A cell rolling cytometer reveals the correlation between mesenchymal stem cell dynamic adhesion and differentiation state†
Sungyoung
Choi
ab,
Oren
Levy
c,
Mónica B.
Coelho
c,
Joaquim M. S.
Cabral
d,
Jeffrey M.
Karp
*c and
Rohit
Karnik
*a
aDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: karnik@mit.edu
bDepartment of Biomedical Engineering, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Gyeonggi-do, Yongin-si, 446-701, Republic of Korea
cDivision of Biomedical Engineering, Department of Medicine, Center for Regenerative Therapeutics, Brigham and Women's Hospital, Harvard Medical School, Harvard Stem Cell Institute, Harvard-MIT Division of Health Sciences and Technology, 65 Landsdowne St., Cambridge, MA 02139, USA. E-mail: jkarp@rics.bwh.harvard.edu
dDepartment of Bioengineering, Instituto Superior Técnico, Technical University of Lisbon, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
First published on 21st October 2013
Abstract
This communication presents quantitative studies of the dynamic adhesion behavior of mesenchymal stem cells (MSCs) enabled by the combination of cell-surface receptor–ligand interactions and three-dimensional hydrodynamic control by microtopography.
Cell adhesion plays an essential role in many cellular processes, and many cell types exhibit focal adhesions on substrates such as the extracellular matrix. Corresponding micro-patterning techniques have been developed for in vitro control of cell adhesion, including that of MSCs.1,2 In contrast to these focal adhesions, some types of cells exhibit dynamic adhesion called cell rolling that is mediated by transient ligand–receptor interactions that form and break under the influence of hydrodynamic fluid shear.3 In particular, migration of circulating cells (i.e. leukocytes, tumor cells, and stem cells) from vascular to extravascular sites is initiated by these weak, transient interactions between carbohydrate ligands on the cells and selectin molecules (i.e. P- and E-selectin) on the vascular endothelium, which results in the characteristic rolling behavior followed by firm adhesion and transmigration.3–6 These interactions are also implicated in the homing of MSCs, which are self-renewing, multipotent cells that offer significant therapeutic potential due to their regenerative and immunomodulatory capacity, lack of ethical issues, and the ability to transplant allogeneic MSCs without immunosuppressive therapy.7–9 According to the FDA clinical trial database, MSCs are being explored in more than 350 clinical trials worldwide10 and a significant portion of these trials involve systemic infusion, where homing to diseased or damaged tissue is presumed to be important for maximizing therapeutic benefit.11–13 However, while the adhesive interactions that mediate homing have been well described for leukocytes,3 the degree of adhesive interactions and the molecules involved remain unclear for MSCs.14–18 Since insufficient homing of systemically infused culture expanded MSCs is a significant obstacle for effective therapy,11–13 understanding the adhesion dynamics of MSCs is crucial not only for extending our knowledge of fundamental stem cell biology, but also for developing new approaches to enhance MSC homing.
Parallel-plate flow chambers coated with adhesion molecules or activated endothelial cells have been previously employed for in vitro rolling adhesion assays of MSCs14–18 as well as leukocytes,19 leukemic cell lines,20 cancer cell lines,21 CD34+ bone marrow cells,22 and CD34+ hematopoietic stem cells.23 This platform has contributed to advancing our understanding of the dynamics of cell rolling adhesion. However, a significant barrier to the quantitative implementation of this assay, especially for weakly interacting cells like MSCs,14,15 is the inability to initiate cell rolling (known as tethering) in the flow chambers and the difficulty in maintaining rolling interactions under dynamic flow conditions. Within the flow chambers, settling of cells prior to applying shear stress can enhance adhesion, but this approach is non-physiological and typically insufficient in the case of weak and non-robust adhesive interactions where hydrodynamic lift forces in a channel with uniform cross-section can push the cells away from the surface.24,25 Microfluidic devices have recently employed mixing approaches using surface grooves to create circulating streamlines that enhance cell-surface interactions,26 resulting in higher cell capture efficiencies.27,28 These approaches, however, are inadequate for characterizing adhesion dynamics at the single cell level because cell capture is distributed along the length of the channels and only a biased fraction of the cell population that exhibits stronger adhesive interactions can be interrogated. Current approaches are, thus, not suitable to quantitatively examine weakly interacting MSCs. For understanding the adhesion dynamics of MSCs, efficient methods to promote adhesion interactions in dynamic flow and enable quantitative analysis of the rolling phenotype need to be developed.
Herein, we report a cell rolling cytometer (CRC) for forced tethering and directed transporting of cells in suspension using a three-dimensional microtopography coated with adhesion molecules, which enables quantification of cell-surface adhesion dynamics via transit time and lateral position at the single cell level (Fig. 1). The device operation is based on “deterministic cell rolling”29,30 wherein three-dimensional adhesion ridges (AR) create rotational flow patterns and induce effective contact or tethering (i.e. initialization of molecular interactions) of cells with surfaces functionalized with adhesion molecules that support cell rolling. The device comprises a narrow focusing channel where the high shear stress prevents cell rolling, even though all channel surfaces are functionalized with adhesion molecules. The focusing channel is followed by a sudden increase in the channel width that lowers the level of shear stress and forces each incoming cell to interact with the AR. The AR focus non-interacting cells to one side of the device, and slow down and laterally displace the trajectories of interacting cells (including those that typically display weak interactions) into the adjacent gutter region. The adhesion channel is designed with small dimensions (w × l = 200 μm × 2000 μm) to fit within a microscope field of view, which enables observation and characterization of the adhesive interactions of every cell flowing through the channel. While we have previously used this effect to alter rolling trajectories for cell separation,29 the CRC is specifically designed to examine the adhesion characteristics of every cell in a given cell sample at the single cell level.
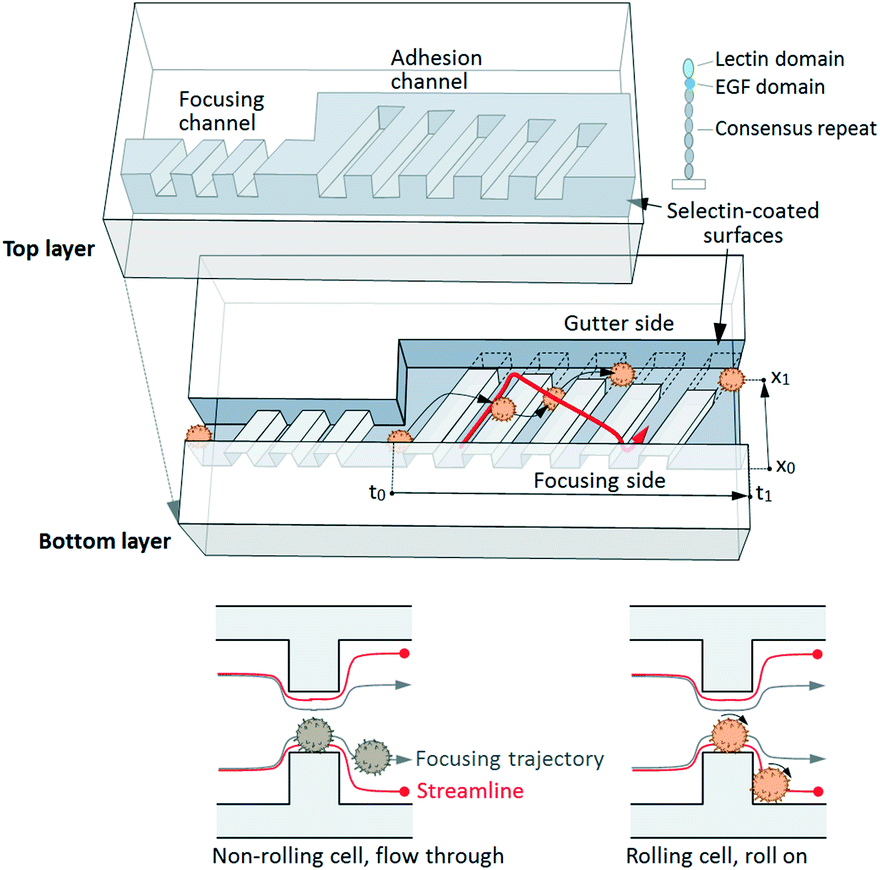 | ||
| Fig. 1 Cell rolling cytometer. (Top) Schematic of the microfluidic cell rolling cytometer (CRC), in which cells are forced into contact with adhesion molecule-coated ridges. Adhesion of single cells is quantified via transit time, tt, and rolling trajectory, xr. The red arrow indicates a schematic helical streamline. (Bottom) Cross-section views of the CRC. Without specific interactions a cell quickly travels through the channel, following the focusing trajectory. Specific adhesion interactions retard the cell and change its trajectory. | ||
As a model system, we first tested a leukemic cell line, HL60, that expresses high levels of P-selectin glycoprotein ligand-1 (PSGL-1, CD62P) (Fig. S2†) and exhibits rolling on P-selectin, mediated primarily by PSGL-1.31 The cells were introduced into the CRC functionalized with P-selectin at an incubation concentration (cs) of 1.5 μg mL−1 or passivated with 1% BSA as a negative control. The trajectories of HL60 cells that were subjected to a wall shear stress of 3.5 dyn cm−2 are shown in Fig. 2. This nominal shear stress (τ) denotes the minimum shear stress on the top surfaces of the AR where cells can contact and tether. The shear stress was determined by computational fluid dynamics simulation (Fig. 2a, bottom) based on the flow rate set by the syringe pump. In the absence of adhesion interactions (BSA control), the physical interactions between the cells and the ridges force the cells to remain above the ridges where the flow converges toward the focusing side of the channel (Fig. 2a and S3,† top).29,30,32 Transient ligand–receptor interactions occur when the surfaces are coated with P-selectin, which mediates cell rolling along the AR, until they are detached in the gutter region (Fig. 2a). In this case, the cell trajectories follow similar paths as the streamlines inside the trenches. This clearly indicates that the flow circulation not only enhances cell-surface interactions by inducing repeated collisions with the ridges, but also directs the flow of cells differently according to their affinity to adhesion ligands (Fig. 1 and 2a). Transit-time and position measurements are represented in scatter plots (Fig. 2b). The scatter plot was divided into quadrants such that nearly all of the cells in the control experiment with passivated channels were in the lower left quadrant. With this division, non-interacting cells were in the left lower quadrant and rolling, interacting cells were mostly in the right upper quadrant (Fig. 2b and S4†). The left upper quadrant indicates weakly interacting cells that can tether to the surface and undergo lateral displacement towards the gutter region, but are readily detached from the surface and therefore flow through quickly. The right lower quadrant represents a few interacting cells that undergo lateral displacement, but go back to the focusing side following the rotational flow (Fig. 1). With the CRC, we observed that the adhesion interactions in the P-selectin-coated channel slowed down HL60 cells (88.3% of the total incoming cells) to a velocity of 7.4 ± 3.5 μm s−1 (n = 39) and prolonged their transit time to transverse the adhesion channel, compared to the BSA control where the cells were flowing fast at 1.5 ± 0.6 mm s−1 (n = 19) (Fig. 2). Since the cells rolled only for a fraction of the distance in the adhesion channel in the P-selectin-coated device, they required ~35 s to traverse the adhesion channel compared to ~1.5 s for the BSA control (Fig. 2b). Transit time can be a gauge to determine whether a cell interacts with adhesion molecules exhibiting a rolling response, while lateral position can be a gauge to determine the potential sorting efficiency29 of the rolling phenotype.
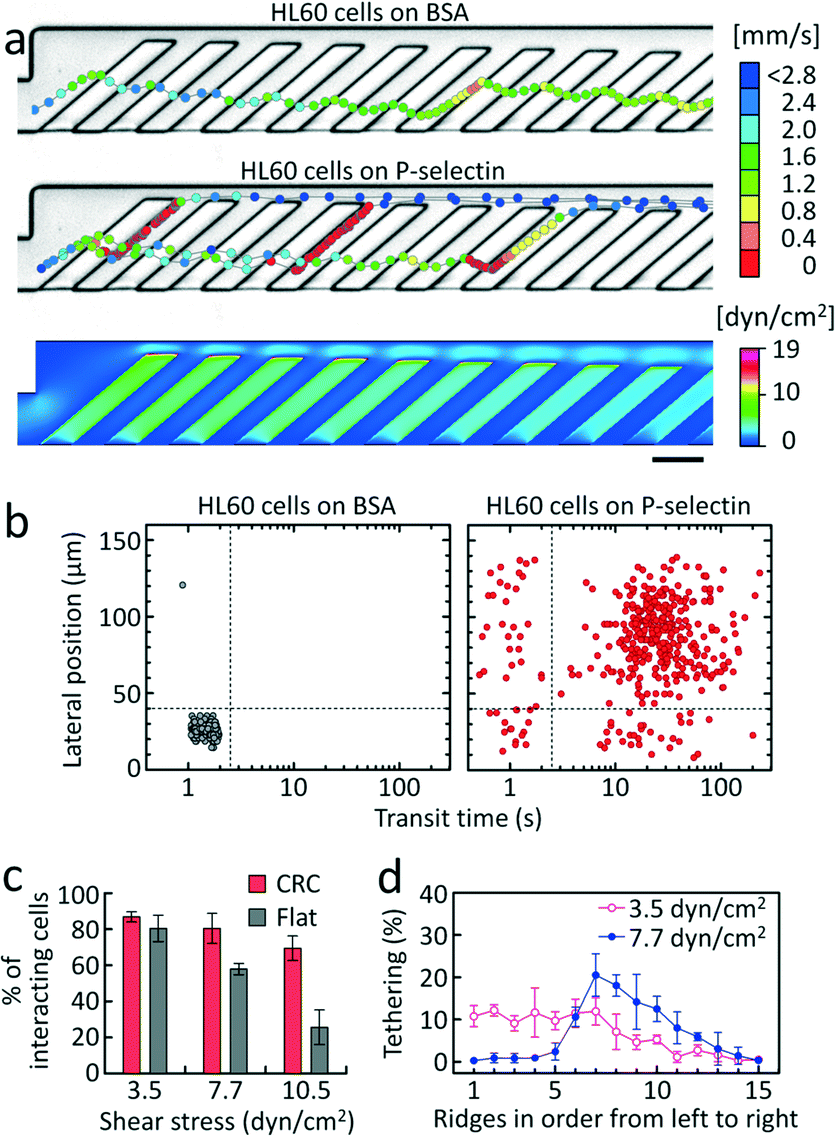 | ||
| Fig. 2 Characterization of the CRC using HL60 cells that exhibit robust rolling on P-selectin. a) Examples of trajectories of HL60 cells in (top) BSA-passivated and (middle) P-selectin-coated channels. Each circle represents the position of a cell and is colored by the magnitude of its velocity. (Bottom) Simulation of the wall shear stress on the channel surfaces. Scale bar, 100 μm. b) Scatter plots of the transit time and lateral position of 394 cells in (left) BSA-passivated and (right) P-selectin-coated channels. c) Cell adhesion efficiency of HL60 cells in the CRC (n = 4) compared to a control device with a flat chamber (n = 3) at the same shear stress where cells were first allowed to settle. Adhesion efficiency is defined as the fraction of cells that exhibit a rolling response, i.e. are in the right two quadrants. d) Tethering frequency of HL60 cells on each ridge (n = 4), calculated as the number of cells tethering at each ridge divided by the total number of rolling cells. Error bars show one standard deviation. | ||
Next, we examined how the shear stress affects the cell adhesion efficiency of the CRC, and compared it with that of a conventional flat flow chamber (w × h = 1000 μm × 98 μm). We define the cell adhesion efficiency as the fraction of cells in the right two quadrants, i.e. cells that slow down and exhibit a rolling response, but that are not necessarily displaced to the gutter side. In the flat chamber, cells were allowed to settle on the bottom surface, and were then subjected to a shear stress corresponding to the minimum shear stress that cells can experience during tethering (i.e. initialization of molecular interactions on the top surfaces of the AR) in the CRC with spatial shear stress gradients (Fig. 2a). All the devices for experiments with HL60 cells were functionalized with P-selectin at cs = 1.5 μg mL−1. At a shear stress of 3.5 dyn cm−2, 86.9 ± 2.8% of cells in the CRC were in the right two quadrants (n = 3). At shear stresses of 7.7 and 10.5 dyn cm−2, the adhesion efficiency of the flat chamber was significantly compromised, with only ~26% of the cells exhibiting a rolling response at 10.5 dyn cm−2 while the remaining cells were washed away (Fig. 2c). In contrast, the CRC exhibited high efficiency in capturing cells and supporting stable rolling in the range of shear stress (3.5 to 10.5 dyn cm−2). With increasing shear stress from 3.5 dyn cm−2 to 7.7 dyn cm−2, the location where cells initiate rolling shifted to the seventh ridge where the level of shear stress is ~1.5 times lower than that at the first ridge, although there was no significant difference in cell-capture efficiency (Fig. 2c and d). Since the level of shear stress and the number of ligand–receptor bonds affect the cell's ability to initiate and sustain rolling, the tethering profile and the fraction of interacting cells in the CRC is consequently modulated, reflecting the diminished ability of cells to tether at higher shear stresses. The results demonstrate that compared to a flat flow chamber, the CRC can more readily achieve sufficiently high cell adhesion efficiency for quantitative analysis of cell adhesion. Under an optimal condition, most of the cells capable of a rolling response were observed to roll in the adhesion channel (~86.9% of HL60 cells), indicating that the CRC is capable of interrogating the rolling behavior of almost every cell that flows through the device.
After characterization of the CRC with HL60 cells, we performed cell adhesion cytometry on human MSCs derived from human bone marrow (Lonza, Walkersville, MD) to examine their adhesion phenotype. Since it has been suggested that human MSCs exhibit a rolling response on activated endothelial cells,14 we first examined adhesion to E- and P-selectin which are expressed on the surface of activated endothelium and mediate rolling of circulating cells.3 The CRC was functionalized with P- or E-selectin at a concentration cs = 30 μg mL−1. Following each MSC experiment, the surface functionalization of the adhesive channel with P- or E-selectin was verified using HL60 cells that interact with both selectins1 as positive control. As shown in Fig. 3 and S5,† the majority of MSCs (85.3 ± 4.0%, n = 3) exhibited a rolling response (i.e., were in the right two quadrants) on E-selectin at τ = 1.7 dyn cm−2 and cs = 30 μg mL−1. The rolling response decreased with increasing shear stress, and increased with cs (Fig. 3b and S5†). An optimal condition τ = 1.7 dyn cm−2 and cs = 30 μg mL−1 was used for subsequent rolling assays, unless otherwise specified. For comparison, the flat chamber was also functionalized with E-selectin at the same concentration cs = 30 μg mL−1. As expected, the CRC was highly effective at capturing cells, whereas significantly fewer cells were captured with the flat device (p < 0.01 using two-tailed unpaired t-test for all shear stress conditions tested), suggesting that forced cell tethering is crucial for initiation of rolling adhesion. There were no observable adhesion events in P-selectin-coated channels (Fig. 3a). Our results suggest that MSCs exhibit specific transient adhesion on E-selectin-coated surfaces.
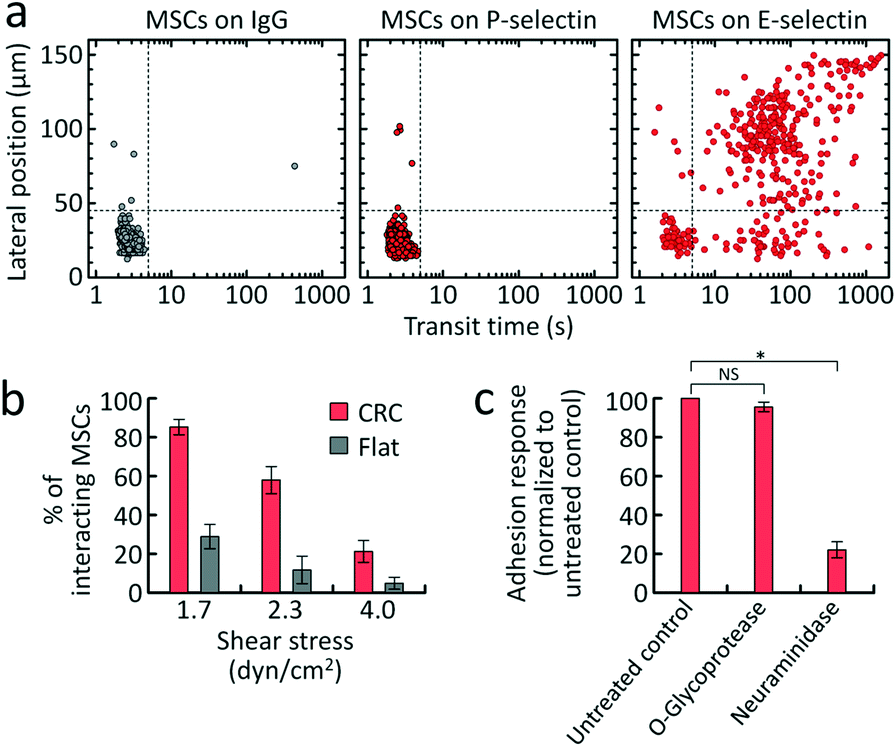 | ||
| Fig. 3 Quantification of the dynamic adhesion phenotype of MSCs. a) Scatter plots of the transit time and lateral position of 400 MSCs in (left) IgG-passivated, (middle) P-selectin-coated, and (right) E-selectin-coated CRCs. b) Efficiency of MSC adhesion in the E-selectin-coated CRC (n = 4) and flat flow chamber (n = 3). c) Effects of enzyme treatments on MSC rolling adhesion (n = 3). The adhesion responses of enzyme-treated MSCs were normalized to untreated control cells. Significant difference in the rolling response between control and enzyme-treated MSCs was observed for neuraminidase (*, p < 0.001 using two-tailed unpaired t-test) but not O-glycoprotease (p = 0.24 using two-tailed unpaired t-test). NS denotes no significant difference. Error bars indicate one-standard deviation. | ||
This result is especially significant given that inflammatory cytokines such as tumor necrosis factor-α up-regulate the expression of E-selectin, but not of P-selectin, in human endothelial cells.33,34 The stronger adhesion to E-selectin observed here suggests that E-selectin may play a major role in recruitment of MSCs in humans and its precise role in vivo warrants further investigation. It is noteworthy that MSCs have been previously shown to exhibit poor rolling properties on selectin-coated flat surfaces.14,17 The increased rolling response in this study likely resulted from synergy between forced cell contact by rotational flow control which can facilitate tethering and rolling and the high concentration of selectins (30 μg mL−1), compared with the low concentration of selectins (5–10 μg mL−1) and weak contact by gravitational settling in previous studies.17 Given the difficulty of studying MSC adhesion in flat flow chambers, the CRC is an effective tool to probe the rolling adhesion of a cell type that exhibits weak, non-robust rolling properties.
Next, we examined the effect of removal of cell surface residues that are known to be associated with rolling adhesion on the rolling response of MSCs. The binding receptors of MSC adhesion still remain unknown,35,36 although one study suggests that human MSCs exhibit weak rolling on human umbilical cord vein endothelial cells mediated by P-selectin and VCAM-1.14 As determined by flow cytometry, MSCs lacked E-selectin ligands such as CD15s, CD18, CD24, CD43, CD65, and CD162 (Fig. S6a†). We therefore used the CRC to test the susceptibility of the unknown binding receptors on MSCs for E-selectin to treatment with O-sialoglycoprotein endopeptidase (O-glycoprotease) or neuraminidase. Neuraminidase and O-glycoprotease are enzymes that cleave neuraminic acids (including O- and N-substituted derivatives called sialic acids) and O-linked sialoglycoproteins that support selectin-mediated rolling, respectively.37,38 The CRC was functionalized with E-selectin at cs = 30 μg mL−1. The shear stress was adjusted between 1.1 dyn cm−2 and 1.7 dyn cm−2 to achieve sufficiently high rolling response (73.0 ± 7.2%) for the untreated control MSCs. The ability of MSCs to form rolling adhesion was significantly blocked by treatment with neuraminidase, while treatment with O-glycoprotease failed to inhibit the rolling response (Fig. 3c). Interestingly, flow cytometry revealed that the enzymes cleave the E-selectin ligands CD15s and CD43, respectively, on HL60 cells (Fig. S8†). The results implicate sialic acid residues in MSC rolling on E-selectin.
Finally, we examined the ability of the CRC to detect alterations in the rolling phenotype of MSCs (Fig. 4). Given that MSCs undergo dramatic phenotypic changes in morphology and cell-surface adhesive interactions during differentiation,39,40 we tested the hypothesis whether the rolling adhesion phenotype of MSCs correlates with their differentiation state. For induction of differentiation into adipocytes or osteoblasts, the medium was replaced with either an adipogenic differentiation medium or the MSC culture medium supplemented with 10 nM dexamethasone,41 respectively. The cells were harvested on day 14 after induction of adipogenic and osteogenic differentiation, and the differentiation was confirmed by histochemical staining. Adipocytes differentiated from MSCs stained positive for oil-red-O and osteogenic cells differentiated from MSCs were positive for alkaline phosphatase (Fig. 4b). Analysis showed that the rolling response in both differentiated populations was significantly decreased relative to non-induced MSC control group, supporting the hypothesis that MSC differentiation leads to a quantifiable change in the adhesion phenotype (Fig. 4a and S7†). This decrease in cell rolling adhesion upon differentiation is not unexpected, as cell rolling is involved in trafficking of cells and we speculate that it may not be biologically useful once the MSCs are differentiated. To verify that differentiated cells lose their homing ability via adhesive interactions, we examined whether human adipocytes – obtained from adipogenic differentiation of primary human preadipocytes (PACs) (Fig. 4c)42,43 – and primary human osteoblasts44 exhibit cell rolling. As shown in Fig. 4a and S9,† both adipocytes differentiated from PACs and osteoblasts exhibited weak rolling response of 12.0 ± 6.3% and 25.3 ± 6.4%, respectively (n = 3, Fig. 4a), indicating that these cells do not exhibit robust cell rolling. Interestingly, PACs exhibited a rolling response (76.7 ± 15.8%, n = 3) similar to MSCs (Fig. 4a and S9†), showing that rolling appears to be lost at terminal differentiation, but more work is required to identify the specific point in the differentiation process where this transition occurs. The results demonstrate that the CRC is able to detect a statistically significant decrease in the rolling adhesion of MSCs upon differentiation that is consistent with the adhesion behavior of the corresponding mature cell types.
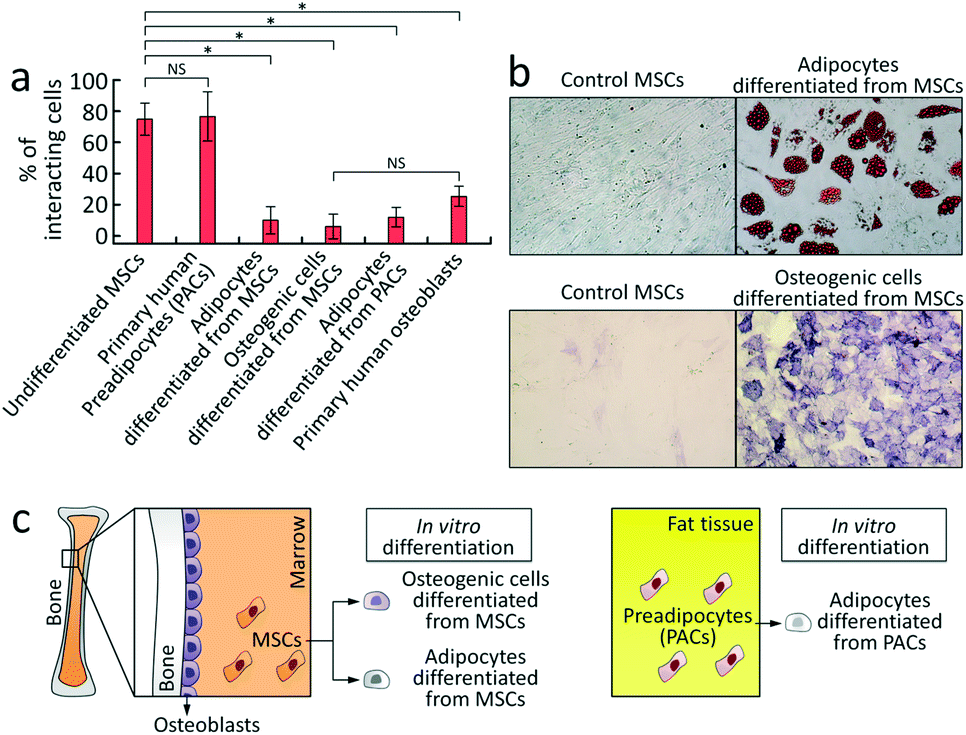 | ||
| Fig. 4 Effect of differentiation on the dynamic adhesion phenotype of MSCs. a) The rolling response of MSCs decreased after induction of adipogenic and osteogenic differentiation. The corresponding mature cell types (adipocytes differentiated from primary human preadipocytes (PACs) and primary human osteoblasts) exhibited weak rolling responses. *, p < 0.001 using one-way ANOVA followed by Tukey's post hoc test; the results for the cells differentiated from MSCs, adipocytes differentiated from PACs, and osteoblasts (n = 3) are significantly different from the results for undifferentiated MSC control (n = 4). No statistically significant differences were observed between the cells differentiated from MSCs, adipocytes differentiated from PACs, and osteoblasts (p > 0.05 using one-way ANOVA followed by Tukey's post hoc test). Error bars show one standard deviation. NS denotes no significant difference. b) Histochemical staining was performed to assess adipogenic (upper, oil red O stain) and osteogenic (lower, alkaline phosphatase stain) differentiation of MSCs. c) (left) MSCs were derived from human bone marrow and their differentiated progeny were obtained by in vitro differentiation. Human primary osteoblasts were derived from human bone tissues. (Right) Adipocytes were obtained from in vitro differentiation of human primary PACs isolated from subcutaneous fat tissue. | ||
The quantitative analysis of transient cell-surface molecular interactions is limited by poor tethering efficiency of cells in conventional parallel-plate chambers.17,24 Forced cell tethering and directed rolling in the CRC can promote a robust rolling response even in dynamic flow and enable observation of adhesion events at the single cell level. The absence of significant tethering and rolling upon sedimentation on a flat surface suggests that the forced cell-surface contact by 3D streamline manipulation was critical to initiate MSC tethering and rolling in the CRC. The CRC also allowed the rolling response to be examined in a continuous flow as opposed to the batch analysis in conventional flow chambers. More significantly, the results demonstrate that enhancing the rolling response is critical to probe the adhesion of MSCs that exhibit relatively weak, non-robust rolling properties. The CRC enabled quantification of MSC rolling interactions and provided insight into the transient adhesion process of human MSCs, revealing that 1) E-selectin is an adhesion molecule that supports rolling adhesion of human MSCs, 2) the rolling response is mediated by sialic acid residues on MSCs, 3) a significant, quantifiable loss of rolling response occurs upon differentiation of MSCs, and 4) compared to leukocytes, MSCs may require greater contact force against the vascular endothelium to initiate tethering and rolling response in vivo. The loss of rolling ability upon differentiation suggests that differentiated MSCs may lose their homing ability in vivo. Importantly, the CRC was used to quantify changes in the rolling phenotype arising from weak adhesions in the absence of known ligands, where analysis by flow cytometry is not possible. The CRC is a potential quantitative tool for detecting changes in MSC phenotype, which could be useful for ensuring MSC quality control that is critical for cell-based therapy.
Acknowledgements
This work was supported by National Institutes of Health grants HL-095722 and HL-097172 (J.M.K.), and in part by NSF CAREER award 0952493 through the Chemical and Biological Separations program (R.K.) and the Deshpande Center for Technological Innovation at MIT. M.B.C. acknowledges support from Fundação para a Ciência e a Tecnologia, Portugal (SFRH/BD/33723/2009), through the MIT-Portugal Program's Bioengineering Systems Focus Area. S.C. acknowledges support by the Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2013M3C1A3064777) and the framework of international cooperation program managed by National Research Foundation of Korea (2013K2A1A2053078). We thank Yagnaseni Roy for assistance with data analysis. Devices were fabricated in the Microsystems Technology Laboratory at MIT.Notes and references
- R. Peng, X. Yao and J. Ding, Biomaterials, 2011, 32, 8048 CrossRef CAS PubMed.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745 CrossRef CAS PubMed.
- R. P. McEver and C. Zhu, Annu. Rev. Cell Dev. Biol., 2010, 26, 363 CrossRef CAS PubMed.
- J. M. Fox, G. Chamberlain, B. A. Ashton and J. Middleton, Br. J. Haematol., 2007, 137, 491 CrossRef CAS PubMed.
- E. F. Srour, A. Jetmore, F. M. Wolber, P. A. Plett, R. Abonour, M. C. Yoder and C. M. Orschell-Traycoff, Leukemia, 2001, 15, 1681 CrossRef CAS.
- K. Konstantopoulos and S. N. Thomas, Annu. Rev. Biomed. Eng., 2009, 11, 177 CrossRef CAS PubMed.
- C. Nombela-Arrieta, J. Ritz and L. E. Silberstein, Nat. Rev. Mol. Cell Biol., 2011, 12, 126 CrossRef CAS PubMed.
- C. M. Kolf, E. Cho and R. S. Tuan, Arthritis Res. Ther., 2007, 9, 204 Search PubMed.
- S. Aggarwal and M. F. Pittenger, Blood, 2005, 105, 1815 CrossRef CAS PubMed.
- ClinitalTrials.gov: A service of the U.S. National Institutes of Health, http://www.clinicaltrials.gov Search PubMed.
- J. Ankrum and J. M. Karp, Trends Mol. Med., 2010, 16, 203 CrossRef PubMed.
- J. M. Karp and G. S. Leng Teo, Cell Stem Cell, 2009, 4, 206 CrossRef CAS PubMed.
- S. K. Kang, I. S. Shin, M. S. Ko, J. Y. Jo and J. C. Ra, Stem Cells Int., 2012, 2012, 342968 Search PubMed.
- B. Rüster, S. Göttig, R. J. Ludwig, R. Bistrian, S. Müller, E. Seifried, J. Gille and R. Henschler, Blood, 2006, 108, 3938 CrossRef PubMed.
- V. Aldridge, A. Garg, N. Davies, D. C. Bartlett, J. Youster, H. Beard, D. P. Kavanagh, N. Kalia, J. Frampton, P. F. Lalor and P. N. Newsome, Hepatology, 2012, 56, 1063 CrossRef CAS PubMed.
- R. Sackstein, J. S. Merzaban, D. W. Cain, N. M. Dagia, J. A. Spencer, C. P. Lin and R. Wohlgemuth, Nat. Med., 2008, 14, 181 CrossRef CAS PubMed.
- H. Cheng, M. Byrska-Bishop, C. T. Zhang, C. J. Kastrup, N. S. Hwang, A. K. Tai, W. W. Lee, X. Xu, M. Nahrendorf, R. Langer and D. G. Anderson, Biomaterials, 2012, 33, 5004 CrossRef CAS PubMed.
- D. Sarkar, J. A. Spencer, J. A. Phillips, W. Zhao, S. Schafer, D. P. Spelke, L. J. Mortensen, J. P. Ruiz, P. K. Vemula, R. Sridharan, S. Kumar, R. Karnik, C. P. Lin and J. M. Karp, Blood, 2011, 118, e184 CrossRef CAS PubMed.
- S. Hong, D. Lee, H. Zhang, J. Q. Zhang, J. N. Resvick, A. Khademhosseini, M. R. King, R. Langer and J. M. Karp, Langmuir, 2007, 23, 12261 CrossRef CAS PubMed.
- R. Karnik, S. Hong, H. Zhang, Y. Mei, D. G. Anderson, J. M. Karp and R. Langer, Nano Lett., 2008, 8, 1153 CrossRef CAS PubMed.
- J. H. Myung, K. A. Gajjar, R. M. Pearson, C. A. Launiere, D. T. Eddington and S. Hong, Anal. Chem., 2011, 83, 1078 CrossRef CAS PubMed.
- A. W. Greenberg and D. A. Hammer, Biotechnol. Bioeng., 2001, 73, 111 CrossRef CAS PubMed.
- J. C. Wojciechowski, S. D. Narasipura, N. Charles, D. Mickelsen, K. Rana, M. L. Blair and M. R. King, Br. J. Haematol., 2008, 140, 673 CrossRef PubMed.
- C. K. W. Tam and W. A. Hyman, J. Fluid Mech., 1973, 59, 177 CrossRef.
- S. C. Hur, N. K. Henderson-MacLennan, E. R. McCabe and D. Di Carlo, Lab Chip, 2011, 11, 912 RSC.
- A. D. Stroock, S. K. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647 CrossRef CAS PubMed.
- S. Wang, K. Liu, J. Liu, Z. T.-F. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y.-K. Lee, L. W. K. Chung, J. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K.-F. Shen and H.-R. Tseng, Angew. Chem., Int. Ed., 2010, 50, 3084 CrossRef PubMed.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd Jr., A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392 CrossRef CAS PubMed.
- S. Choi, J. M. Karp and R. Karnik, Lab Chip, 2012, 12, 1427 RSC.
- S. Choi, O. Levy, M. B. Coelho, J. M. Karp and R. Karnik, 16th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Japan, 2012 Search PubMed.
- K. E. Norman, K. L. Moore, R. P. McEver and K. Ley, Blood, 1995, 86, 4417 CAS.
- S. Choi and J.-K. Park, Small, 2009, 19, 2205 CrossRef PubMed.
- L. Yao, H. Setiadi, L. Xia, Z. Laszik, F. B. Taylor and R. P. McEver, Blood, 1999, 94, 3820 CAS.
- Z. Liu, J. J. Miner, T. Yago, L. Yao, F. Lupu, L. Xia and R. P. McEver, J. Exp. Med., 2010, 207, 2975 CrossRef CAS PubMed.
- G. Brooke, H. Tong, J. P. Levesque and K. Atkinson, Stem Cells Dev., 2008, 17, 929 CrossRef CAS PubMed.
- D. Sarkar, J. A. Spencer, J. A. Phillips, W. Zhao, S. Schafer, D. P. Spelke, L. J. Mortensen, J. P. Ruiz, P. K. Vemula, R. Sridharan, S. Kumar, R. Karnik, C. P. Lin and J. M. Karp, Blood, 2011, 118, e184 CrossRef CAS PubMed.
- R. A. Clark, H. P. Erickson and T. A. Springer, J. Cell Biol., 1997, 137, 755 CrossRef CAS.
- B. Byrne, G. G. Donohoe and R. O'Kennedy, Drug Discovery Today, 2007, 12, 319 CrossRef CAS PubMed.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143 CrossRef CAS.
- P. O. Bagnaninchi and N. Drummond, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6462 CrossRef CAS PubMed.
- S. E. Cross, Y. S. Jin, J. Rao and J. K. Gimzewski, Rheumatology, 2001, 40, 74 CrossRef.
- A. Sorisky, Crit. Rev. Clin. Lab. Sci., 1999, 36, 1 CrossRef CAS PubMed.
- Q. Q. Tang and M. D. Lane, Annu. Rev. Biochem., 2012, 81, 715 CrossRef CAS PubMed.
- P. Ducy, T. Schinke and G. Karsenty, Science, 2000, 289, 1501 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; two movies illustrating adhesion and flowing behaviors of human MSCs. See DOI: 10.1039/c3lc50923k |
| This journal is © The Royal Society of Chemistry 2014 |