A microdevice for rapid optical detection of magnetically captured rare blood pathogens
Ryan M.
Cooper
ab,
Daniel C.
Leslie
abc,
Karel
Domansky
a,
Abhishek
Jain
a,
Chong
Yung
ab,
Michael
Cho
a,
Sam
Workman
a,
Michael
Super
a and
Donald E.
Ingber
*abc
aWyss Institute for Biologically Inspired Engineering at Harvard University, 3 Blackfan Circle, CLSB 517, Boston, MA 02118, USA. E-mail: don.ingber@wyss.harvard.edu; Fax: +1 617 432 7068; Tel: +1 617 432 7044
bVascular Biology Program, Boston Children's Hospital and Harvard Medical School, Boston, MA 02115, USA
cHarvard School of Engineering and Applied Sciences, Cambridge, MA 02139, USA
First published on 3rd October 2013
Abstract
Sepsis diagnosis requires development of methods to identify rare pathogen cells in small samples of human blood. Magnetic beads functionalized with pathogen-binding ligands have been used to rapidly isolate microbes from blood; however, it is commonly difficult to optically detect the captured species because the excess numbers of beads required for pathogen binding physically interfere with light transmission after they have been concentrated. Here we describe a microdevice that uses microfluidics combined with optimized magnetic field concentrators and magnetic beads coated with a generic blood opsonin to efficiently capture unknown blood pathogens and spread them into a thin layer suitable for automated optical detection. Using this device, we have been able to detect fungal pathogens in less than three hours after sample collection compared to days with current technology, and with an extremely high sensitivity (<1 cell mL−1 of human blood).
Introduction
Sepsis is a systemic inflammatory reaction caused by the uncontrolled spread of pathogens through the body via the bloodstream. Survival rates decrease as much as 9% for every hour that correct treatment is delayed.1–3 Unfortunately, it can often take days to identify the pathogen using blood cultures, which remains the gold standard for clinical diagnosis. Molecular detection techniques (e.g., PCR) offer a faster alternative, but currently lack the sensitivity to replace blood cultures. This is especially true for fungi, whose tough cell walls make DNA extraction difficult.4,5 Sepsis diagnosis is a difficult challenge because the concentration of pathogens in the blood of septic patients is typically very low (1 to 100 cells mL−1), which makes direct detection difficult.6–10 We previously showed that living bacteria and fungi can be rapidly isolated from human blood samples flowing through microfluidic channels using magnetic beads coated with pathogen-specific antibodies in combination with applied magnetic field gradients.11,12 More recently, we developed a single ligand that when coated on magnetic beads makes them capable of binding a wide range of pathogenic bacteria and fungi.13 We therefore explored whether we could adapt this magnetic capture approach to develop a rapid method to isolate unknown pathogens from human blood to enable their rapid detection. Moreover, because this simple method offers a way to capture both living and dead pathogens, it also could potentially enable rapid antibiotic susceptibility testing in the future, which is not possible using molecular diagnostic methods.4,5Pathogen detector design
Our concept for a device to carry out magnetic isolation and optical detection of rare blood pathogens is to add ligand-coated magnetic beads to blood that bind specifically to pathogens, and to isolate the bound beads from the blood when flowing through a microfluidic channel using an external magnet to pull the beads up to the ceiling of the channel (Fig. 1a and b). In this manner, all of the magnetic beads and rare pathogens magnetically captured from a large fluid volume would be concentrated in a small area that should be well suited for optical detection. As the identity of the pathogen is unknown in patients presenting with sepsis, we could not use magnetic beads coated with specific antibodies for a meaningful clinical diagnostic. Instead, we coated magnetic beads (1 μm diameter, MyOne Dynabeads Streptavidin T1, Invitrogen) with a modified form of mannose binding lectin (MBL),13 which is a calcium-dependent lectin normally found in human blood that is able to recognize and bind terminal mannose and fucose residues that are expressed on the surface of over 90 different bacteria, fungi, protozoa and viruses, but not on mammalian cells.14–18 The genetically engineered version of MBL we used has similar binding properties, but it lacks domains that could promote blood coagulation and can be easily isolated with high efficiency.13 Microfluidic flow cell prototypes containing a single channel (3 mm wide × 35 mm long × 280 μm high) were fabricated from polydimethylsiloxane (PDMS) using standard soft lithography techniques. In initial studies using whole human blood spiked with fluorescently labelled C. albicans fungal pathogens (10–100 cells mL−1), we were able to reliably capture virtually all of the magnetic bead-bound pathogens from blood, which we verified by checking the outlet fluid for pathogens using both flow cytometry and quantitative plating (Fig. 1c). However, to obtain optimal binding, we needed to use over a thousand-fold excess of magnetic beads relative to pathogens. As a result, the magnetic field created by placing the magnet directly on top of the device caused the bound fluorescently labelled pathogens and excess magnetic beads to form dense piles that made it impossible to visualize the majority of the pathogens (Fig. 1c and d).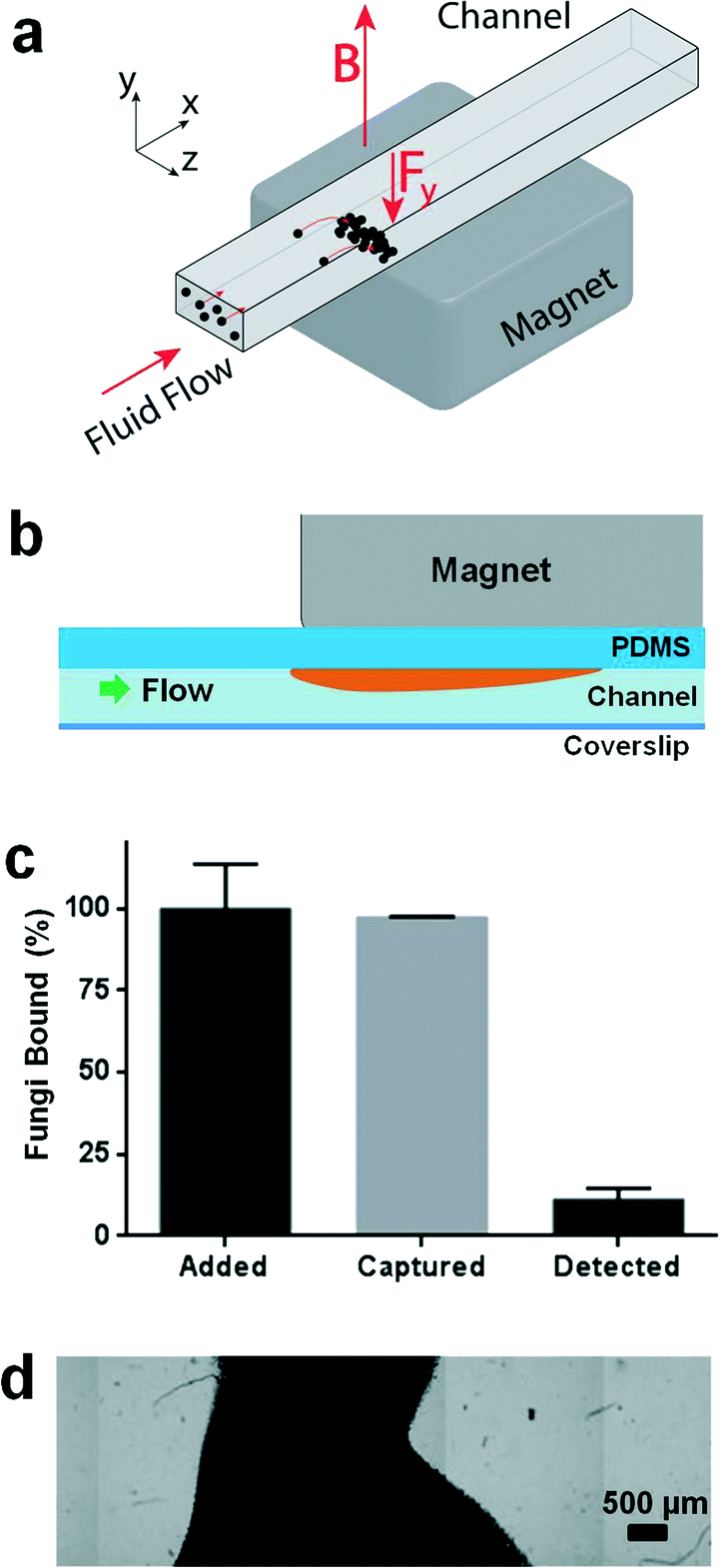 | ||
| Fig. 1 Limits of optical detection with captured magnetic beads. a) Permanent magnet on the outside of the device creates a magnetic field (B) that induces a force (Fy) on the magnetic bead and tagged pathogen, concentrating them for optical detection. b) Device cross section parallel to flow path showing beads piling beneath the magnet in the initial prototype. c) Mass balance study with initial prototype revealed that 98% of pathogens added to the sample could be captured, but only 13% were detected optically. d) Image composite showing a dense (optically black) pile of beads in the channel near the edge of the magnet, which buries most of the captured pathogens and makes optical detection impossible. | ||
To solve the excess bead problem, we modified the microdevice design to better balance the force of the applied magnetic field gradient on the beads with fluidic drag forces so that the collected beads could be spread thinly enough to visualize the pathogens using fluorescence microscopy (Fig. 2b). We fabricated a polysulfone holder to position a magnetic flux concentrator (MFC) directly beneath a 3/4′′ neodymium cube magnet (K&J Magnetics, grade N42 magnet, magnetization 875![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 352 A m−1) and placed it directly above the ceiling of a microfluidic channel with the same dimensions as the channel described above (Fig. 2a and b). The ceiling was made thin (300 μm) to minimize dissipation of the generated magnetic field. The shape of the MFC (5 mm wide × 1.55 mm thick × 25 mm long) was optimized in COMSOL using finite element modelling, and it was CNC-machined from a single bar of EFI alloy 79 (Ed Fagan, Inc.). The front end of the MFC was tapered to a wedge to decrease the initial magnetic field gradient spike produced by the magnet, and the remaining area of the MFC was covered by rows of parallel grooves (400 μm deep and 400 μm wide, with 800 μm center-to-center spacing) (Fig. 2c). The trailing edge of the MFC has a sharp, untapered edge to ensure capture of the magnetic beads with a strong magnetic field gradient spike. The MFC was designed to be wider than the channel width to produce an even magnetic field gradient over the entire channel width to reduce clumping on the sides of the channel. We tested a range of periodic groove sizes in the MFC and found that use of 800 μm wide grooves provided only a minimal advantage because there were relatively few places where gradient spikes formed along the length of the device and the beads still formed relatively dense piles. With the 200 μm spacing, we found that the bead capture efficiency was poor and finite element model simulations showed that the beneficial gradient spikes were smaller with the shallower grooves and dropped to low levels before penetrating the height of the channel. Thus, the 400 μm spacing was the best compromise to create an even bead distribution, while still ensuring bead capture in the channel.
352 A m−1) and placed it directly above the ceiling of a microfluidic channel with the same dimensions as the channel described above (Fig. 2a and b). The ceiling was made thin (300 μm) to minimize dissipation of the generated magnetic field. The shape of the MFC (5 mm wide × 1.55 mm thick × 25 mm long) was optimized in COMSOL using finite element modelling, and it was CNC-machined from a single bar of EFI alloy 79 (Ed Fagan, Inc.). The front end of the MFC was tapered to a wedge to decrease the initial magnetic field gradient spike produced by the magnet, and the remaining area of the MFC was covered by rows of parallel grooves (400 μm deep and 400 μm wide, with 800 μm center-to-center spacing) (Fig. 2c). The trailing edge of the MFC has a sharp, untapered edge to ensure capture of the magnetic beads with a strong magnetic field gradient spike. The MFC was designed to be wider than the channel width to produce an even magnetic field gradient over the entire channel width to reduce clumping on the sides of the channel. We tested a range of periodic groove sizes in the MFC and found that use of 800 μm wide grooves provided only a minimal advantage because there were relatively few places where gradient spikes formed along the length of the device and the beads still formed relatively dense piles. With the 200 μm spacing, we found that the bead capture efficiency was poor and finite element model simulations showed that the beneficial gradient spikes were smaller with the shallower grooves and dropped to low levels before penetrating the height of the channel. Thus, the 400 μm spacing was the best compromise to create an even bead distribution, while still ensuring bead capture in the channel.
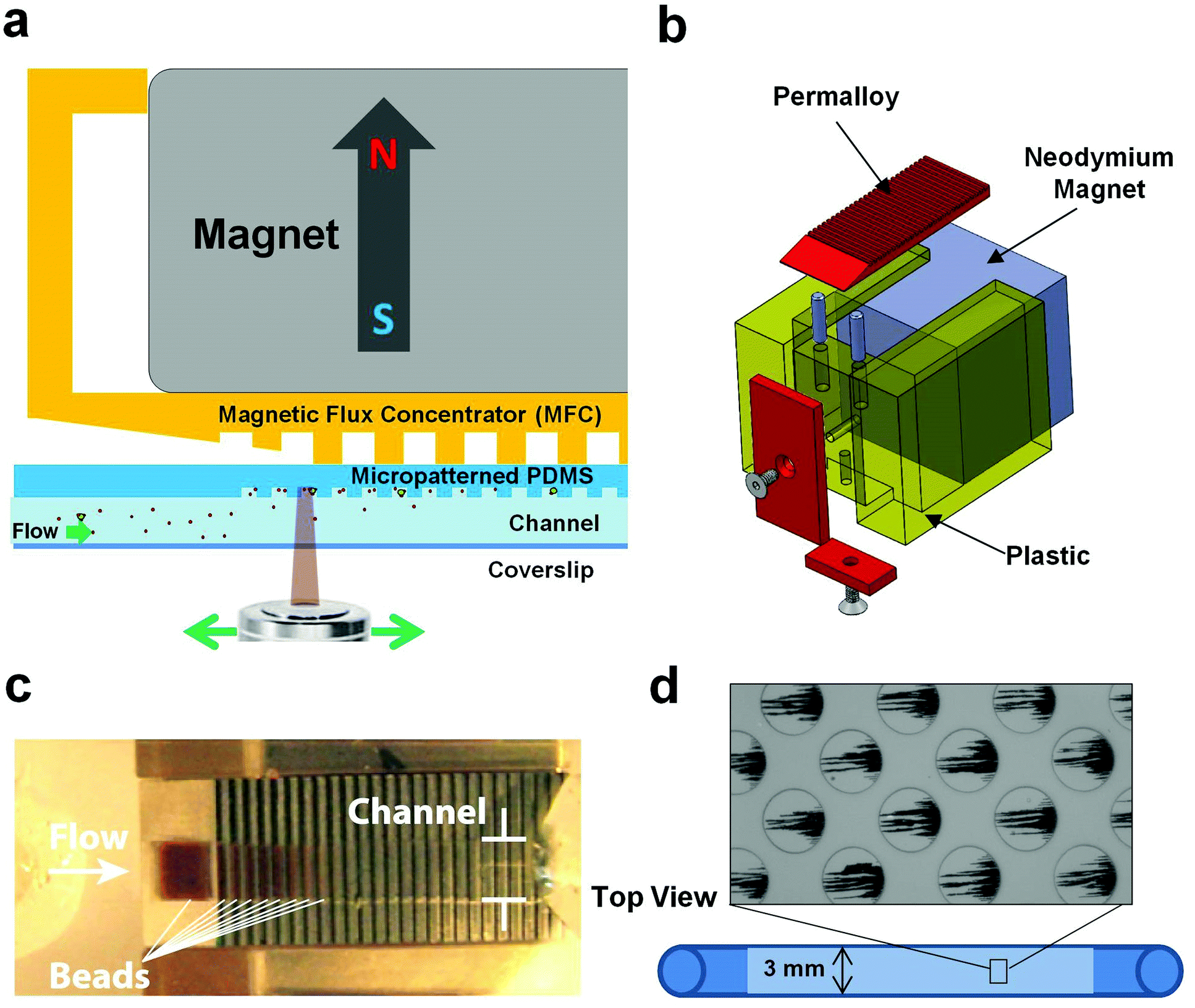 | ||
| Fig. 2 Design of the magnetic capture microfluidic device. a) Cross section of the entire device, showing orientation of the magnet, magnetic flux concentrator (MFC) and microfluidic channel (not to scale). b) MFC made out of permalloy (red), neodymium magnet (grey) and plastic casing (yellow), forming a single, simple to use unit for capturing and displaying magnetic beads and tagged pathogens. c) Photograph of beads captured in the device and spread into a thin layer by the presence of the MFC. d) Higher magnification view of the microfluidic channel showing aligned chains of magnetic beads that were magnetically captured in the microwells moulded into the ceiling of the PDMS microchannel. | ||
We also modified the internal surface of the middle 25 mm section of the microfluidic channel by micromolding a repeating array of closed packed, circular wells (50 μm diameter × 20 μm deep, spaced 100 μm apart with each subsequent row 50 μm offset from the other) (Fig. 2d). We tested a range of well depths (10 through 80 μm deep) and found that bead capture was the most reliable with the 20 μm deep wells. The MFC created places where the magnetic field lines diverged outwards periodically, resulting in production of high magnetic field gradient zones that promoted efficient bead capture and alignment within the circular microwells (Fig. 2d).
The magnetic force acting on the superparamagnetic beads is proportional to the local magnetic field gradient, so we produced a finite element model (COMSOL) of the magnetic field (∇B) in the various device configurations (magnet with or without MFC) (Fig. 3a). This analysis revealed that when the magnet is directly apposed to the channel without a MFC, it produces a large field gradient spike near its upstream and downstream edges. In contrast, the simulation predicted that the presence of a MFC with a tapered leading edge and a grooved washboard structure along its length will produce multiple smaller gradient spikes distributed along the entire length of the magnet, which should create a more uniform bead distribution (Fig. 3a).
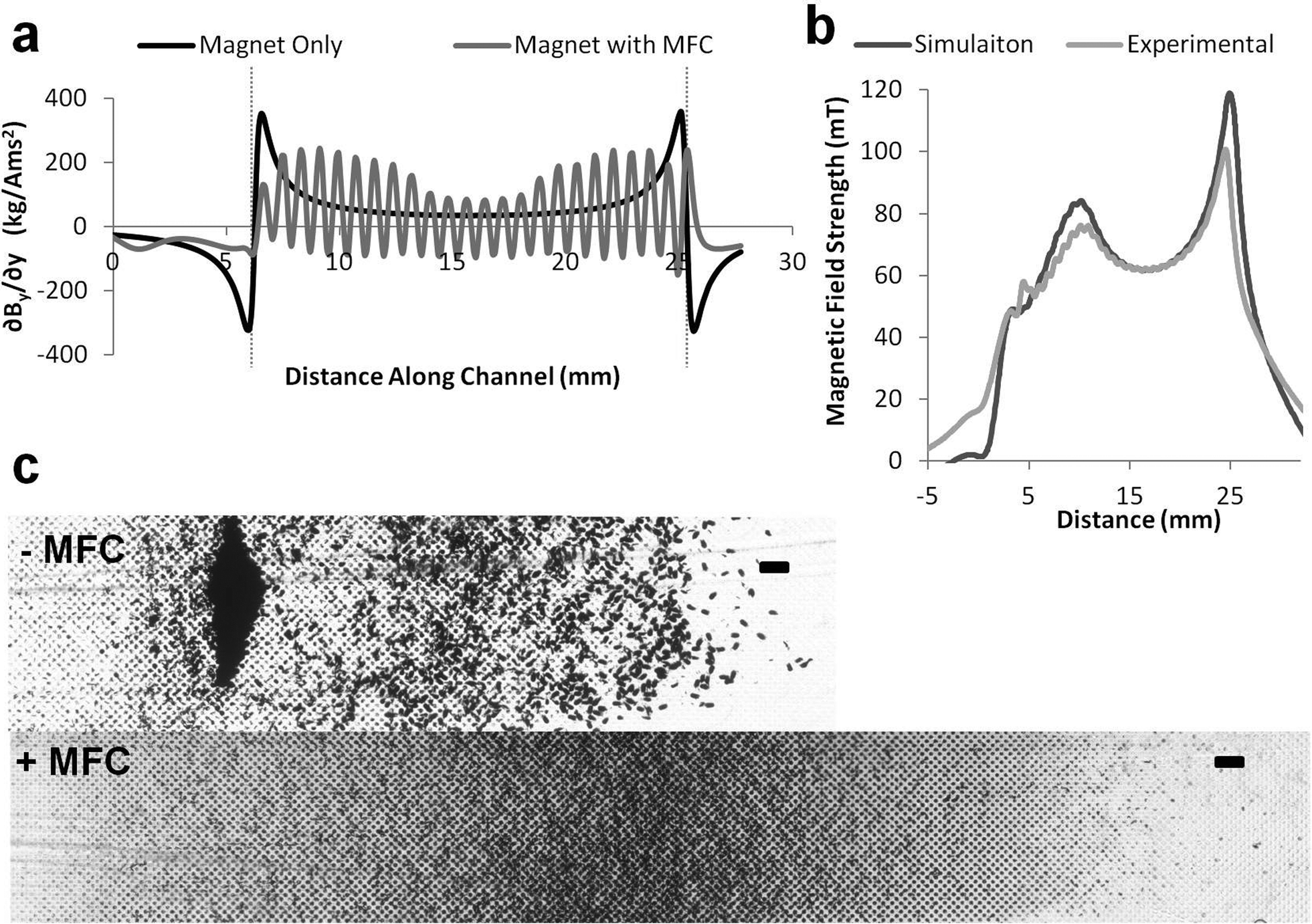 | ||
| Fig. 3 Characterization of the magnetic flux concentrator (MFC). a) Magnetic field gradient calculated in the microchannel in the presence of magnet alone (black) versus magnetic plus the MFC (gray). Note the presence of a large gradient spike with magnet alone that causes the beads to pile up at the upstream edge of the magnet, whereas there is much more even distribution of the magnetic gradient with the MFC. The peaks of the sinusoidal pattern correspond to the teeth of washboard in the MFC. kg A−1m−1s−2 is the SI unit for the magnetic field gradient (kilograms per amp meter second squared). b) Comparison of calculated and experimentally measured magnetic field with the MFC showing close similarity between the predictions and experimental results. c) Photomicrographs showing distribution of magnetically captured beads in the absence (−MFC) or presence (+MFC) of the magnetic flux concentrator. Only the first half of the channel is shown in each device to better highlight the areas where the majority of the magnetic beads were captured. Note that large number of beads piled up at the forward edge of the edge regardless of the micropatterned wells with the magnet alone, whereas there was much better spread of beads with the MFC in place (scale bar, 250 μm). | ||
To validate the simulation, we used a magnetometer (Bell 5080 gaussmeter with STD58-0404 transverse probe) to scan over the surface of the magnet with and without the MFC in place. These results revealed a close correlation between experimental measurements and predictions from the simulation (Fig. 3b). Furthermore, when we flowed magnetic beads through a microfluidic channel positioned in the device, a dense pile of beads formed at the upstream edge of the magnet when only the magnet was used, whereas there was much more uniform spread of the captured beads when the MFC was in place (Fig. 3c).
These results confirm that modulating the magnetic gradient can greatly improve the bead distribution in the capture chamber. More specifically, altering the magnetic field gradient distributions with the MFC allowed us to spread the beads within microwells distributed along the channel much more effectively than was possible with the magnet alone using the same channel dimensions (Fig. 2d and 3c). By experimenting with different channel sizes, we determined that a channel 280 μm high by 3 mm wide produced optimal spreading of the magnetic beads and tagged pathogens in a thin layer, while allowing us to operate at a flow rate of 10 ml h−1 with minimal variation across the width of the channel.
Optical detection of isolated pathogens
By using the MFC to spread the beads out more evenly in the channel, we were able to place all the pathogens in a single focal plane, greatly facilitating automated optical scanning of the captured pathogens. Importantly, this enabled us to optically detect rare fluorescently labelled pathogens among a large excess of unbound beads, while still ensuring removal of more than 99% of the magnetic particles from the flow. For staining of the magnetically captured fungi, we employed 25 μM calcofluor white that stains fungal cell walls brightly and is easy to detect when cells are captured in individual wells on the upper ceiling of the channel (Fig. 4a). Similar results were obtained in control studies using GFP-labelled pathogens or carrying out immunofluorescence staining with pathogen-specific antibodies.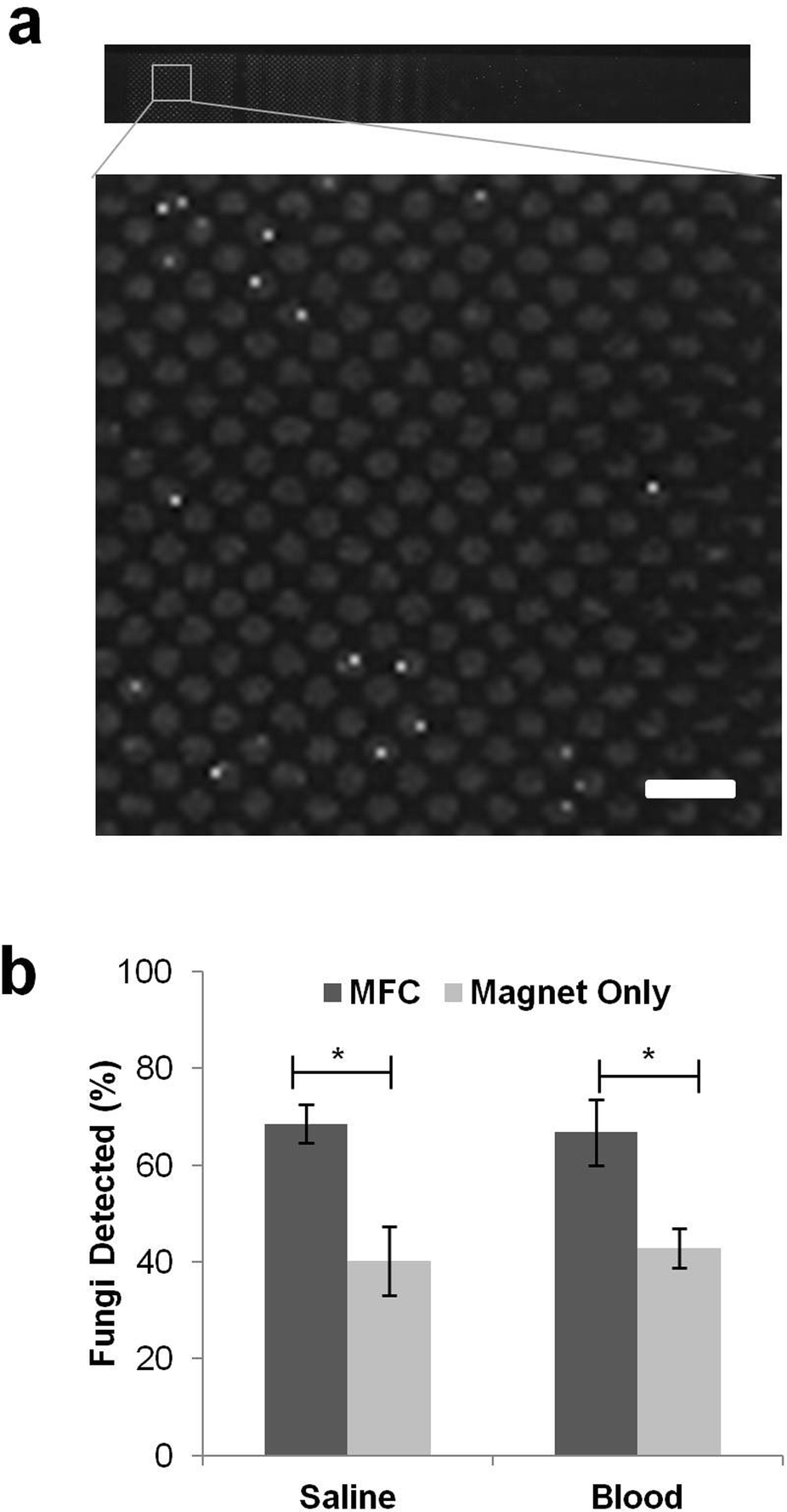 | ||
| Fig. 4 Optical detection of C. albicans magnetically captured from saline and blood. a) Composite fluorescence micrograph showing C. albicans cells (bright white dots) captured and visualized in the device after immunofluorescence staining. Scale bar is 150 μm. b) Comparison of the fungal cells detected with and without MFC in both saline and blood (* indicates p < 0.05 using a Student's t-test). | ||
Fluorescent images obtained from this automatic scanning were stitched back together and analyzed using an automated image capture and analysis protocol (Zeiss Axio 2 microscope with a Zeiss EC-Plan NEOFLUAR 10× objective, numerical aperture of 0.3, depth of field 8 μm, Metamorph for image acquisition and stitching to form composite image, ImageJ for thresholding and image registration, MATLAB for cell quantification). Pathogen counts obtained by manually scanning the entire device on the microscope were compared with results obtained in the same device using the automated image capture and analysis protocol. After setting thresholding values appropriately, it was possible to correctly quantify the captured pathogens within ±10% of the manual count, and to consistently detect when no fungi were present in the device. Red blood cells were lysed during washing process with 1% Triton X-100 to minimize interference because they tended to settle in the bottom of the device, making it difficult to image the magnetically tagged pathogens on the ceiling of the chamber.
Using the improved detector design and pathogen counting methods, we were able to detect significantly (p < 0.05) more C. albicans fungi in saline with the MFC compared to the magnet alone (Fig. 4b). With the magnet directly adjacent to the PDMS microdevice, the beads formed a pile under the upstream edge that buried a large fraction of the bead-bound fungal cells (Fig. 3c) so that we could only detect 47% of the 850 fungal cells that were initially seeded into the 10 mL saline fluid sample. Adding the MFC between the channel and the magnet boosted the detection sensitivity significantly (p < 0.05), resulting in detection of 68% of the pathogens (Fig. 4b). Inclusion of the MFC also significantly (p < 0.05) improved the detection of fungi captured from human blood samples: using pre-bound C. albicans, we could detect up to 67% of the cells added to a 5 mL blood sample (diluted 1:1 with saline to bring total volume to 10 mL), which was nearly equivalent to our detection rate in saline, whereas only 43% of the spiked pathogens could be detected in blood using the magnet alone (Fig. 4b). The spread of the beads in blood was similar to that seen in saline, with piling of beads at the leading edge only being observed in the absence of the MFC and the captured pathogens were localized almost exclusively within the micropatterned wells in the presence of the MFC.
After optical identification of the pathogens, the magnet and MFC can be removed from the device, which allows the magnetically tagged pathogens to drop from the ceiling and to be flushed out of the device with a 1 ml saline wash. The captured beads and pathogens were then spread on blood agar plates and cultured for 36 hours in a 30 °C incubator; colonies of the original C. albicans can then be used antibiotic susceptibility testing.
Discussion
We previously created a combined microfluidic–micromagnetic separator device that can continuously isolate magnetically tagged pathogens out of flowing blood for sepsis therapy applications.11,12 But when we explored whether we can visualize the captured pathogens using optical microscopy as a new approach for sepsis diagnosis, no pathogens were detected. Thus, in the present study, we set out to develop a device that would maintain high magnetic pathogen capture efficiencies, but position the beads and bound pathogens so that they could be detected optically. Here we described a rapid pathogen detector device that is able to efficiently capture pathogens from saline and blood, as well as detect the pathogens optically, within three hours after sample collection using superparamagnetic beads coated with broad-spectrum opsonins in combination with a microengineered MFC. In our early prototype, we could detect down to pathogen concentrations as low as 0.4 fungal cells mL−1 in saline and the refinements to the design have allowed us to boost detection of magnetically tagged C. albicans in blood to a similar level seen in saline (Fig. 4b). We therefore expect that this system should have the sensitivity to detect clinically relevant fungal loads in blood and plan to begin testing is with patient samples in the near future.Our early device prototypes captured more than 99% of bound pathogens from flowing blood, but the unmodulated magnetic field caused the magnetically tagged pathogens and excess beads to form a dense pile in which only 15% of the captured cells were detectable. To overcome this limitation, we balanced the magnetic pull on the beads and the downstream fluidic drag by inserting a microengineered ferromagnetic flux concentrator (MFC) between the magnet and the upper surface of the channel, and by adjusting the channel geometry and surface topography. This caused the captured beads to spread out evenly into a thin layer through which we could easily visualize the fluorescently labelled pathogens. Finite element models of the magnetic gradient allowed us to determine the MFC shape that would create a high, relatively uniform gradient along the entire length of the channel so that the force on the beads was more uniform to make them spread out in the microwells in the ceiling of the device rather than piling up at the upstream edge of the magnet. The channel dimensions were adjusted so that the beads would only stop in one of the micropatterned wells where they were sheltered from the fluidic shear that tries to push them downstream. Moreover, if one microwell fills, subsequent beads and magnetically tagged pathogens are pushed downstream and collected in the next open well, rather than forming a dense pile. The main problem with the magnet alone was that there was no way to prevent the beads from piling up regardless of the micropatterned wells in the region of the gradient spike. Inserting the MFC between the magnet and the microchannel allowed us to decrease this initial spike and to boost the gradient across the rest of the device so that the bead spread was far more uniform and suitable for optical detection
The ability to capture and concentrate the beads and bound pathogens from a 10 mL fluid sample, and to spread them into a thin layer inside our microdevice, made it possible to detect most of the bound pathogens when stained with fluorescent dyes. Using this approach, we could detect more than two-thirds of the pathogen cells spiked into a 10 mL volume of saline using the magnet with the MFC, whereas most of the captured pathogens were buried by beads (and hence, undetectable) with the magnet alone. The pathogens were labelled with three fluorophores, calcofluor as a general fungal stain, GFP and a Cy3 anti-C. albicans antibody. The calcofluor and GFP stains were used for manually counting the captured pathogens and immunofluorescent Cy3 stain (Fig. 4a) was used for our automated detection protocol.
Importantly, inclusion of the MFC in the device significantly increased detection of pathogens in both saline and blood, when compared with magnet alone. By focusing on pathogens pre-bound with beads in blood, we were able to probe only the pathogen detection sensitivity, rather than having to assess the cumulative effects of bead–pathogen binding in blood and detection simultaneously. Using this approach, we obtained similar detection sensitivities in blood and saline using the MFC. But even with this improved sensitivity, about a third of the captured pathogens could not be visualized in the device. There are two possible explanations for this: 1) some of the bead-bound pathogens might be surrounded by multiple beads, which still obscure their fluorescent signature, or 2) some of the bead piles are still deep enough to bury the pathogens since the wells are 20 μm deep. Shallower wells were not sufficient to arrest the beads and bound pathogens, which resulted in a decrease in bead capture, causing them to be lost downstream in the waste flow. The 50 μm diameter of the wells was determine by the limit of our photolithographic transparency mask; however, mirror chrome masks which can produce smaller and shallower wells could be used in the future. This would give the wells similar dimensions to a single pathogen, which should lower the chance of any pile being deep enough to completely obscure pathogens.
An advantage of this method over rapid molecular diagnostic techniques is that pathogen viability is not significantly affected by the magnetic capture and concentration process, so viable pathogens can be recovered after imaging for conventional antibiotic susceptibility testing. This could allow clinicians to rapidly diagnose sepsis and remove the need for blood culture before performing sub-culturing for antibiotic susceptibility testing, cutting approximately 24 hours off the total processing time.
In summary, we have shown that our technology can detect the presence of dilute pathogens in blood using a simple optical readout that can be detected by optical microscopy. Using this approach, we were able to detect C. albicans fungal cells in human blood at concentrations down to 1 fungal cell mL−1 in less than three hours. We believe that this platform could form the basis of a rapid diagnostic device because the beads are coated with a generic opsonin and thus, the identity of the pathogen does not need to be known for capture to be carried out. Moreover, it has the ability to concentrate rare pathogens from a large fluid volume (e.g., 20–30 mL clinical blood sample) for rapid optical detection. While our results were obtained with fungal cells, it should be possible to create a version of the device to reliably detect bacteria as well by modifying flow rate, magnetic field and channel geometry. The ability to detect rare pathogens directly within human blood within a few hours after collection would represent a major advance relative to current pathogen detection methods that often require a day or more to complete.
Acknowledgements
Funded by Center for Integration of Medicine and Innovative Technology (CIMIT) Grant 09-303, the Defense Advanced Research Projects Agency (DAPRA) Grant N66001-11-1-4180 and the Wyss Institute for Biologically Inspired Engineering.Notes and references
- J. Garnacho-Montero, T. Aldabo-Pallas, C. Garnacho-Montero, A. Cayuela, R. Jimenez, S. Barroso and C. Ortiz-Leyba, Crit. Care, 2006, 10, 12 CrossRef PubMed.
- B. Guery, M. Arendrup, G. Auzinger, E. Azoulay, M. B. Sa, E. Johnson, E. Muller, C. Putensen, C. Rotstein, G. Sganga, M. Venditti, R. Z. Crespo and B. Kullberg, Intensive Care Med., 2009, 35, 206–214 CrossRef CAS PubMed.
- D. W. Wilmore, Ann. Surg., 1998, 227, 10–11 CrossRef CAS PubMed.
- M. Pammi, A. Flores, M. Leeflang and J. Versalovic, Pediatrics, 2011, 128, e973–e985 CrossRef PubMed.
- M. Venkatesh, A. Flores, R. A. Luna and J. Versalovic, Expert Rev. Anti-Infect. Ther., 2010, 8, 1037–1048 CrossRef PubMed.
- D. C. Angus, W. T. Linde-Zwirble, J. Lidicker, G. Clermont, J. Carcillo and M. R. Pinsky, Crit. Care Med., 2001, 29, 1303–1310 CrossRef CAS PubMed.
- M. Bassetti, E. M. Trecarichi, E. Righi, M. Sanguinetti, F. Bisio, B. Posteraro, O. Soro, R. Cauda, C. Viscoli and M. Tumbarello, Diagn. Microbiol. Infect. Dis., 2007, 58, 325–331 CrossRef PubMed.
- R. S. Hotchkiss and I. E. Karl, N. Engl. J. Med., 2003, 348, 138–150 CrossRef CAS PubMed.
- L. G. Reimer, M. L. Wilson and M. P. Weinstein, Clin. Microbiol. Rev., 1997, 10, 444–465 CAS.
- A. Viudes, J. Peman, E. Canton, P. Ubeda, J. L. Lopez-Ribot and M. Gobernado, Eur. J. Clin. Microbiol. Infect. Dis., 2002, 21, 767–774 CrossRef CAS PubMed.
- N. Xia, T. P. Hunt, B. T. Mayers, E. Alsberg, G. M. Whitesides, R. M. Westervelt and D. E. Ingber, Biomed. Microdevices, 2006, 8, 299–308 CrossRef CAS PubMed.
- C. W. Yung, J. Fiering, A. J. Mueller and D. E. Ingber, Lab Chip, 2009, 9, 1171–1177 RSC.
- J. H. Kang, M. Super, C. W. Yung, R. M. Cooper, K. Domansky, A. R. Graveline, T. Mammoto, J. B. Berthet, H. Tobin, M. J. Cartwright, A. L. Watters, M. Rottman, A. Waterhouse, A. Mammoto, N. Gamini, M. J. Rodas, A. Kole, A. Jiang, T. M. Valentin, A. Diaz and D. E. Ingber, 2013, in review.
- D. L. Jack, N. J. Klein and M. W. Turner, Immunol. Rev., 2001, 180, 86–99 CAS.
- K. Gupta, R. K. Gupta and K. Hajela, Indian J. Med. Res., 2008, 127, 431–440 CAS.
- M. W. Turner, Immunol. Today, 1996, 17, 532–540 CAS.
- M. Gadjeva, K. Takahashi and S. Thiel, Mol. Immunol., 2004, 41, 113–121 CrossRef CAS PubMed.
- K. Takahashi, W. K. E. Ip, I. C. Michelow and R. A. B. Ezekowitz, Curr. Opin. Immunol., 2006, 18, 16–23 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |