UV activation of polymeric high aspect ratio microstructures: ramifications in antibody surface loading for circulating tumor cell selection†
Joshua M.
Jackson‡
a,
Małgorzata A.
Witek‡
b,
Mateusz L.
Hupert
b,
Charles
Brady
b,
Swathi
Pullagurla
c,
Joyce
Kamande
c,
Rachel D.
Aufforth
d,
Christopher J.
Tignanelli
e,
Robert J.
Torphy
f,
Jen Jen
Yeh
defgh and
Steven A.
Soper
*abi
aDepartment of Chemistry, UNC-Chapel Hill, NC, USA. E-mail: ssoper@unc.edu
bDepartment of Biomedical Engineering, UNC-Chapel Hill, NCSU, Raleigh, NC, USA
cDepartment of Chemistry, Louisiana State University, Baton Rouge, LA, USA
dDivision of Surgical Oncology, UNC, Chapel Hill, NC, USA
eDepartment of Surgery, UNC, Chapel Hill, NC, USA
fSchool of Medicine, UNC, Chapel Hill, NC, USA
gDepartment of Pharmacology, UNC, Chapel Hill, NC, USA
hLineberger Comprehensive Cancer Center, UNC, Chapel Hill, NC, USA
iUlsan National Institute of Science and Technology, South Korea
First published on 12th July 2013
Abstract
The need to activate thermoplastic surfaces using robust and efficient methods has been driven by the fact that replication techniques can be used to produce microfluidic devices in a high production mode and at low cost, making polymer microfluidics invaluable for in vitro diagnostics, such as circulating tumor cell (CTC) analysis, where device disposability is critical to mitigate artifacts associated with sample carryover. Modifying the surface chemistry of thermoplastic devices through activation techniques can be used to increase the wettability of the surface or to produce functional scaffolds to allow for the covalent attachment of biologics, such as antibodies for CTC recognition. Extensive surface characterization tools were used to investigate UV activation of various surfaces to produce uniform and high surface coverage of functional groups, such as carboxylic acids in microchannels of different aspect ratios. We found that the efficiency of the UV activation process is highly dependent on the microchannel aspect ratio and the identity of the thermoplastic substrate. Colorimetric assays and fluorescence imaging of UV-activated microchannels following EDC/NHS coupling of Cy3-labeled oligonucleotides indicated that UV-activation of a PMMA microchannel with an aspect ratio of ∼3 was significantly less efficient toward the bottom of the channel compared to the upper sections. This effect was a consequence of the bulk polymer's damping of the modifying UV radiation due to absorption artifacts. In contrast, this effect was less pronounced for COC. Moreover, we observed that after thermal fusion bonding of the device's cover plate to the substrate, many of the generated functional groups buried into the bulk rendering them inaccessible. The propensity of this surface reorganization was found to be higher for PMMA compared to COC. As an example of the effects of material and microchannel aspect ratios on device functionality, thermoplastic devices for the selection of CTCs from whole blood were evaluated, which required the immobilization of monoclonal antibodies to channel walls. From our results, we concluded the CTC yield and purity of isolated CTCs were dependent on the substrate material with COC producing the highest clinical yields for CTCs as well as better purities compared to PMMA.
Introduction
While many chip-based systems are built using glass or silicon substrates because their well-defined fabrication modalities and established chemistries allow the facile attachment of biologics to their surfaces, thermoplastics have been explored as alternatives to glass or silicon.1,2 The development of suitable polymer manufacturing techniques, such as hot embossing and injection molding, can generate high quantities of microfluidic chips at low cost. Therefore, devices made from thermoplastics can be particularly attractive for in vitro diagnostics due to the ability to accommodate the need for one-time-use operation.2–20 This is particularly true for circulating tumor cell (CTC) analyses, where whole blood clinical samples serve as the input and rare cells are selected and enumerated directly within the device. The rarity of CTCs makes potential sample carryover issues problematic; CTC analysis demands one-time use devices that can be produced in large quantities and with high fidelity.For microfluidic systems designed for in vitro diagnostics, surface functionalization and immobilization of biologics must, in most cases, be undertaken. For many non-functional surfaces, passive adsorption of the biologic to the surface is used, which can result in high loss of activity of the adsorbate.21,22 Alternatively, activity may be retained using covalent coupling chemistry, which requires surface functional groups on the substrate. An example would be a substrate containing surface confined carboxylic acids and reacting these with EDC/NHS reagents forming an ester intermediate that subsequently reacts with primary amine bearing biologics.22–24
Many thermoplastics do not contain surface functional groups, and therefore, activation protocols are employed to create the appropriate surface scaffolds. For example, devices using positive selection of CTCs require the attachment of monoclonal antibody (mAb) to appropriately prepared surfaces. A typical thermoplastic chip production and assembly pipeline using thermoplastics involves: (i) Forming the fluidic network on the appropriate substrate by molding; (ii) UV irradiation of exposed surfaces (cover plate and substrate) to generate functional scaffolds via photo-oxidation reactions; (iii) thermal fusion bonding the irradiated cover plate to the substrate to enclose microfluidic channels; and (iv) covalent coupling of biologics to the surfaces of the enclosed channels.25,26
The UV activation process is more accurately described as UV/O3 treatment using a quartz Hg lamp, which continually generates and destroys O3 yielding a steady-state concentration of strongly oxidizing atomic O. At sufficiently high energy, both UV exposure and oxidative stress can generate radicals within the polymer, which may break or scission polymer chains into smaller fragments, crosslink polymer chains, cause intramolecular rearrangements, and/or react with water or oxidative species to form carboxyls or other O-containing species.27–39 Thus, polymer surfaces are exposed to both intense UV light and highly reactive oxidizing species; we will refer to this UV/O3 process as UV activation in this manuscript.40
Even though there has been extensive work characterizing UV-induced functionalization of thermoplastics, most of these studies assessed the activation performance using planar substrates or thin films as models.27–37 For microfluidics, microchannels of various aspect ratios require surface functionalization, and it is not clear if the observations made for planar surfaces are applicable to microchannel surfaces.38,39,41 Several variables are implicitly different between planar surfaces and microstructured surfaces: (i) The bulk polymer's optical transmissivity may affect the flux of radiation reaching the interior of microchannel surfaces; and (ii) the channel's geometry may determine the uniformity of the activation protocol throughout the channel's cross-section, especially for high aspect ratio structures.
UV irradiation of polymer surfaces may result in the formation of photo-fragments due to scissioning of the polymer backbone and these photo-fragments can be more thermally mobile compared to the native polymer due to their lower molecular mass. Thus, thermal fusion bonding, which is accomplished near the glass transition temperature (Tg) of the polymer, may bury generated surface functional groups.42 Such effects have been indirectly observed for the long-term aging of appropriately modified thermoplastics.38 Therefore, the surface accessibility of functional groups could differ dramatically before and after thermal processing depending on the polymer substrate's tendency to fragment upon irradiation.
In this manuscript, we determined the extent and uniformity of the UV-induced generation of surface-confined carboxyl groups within thermally fusion bonded microchannels of different aspect ratios using UV-Vis spectroscopy, a uniquely adapted colorimetric assay, and imaging fluorescent dye-labeled oligonucleotides covalently immobilized to surface-generated functional groups. We also employed an array of surface characterization tools, including water contact angle measurements, atomic force microscopy (AFM), attenuated total reflectance Fourier-transform infrared (ATR-FTIR), Raman, and X-ray photoelectron (XPS) spectroscopies to assess thermal processing effects of UV-modified plastics. Poly(methyl methacrylate), PMMA, cyclic olefin copolymer, COC, and polycarbonate, PC, were evaluated. PC’s poor optical properties did not permit CTC evaluation via fluorescence imaging.43 Therefore, the CTC assay was abandoned using this polymer; however, surface characterizations of functionalized PC are presented in the ESI.†
The importance of high surface loading of a biologic to the appropriate high aspect ratio micro-structured thermoplastic device was demonstrated using the covalent attachment of mAbs directed against the epithelial cell adhesion molecule (EpCAM) to UV activated, high aspect ratio thermoplastic microchannels. Anti-EpCAM has been used extensively for the positive selection of CTCs from whole blood using microfluidics.23,24,44–53 Performance metrics such as the recovery and purity of isolated CTCs were evaluated, both of which depend intimately on the proper surface activation protocol of microchannel walls. Comparison of PMMA and COC for the isolation of CTCs was evaluated using clinical samples to select the appropriate thermoplastic substrate for maximizing the CTC clinical yield and purity.
Experimental methods
Reagents and chemicals
PMMA and 6013S-04 COC were purchased from Plaskolite (Columbus, OH) and TOPAS Advanced Polymers (Florence, KY), respectively. Frame-sealed incubation chambers were purchased from Bio-Rad (Hercules, CA). Chemicals and reagents used in these studies included: Micro-90, sodium dodecyl sulfate (SDS), sodium hydroxide, sodium carbonate and bicarbonate (Fisher Scientific, Houston, TX); reagent-grade isopropyl alcohol (IPA), 2-(4-morpholino)-ethane sulfonic acid (MES), bovine serum albumin (BSA), Triton X-100, and paraformaldehyde solution (PFA) (Sigma-Aldrich, St. Louis, MO); toluidine blue O (TBO) (Carolina Biological Supply, Burlington, NC); phosphate-buffered saline (PBS, pH = 7.4) (Life Technologies, Carlsbad, CA); 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) (Pierce, Rockford, IL); Cy3-labeled oligonucleotides (5′NH2-C6-TTT-TTT-TTT-TTC-CGA-CAC-TTA-CGT-TTT-TTT-T-Cy3-3′; Integrated DNA Tech., Coralville, IA); NorthernLights 493-streptavidin (NL493-streptavidin), monoclonal anti-EpCAM Abs, and Fc blocker IgG (R&D Systems, Minneapolis, MN); and 4′,6-diamidino-2-phenylindole (DAPI), and CD45-FITC and cytokeratin 8 and 19 (CK8/19-PK) Abs (eBioscience, San Diego, CA). Nuclease-free water and microtubes (Ambion, Foster City, CA) were used for preparation and storage of all samples and reagents unless noted otherwise.Fabrication and assembly of the microfluidic chips
Polymer microfluidic chips were hot embossed from a metal mold master. Fabrication of the CTC chip followed steps previously reported.23 Unless noted otherwise, microfluidic chips and planar substrates were sonicated in 10% Micro-90 for 10 min, rinsed with IPA and DI water, dried at 70 °C, and UV-modified for 15 min (22 mW cm−2) in a home-built UV activation chamber equipped with a quartz, low-pressure Hg lamp. Chips were thermally fusion bonded for 20 min at temperatures between 98 °C–102 °C for PMMA and 132–134 °C for COC.Water contact angle measurements
Sessile water contact angle results were obtained using a VCA Optima instrument (AST Products). The measurements entailed depositing 2.0 μL of DI water onto the appropriate substrate followed by collecting the image and measuring the contact angle using the manufacturer's software.Quantification of carboxylic acid surface densities
An in situ incubation chamber (BioRad) was attached to the substrate's surface and filled with 0.1% (w/v) TBO in carbonate buffer (50 mM, pH = 10.5). After 15 min, the substrate was submersed in the same buffer for 15 min and air dried. TBO was desorbed using 40% acetic acid (d = 1.0196 g mL−1), collected in a pre-weighed microfuge tube, and analyzed with an Ultrospec 4000 UV/Vis spectrophotometer (Pharmacia Biotech) against a 40% acetic acid blank. All TBO-determined carboxy surface densities were corrected for surface roughness as determined by AFM imaging.Spectroscopies
AFM images were acquired with an MFP3D AFM instrument (Asylum Research) in repulsive tapping mode at a rate of 0.40 Hz over a 100.0 μm2 area with 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 536 points per image. The Tap300A1-G cantilever tips (Ted Pella) had a frequency of 300 kHz and force constant of 40 N m−1. The manufacturer's software was used for image analysis. For XPS measurements, C 1s and O 1s photoelectron signals were acquired using an Axis Ultra DLD X-ray photoelectron spectrometer (Kratos Analytical) under ultra-high vacuum conditions (10−8 to 10−10 Torr) with a monochromatic Al Kα X-ray source, 20 eV pass energy, 370 s acquisition time, 1600 ms dwell time, and 20° electron take-off angle. Given an inelastic mean free path of 3–4 nm, ∼95% of the resultant signal originated 9–12 nm from the surface.54–56 ATR-FTIR spectra were scanned from 375–4000 cm−1 using an ALPHA FTIR spectrometer and a Platinum ATR module (Bruker Optics). Raman spectra were recorded using an inVia Raman Confocal microscope (Renishaw) excited using the 457.9 nm output of a Stabilite 2017 Ar ion laser (Spectra-Physics). A neutral density filter was used to produce 10 mW of laser power at the sample. Spectra were scanned from 610–2300 cm−1 with 10 s exposure averaged over 3 scans. For SEM, substrates coated with a 3–4 nm layer of Au/Pd were imaged under high vacuum with an FEI Quanta 200 field emission gun at a 10 kV accelerating voltage and an Everhart-Thornley detector.
536 points per image. The Tap300A1-G cantilever tips (Ted Pella) had a frequency of 300 kHz and force constant of 40 N m−1. The manufacturer's software was used for image analysis. For XPS measurements, C 1s and O 1s photoelectron signals were acquired using an Axis Ultra DLD X-ray photoelectron spectrometer (Kratos Analytical) under ultra-high vacuum conditions (10−8 to 10−10 Torr) with a monochromatic Al Kα X-ray source, 20 eV pass energy, 370 s acquisition time, 1600 ms dwell time, and 20° electron take-off angle. Given an inelastic mean free path of 3–4 nm, ∼95% of the resultant signal originated 9–12 nm from the surface.54–56 ATR-FTIR spectra were scanned from 375–4000 cm−1 using an ALPHA FTIR spectrometer and a Platinum ATR module (Bruker Optics). Raman spectra were recorded using an inVia Raman Confocal microscope (Renishaw) excited using the 457.9 nm output of a Stabilite 2017 Ar ion laser (Spectra-Physics). A neutral density filter was used to produce 10 mW of laser power at the sample. Spectra were scanned from 610–2300 cm−1 with 10 s exposure averaged over 3 scans. For SEM, substrates coated with a 3–4 nm layer of Au/Pd were imaged under high vacuum with an FEI Quanta 200 field emission gun at a 10 kV accelerating voltage and an Everhart-Thornley detector.
Conjugation reactions and fluorescence imaging
Cy3-labeled oligonucleotides (40 μM) suspended in PBS buffer (100 mM, pH = 7.4) containing EDC (10 mg mL−1) were either spotted (0.25 μL) on planar substrates or filled into assembled microfluidic devices. NL493-streptavidin was conjugated by functionalizing an activated surface with EDC (20 mg mL−1) and NHS (2 mg mL−1) in MES buffer (pH = 4.8) for 15 min, air drying, spotting (0.25 μL) with NL493-streptavidin (1 mg mL−1) in 150 mM PBS (pH = 7.4). These conjugation reactions proceeded overnight at 4 °C in the dark in a humidified incubation chamber. Planar substrates were incubated in 0.1% SDS overnight in the same manner. Microchannels were rinsed with >2 mL of 150 mM PBS (pH = 7.4) and >2 mL of 0.1% SDS in DI water. Microchannels were cut using a saw and were sonicated in DI water for 15 min prior to imaging. All images were 16-bit resolution without binning and were acquired for 230 ms using a 200M inverted microscope (Zeiss) that contained an XBO 75 Xe arc lamp, a single band filter set (Omega Optical), and a Cascade 1 K CCD camera (Photometrics) with Micro-Manager software. Final images were analyzed using the Image-J software.Microfluidic chips for CTC selection
CTC microfluidic chips were fabricated in PMMA or COC substrates. The chip design,57 which consisted of a z-configuration and was a second generation chip compared to our reported design,23,24 consisted of a 26.3 × 20.5 mm footprint with inlet and outlet leading channels (20.5 mm long, 400 μm wide, and 150 μm deep) connecting a series of 50-curvilinear channels that in concert formed the cell selection bed. Fig. 5 shows the CTC selection chip design and an SEM image of a section of the mold master with microchannels. Each curvilinear selection channel was 30.6 mm long, 150 μm deep, and 30 μm wide. The surface area of the CTC selection bed was 596 mm2 (11 mm2 per channel) with 45.1 mm2 of that surface area in the lead channels. The chip's total volume was 9.4 μL (138 nL per channel) with 2.5 μL volume for the lead channels. Monoclonal anti-EpCAM Abs were immobilized onto the chip's UV photo-activated surfaces following a procedure outlined in the ESI.† Computational modeling of the device's fluid dynamics is described in the ESI.†Patient derived xenografts (PDX) models for pancreatic ductal adenocarcinoma (PDAC) and clinical sample processing
Patient derived xenograft (PDX) models were generated from PDAC patient tumors and were grafted into the subcutaneous tissue of NSG/NOD or Nude mice under approval by the University of North Carolina's IRB following protocols set by the University of North Carolina School of Medicine Institutional Animal Care and Use Committee. Tumors in the mice were grown to a size of ∼2 cm3 for a period of 1 to 6 months. Further details are in the ESI.† Cardiac puncture was determined as the optimal route for collection of blood to minimize contamination by epithelial cells.58 A total of 9 PDX models were used for this study.Two patients with advanced melanoma and two with colorectal cancer were recruited according to a protocol approved by the University of North Carolina's IRB. All blood specimens were collected into BD Vacutainer® (Becton-Dickinson, Franklin Lakes, NJ) tubes containing the anticoagulant EDTA and were processed within 3 h of the blood draw.
For CTC analysis from patient derived xenografts, a 0.5–0.85 mL volume of whole blood was infused, and for clinical samples, 2 mL of blood was infused directly into the CTC chip at 1.6 mL h−1, or a linear velocity of ∼2.5 mm s−1.23 Following infusion of the blood sample, a post-selection rinse was performed with 2 mL of 150 mM PBS/0.5% BSA (pH = 7.4) at 3.2 mL h−1 (∼5.0 mm s−1). Selected cells were analyzed and identified via immunostaining by: (i) treating with Fc blocker (IgG); (ii) incubation with anti-mouse or anti-human CD45-FITC Abs for 30 min; (iii) cell fixation with 2% PFA; (iv) poration with 0.1% Triton-X100; and (v) incubation with CK8/19-PK Abs and the nuclear dye, DAPI. Images of stained cells were obtained using an Olympus IX71-DSU Spinning Disk Confocal inverted microscope controlled via MetaMorph software and furnished with 10x, 20x, and 40x dry objectives, a mercury arc lamp illumination source, two cameras (high sensitivity Hamamatsu EMCCD and high resolution Hamamatsu ORCA-03G CCD), and DAPI, FITC, TRITC, and Cy5 filter sets. CTCs from clinical samples were also enumerated with an impedance sensor. As the CTCs traversed through the impedance sensor, an electrical signal was recorded for single cells using electronics designed and built in-house as described previously.23 The raw output data was subjected to a 1000 point adjacent averaging algorithm to establish the baseline for the measurement without generating signal bias. Baseline was then subtracted from the data in order to correct for signal drift. Impedance responses were counted as CTCs when the signal-to-noise threshold exceeded 3:1.
Results and discussion
Herein, we investigated issues associated with UV activation of microstructured polymers to form surface-confined functional groups for biologic attachment. Specifically, we seek to address the following questions: What is the surface coverage uniformity of functionalities (i.e., carboxyl groups) in high-aspect ratio microchannel surfaces following UV-activation? Does the surface coverage depend on the channel's geometry and the polymer's optical properties? Does the thermal fusion bonding process affect surface functional group densities generated by UV irradiation? We will finally demonstrate how these issues impact the performance of a microfluidic device for CTC analysis.It has been documented that the surface density of mAbs on selection channel walls determines the adhesion force between the mAbs and a CTC. This adhesion force (FA) can be calculated from;
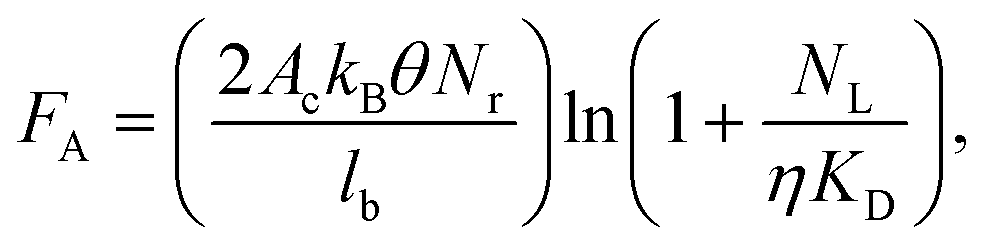 | (1) |
Assessing UV activation efficiency and uniformity within microfluidic channels
The polymer's optical properties and the channel's dimensions are two primary factors that may affect the efficiency of UV activation within microfluidic channels. If the polymer substrate's optical transmissivity is poor, the flux of modifying radiation reaching microchannel surfaces could be reduced, and this would be especially true for high aspect ratio microchannels, which enable high throughput processing of clinical specimens without sacrificing yield.59We first evaluated the transmissivity of 250 μm thick polymer films before and after UV activation (Table S1, ESI†). Native PMMA's 1.5% transmissivity at 254 nm was reduced to 0.5% after UV activation. Native COC showed a transmissivity of 53.8% that changed to 36.8% after 15 min UV irradiation. Due to the low transmissivity of PMMA, the flux of the UV radiation reaching surfaces of high aspect ratio structures will be damped, whereas this is less likely for COC microstructures.
We determined the carboxyl group surface densities for various aspect ratio microchannels using a colorimetric assay with the cationic TBO dye that binds electrostatically (1:1 ratio) to deprotonated carboxylic acid functionalities.60,61 Thermally fusion bonded microchannels were incubated with a TBO solution, then washed, and the remaining TBO molecules were released using acetic acid and the effluent evaluated spectrophotometrically. As a note, the TBO assay employed not only probed surface functional groups, but also could probe molecules in underlying layers due to photo-fragmentation of surfaces. Photo-fragmented surfaces were essentially porous to the small TBO molecules. Consequently, absolute carboxyl surface densities were biased by the extent of surface photo-fragmentation.
As the microchannel's aspect ratio increased, the carboxyl surface densities in both PMMA and COC microchannels decreased (Fig. 1). Even with an aspect ratio of 0.5, the carboxyl surface densities for PMMA decreased from 12.4 ± 1.8 nmol cm−2 to 3.5 ± 0.1 nmol cm−2 relative to UV-activated and thermally treated, planar substrates. Similarly, the respective values were 9.5 ± 2.3 nmol cm−2 to 2.6 ± 0.3 nmol cm−2 for COC microchannels. Further decreases in these signals due to increasing aspect ratio were likely caused by optical filtering of UV light, even in the more transparent COC substrate; however, it is unknown whether decreased UV flux would lead to less surface carboxyl formation or reduced efficiency of photo-fragmentation processes.
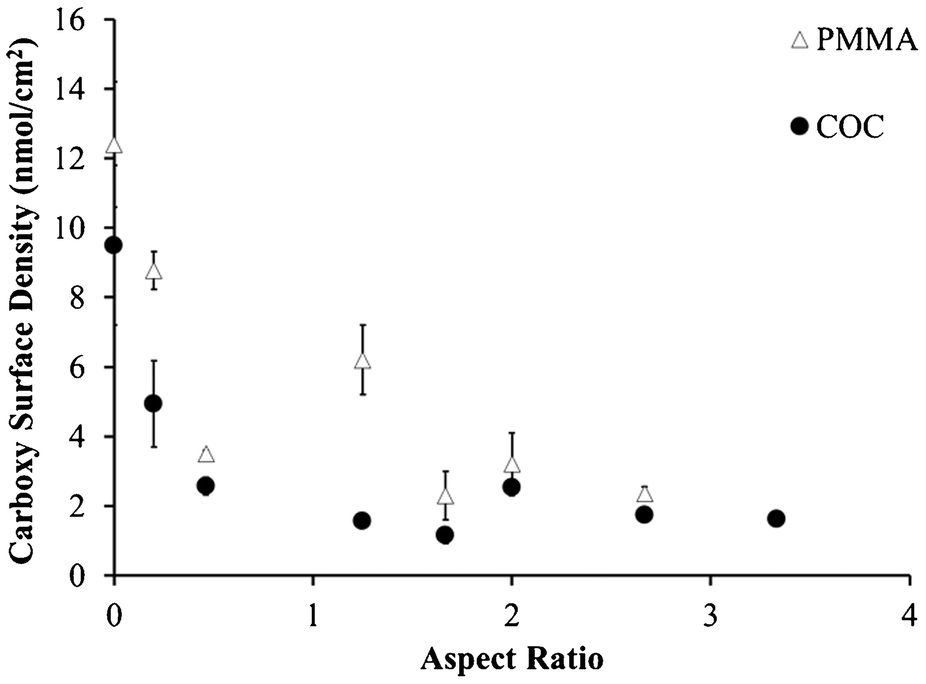 | ||
| Fig. 1 Carboxy surface densities determined by collecting TBO solution from UV-modified and thermal fusion bonded PMMA and COC microchannels with several different aspect ratios. Zero aspect ratio indicates UV/thermal, planar substrates. | ||
To specifically assess the surface densities of carboxyl groups that are accessible to biologic macromolecules on the microstructures, we coupled fluorescently-labeled (3′ end) oligonucleotides containing a pendant amino group on their 5′ end to accessible carboxyl groups in microchannels. Results indicated that PMMA's background corrected fluorescence was 25% of COC's fluorescence intensity, 503 ± 72 cps vs. 2357 ± 218 cps, respectively (Fig. 4G–H). These observations match spectroscopic evidence (see below) that show more efficient carboxyl formation on COC surfaces, likely due to highly competitive scissioning pathways leading to more photo-fragmentation for PMMA.
In microchannels cut along their lengths to expose oligonucleotides immobilized onto the sidewalls, we observed non-uniform coverage of Cy3-labeled oligonucleotides on the PMMA channel wall (Fig. 2), where the top third of the microchannel showed significantly higher levels of fluorescence (384 ± 81 cps) compared to the bottom two thirds of the same channel (112 ± 55 cps). However, this was not observed for COC microchannels, which showed both higher oligonucleotide loading based on higher fluorescence intensity irrespective of the vertical position along the channel wall (2233 ± 310 cps over entire depth).
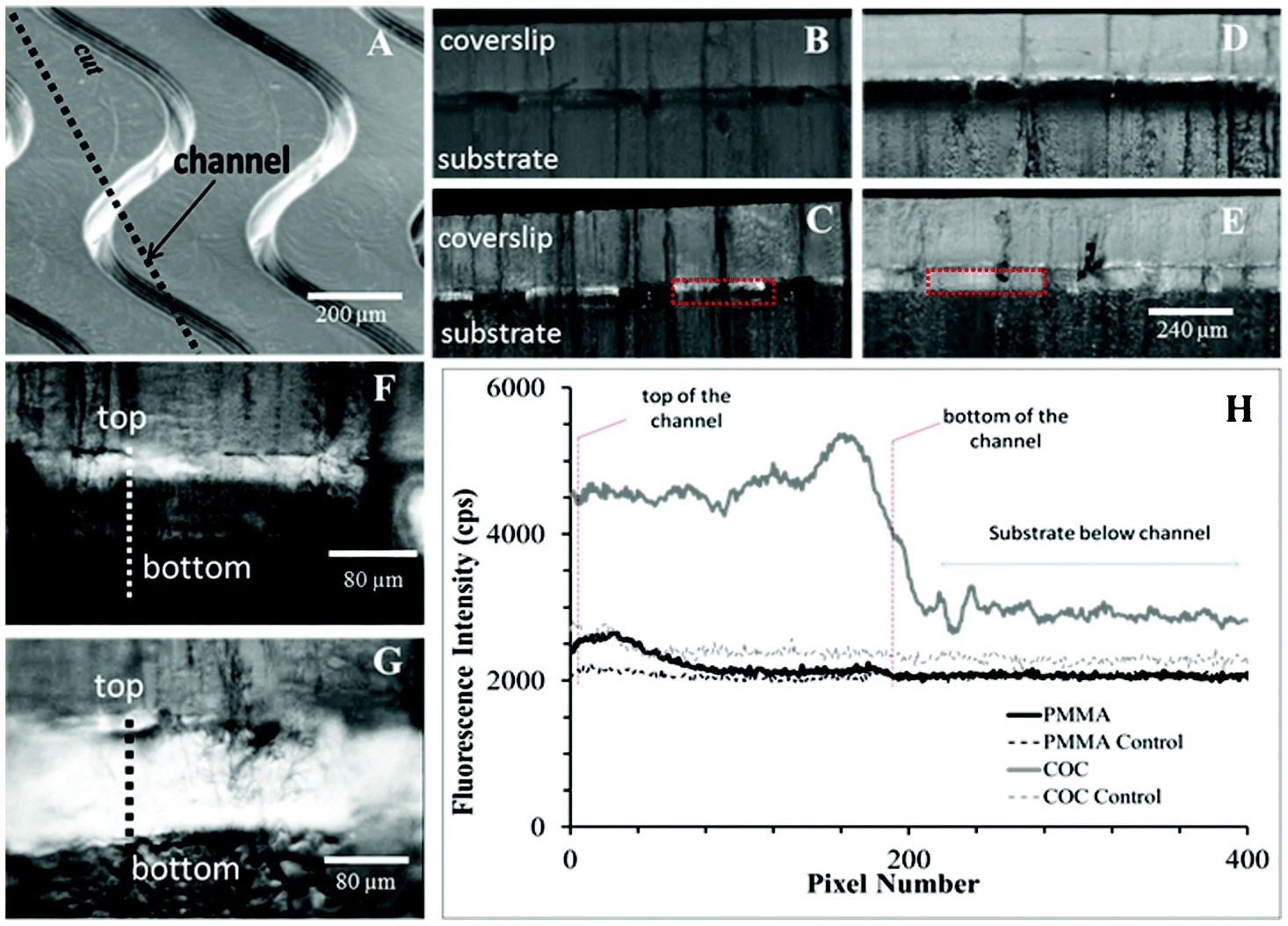 | ||
| Fig. 2 (A) Curvilinear channels (30 × 80 μm, w × d) were cut along their length to expose Cy3-labeled oligonucleotides immobilized along sidewalls. 5× fluorescence images of controls and Cy3-labeled oligonucleotides are shown for (B,C) PMMA and (D,E) COC, respectively. 20× fluorescence images for (F) PMMA and (G) COC are presented along with (H) line plots as indicated by the thick, dotted lines. Controls were Cy3-labeled oligonucleotides immobilized without the EDC coupling agent and measure nonspecific adsorption and autofluorescence. Only control images are scaled to the same intensity as their counterparts. All other fluorescence images are not normalized. | ||
Thermal effects on UV-activated thermoplastics
Following micro-replication and UV-activation of the micro-structured substrate, a cover plate is thermal fusion bonded to the substrate to enclose the fluidic network.23,24,45 In this section, we were interested to understand the effects of thermal processing on the accessibility of the functional groups resulting from the UV activation step. To understand these effects, we examined planar sheets of the appropriate thermoplastic with a variety of spectroscopic techniques to determine the surface chemistry resulting after thermal treatment.The water contact angles (WCAs) for native PMMA and COC substrates were 76.4 ± 1.4° and 95.5 ± 1.9°, respectively. Because COC is composed entirely of saturated hydrocarbons, this polymer is more hydrophobic than PMMA, which contains ester moieties producing a lower water contact angle for native PMMA. Neither of these native polymers should generate significant TBO colorimetric signals because the positively charged TBO molecules electrostatically bind only to negatively charged functional groups (see ESI† for a thorough description of the TBO assay).60,61 At pH = 10.5, only carboxylic acids are deprotonated and thus available for TBO association. The small signal (0.2–0.3 nmol cm−2) observed for the native surfaces (Table 1) was most likely due to non-specific adsorption to the surface.62 All values discussed henceforth have been corrected for this background signal.
| Substrate | Treatment | Water contact angle (°) | RMS roughness (nm) | COOH surface density (nmol cm−2) | O/C ratio | COOH C 1s (%)a | FTIR peak area (a.u. cm−1) | |
|---|---|---|---|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
O–H | |||||||
| a % COOH of total C 1s signal. | ||||||||
| PMMA | Native | 76.4 ± 1.4 | 1.4 | 0.2 ± 0.1 | 0.32 | 0.0 | 21.4 | 3.1 |
| UV | 36.7 ± 0.9 | 8.6 | 14.7 ± 2.6 | 0.38 | 1.9 | 25.2 | 13.9 | |
| UV + 102 °C | 63.9 ± 1.2 | 9.0 | 12.4 ± 1.8 | 0.32 | 1.0 | 24.8 | 8.5 | |
| COC | Native | 95.5 ± 1.8 | 4.8 | 0.3 ± 0.2 | 0.01 | 0.0 | 0.0 | 0.0 |
| UV | 43.1 ± 1.9 | 17.9 | 19.0 ± 2.9 | 0.27 | 8.9 | 11.2 | 13.4 | |
| UV + 130 °C | 80.0 ± 3.0 | 12.0 | 9.5 ± 2.3 | 0.10 | 3.0 | 10.5 | 7.8 | |
Upon UV irradiation, activation of the surfaces was apparent as the wettability increased; water contact angles decreased by 52.0 ± 1.6% (from 76.4 ± 1.4° to 36.7 ± 0.9° after modification) and 52.8 ± 2.5% (95.5 ± 1.8° to 43.1 ± 1.9° after modification) for UV-modified PMMA and COC, respectively (Table 1). The TBO signals for these surfaces corresponded to carboxyl functional group densities (see calibration curve in Fig. S1, ESI†) of 14.5 ± 2.6 nmol cm−2 and 18.7 ± 2.9 nmol cm−2, respectively. As noted, while these TBO signals are indicative of relative changes in the degree of activation through generation of carboxyl groups, they cannot be interpreted as absolute surface densities.
The carboxyl group densities on UV-treated polymers determined by the TBO assay were higher than theoretically possible for a carboxylic acid monolayer on either surface (0.83 nmol cm−2).63 This anomaly can be explained by scissioning and fragmentation of the surface polymer chains upon UV irradiation.37,64 The photo-damaged surface may be porous to the TBO molecules, increasing the apparent probed surface area several-fold.37 We provide thorough evidence for this photo-fragmentation in the ESI† including microscopy images of ablated surfaces (Fig. S3, ESI†).
After the UV-activated PMMA surface was washed with IPA, the RMS roughness decreased by 90% (Fig. S2, ESI†), and the TBO signal decreased to 1.5 ± 0.5 nmol cm−2, which agreed with previously reported values.37,65 These changes likely occurred due to dissolution and removal of carboxylated surface photo-fragments (Fig. S2, ESI†).66 Consequently, these surface fragments can confound the TBO results. For example, even though ablation and fragmentation were substantial for UV-irradiated PMMA (Fig. S3, ESI†), the UV-COC surface gave the greatest TBO signal (18.7 ± 2.9 nmol cm−2). This signal is likely due to more efficient carboxylic acid formation rather than increased fragmentation of the UV-activated COC surface, which is in accordance with our spectroscopic observations (see below).
These carboxylated photo-fragments have a lower molecular mass and higher thermal mobility than the native material, making them critical effectors of thermally-induced changes on the surfaces' carboxyl functional group densities.42 With this in mind, it is not surprising that the UV-activated PMMA surfaces would remain porous after heating, as evident by only a slight decrease in its TBO signal, whereas the less fragmented UV-activated COC surface showed larger decreases in its TBO signal. Accompanying these losses, the water contact angles for these surfaces returned to near their native values following thermal treatment (see Table 1),67 and there was a change in the UV-activated PMMA surface's morphology (see AFM images, Fig. S4, ESI†) that was not observed for the UV-activated COC surface. These observations may be attributed to rearrangement of hydrophilic functional groups away from the surface due to the interfacial energy with hydrophobic air, thereby becoming inaccessible.38 This rearrangement would explain the losses in wettability for the thermally treated UV-activated PMMA and COC substrates and should be independent of the starting material's molecular weight and purity when substantial fragmentation occurs (Fig. S5, ESI†).
ATR-FTIR, Raman, and XPS analyses of thermally treated, UV-activated polymers
For the native substrates, all spectra agreed with referenced spectra.38,68–70 For semi-quantitative analysis, ATR-FTIR signatures were integrated over the CO and O–H stretching regions. XPS C 1s spectra were deconvoluted to calculate the percent of the C 1s signal from carboxyl groups (see ESI†). The XPS and ATR-FTIR data are presented in Table 1, and the C 1s spectra and relevant regions of the FTIR spectra are presented in Fig. 3. Deconvoluted peaks for the native and UV-modified polymers are presented in Fig. S6, ESI,† and a detailed set of deconvoluted C 1s peak percentages are available in Table S2, ESI.†
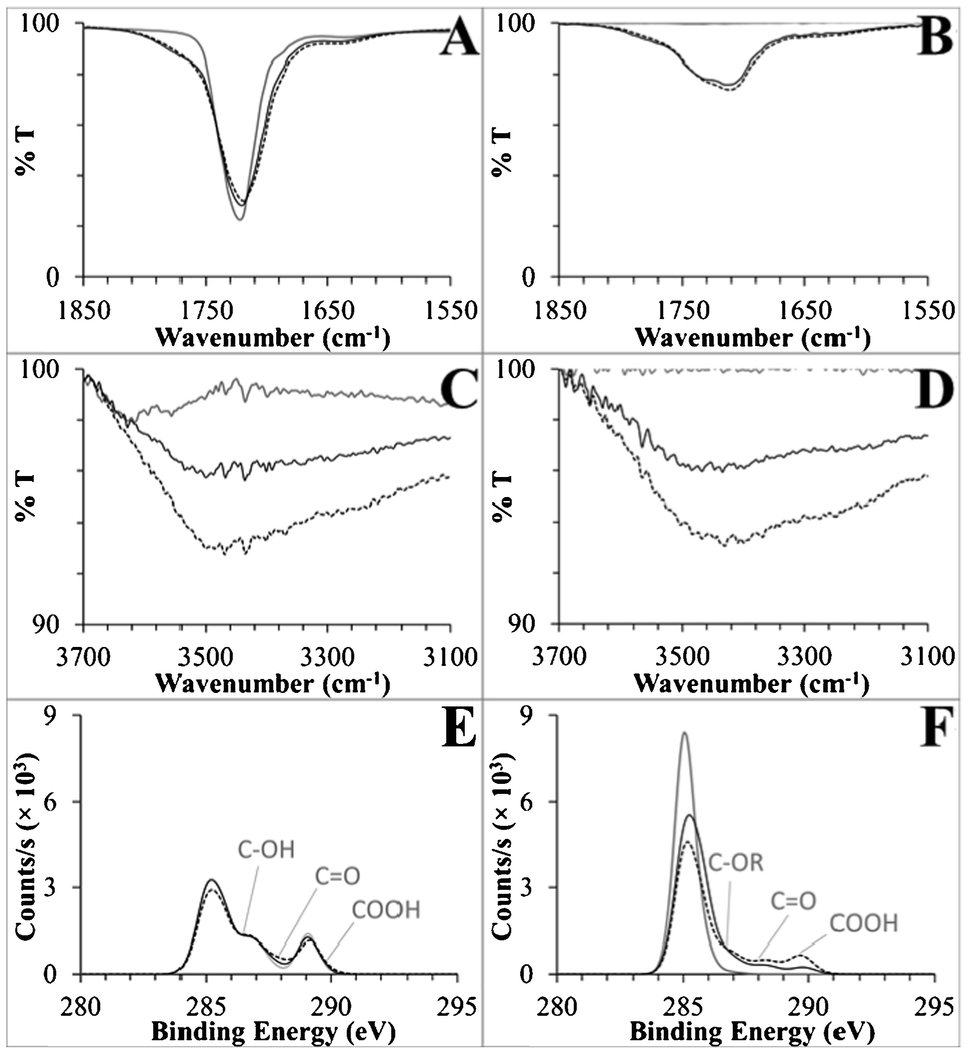 | ||
| Fig. 3 (A,B) Carbonyl regions of the ATR-FTIR spectra, (C,D) hydroxyl regions of the same spectra, and (E,F) C 1s XP spectra for PMMA and COC, respectively. Shown are spectra for native substrates (solid grey lines), UV-activated (dotted black lines), and UV/thermal (solid black lines) surfaces. General peak positions are labeled on the XP spectra corresponding to deconvoluted functional groups, where R generically represents carbon or hydrogen. See the ESI† for detailed XPS deconvolution methods and data. | ||
There were changes in the carbonyl region of the UV-activated PMMA ATR-FTIR spectra (Fig. 3A). The native substrate's ester CO stretch at 1719 cm−1 broadened to include stretches with higher and lower energy (an increase in peak area of 3.8 au cm−1 integrated from 1650–1850 cm−1) that corresponded to the formation of carboxyls and aldehydes/ketones, which comprised only 1.9% and 7.0% of the XPS C 1s signal, respectively.71 Hydroxyl groups were abundant in the XPS data, 8.5% of UV-activated PMMA's total C 1s signal (Fig. 3C, Table S2, ESI†). Taken together, these data strongly suggest that carboxyl formation is only a minor product of PMMA's UV activation, with scissioning and aldehyde, ketone, and hydroxyl formation serving as competing reactions.
Strong signals from carboxyl containing functionalities were observed spectroscopically for UV-activated COC. Native COC did not contain either carbonyl or hydroxyl peaks, but after UV activation, a CO peak at 1711 cm−1 and a 3430 cm−1 hydroxyl peak were observed with relatively large peak areas of 11.2 au cm−1 (1520–1850 cm−1) and 13.4 au cm−1 (3075–3700 cm−1), respectively. In an effort to deconvolute the CO peak and isolate carboxyl formation, we first disproved the supposition that the small shoulder at 1637 cm−1 (Fig. 3B) was due to alkene formation after scissioning of COC's norbornane ring because no changes were observed in the Raman spectra of UV-modified COC (Fig. S7, ESI†). Rather, aldehydes/ketones generated this stretch along with 7.1% of the deconvoluted C 1s spectrum, as compared to 8.9% carboxyl (Fig. 3F, Table S2, ESI†). These data are the first detailed spectral information to be published with regards to the UV/O3 activation of COC and showed that carboxyl groups can be directly generated on a COC surface by this simple activation modality.40,72 Moreover, this activation method is clearly more efficient at producing carboxyl groups on COC than PMMA.
Besides quantification of the formation of surface-confined carboxylic acids, of great interest was whether the ATR-FTIR and XPS results support or reject the hypothesis that photo-fragments have a propensity to thermally rearrange and thus bury carboxyl moieties into the bulk polymer. In general, ATR evanescent waves penetrate from hundreds of nanometers to tens of micrometers, but penetration depth is linearly dependent on the wavelength of light passing through the ATR crystal and inversely dependent on the wavenumber.73 Therefore, hydroxyl peaks are more surface specific, with a penetration depth roughly half that of carbonyl signals. Decreases in the UV and thermally treated PMMA and COC surface CO peak areas were only 11% and 6.2% as compared to larger change closer to the surface in the O–H peak areas, 50% and 42%, respectively. Secondly, the carboxyl C 1s signals, which are highly surface specific probing only 9 to 12 nm into the bulk,54–56 decreased by 47% and 66% after heating UV-activated PMMA and COC, respectively, and the corresponding decreases in the O/C ratios were 100% and 63%. This data supports the generation of hydrophobic surfaces by thermal migration of hydrophilic functional groups present on photo-fragments, including carboxylic acids, away from the surface during thermal processing.
Fluorescence images of immobilized Cy3-labeled oligonucleotides to UV treated thermoplastics
Selection of a polymer for a fluidic device based on efficient coupling of biological macromolecules, such as mAbs, to its surface-confined carboxylic acid groups depend on both the degree of UV-induced carboxyl formation and fragmentation during UV exposure. Even though carboxyl surface densities are generally reduced during thermal processing, we have not shown that this will completely inhibit the coupling of biologics to these functional scaffolds. As an example, biologic monolayers are roughly three orders of magnitude less dense than carboxyl monolayer.63To illustrate the consequences of the heating effects detailed above, we covalently tethered Cy3-labeled oligonucleotides to substrates that were UV-activated and subsequently thermally treated (see Fig. 4). The background-subtracted fluorescence intensity on UV-activated COC presented higher signal (1978 ± 229 cps) compared to UV-activated PMMA (282 ± 98 cps). The oligonucleotide load on UV-activated COC was ∼5 times greater than on UV-activated PMMA and indicated that surface-accessible carboxylic acids generated on the UV-activated PMMA surface must be less than an oligonucleotide monolayer. After heating the UV-activated PMMA surface, the fluorescence of PMMA's oligonucleotide load decreased substantially (61 ± 98 cps), matching the substantial losses after heating observed in the FTIR and XPS data.
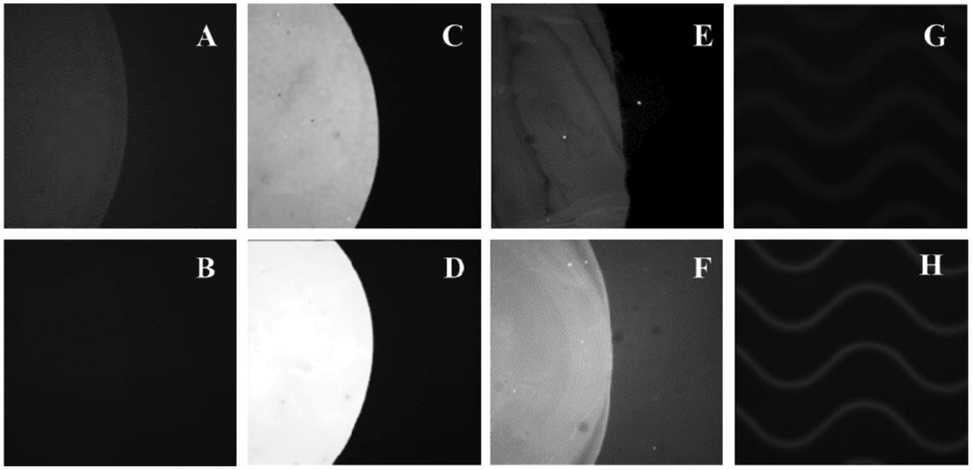 | ||
| Fig. 4 Images of Cy3-labeled oligonucleotides immobilized on UV and UV/thermal planar substrates for (A,B) PMMA and (C,D) COC, respectively. (E,F) NL493-streptavidin was immobilized on UV and UV/thermal COC, respectively. Images of Cy3-labeled oligonucleotides immobilized within UV-modified and thermal fusion bonded (G) PMMA and (H) COC microchannels. All fluorescence images of Cy3-labeled oligonucleotides are scaled to same intensity. NL493-streptavidin images are scaled independently. | ||
The oligonucleotide load on UV-activated COC increased after thermal treatment (Fig. 4D). However, we speculate that this observation was an artifact of the surface's increased hydrophobicity after heating, which could have improved the kinetics of oligonucleotide immobilization by hydrophobic interaction with the partially unfolded oligonucleotide.74,75 This is supported by the fluorescence of immobilized NL493-streptavidin on both UV-activated COC and thermally treated UV-activated COC surfaces (Fig. 4E–F), which yielded signals that were not statistically difference at the 95% confidence level.
Polymer choice and UV-activation scheme for efficient CTC clinical yield and purity
Thermoplastics are attractive substrates for in vitro diagnostics, such as CTC selection assays, due to the ability to manufacture devices at low cost and in a high production mode, and UV activation is a simple method to prepare these substrates for the covalent attachment of positive selection moieties such as anti-EpCAM mAbs.57,76 In light of the results discussed above, the choice of thermoplastic (PMMA or COC) may be guided by the polymer's response to UV radiation. In this section, we evaluated UV-activated and thermally fusion bonded PMMA and COC devices for the positive selection of CTCs. In particular, we were interested in comparing clinical yields and purity of the selected CTC fractions from undiluted, unfixed, and unfractionated blood.The CTC selection device utilized herein employed an array of 50, curvilinear, high-aspect ratio microchannels with nominal dimensions similar to ones previously reported by our group (30 μm × 150 μm × 30.6 mm, w × d × l).23,24 The depth of these channels increased the throughput as well as provided reduced pressure drop throughout the selection channels, especially when occupied by captured CTCs.23,57 SEM indicated that the widths of the top and bottom of the channels were 27.8 ± 1.0 μm and 19.7 ± 0.5 μm, respectively (aspect ratio of ∼6.3).77 On average, the widths of these microchannels are only slightly larger than the average CTC diameter (12–20 μm) but much larger than the average leukocyte diameter (7–15 μm). Channel width plays a critical role in maximizing the probability of cell/wall interactions and allows for achieving high CTC yield but lower probability of interactions with smaller cells.23 For even smaller cells, such as erythrocytes, the likelihood of approaching the channel wall is very limited due in part to the formation of a marginal cell-free layer.78,79
Fluidic addressing of the microchannel array was achieved using a z-geometry, in which large cross-section inlet and outlet channels were poised orthogonal to the curvilinear microchannels (see Fig. 5). This geometry was recently introduced by our group as a replacement for our previous CTC isolation device and offers a smaller overall footprint, retains uniformity of the flow between all selection channels thus ensuring optimal recovery throughout the device,23 and demonstrates a reduction in the formation and persistence of air bubbles formed during blood infusion. Additionally, the z-geometry addressing is easily scalable to isolation beds comprised of larger number of microchannels, which allows for high throughput processing of relatively large input volumes (<45 min for 500 selection channels and 7.5 mL input volumes).57
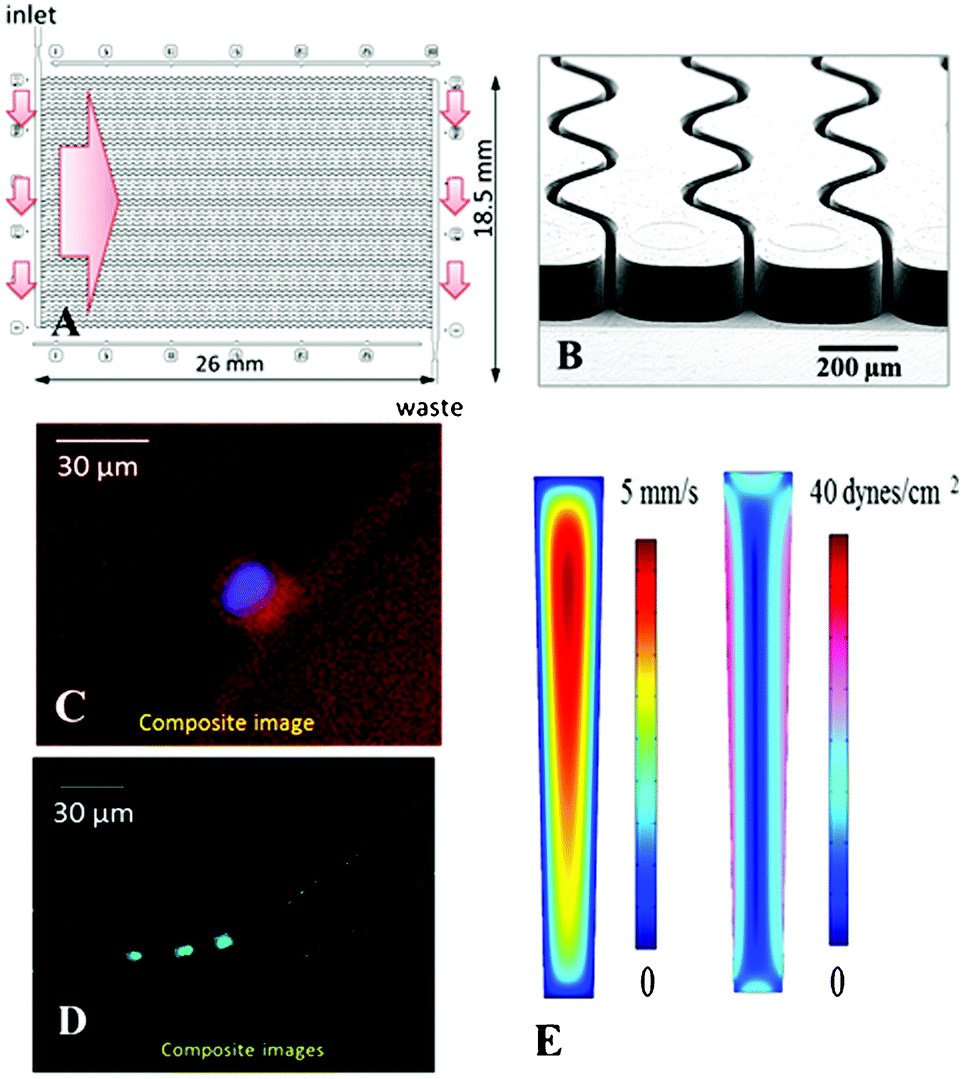 | ||
| Fig. 5 (A) Schematic operation of the CTC selection module with 50 parallel, sinusoidal microchannels and inlet/outlet channels arranged in the z-configuration. The large arrow indicates sample flow direction through the selection channels. (B) SEM of the selection bed showing high-aspect ratio (30 × 150 μm, w × d) sinusoidal microchannels and the output channel. (C) Composite fluorescence images of a CTC stained with DAPI(+), CK8/19(+), and not with CD45(−). (D) Four WBCs staining positively for DAPI(+) and CD45(+) and negatively for CD8/19(−). (E) Fluid dynamics simulation results showing the distribution of flow velocities and shear stress in microfluidic selection channels. | ||
For both PMMA and COC substrates, hot-embossed microfluidic devices and cover plates were UV-irradiated and thermal fusion bonded prior to EDC/NHS coupling of mAbs to the microchannel surfaces. Between 500 and 850 μL of whole blood, acquired from PDX models that were engrafted with tumor tissue biopsied from human patients with metastatic pancreatic ductal adenocarcinoma (PDAC), were infused through UV-activated PMMA (n = 5) and UV-activated COC (n = 4) devices. After cell selection and rinsing, cells were stained on-chip and counted, and the devices' figures-of-merits were evaluated. White blood cells were scored as DAPI(+) and FITC-CD45(+), but PE-CK8/19(−). CTCs were DAPI(+), PE-CK8/19(+), and FITC-CD45(−) (see Fig. 5C–D). Purity was defined as the ratio of PE-CK8/19(+) cells to the total number of nucleated cells (PE-CK8/19(+) and FITC-CD45(+)) captured within a device.80Fig. 6 provides a summary of the CTC results for PMMA and COC devices.
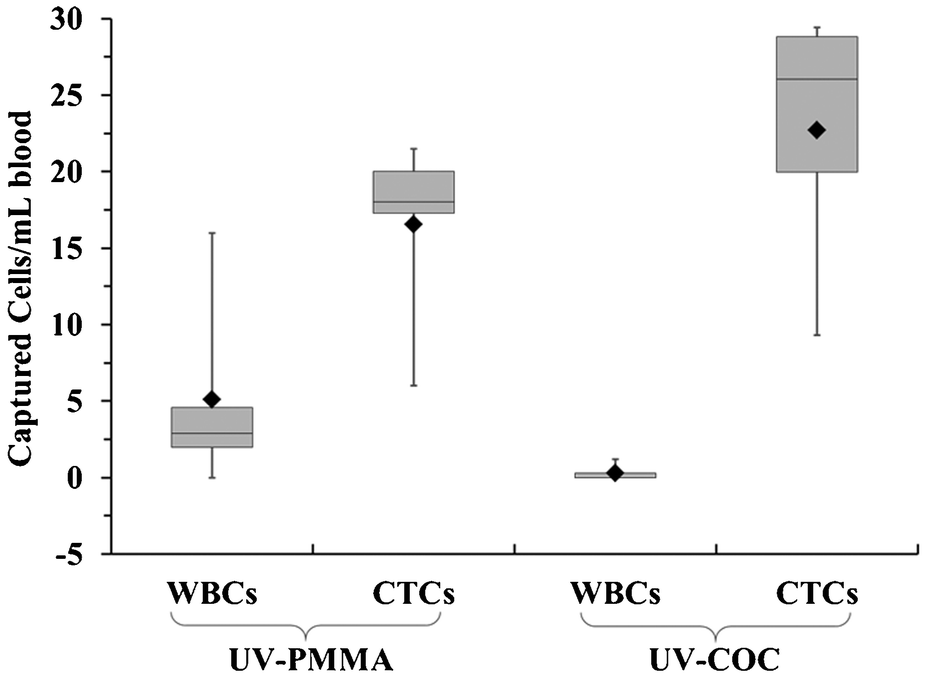 | ||
| Fig. 6 (A) Box plots presenting counts of CTCs and WBCs selected in UV-PMMA (5 PDX) and UV-COC (4 PDX) chips from mice blood samples. Data are normalized to 1 mL. Lower and upper edge of box shows 25th and 75th percentiles, respectively. Solid line in box represents median, and solid diamond represents mean. Error bars show maximum and minimum values. | ||
COC devices provided higher clinical yields of CTCs compared to PMMA devices. We observed a 37 ± 20% increase in the number of selected CTCs from PDX models for UV-activated COC versus UV-activated PMMA devices. For the high aspect ratio channels, higher loads of covalently coupled mAbs throughout the channel wall depth were observed for the COC device compared to the PMMA device. In the PMMA device, a portion of mAbs may have been physisorbed to the selection surfaces, especially towards the bottom of the channel, leading to decreased antigen-binding activity.22 The higher load of active mAbs to the COC microchannel surfaces (effective increase of NL in Eq. (1)) would improve the adhesion force of selected CTCs, especially for CTCs with low EpCAM expression that are expected in clinical samples, and provide higher yields consistent with Eq. (1).57 Two melanoma and two colorectal cancer patients blood were also analyzed using UV-activated and mAb functionalized COC selection devices using anti-EpCAM mAbs; we detected an average of 29 ± 17 CTCs and 30 ± 6 CTCs in 2 mL of blood (negative control CTCs = 3 ± 3 in 2 mL).
We determined the purity level of the selected fractions from PDX mouse model samples and found these to be 78.3 ± 19.7% and 98.8 ± 2.4% for the UV-activated PMMA and UV-activated COC devices, respectively. Furthermore, the purities of the isolated CTCs from clinical samples were >90%. Studies using the CellSearch® system have reported much lower purities, approximately 0.01 to 0.1%.81
The improvement in purity for COC devices can be explained by more efficient surface activation and mAb immobilization. Interfering leukocytes can be activated by various properties of a surface, including surface polymer chain mobility, surface chemical composition, hydrogen bonding properties, charge density, and hydrophobicity/hydrophilicity.83,84 Within short periods of time, a polymer's hydrophobic domain can lead to leukocyte adsorption. Due to less efficient activation, the presence of these hydrophobic domains was likely more prevalent in UV-activated and thermally fusion bonded PMMA devices as compared to COC devices.
We conducted three-dimensional, computational simulations of the dynamics of fluid flow through the microfluidic selection channels (details given in the ESI† and Fig. S10, ESI†) and observed average shear stresses of 3.4 dynes cm−2 for buffer and 13.3 dynes cm−2 for blood, which matches well with analytic expressions and is approximately an order of magnitude larger than comparable devices.43,85–87 In such flow conditions, the high shear stress along the high aspect ratio microchannels (see Fig. 5E) can disrupt associations with low FA (Eq. (1)). Thus, a high percentage of weakly associated leukocytes should be removed.23,85 This may not be the case for the designs utilizing microposts or herringbones as they possess low velocity regions behind posts and within herringbone grooves.44,87 Leukocytes in these regions will likely not experience strong shear forces to remove them from the surface. Even though we observed an increase in purity of isolated CTC fractions for UV-activated COC devices, the purities for both materials are the highest reported for CTC analysis.44,80,88
Conclusion
We have provided evidence elucidating two previously unreported effects that occur during the UV-activation of polymer microfluidic devices that are subjected to thermal fusion bonding for assembly and are also comprised of high aspect ratio microchannels, especially those applied to CTC analysis. Our results indicated that thermal fusion bonding of microfluidic channels reduces carboxylic acid surface densities because of photo-fragmentation occurring during UV irradiation. This phenomenon is much more pronounced for PMMA as compared to COC. Furthermore, the extent and uniformity of microchannel activation was highly dependent on the polymer's optical properties. For COC, its excellent optical properties enabled uniform functional group generation throughout the microchannel's depth, even for high aspect ratio structures, that translated into a more uniform coverage of mAbs attached to its walls. We applied this knowledge to guide the choice of proper material for a microfluidic device to select CTCs directly from whole blood with high recoveries and purities. The COC-based device provided both higher clinical CTC yields and better purity of the selected fractions compared to PMMA.We are currently in the process of evaluating CTCs using our UV-activated COC CTC device from other epithelial cancers as well, such as ovarian, prostate, and pancreatic cancers, and will be reported in subsequent work.57 In addition, a critical comparison of the performance metrics using the CellSearch® system and the CTC technology reported herein, such as capture efficiency, purity, and the ability to enrich cells with low EpCAM expression levels, are on-going and will be the subject of a future publication.82
Acknowledgements
Authors would like to thank the UNC's the University Cancer Research Fund (UCRF) for financial support of this work. We also acknowledge Dr. Carrie Donley, Director of the UNC Chapel Hill Analytical and Nanofabrication Laboratory, for help in acquiring and analyzing XPS data. We thank the UNC-Olympus Imaging Research Center for access to the Olympus IX71-DSU Spinning Disk Confocal inverted microscope. For Raman analysis, the authors acknowledge support for the purchase of instrumentation from the UNC Energy Frontier Research Center for Solar Fuels funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001011 and from UNC Solar Energy Research Center (SERC) Instrumentation Facility funded by the US Department of Energy – Office of Energy Efficiency & Renewable Energy under Award Number DE-EE0003188.References
- A. de Mello, Lab Chip, 2002, 2, 31N–36N RSC.
- S. A. Soper, S. M. Ford, S. Qi, R. L. McCarley, K. Kelly and M. C. Murphy, Anal. Chem., 2000, 72, 642A–651A CrossRef.
- M. Hashimoto, M. L. Hupert, M. C. Murphy, S. A. Soper, Y. W. Cheng and F. Barany, Anal. Chem., 2005, 77, 3243–3255 CrossRef CAS.
- F. Xu, P. Datta, H. Wang, S. Gurung, M. Hashimoto, S. Wei, J. Goettert, R. L. McCarley and S. A. Soper, Anal. Chem., 2007, 79, 9007–9013 CrossRef CAS.
- A. F. Sauer-Budge, P. Mirer, A. Chatterjee, C. M. Klapperich, D. Chargin and A. Sharon, Lab Chip, 2009, 9, 2803–2810 RSC.
- K. Han, T. Y. Lee, D. E. Nikitopoulos, S. A. Soper and M. C. Murphy, Anal. Biochem., 2011, 417, 211–219 CrossRef CAS.
- A. Kotani, M. A. Witek, J. K. Osiri, H. Wang, R. Sinville, H. Pincas, F. Barany and S. A. Soper, Anal. Methods, 2012, 4, 58–64 RSC.
- H. Wang, H.-W. Chen, M. L. Hupert, P.-C. Chen, P. Datta, T. L. Pittman, J. Goettert, M. C. Murphy, D. Williams, F. Barany and S. A. Soper, Angew. Chem., Int. Ed., 2012, 51, 4349–4353 CrossRef CAS.
- Y. W. Chen, H. Wang, M. Hupert and S. A. Soper, Analyst, 2013, 138, 1075–1083 RSC.
- A. Alrifaiy, O. A. Lindahl and K. Ramser, Polymers, 2012, 4, 1349–1398 CrossRef CAS.
- U. M. Attia and J. R. Alcock, Int. J. Adv. Manuf. Technol., 2010, 48, 973–991 CrossRef.
- A. Bhattacharyya and C. M. Klapperich, Anal. Chem., 2006, 78, 788–792 CrossRef CAS.
- J. Do, S. Lee, J. Y. Han, J. H. Kai, C. C. Hong, C. A. Gao, J. H. Nevin, G. Beaucage and C. H. Ahn, Lab Chip, 2008, 8, 2113–2120 RSC.
- K. Y. Hwang, J. H. Kim, K. Y. Suh, J. S. Ko and N. Huh, Sens. Actuators, B, 2011, 155, 422–429 CrossRef CAS.
- J. Rossier, F. Reymond and P. E. Michel, Electrophoresis, 2002, 23, 858–867 CrossRef CAS.
- M. W. Royal, N. M. Jokerst and R. B. Fair, IEEE Photonics J., 2012, 4, 2126–2135 CrossRef.
- A. F. Sauer-Budge, P. Mirer, A. Chatterjee, C. M. Klapperich, D. Chargin and A. Sharon, Lab Chip, 2009, 9, 2803–2810 RSC.
- S. Schumacher, J. Nestler, T. Otto, M. Wegener, E. Ehrentreich-Forster, D. Michel, K. Wunderlich, S. Palzer, K. Sohn, A. Weber, M. Burgard, A. Grzesiak, A. Teichert, A. Brandenburg, B. Koger, J. Albers, E. Nebling and F. F. Bier, Lab Chip, 2012, 12, 464–473 RSC.
- S. Song and K. Y. Lee, Macromol. Res., 2006, 14, 121–128 CrossRef CAS.
- Z. Zhao, R. Peytavi, G. A. Diaz-Quijada, F. J. Picard, A. Huletsky, E. Leblanc, J. Frenette, G. Boivin, T. Veres, M. M. Dumoulin and M. G. Bergeron, J. Clin. Microbiol., 2008, 46, 3752–3758 CrossRef CAS.
- T. M. Herne and M. J. Tarlov, J. Am. Chem. Soc., 1997, 119, 8916–8920 CrossRef CAS.
- J. E. Butler, L. Ni, W. R. Brown, K. S. Joshi, J. Chang, B. Rosenberg and E. W. Voss Jr, Mol. Immunol., 1993, 30, 1165–1175 CrossRef CAS.
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Gottert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS.
- U. Dharmasiri, S. Balamurugan, A. A. Adams, P. I. Okagbare, A. Obubuafo and S. A. Soper, Electrophoresis, 2009, 30, 3289–3300 CrossRef CAS.
- J. M. Goddard and J. H. Hotchkiss, Prog. Polym. Sci., 2007, 32, 698–725 CrossRef CAS.
- C.-W. Tsao and D. DeVoe, Microfluid. Nanofluid., 2009, 6, 1–16 CrossRef CAS.
- A. A. Miller, E. J. Lawton and J. S. Balwit, J. Polym. Sci., 1954, 14, 503–504 CrossRef CAS.
- R. B. Fox, L. G. Isaacs and S. Stokes, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 1079–1086 CrossRef CAS.
- M. Dole, Radiation chemistry of macromolecules, Academic Press, New York, 1973 Search PubMed.
- J. A. Moore and O. Choi Jin, in Radiation Effects on Polymers, American Chemical Society, 1991, vol. 475, ch. 11, pp. 156–192 Search PubMed.
- B. G. Rånby and J. F. Rabek, Photodegradation, photo-oxidation, and photostabilization of polymers, principles and applications, Wiley, London, New York, 1975 Search PubMed.
- A. Reiser, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 679–679 CrossRef.
- M. Diepens and P. Gijsman, Polym. Degrad. Stab., 2007, 92, 397–406 CrossRef CAS.
- J. S. Humphrey Jr, Polym. Prepr., Amer. Chem. Soc., Div. Polym. Chem., 1968, 9, 453–460 Search PubMed.
- A. Rivaton, Polym. Degrad. Stab., 1995, 49, 163–179 CrossRef CAS.
- N. Nagai, H. Okumura, T. Imai and I. Nishiyama, Polym. Degrad. Stab., 2003, 81, 491–496 CrossRef CAS.
- S. Wei, B. Vaidya, A. B. Patel, S. A. Soper and R. L. McCarley, J. Phys. Chem. B, 2005, 109, 16988–16996 CrossRef CAS.
- S. Roy, C. Y. Yue, Y. C. Lam, Z. Y. Wang and H. Hu, Sens. Actuators, B, 2010, 150, 537–549 CrossRef CAS.
- S. Roy and C. Y. Yue, Plasma Processes Polym., 2011, 8, 432–443 CrossRef CAS.
- C. W. Tsao, L. Hromada, J. Liu, P. Kumar and D. L. DeVoe, Lab Chip, 2007, 7, 499–505 RSC.
- C. H. Ahn, C. Jin-Woo, G. Beaucage, J. H. Nevin, L. Jeong-Bong, A. Puntambekar and J. Y. Lee, Proc. IEEE, 2004, 92, 154–173 CrossRef CAS.
- G. S. Y. Yeh, R. Hosemann, J. Loboda-Čačković and H. Čačković, Polymer, 1976, 17, 309–318 CrossRef CAS.
- H. Shadpour, H. Musyimi, J. Chen and S. A. Soper, J. Chromatogr., A, 2006, 1111, 238–251 CrossRef CAS.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS.
- U. Dharmasiri, S. K. Njoroge, M. Witek, M. G. Adebiyi, J. W. Kamande, M. L. Hupert, F. Barany and S. A. Soper, Anal. Chem., 2010, 83, 2301–2309 CrossRef.
- S. Maheswaran, L. V. Sequist, S. Nagrath, L. Ulkus, B. Brannigan, C. V. Collura, E. Inserra, S. Diederichs, A. J. Lafrate, D. W. Bell, S. Digumarthy, A. Muzikansky, D. Irimia, J. Settleman, R. G. Tompkins, T. J. Lynch, M. Toner and D. A. Haber, N. Engl. J. Med., 2008, 359, 366–377 CrossRef CAS.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS.
- S. L. Stott, R. J. Lee, S. Nagrath, M. Yu, D. T. Miyamoto, L. Ulkus, E. J. Inserra, M. Ulman, S. Springer, Z. Nakamura, A. L. Moore, D. I. Tsukrov, M. E. Kempner, D. M. Dahl, C. L. Wu, A. J. Iafrate, M. R. Smith, R. G. Tompkins, L. V. Sequist, M. Toner, D. A. Haber and S. Maheswaran, Sci. Transl. Med., 2010, 2 CrossRef CAS.
- G. E. Owens, S. Wang, H. Wang, J. Jiao, K. Chen, J. Huang, H. Pantuck and H. Tseng, Journal of Investigative Medicine, 2010, 58, 130–131 Search PubMed.
- S. Wang, H. Wang, J. Jiao, K.-J. Chen, G. E. Owens, K. Kamei, J. Sun, D. J. Sherman, C. P. Behrenbruch, H. Wu and H.-R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 8970–8973 CrossRef CAS.
- S. T. Wang, K. Liu, J. A. Liu, Z. T. F. Yu, X. W. Xu, L. B. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, L. W. K. Chung, J. T. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K. F. Shen and H. R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS.
- F. C. Bidard, A. E. Saliba, L. Saias, A. Degeorges, P. de Cremoux, J. L. Viovy, A. Vincent-Salomon, C. Mathiot, J. Y. Pierga and A. de Gramont, Bulletin Du Cancer, 2009, 96, 73–86 Search PubMed.
- A. E. Saliba, L. Salas, E. Psychari, N. Minc, D. Simon, F. C. Bidard, C. Mathiot, J. Y. Pierga, V. Frasier, J. Salamero, V. Saada, F. Farace, P. Vieh, L. Malaquin and J. L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14524–14529 CrossRef CAS.
- M. P. Seah and W. A. Dench, Surf. Interface Anal., 1979, 1, 2–11 CrossRef CAS.
- D. F. Mitchell, K. B. Clark, J. A. Bardwell, W. N. Lennard, G. R. Massoumi and I. V. Mitchell, Surf. Interface Anal., 1994, 21, 44–50 CrossRef CAS.
- C. J. Powell, A. Jablonski, S. Tanuma and D. R. Penn, J. Electron Spectrosc. Relat. Phenom., 1994, 68, 605–616 CrossRef CAS.
- J. W. Kamande, M. L. Hupert, M. A. Witek, H. Wang, R. J. Torphy, U. Dharmasiri, S. K. Njoroge, J. M. Jackson, R. D. Authforth, A. Snavely, J. J. Yeh and S. A. Soper, Anal. Chem., 2013 Search PubMed , submitted for publication.
- J.-P. Eliane, M. Repollet, K. E. Luker, M. Brown, J. M. Rae, G. Dontu, A. F. Schott, M. Wicha, G. V. Doyle, D. F. Hayes and G. D. Luker, Cancer Res., 2008, 68, 5529–5532 CrossRef CAS.
- U. Dharmasiri, A. A. Adams, M. Witek and S. A. Soper, Annu. Rev. Anal. Chem., 2010, 3, 409–431 CrossRef CAS.
- Y. Wang, M. Bachman, C. E. Sims, G. P. Li and N. L. Allbritton, Langmuir, 2006, 22, 2719–2725 CrossRef CAS.
- E. Uchida, Y. Uyama and Y. Ikada, Langmuir, 1993, 9, 1121–1124 CrossRef CAS.
- A. Tiraferri and M. Elimelech, J. Membr. Sci., 2012, 389, 499–508 CrossRef CAS.
- J. R. Rasmussen, E. R. Stedronsky and G. M. Whitesides, J. Am. Chem. Soc., 1977, 99, 4736–4745 CrossRef CAS.
- C. Wochnowski, M. A. Shams Eldin and S. Metev, Polym. Degrad. Stab., 2005, 89, 252–264 CrossRef CAS.
- This value agrees well with Wei, et al.'s results of 1.312 ± 0.093 nmol cm−2 COOH considering that UV activation protocols differed (22 mW cm−2 for 15 min vs. 15 mW cm−2 for 30 min by Wei, et al.). Furthermore, their method was multi-step, necessarily assumed 100% efficiency in each reaction step, and intrinsically underestimated COOH surface densities.
- A. A. Tseng, C. Kuan, C. D. Chen and K. J. Ma, IEEE Trans. Electron. Packag. Manuf., 2003, 26, 141–149 CrossRef CAS.
- We could find no statistically significant difference between the WCAs of these surfaces and heated, native controls (data not shown).
- I. Lipschitz, Polym.-Plast. Technol. Eng., 1982, 19, 53–106 CrossRef CAS.
- S. Ben Amor, G. Baud, M. Jacquet, G. Nansé, P. Fioux and M. Nardin, Appl. Surf. Sci., 2000, 153, 172–183 CrossRef CAS.
- C. Seidel, H. Kopf, B. Gotsmann, T. Vieth, H. Fuchs and K. Reihs, Appl. Surf. Sci., 1999, 150, 19–33 CrossRef CAS.
- D. L. Pavia, Introduction to spectroscopy, Brooks/Cole, Cengage Learning, Belmont, CA, 2009 Search PubMed.
- P. S. Nunes, P. D. Ohlsson, O. Ordeig and J. P. Kutter, Microfluid. Nanofluid., 2010, 9, 145–161 CrossRef CAS.
- B. C. Smith, Fundamentals of Fourier transform infrared spectroscopy, CRC Press, Boca Raton, 1996 Search PubMed.
- J. M. Martinez, S. K. C. Elmroth and L. Kloo, J. Am. Chem. Soc., 2001, 123, 12279–12289 CrossRef CAS.
- J. H. Monserud and D. K. Schwartz, Biomacromolecules, 2012, 13, 4002–4011 CAS.
- M. Hupert, W. J. Guy, S. Llopis, H. Shadpour, S. Rani, D. Nikitopoulos and S. Soper, Microfluid. Nanofluid., 2007, 3, 1–11 CrossRef CAS.
- Similar dimensions were observed for our previous CTC selection device (data not shown), indicating that the previously reported optimal volumetric flow rate of 1.6 mL h−1 more accurately corresponds to the linear velocity reported herein rather than 2 mm s−1 (see ref. 23 and 24).
- M. Sharan and A. S. Popel, Biorheology (Oxford), 2001, 38, 415–428 CAS.
- R. D. a. R. L. Valdemar Garcia, ed., In Vitro Blood Flow Behaviour in Microchannels with Simple and Complex Geometries, 2012 Search PubMed.
- J. P. Gleghorn, E. D. Pratt, D. Denning, H. Liu, N. H. Bander, S. T. Tagawa, D. M. Nanus, P. A. Giannakakou and B. J. Kirby, Lab Chip, 2010, 10, 27–29 RSC.
- D. A. Smirnov, D. R. Zweitzig, B. W. Foulk, M. C. Miller, G. V. Doyle, K. J. Pienta, N. J. Meropol, L. M. Weiner, S. J. Cohen, J. G. Moreno, M. C. Connelly, L. W. M. M. Terstappen and S. M. O'Hara, Cancer Res., 2005, 65, 4993–4997 CrossRef CAS.
- J. Lu, T. Fan, Q. Zhao, W. Zeng, E. Zaslavsky, J. J. Chen, M. A. Frohman, M. G. Golightly, S. Madajewicz and W.-T. Chen, Int. J. Cancer, 2010, 126, 669–683 CrossRef CAS.
- H. Bi, S. Meng, Y. Li, K. Guo, Y. Chen, J. Kong, P. Yang, W. Zhong and B. Liu, Lab Chip, 2006, 6, 769–775 RSC.
- S. Sagnella and K. Mai-Ngam, Colloids Surf., B, 2005, 42, 147–155 CrossRef CAS.
- W. Zhao, C. H. Cui, S. Bose, D. Guo, C. Shen, W. P. Wong, K. Halvorsen, O. C. Farokhzad, G. S. L. Teo, J. A. Phillips, D. M. Dorfman, R. Karnik and J. M. Karp, Proc. Natl. Acad. Sci. U. S. A., 2012 Search PubMed.
- V. Garcia, R. Dias and R. Lima, in Applied Biological Engineering – Principles and Practice, ed. G. R. Naik, InTech, Rijeka, Croatia, 2012, ch. 17, pp. 393–416 Search PubMed.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS.
- M. Yu, D. T. Ting, S. L. Stott, B. S. Wittner, F. Ozsolak, S. Paul, J. C. Ciciliano, M. E. Smas, D. Winokur, A. J. Gilman, M. J. Ulman, K. Xega, G. Contino, B. Alagesan, B. W. Brannigan, P. M. Milos, D. P. Ryan, L. V. Sequist, N. Bardeesy, S. Ramaswamy, M. Toner, S. Maheswaran and D. A. Haber, Nature, 2012, 487, 510–513 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50618e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |