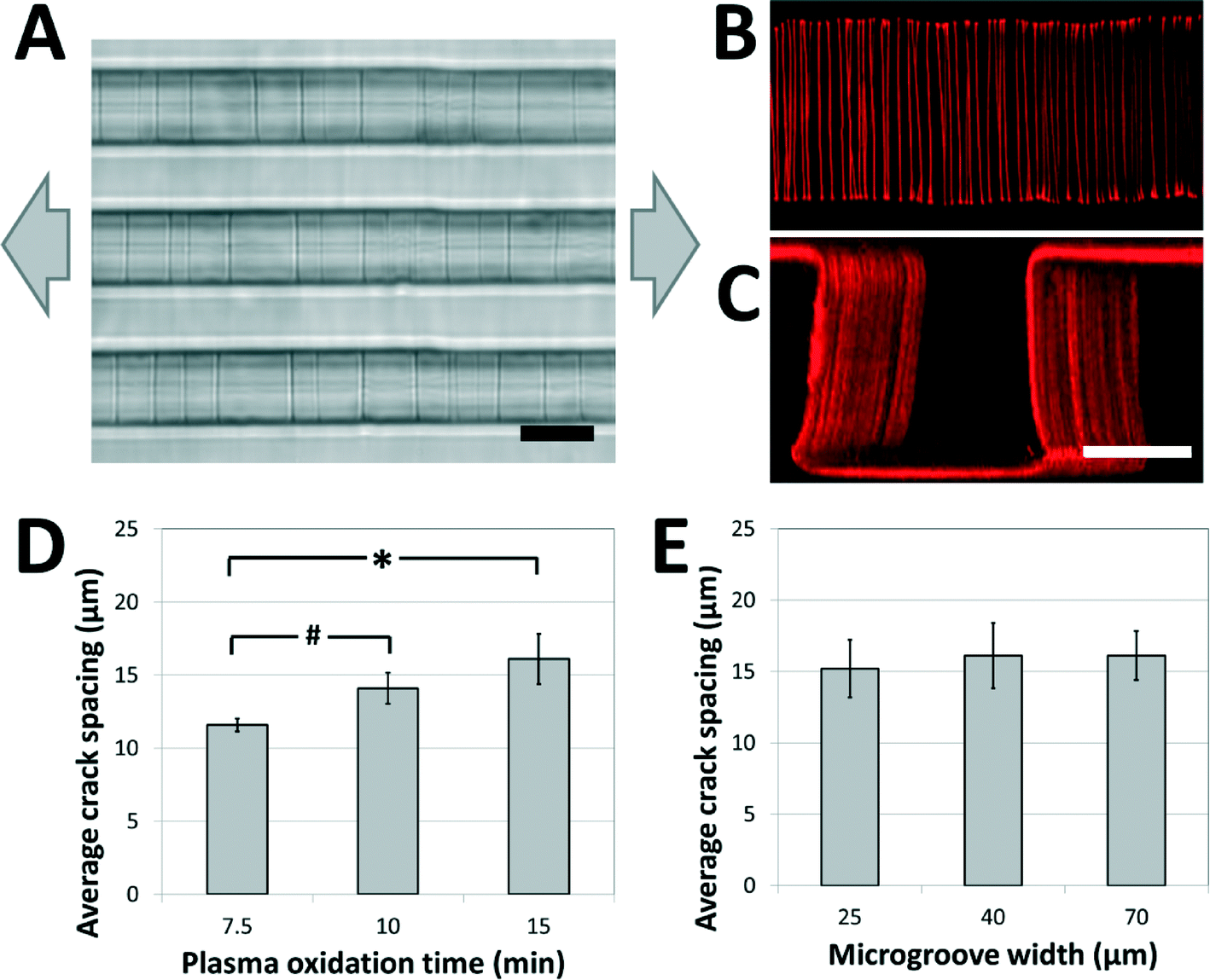 | ||
| Fig. 2 Crack structures fabricated on three-dimensional surfaces. (A) Representative bright-field image of cracks generated in topologically complex PDMS substrates (scale bar = 20 μm; arrows represent direction of applied strain). (B, C) Fluorescent visualization of candidate matrix proteins (TRITC-BSA) adsorbed to crack structures in (B) the bottom of the microgroove (top view), and (C) across the surfaces of the microgroove (perspective view; scale bars = 30 μm). These images and the ESI† movies 1A and B demonstrate that cracks are continuous across the microgroove structure. (D) The crack spacing depends on plasma oxidation treatment times (# p < 0.05, * p < 0.01 by one-tailed t-test). (E) The crack spacing is relatively robust to changes in microgroove widths (no significant differences, p > 0.8). The applied strain in (D) and (E) was 5%. | ||
In order to investigate the range of adhesive patterns possible with this system, and to confirm that cracks generated on topologically complex substrates behave in a similar manner to cracks generated on flat surfaces,38 the effects of varying the processing parameters on crack features were characterized. Under applied strains of 5% the crack spacing on the microgrooved substrates increased with the thickness of the brittle oxidized layer, and could be manipulated between 10 and 20 μm by controlling the duration of plasma oxidation (Fig. 2D). Hence, the present system is best suited to recapitulate biomaterials having fibers spaced within this range, such as electrospun scaffolds of nanofibrous materials. Increasing the protein spacing beyond this range using plasma oxidation is challenging, as the thicker brittle layer required for more widely spaced cracks is more prone to uncontrolled fracture caused by sample heating and the mismatch between the thermal coefficients of PDMS and oxidized PDMS.39 A smaller protein spacing can be achieved by replacing the Sylgard 184 PDMS used in these studies with a silicone material that has a greater ultimate tensile strain, such as those of the Silastic™ range of products, and applying larger deformations to generate a higher density of cracks. The applied tension can then be reduced to control the width of the fibers while maintaining the density.
Finally, it was observed that the crack spacing was independent of the lateral microgroove dimensions used in this study. Specifically, for microgroove heights of 30 μm, the spacing was independent of microgroove width (Fig. 2E), demonstrating that spacing between adhesive sites and topology of the fibers could be independently manipulated for carefully controlled biological experiments. Although this initial work demonstrates the fabrication of fiber-like patterns on microgrooved substrates, these findings suggest that the technique can be applied to more complex substrate microfeatures (sawteeth, overhangs, curves, etc.), and thus enables high-resolution, inexpensive and accessible adhesive patterning on a variety of topologically complex samples.
Control of cell shape in 3D protein matrices
Fibronectin adsorbed to the crack structures enables the adhesion of cells to these fiber-like patterns. In this work, we used C2C12 myoblasts and NIH 3T3 fibroblasts as model cell lines, and demonstrated that cells develop morphologies aligned with the crack direction.20,34 When the cracks were patterned perpendicular to the long axis of the microgrooves, cells spread across the width of the microgrooves, and adhered to the patterned structures on the side walls of the groove. As demonstrated in Fig. 3A, C2C12 cells cultured on fiber-like patterns in narrow grooves of 25 to 70 μm were limited in their ability to elongate, and hence spanned the microgrooves from wall to wall, while cells cultured on fiber-like patterns on flat surfaces were free to elongate along the crack pattern to a greater degree. We expect that the largest microgroove width across which cells can span is dependent on cell type, cell size and adhesive characteristics. In cases where the cells spread across the microgroove, confocal microscopy indicated that cells had attached to the side walls along the adhesive lines (Fig. 3B, ESI† movie 2), demonstrating the ability to micropattern adhesive structures in three dimensions. When cells in this configuration adhered to a single fiber-like pattern, they typically adopted a “boat-like” morphology, with a sharp prow and stern, and a bulging middle owing to the relatively stiff structure of the nucleus.40,41 Similar observations were made for NIH 3T3 cells. | ||
| Fig. 3 Cell culture on fracture-patterned topologically complex surfaces. (A) C2C12 myoblast cells were used as a model cell line and cultured on crack-generated adhesive structures traversing microgrooves of width 25 and 70 μm. For comparison purposes, adhesive cracks are generated on flat surfaces, and cells are allowed to spread on these patterns. When cultured in patterned microgrooves of small enough widths, cells do not spread to their maximum possible lengths. (B) Fluorescent confocal imaging was used to reconstruct a side-view of a cultured cell in a patterned 40 μm microgroove (green = actin; blue = nucleus; red = fibronectin matrix protein). These images and ESI† movie 2 demonstrate that cells adhere to the topologically complex linear adhesive patterns, and remain suspended across the width of the groove, indicating that cell shape can be controlled in 3D (scale bars = 20 μm). This technique can be applied to a broad variety of adhesive cells, and similar morphologies were observed when culturing NIH 3T3 fibroblasts on these patterns. | ||
Not surprisingly, the characteristics of the adhesive patterns presented by the cracks play a critical role in directing cell morphology. The width of the adhesive crack is an important parameter that dictates cell spreading,34 and can be controlled by reducing the strain after the cracks have been formed.34Fig. 4A shows how the crack width varies at increasing crack generation strains. For this system under these conditions, the crack width was significantly reduced at strains lower than about 5%; at higher strains, increases in crack density (Fig. 4B) allow crack width to remain relatively constant. On the narrower cracks, a majority of cells adopted a circular morphology, similar to morphologies of cells in suspension (Fig. 4C). On cracks that were wide enough to support cell spreading, most cells adhere to a single crack and elongate along the crack direction to span the microgroove (Fig. 4D). The switch between elongated and circular morphologies is similar to previous observations when varying crack widths on flat surfaces.34 Crack density also plays a significant role in cell morphology. The crack spacing decreased with increasing strain used to generate them (Fig. 4B), as expected for these systems. At still greater strains, the crack spacing was reduced such that the majority of cells spread across multiple adhesive cracks (Fig. 4E). The fibroblasts project filopodia along the adhesive cracks, and do not span the microgroove structure. Hence, the width of individual fiber-like patterns and the spacing between these patterns can be controlled to prompt distinct morphological responses from cultured cells.
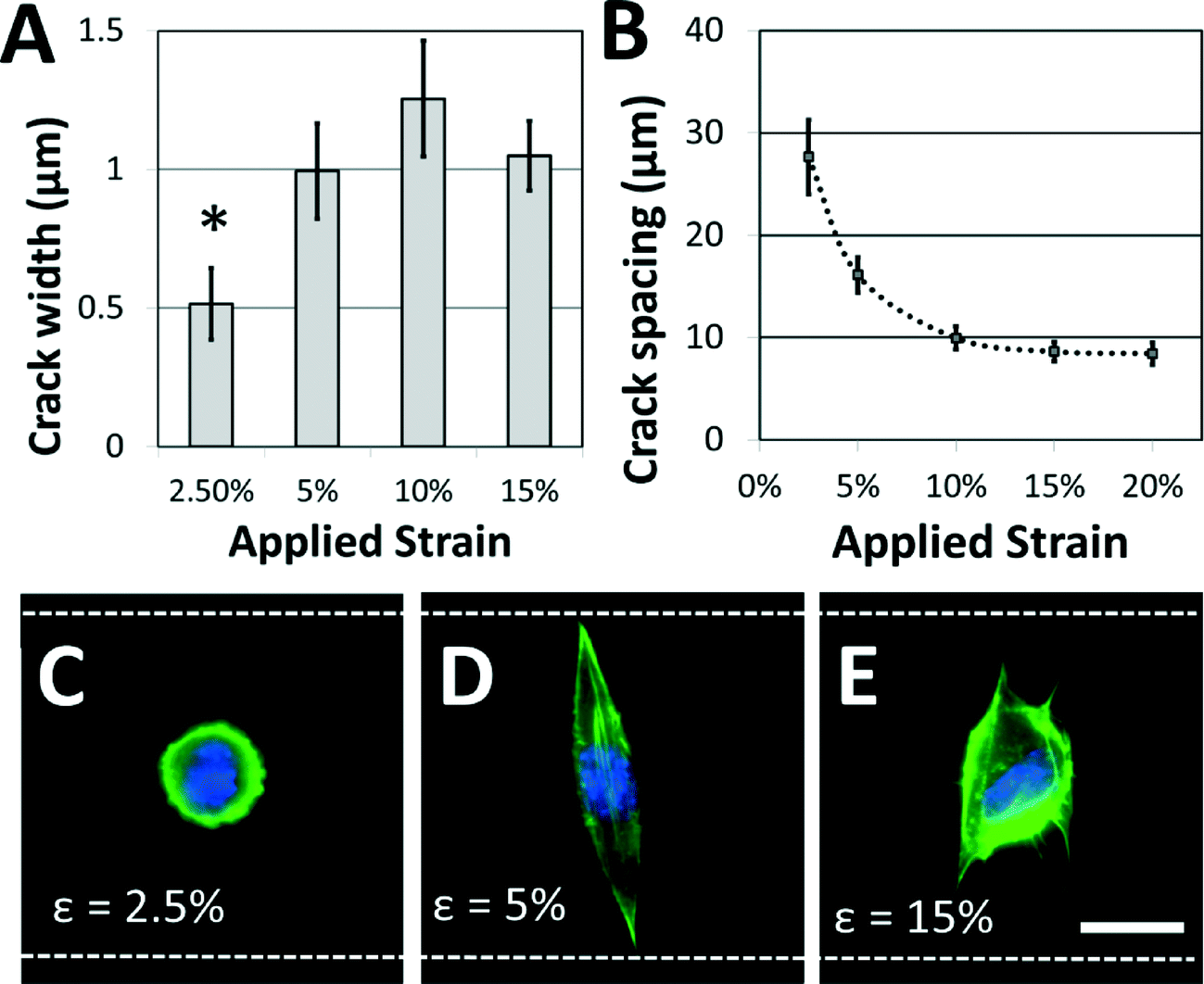 | ||
| Fig. 4 Adhesive fiber-like pattern dimensions and spacing influence cell shape. (A) Crack widths vary with applied strain (* p < 0.01 compared to all other conditions) while (B) crack spacing decreases with applied strain. These dimensions play a critical role in dictating cell morphology. (C) If the applied strain is low, the adhesive patterns are too narrow to support cell spreading, and a majority of NIH 3T3 cells retain a rounded morphology. (D) At increased strains, adhesive pattern width is sufficient to support spreading, and a majority of cells assume an elongated morphology that traverses the microgroove. (E) At still higher strains, crack density is increased and cells tend to span multiple adhesive lines. Filopodia are typically observed along adhesive lines as the cell spreads. (green = actin; blue = nucleus; white dotted lines = edges of the microgroove PDMS structures; scale bar = 15 μm). | ||
While the crack widths can be varied by changing the applied strain, an inherent limitation in this system is the relatively wide range of crack widths (typically ±200 nm) associated with a given strain (Fig. 4A). This is likely caused by variations in the crack spacing, which affects the crack width.38 It is possible that this issue could be addressed by carefully controlling the crack position using micropatterned notch-shaped crack-initiating features, as has previously been developed by our lab.38,42
3D adhesive matrix architecture directs nuclear morphology
As a first use of this fracture-based patterning technique, we applied it to study the effects of fiber structure on nuclear morphology. Nuclear morphology is regulated by the mechanics of the microenvironment, and by the morphology of spread cells.43–45 Nuclear deformation can play a significant role in modulating cell function, particularly in the context of disease mechanisms.46–48 It could, therefore, be an important comparative parameter in assessing the relevance of in vitro model systems to in vivo studies. In previous studies of nuclear morphology in a 3D fibrous environment, Nathan et al. generated electrospun nanofibrous poly(ε-caprolactone) biomaterial scaffolds, and demonstrated that prolonged application of mechanical strain causes a distinct elongation of the nuclei of encapsulated human mesenchymal stem cells.49 It is likely that the strain increases the average length of aligned fiber bundles in the nanofibrous matrix, enabling cells to spread along longer fibers without the interference of non-aligned fibers. It may also affect the matrix by changing the spacing between the fibers, or by anisotropically stiffening the matrix by local alignment of the fibers. It is also possible that the strain may directly influence cells by mechanotransduction effects. By generating precisely-patterned fiber-like adhesive sites we can create a model of the 3D adhesive environment presented by electrospun nanofibrous biomaterials, allowing us to explore the independent role of the adhesive-matrix architecture on nuclear morphology, while controlling for other parameters that may confound the interpretation of experimental results.Microgrooves decorated with crack-generated adhesive structures present well-aligned fibers along the bottom and side walls. In the general 3-D context of fibrous matrices, we define the ‘coherent length’ as the characteristic distance along which a cell may attach, without meeting an intersecting fiber. In the model 3-D environment presented in this work, the ‘coherent length’ can be considered to be the width of the microgrooves along which an adhesive line has been patterned. It can be controlled by varying the geometry of the microgrooves.
It was observed that cells attached to the adhesive lines, and then spread along them within the confines of the microgrooves. In the experiments described here, NIH 3T3 cells are used as a model mesenchymal-like cell. Adhesive patterns in wider grooves have greater coherent length, and NIH 3T3 cells cultured on these patterns of greater coherent length exhibit significant increases in nuclear length (Fig. 5A, B). The nuclear length of cells cultured in the 70 μm wide channels was comparable to the nuclear length of the cells grown on 1-D adhesive lines formed on a flat substrate. This indicates that 70 μm is about the limit in coherent length for which this cell type may undergo increases in nuclear deformation. At shorter coherent lengths, there are morphological effects caused by adhesion to the intersecting adhesive fiber-like patterns on the microgroove side-walls. These effects are reduced as the microgrooves grow wider, and the aspect ratio of the cell approaches that of a cell cultured on a simple 1-D linear pattern on a flat surface. Once the microgroove is sufficiently wide, the cell is no longer able to span the width of the groove, and the cell only experiences the ‘1-D’ component of the micropattern.
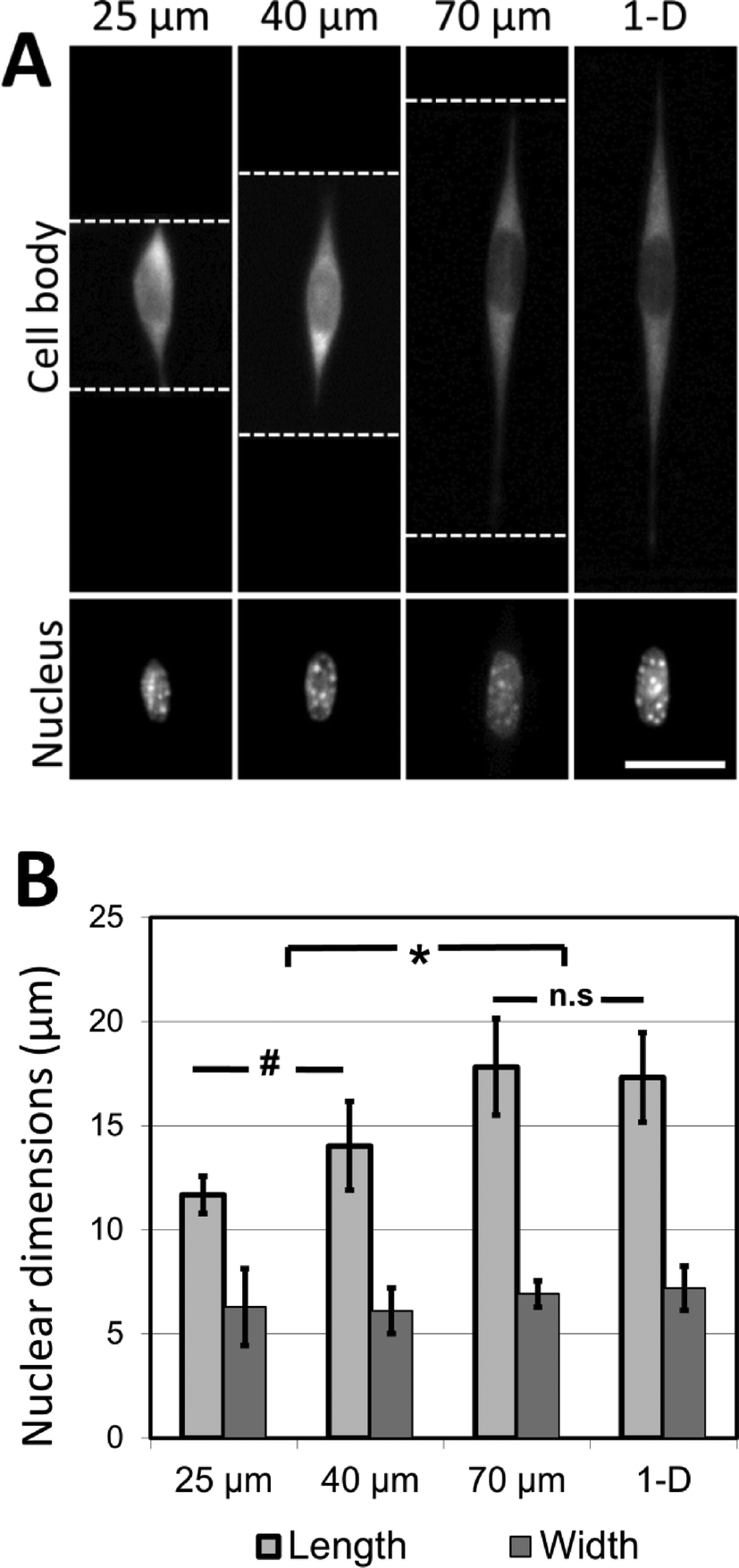 | ||
| Fig. 5 Cell shape directs nuclear morphology in three-dimensional linear adhesive patterns. (A) NIH 3T3 cells are cultured on adhesive fiber-like patterns that either traverse microfabricated grooves 25, 40 and 70 μm wide or exist on flat surfaces (thereby forming linear 1-D patterns). In each case, cells follow the pattern dictated by the crack-generated adhesive patterns (white dotted lines = edge of the micropatterned grooves; scale bar = 15 μm). (B) Nuclear length and width are quantified and demonstrate an increase in nuclear length along the direction of cell spreading (* p < 0.001, # p < 0.1, n.s p > 0.99; results reported as means ± standard deviation; n = 33–60, experiment repeated 3 times). Cells cultured on crack-generated adhesive patterns spanning grooves greater than 70 μm in width do not present nuclear lengths significantly different from cells cultured on crack-generated adhesive patterns on flat surfaces. No significant differences in nuclear width across any of the culture conditions are observed (p > 0.9). | ||
These findings strongly indicate that the coherent lengths of 3D fiber patterns in biomaterial scaffolds are responsible for the nuclear elongation effects observed by other research groups. Hence, the role of mechanical forces on nuclear shape in the 3D biomaterial is most likely caused by the mechanical restructuring of the 3D matrix, rather than through a cell mechanotransduction pathway activated by the direct application of external force. The changes in nuclear shape presented here are also consistent with a model relating nuclear deformation and cell morphology developed by Versaevel et al.,50 who demonstrate that the nuclei of cells cultured on adhesive micropatterns (fabricated by microcontact printing on flat surfaces) deform in proportion to the aspect ratio of the patterned cell. This deformation is mediated by actin fibers which tether the nucleus to the sites of adhesion between the cells and the supporting adhesive substrate. Our results demonstrate that a similar trend of nuclear deformation occurs when the sites of adhesion are no longer restricted to one plane, as the cells are able to attach to the patterned sidewalls of the microgrooves while maintaining extended aspect ratios. More broadly, the experiments presented here demonstrate the applicability of this micropatterning approach in dissecting the physical effects of complex 3D fibrous environments on cell morphology.
3D adhesive matrix architecture directs actin cytoskeletal structure
To further investigate the role of 3D fiber-like adhesive patterns on the cellular cytoskeleton, we examined the structure of the cytoskeletal actin network in NIH 3T3 fibroblasts after one day of culture in the crack-patterned microgrooves. In order to quantify our findings, we classified actin cytoskeletal (CSK) phenotypes into three categories: diffuse, stage 1 and stage 2. No distinct actin fibers were observable in diffuse CSK phenotypes. Actin stress fibers were observed around the edges of the cell in stage 1 phenotypes. Fibers were observed around the edges and in other regions of the cell in stage 2 phenotypes. Representative images of these phenotypes are shown in Fig. 6A. Fibroblasts cultured in the patterned microgrooves displayed distinct differences in actin CSK phenotypic distributions based on the coherent length of the fiber patterns. In all cases, stage 1 actin CSK structures were the most frequent. However, the prevalence of diffuse CSK structures significantly decreased and the prevalence of stage 2 structures significantly increased for cells cultured on fibrous patterns of greater coherent length (Fig. 6B). This is consistent with the well-established finding that actin stress fibers form when cells are well-spread and under endogenously generated mechanical tension.51 These results are consistent with the notion that the structure and orientation of adhesive fibers within a fibrous biomaterial matrix has a significant effect in dictating the formation of stress fibers, which in turn plays a critical role in disease-related mechanisms such as differentiation of fibroblasts towards fibrotic phenotypes.52,53 Hence, this micropatterning approach may provide insight into critical microstructural design considerations for tissue engineered scaffolds, in addition to addressing more fundamental biological questions.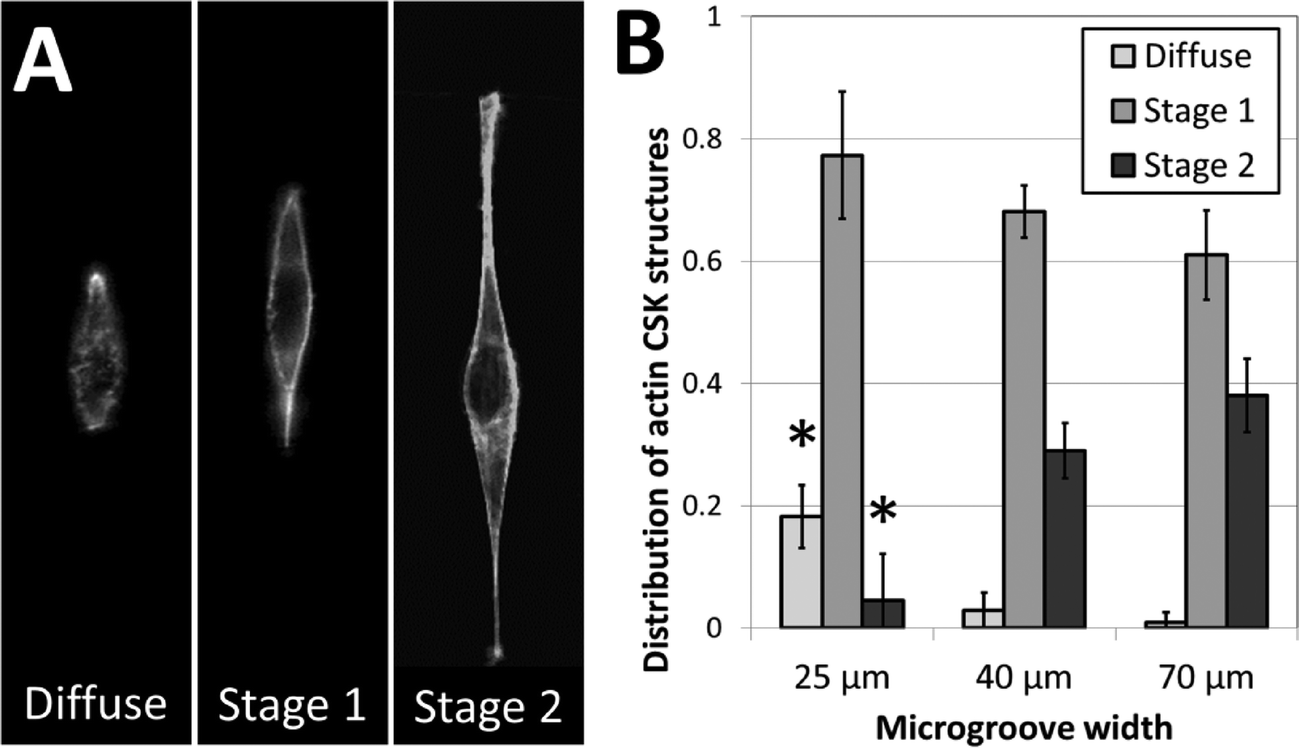 | ||
| Fig. 6 Actin fiber structure in NIH 3T3 cells varies based on 3D cell morphology. (A) Representative images of the three categories into which actin fiber structures were classified. High-resolution imaging reveals that actin phenotypes were either (i) diffuse, (ii) formed fibers along the perimeter of the cell only (stage 1), or (iii) formed fibers along the perimeter and within the cell body (stage 2). (B) The fraction of cell populations displaying these phenotypes varies based on the width of the micropatterned microgroove. The occurrence of diffuse actin phenotypes decreases with increasing microgroove width, while the occurrence of stage 2 actin phenotypes increases with increasing microgroove width (* p < 0.01, as compared to all other fractions of the same phenotype; n = 3; 35–60 cells per sample). | ||
Current limitations and future directions
Several limitations exist in applying this technique to the study of 3D cell-biomaterial interactions. First, this method of generating adhesive micropatterns in 3D is inherently limited in that adhesive patterns only form in connected lines, and hence cannot be considered a ‘complete’ 3D patterning technology in which arbitrary adhesive patterns can be generated in a three-dimensional space. While the system may be used in conjunction with other techniques such as microcontact printing to provide complete control over adhesion sites in 3D, there are alternative (albeit more complex) strategies to completely pattern a 3D environment. The merit of our crack-based patterning approach is in the simplicity of the system: since many biological questions require culture on fibrous mesh-like structures, we believe that the ability to rapidly and easily generate fiber-like adhesive patterns can be an important micropatterning technology.Second, while the system is well-suited to simulating the adhesive environment presented by fibrous mesh structures, it is currently unable to adequately simulate the stiffness and porosity presented in 3D matrices. The PDMS we have used was typically several orders of magnitude stiffer than fibrous biomaterials, and it is known that environmental stiffness plays a key role in directing cell function.6,12,54 Compared with fibrous mesh structures made from collagen, fibrin, or polymer fibers, the 3D patterned substrates generated with our method are mechanically stiffer, but have the advantage of providing well-defined geometric presentation of ECM at cellular length scales. However, it is expected that the stiffness could be reduced to a more appropriate physiological level by using this fracture-based technique with alternative substrate materials such as Sylgard 527 gel55 or UV-modified PDMS.26
Third, local curvatures in individual biomaterial nanofibers may also play a role in driving cell function,56 and microgrooves patterned using conventional soft lithography are unable to provide these curvatures. Fortunately, the described crack patterning process is compatible with non-planar substrates57 including curved surfaces, which can be fabricated using a variety of methodologies. Provided a conformal oxidized layer can be generated on the sample surface, by either chemical or physical means, the approach is adaptable to a wide range of geometric surface structures.
More broadly, the degree to which presenting the cell with 3D adhesive patterns prompts 3D-like cell functionality remains an open question. In addition to changing the adhesive patterns surrounding the cell, 3D environments alter a wide variety of other parameters known to influence cell function.58,59 For example, transport properties are significantly different in 3D culture systems,60,61 as are ligand presentation62 and the transfer of intrinsic12 and applied mechanical forces.63 Although we focused primarily on the 3D adhesive environment in this work, each of these other features may play a role in prompting 3D functionality. Furthermore, while the partial encapsulation of cells in fiber-like adhesive patterns may be sufficient to prompt 3D cell functionality, the extent to which cells need to be encapsulated to replicate 3D functions is currently unknown. The system described in this work is amenable to altering the aspect ratio of the microgroove structures, by changing the thickness of the fabricated SU-8 mold. Although all the experiments described in this work were conducted at a fixed microgroove depth, systematically varying the cavity aspect ratio may be of importance in understanding higher order cell functions such as differentiation or apoptosis.62,64 Understanding the nature of dimensionality in regulating cell function is of critical importance in designing physiologically relevant 3D high-throughput screening systems, and the techniques presented in this work to design and manipulate fibrous 3D adhesive environments may be of significance in addressing these issues.
Conclusions
Crack-based patterning is a viable approach to produce topologically complex patterned adhesive environments for cell culture. Continuous ‘fiber-like’ adhesive patterns can be generated on all surfaces of a microfabricated substrate, and fiber features including spacing, width and topology can be manipulated by appropriately designing the substrate dimensions or by varying the fabrication process conditions. Cell morphology is dictated by pattern width and density, and cells can adhere to the patterned walls of the culture substrate. We have used this technology to understand the decoupled effects of physical structure in nanofibrous biomaterials on encapsulated cells. We have also demonstrated that nuclear morphology is predictively regulated by cohesive fiber length in three-dimensional matrix architectures, and that actin CSK development is strongly influenced by the structure of the supporting matrix. The technique that has been presented enables high-resolution adhesive micropatterning in topologically complex substrates, a capability that is challenging to achieve in a high-throughput manner without highly specialized equipment and expertise. More broadly, this approach may prove useful in dissecting 3D environments to understand how complex microenvironmental structures regulate cell function.Acknowledgements
We gratefully acknowledge support from the Natural Sciences and Engineering Research Council of Canada, and from the Banting postdoctoral fellowship programs to CM. Project support was provided by the NIH (EB003793 & HG004653) and the Biointerfaces Institute.References
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- E. W. Gomez, Q. K. Chen, N. Gjorevski and C. M. Nelson, J. Cell. Biochem., 2010, 110, 44–51 CAS.
- R. A. Desai, S. B. Gopal, S. Chen and C. S. Chen, J. R. Soc., Interface, 2013, 10, 20130717 CrossRef PubMed.
- M. Thery, A. Jimenez-Dalmaroni, V. Racine, M. Bornens and F. Julicher, Nature, 2007, 447, 493–496 CrossRef CAS PubMed.
- C. Moraes, Y. Sun and C. A. Simmons, Integr. Biol., 2011, 3, 959–971 RSC.
- S. Raghavan and C. S. Chen, Adv. Mater., 2004, 16, 1303–1313 CrossRef CAS.
- M. Théry, J. Cell Sci., 2010, 123, 4201–4213 CrossRef PubMed.
- E. Cukierman, Science, 2001, 294, 1708–1712 CrossRef CAS PubMed.
- K. S. M. Smalley, M. Lioni and M. Herlyn, In Vitro Cell. Dev. Biol.: Anim., 2006, 42, 242 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Raynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- J. B. Kim, R. Stein and M. J. O'Hare, Breast Cancer Res. Treat., 2004, 85, 281–291 CrossRef.
- E. Cukierman, R. Pankov and K. M. Yamada, Curr. Opin. Cell Biol., 2002, 14, 633–640 CrossRef CAS.
- A. D. Doyle, F. W. Wang, K. Matsumoto and K. M. Yamada, J. Cell Biol., 2009, 184, 481 CrossRef CAS PubMed.
- T. P. Kraehenbuehl, R. Langer and L. S. Ferreira, Nat. Methods, 2011, 8, 731–736 CrossRef CAS PubMed.
- F. Pampaloni and E. H. K. Stelzer, Biotechnol. Genet. Eng. Rev., 2009, 26, 117–137 CrossRef.
- N. T. Elliott and F. Yuan, J. Pharm. Sci., 2011, 100, 59–74 CrossRef CAS PubMed.
- C. M. Kraning-Rush, S. P. Carey, J. P. Califano, B. N. Smith and C. A. Reinhart-King, Phys. Biol., 2011, 8, 015009 CrossRef PubMed.
- A. R. Dixon, C. Moraes, M. E. Csete, M. D. Thouless, M. A. Philbert and S. Takayama, J. Biomed. Mater. Res., Part A, 2013 DOI:10.1002/jbm.a.34814.
- W. C. Ruder, E. D. Pratt, S. Bakhru, M. Sitti, S. Zappe, C.-M. Cheng, J. F. Antaki and P. R. LeDuc, Lab Chip, 2012, 12, 1775 RSC.
- W. C. Ruder, E. D. Pratt, N. Z. D. Brandy, D. A. LaVan, P. R. LeDuc and J. F. Antaki, Sci. Rep., 2012, 2, 554 Search PubMed.
- S. A. Ruiz and C. S. Chen, Soft Matter, 2007, 3, 168–177 RSC.
- J. E. Petrzelka and D. E. Hardt, J. Micromech. Microeng., 2012, 22, 075015 CrossRef.
- H.-Y. Chen, J.-M. Rouillard, E. Gulari and J. Lahann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11173–11178 CrossRef CAS PubMed.
- Y. Sun, L.-T. Jiang, R. Okada and J. Fu, Langmuir, 2012, 28, 10789–10796 CrossRef CAS PubMed.
- A. M. Greiner, B. Richter and M. Bastmeyer, Macromol. Biosci., 2012, 12, 1301–1314 CrossRef CAS PubMed.
- D. S. Ginger, H. Zhang and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 30–45 CrossRef PubMed.
- B. C. Kim, C. Moraes, J. Huang, M. D. Thouless and S. Takayama, Biomater. Sci., 2014, 2, 288 RSC.
- D. Huh, K. L. Mills, X. Zhu, M. A. Burns, M. D. Thouless and S. Takayama, Nat. Mater., 2007, 6, 424–428 CrossRef CAS PubMed.
- T. Matsuoka, B. C. Kim, J. Huang, N. J. Douville, M. D. Thouless and S. Takayama, Nano Lett., 2012, 12, 6480–6484 CrossRef CAS PubMed.
- T. Matsuoka, B. Choul Kim, C. Moraes, M. Han and S. Takayama, Biomicrofluidics, 2013, 7, 041301 CrossRef PubMed.
- H.-N. Kim, S.-H. Lee and K.-Y. Suh, Lab Chip, 2011, 11, 717–722 RSC.
- X. Zhu, K. L. Mills, P. R. Peters, J. H. Bahng, E. H. Liu, J. Shim, K. Naruse, M. E. Csete, M. D. Thouless and S. Takayama, Nat. Mater., 2005, 4, 403–406 CrossRef CAS PubMed.
- S. Oh, K. S. Brammer, Y. S. J. Li, D. Teng, A. J. Engler, S. Chien and S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2130–2135 CrossRef CAS PubMed.
- O. F. Zouani, C. Chanseau, B. Brouillaud, R. Bareille, F. Deliane, M.-P. Foulc, A. Mehdi and M.-C. Durrieu, J. Cell Sci., 2012, 125, 1217–1224 CrossRef CAS PubMed.
- D.-H. Kim, E. A. Lipke, P. Kim, R. Cheong, S. Thompson, M. Delannoy, K.-Y. Suh, L. Tung and A. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2009, 107, 565–570 CrossRef PubMed.
- J. Huang, B. C. Kim, S. Takayama and M. D. Thouless, J. Mater. Sci., 2014, 49, 255–268 CrossRef CAS.
- W. W. Tooley, S. Feghhi, S. J. Han, J. Wang and N. J. Sniadecki, J. Micromech. Microeng., 2011, 21, 054013 CrossRef.
- K. N. Dahl, A. J. Engler, J. D. Pajerowski and D. E. Discher, Biophys. J., 2005, 89, 2855–2864 CrossRef CAS PubMed.
- K. N. Dahl, A. J. S. Ribeiro and J. Lammerding, Circ. Res., 2008, 102, 1307–1318 CrossRef CAS PubMed.
- B. C. Kim, T. Matsuoka, C. Moraes, J. Huang, M. D. Thouless and S. Takayama, Sci. Rep., 2013, 3, 3027 Search PubMed.
- S. B. Khatau, C. M. Hale, P. J. Stewart-Hutchinson, M. S. Patel, C. L. Stewart, P. C. Searson, D. Hodzic and D. Wirtz, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19017–19022 CrossRef CAS PubMed.
- K. N. Dahl and A. Kalinowski, J. Cell Sci., 2011, 124, 675–678 CrossRef CAS PubMed.
- Z. Pan, C. Yan, R. Peng, Y. Zhao, Y. He and J. Ding, Biomaterials, 2012, 33, 1730–1735 CrossRef CAS PubMed.
- S. B. Khatau, D.-H. Kim, C. M. Hale, R. J. Bloom and D. Wirtz, Nucleus, 2010, 1, 337–342 CrossRef PubMed.
- M. Zwerger, C. Y. Ho and J. Lammerding, Annu. Rev. Biomed. Eng., 2011, 13, 397–428 CrossRef CAS PubMed.
- Y. Fu, L. K. Chin, T. Bourouina, A. Q. Liu and A. M. J. VanDongen, Lab Chip, 2012, 12, 3774–3778 RSC.
- A. S. Nathan, B. M. Baker, N. L. Nerurkar and R. L. Mauck, Acta Biomater., 2011, 7, 57–66 CrossRef CAS PubMed.
- M. Versaevel, T. Grevesse and S. Gabriele, Nat. Commun., 2012, 3, 671 CrossRef PubMed.
- D. E. Discher, P. Janmey and Y. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- B. Hinz, J. Biomech., 2010, 43, 146–55 CrossRef PubMed.
- C. Moraes, M. Likhitpanichkul, C. J. Lam, B. M. Beca, Y. Sun and C. A. Simmons, Integr. Biol., 2013, 5, 673–680 RSC.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- R. N. Palchesko, L. Zhang, Y. Sun and A. W. Feinberg, PLoS One, 2012, 7, e51499 CAS.
- C. Shen, Q. Meng and G. Zhang, Biotechnol. Bioeng., 2013, 110, 2173–2183 CrossRef CAS PubMed.
- T. Uchida, K. L. Mills, C.-H. Kuo, W. Roh, Y.-C. Tung, A. L. Garner, K. Koide, M. D. Thouless and S. Takayama, Langmuir, 2009, 25, 3102–3107 CrossRef CAS.
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CrossRef CAS PubMed.
- M. A. Schwartz and C. S. Chen, Science, 2013, 339, 402–404 CrossRef CAS PubMed.
- S. Raghavan, C. J. Shen, R. A. Desai, N. J. Sniadecki, C. M. Nelson and C. S. Chen, J. Cell Sci., 2010, 123, 2877–2883 CrossRef CAS PubMed.
- C. Moraes, A. B. Simon, A. J. Putnam and S. Takayama, Biomaterials, 2013, 34, 9623–9631 CrossRef CAS PubMed.
- J. Lee, A. A. Abdeen, D. Zhang and K. A. Kilian, Biomaterials, 2013, 34, 8140–8148 CrossRef CAS PubMed.
- C. Moraes, G. H. Wang, Y. Sun and C. A. Simmons, Biomaterials, 2010, 31, 577–584 CrossRef CAS PubMed.
- K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00122b |
| ‡ Currently at College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, PR China. |
| § Currently at Max Planck Institute for Intelligent Systems, Department of New Materials and Biosystems, Heisenbergstrasse 3, 70569 Stuttgart, Germany. |
| This journal is © The Royal Society of Chemistry 2014 |