Advances and critical concerns with the microfluidic enrichments of circulating tumor cells
Kyung-A
Hyun
and
Hyo-Il
Jung
*
School of Mechanical Engineering, Yonsei University, 50 Yonsei-no Seodaemun-gu, Seoul 120-752, South Korea. E-mail: uridle7@yonsei.ac.kr; Fax: +82-(0)2-312-2159; Tel: +82-(0)2-2123-5814
First published on 7th August 2013
Abstract
Over the past two decades, circulating tumor cells (CTCs) have been widely recognized for their importance in clinical trials. While most enrichment methods for these cells have been conducted through the batch process due to their rarity in blood and the need for large sample volumes, the batch process leads to unavoidable cell loss. Given the heterogenetic features of CTCs, this cell loss may limit the validity of research that relies on the isolation of CTCs; such research includes cancer prognosis, diagnosis of minimal residual diseases, assessment of tumor sensitivity to anticancer drugs, and the personalization of anticancer therapies. Recent advances in microfluidic approaches have made it possible to enrich CTCs with a small degree of cell loss. In this review, we highlight several microfluidic-based positive and negative enrichment methods that are the subject of considerable research interest (e.g. EpCAM-dependent assay and EpCAM-independent assay) and suggest a microfluidic-based single cell analysis platform for the down-stream analysis of CTCs. We also discuss critical concerns and future directions for research.
 Kyung-A Hyun | Kyung-A Hyun is a graduate student in the unified masters and doctoral degrees course at the School of Mechanical Engineering, Yonsei University, South Korea. She received her bachelor’s degree in Biomedical Engineering in 2011 from Gachon University of Medicine and Science. Her current research area is the separation and characterization of circulating rare cells (CRCs) from blood using microfluidic technology. |
 Hyo-Il Jung | Hyo-Il Jung is a professor in the School of Mechanical Engineering, Yonsei University, South Korea, where he leads the Laboratory of Biochip Technology. He obtained BSc and MSc degrees (Biotechnology) from the Korea Advanced Institute of Science and Technology (KAIST) in 1993 and 1995, respectively, and received a PhD degree (Physical Biochemistry) from the University of Cambridge, United Kingdom, in 2001. His current research interests centre on developing microfluidic devices to solve the major technical problems in biomedical sciences. |
1. Introduction
Cancer metastases, which lead to organ malfunction and cause approximately 90% of all cancer-related deaths, are known to be caused by circulating tumor cells (CTCs).1,2 CTCs are rare malignant cells disseminated from the primary tumor that circulate in the peripheral blood.3 When certain conditions arise in the microenvironment at secondary sites, these cells begin to create tumors in distant organs.4 For this reason, CTCs have great potential to serve as biomarkers for prognosis, predictors of therapeutic responses, and companion diagnostics in the development of novel therapeutics. Cancer patients are frequently asked to provide blood samples for various diagnostic and treatment-responses assessments. Thus, using CTCs as a “liquid biopsy” to study the biomarkers of new targeted therapies can be a critical tool for monitoring therapeutic responses while informing clinicians whether a change in the treatment course is indicated.4–6 In this way, the detection of CTCs may facilitate a paradigm shift from treatment based only on primary tumor characteristics to treatment that considers the molecular characteristics of CTCs, as well. However, the most significant hurdle in the isolation of CTCs is their rarity in blood. Approximately 1 to 100 CTCs are found in 1 ml of peripheral blood from a human cancer patient; thus, a very large number of normal hematological cells, such as red blood cells (RBCs) and white blood cells (WBCs), have to be depleted in order to obtain pure CTCs. Another tremendous challenge is investigating the heterogeneous characteristics of the CTCs, such as their morphological alterations and gene expressions, even when they are disseminated from the same primary tumor. In order to realize ideal CTC separation, technologies must be able to obtain high purity and recovery rates while also keeping the CTCs alive and intact for downstream morphological characterization and molecular analyses (purity refers to how many non-target cells are excluded from the analysis. Recovery refers to how many target cells are collected from the input sample.). Moreover, many CTCs are known to have a short half-life (1.0–2.4 h) and to rapidly undergo apoptosis when in circulation.7 Thus, CTCs must be enriched from a large blood volume as quickly as possible (i.e., high throughput isolation should be performed. Throughput refers to the amount of sample volume or the number of cells handled within a given time.).Numerous approaches have been developed to isolate CTCs, but none of them satisfies all of the conditions mentioned above. CellSearchTM (Verdex, USA) is a well-known technique for batch processing and is the only technique approved for the isolation and enumeration of CTCs by the U.S. Food and Drug Administration (FDA). This system uses an immuno-magnetic approach to capture CTCs that are revealed to be epithelial adhesion molecules (EpCAM) by binding with antibody (anti-EpCAM)-coated magnetic particles. Although the CellSearch system has been approved by the FDA, significant cell loss arises from the pretreatment process, including pipetting for the improvement of binding between cells and magnetic particles.
In this review, we shed light on the reasons why microfluidics are being used in CTC enrichment and describe current microfluidic technology-based methods for separating CTCs, with a focus on positive enrichment (i.e. capturing CTCs and eluting blood cells; EpCAM [epithelial cell adhesion molecule]-dependent assay) and negative enrichment (i.e. capturing blood cells and eluting CTCs; EpCAM non-dependent assay). Additionally, we suggest that microfluidic systems have the potential to parlay into a single CTC analysis.
2. Why microfluidics in CTC enrichment?
In the last decade, CTC research has increased dramatically. We investigated the number of papers related to CTCs by using all of the following search terms: “Circulating tumor cell,” “Separation,” “Isolation,” “Enrichment,” “Detection,” “Metastasis,” and “Analysis.” We found approximately 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 130 relevant papers that have been published since the 1930s (http://www.scopus.com). It can be cautiously speculated that this accumulation of research has arisen due to the wide acceptance and recognition of the significance of CTC enrichment for cancer analysis.
130 relevant papers that have been published since the 1930s (http://www.scopus.com). It can be cautiously speculated that this accumulation of research has arisen due to the wide acceptance and recognition of the significance of CTC enrichment for cancer analysis.
The batch process, however, involves multiple steps—such as pipetting, changing of the sample container, and chemical treatments—that may contribute to the risk of losing CTCs and adversely affecting the cells of interest. Due to the risk of CTC loss and the resulting unsatisfactory recovery rate, researchers using CTC isolation platforms have increasingly turned to microfluidic chip technologies. There have been a large number of studies reporting on CTC separation technologies using microfluidics (Keywords: “Circulating tumor cell,” “Microfluidics,” “Separation,” “Isolation,” “Enrichment,” “Detection,” “Metastasis,” and “Analysis”) (Fig. 1 (a)). To support the growing tendency to use microfluidic chip technologies for CTC isolation, we examined the number of papers related to CTCs at Pubmed in the same way as above and found a similar result (Fig. 1 (b)). A microfluidic device manipulates small amounts (10−6 to 10−12 liters) of fluid using 1 μm to 1000 μm channel sizes. The device offers a number of useful capabilities: the ability to use very small quantities of samples and reagents, to carry out separations and detections with high resolution and sensitivity, and to integrate easily with other techniques that improve the efficiency of the device. It also has the advantages of low cost, high throughput, and small footprints for analytical devices.8 Additionally, microfluidics can provide the user with continuity for the entire process (i.e., a one-step process of loading sample–separation–identification), resulting in the reduction of cell loss. For these reasons, microfluidics are now applied in CTC research. As the cells may be lost during transportation of the sample from the macro-components to the micro-components of microfluidic devices, causing considerable deterioration in the cell detection sensitivity, this intrinsic cell loss has been investigated and can be effectively minimized through the following measures: (a) increasing the tubing diameter of the device, connecting the sample storage and the micro-device; (b) applying a hydrodynamic focusing approach for sample delivery in order to reduce cells contacting and adhering to the walls of the micro-channel and chip inlet; (c) optimizing the filter design with a zigzag arrangement of the pillars to prolong the effective filter length; and (e) the use of diamond-shaped pillars instead of the traditionally used rectangular shape, so as to reduce the gap length between any two given pillars (i.e., pressure drop) at the filter region.9
 | ||
| Fig. 1 Number of papers by publication period addressing CTC isolation technology. The number of papers dramatically increased in the last two decades. (a) Scopus results and (b) Pubmed results. | ||
3. Positive versus negative enrichment of CTCs
Many techniques have been explored for enriching CTCs from a large number of hematologic cells (Table 1). These techniques can be classified into two categories: 1) positive enrichment (i.e., capturing target cells and eluting non-target cells) and 2) negative enrichment (i.e., capturing non-target cells and eluting target cells) (Fig. 2). Isolation methodologies with the affinity-based positive enrichment using the epitopes expressed on the cell surface are the most popular; therefore, they have been extensively developed. EpCAM (epithelial cell adhesion molecule) is the most commonly used epitope for all positive enrichment methods. One of the simplest positive enrichments is to capture CTCs in the anti-EpCAM antibody-coated microfluidic channel. In order to increase the capture efficiency, it is important to improve the surface interaction between the cells and the antibody-coated channel surface. However, the laminar flow, which makes the motion of cells very orderly along the straight lines that are parallel to the fluidic channel, is more dominant for the microfluidic channel. In order to overcome this limitation, Stott et al. introduced the ‘Herringbone chip’ (HB-chip).10 The grooves, or the chevron dimensions, of the main straight channel produce transverse flow that facilitates effective contact between EpCAM on the CTCs and anti-EpCAM antibodies on the channel surface.10 Using this device, CTCs were successfully captured from 14 of 15 (93%) patients with metastatic prostate cancer (median 63 CTCs/ml).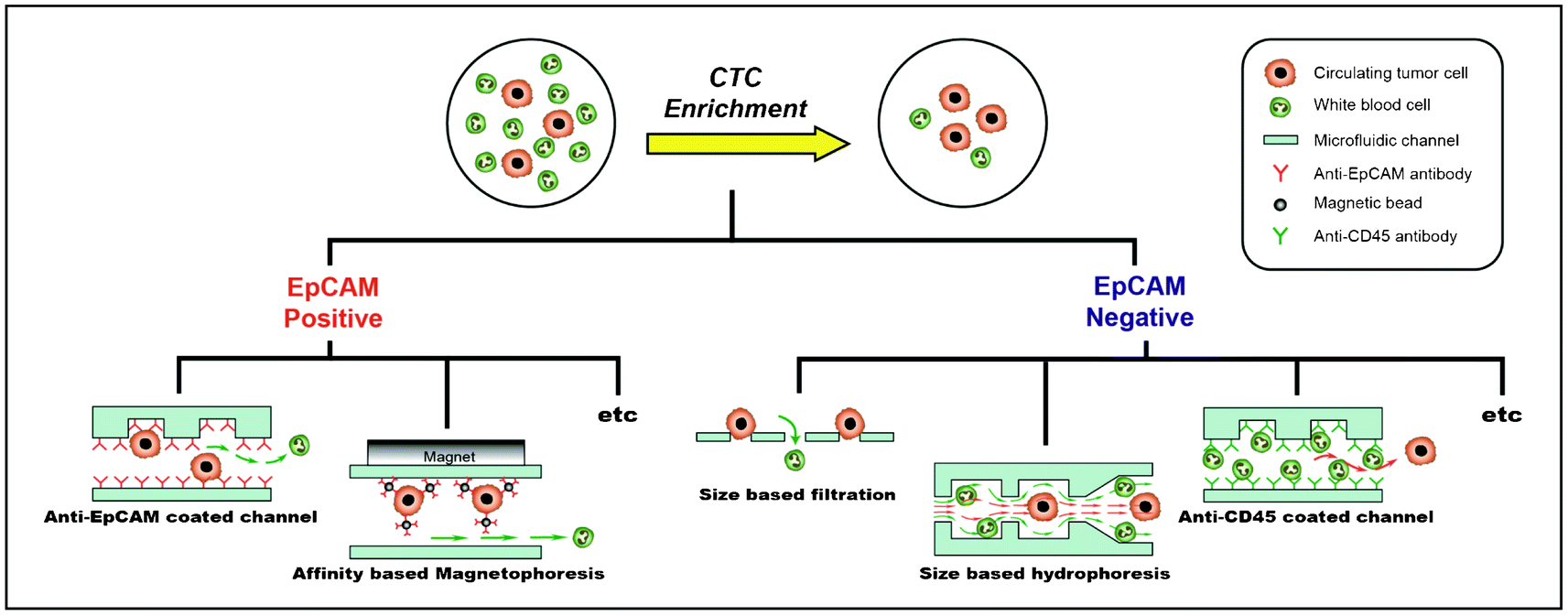 | ||
| Fig. 2 Enrichment of circulating tumor cells (CTCs) from the peripheral blood of cancer patients. CTC enrichment technologies can be classified into two types: EpCAM-dependent enrichment and EpCAM-independent enrichment. The EpCAM-dependent technologies include CTC capture using anti-EpCAM coated microfluidic channels or affinity-based magnetophoresis. The EpCAM-independent technologies include size-based filtration, size-based hydrophoresis, or normal cell depletion using an anti-CD45 antibody-coated microfluidic channel. | ||
| Approach | Technology | Target/Non-target cell line | Clinical trial (number of patients)/Sample volume | Efficiency | Throughput | Reference |
|---|---|---|---|---|---|---|
| Positive enrichment | Microfluidic mixer/EpCAM antibody functionalized chip | PC3 prostate cancer cells/Leukocytes | Metastatic Prostate Cancer patients (15)/∼4 ml | Capture efficiency: 91.8 ± 5.2% (n = 6) | 1.2 ml h−1 | 10 |
| Purity: 14.0 ± 0.1% | ||||||
| Integrated microfluidic system/Integration of size-based and affinity based techniques | MCF-7 breast cnacer cells/10 times diluted blood cells | N/A | Capture efficiency: 90% | 9.6 ml min−1 | 50 | |
| Purity: 50% | ||||||
| Integrated microfluidic system/Positive selection mode (posCTC-iChip) with EpCAM antibody conjugated magnetic beads. | MDA-MB-231, SKBR3 and MCF10A-LBX1 breast cancer, PC3-9 prostate cancer cells/Whole blood | Prostate(19), breast (12), pancreas (6), colorectal (2) and lung (2) cancer patients/6–12 ml | Capture efficiency: 77.8 ± 7.8% for MDA-MB-231, 98.6 ± 4.3% for SKBR3 and 10.9 ± 3.0% for MCF10A-LBX1 | 8 ml h−1 (107 cells s−1) | 51 | |
| Purity: >3.5-log purification | ||||||
| Nanostructured silicon substrate with microfluidic mixer/EpCAM antibody functionalized chip | MCF-7 breast cancer, PC3 prostate cancer and T24 bladder cancer cells/Blood cells | Prostate cancer patients (26)/1 ml | Recovery: ∼95% | 1.0 ml h−1 | 11 | |
| Halloysite nanotube-coated device/E-selectin and antibody molecules against epithelial marker functionalized chip | KG1a leukemic cells/Whole blood | Metastatic breast (6), prostate(3), lung (2) and ovarian cancer (1) patients/3.75 ml | Capture efficiency: 50% | 4.8 ml h−1 | 12 | |
| Purity: ∼50% | ||||||
| Packed bed device/EpCAM antibody coated microbeads loaded chip | MCF-7 breast cancer cells/Whole blood | N/A | Capture efficiency: 35 to 70% (depending on blood samples) | 0.2 ml h−1 | 13 | |
| Flow cytometry/Immuno-magnetic enrichment | MCF-7 breast cancer and PC-9 lung cancer cells/Molt-4 T-lymphoblastoid cells and PBMC (Peripheral blood mononuclear cell) | N/A | Detection efficiency: ∼88% | 10 μl min−1 | 14 | |
| A combined micromagnetic-microfluidic device/Immuno-magnetic enrichment | Mouse metastatic M6C breast cancer cells/Blood cells | Mice bearing implantable 4T1 breast tumors or mammary cancer-bearing transgenic mice (4 to 29 weeks of age)/∼1 ml | Isolation efficiency: ∼90% | 1.2 ml h−1 | 15 | |
| Specificity: ∼0.4% WBC captured | ||||||
| A motion controlled microfluidic system/Immuno-magnetic enrichment | Colo205 colon cancer, PC3 prostate cancer and | Breast (7), prostate(1)and lung (3) cancer patiens/∼10 ml | Capture efficiency: ∼90% | 2.5 ml h−1 | 16 | |
| SkBr3 breast cancer cells/Blood sample | ||||||
| Immunomagnetic nanobeads coated with anti-EpCAM antibodies and lateral magnetophoresis technology | SKBR-3 breast cancer cells/Blood sample | Breast (3) and lung (2) cancer patiens, healty donor (2)/3 ml | Recovery: 90% | 5 ml h−1 | 17 | |
| Purity: 97% | ||||||
| Microfluidic disk integrating immunomagnetic-based, positive selection with detection of fluorescence on a single disk | MCF-7 breast cancer cells/Jurkat T cell lymphoblast like cells | N/A | Detection yield: 80 ± 15% | Few hundred microliter within 30 min | 18 | |
| Selective size amplification (SSA) for target cells and a multi-obstacle architecture (MOA) filter | MCF-7 and MDA-MB-231 breast cancer cells/Human primary leukocytes | N/A | Recovery: 92% | 20 μl min−1 | 20 | |
| Purity: 351 peripheral blood leukocytes(PBL)/ml | ||||||
| Selective sedimentation and microfiltration | MCF-7 breast cancer and DMS-79 small cell lung cancer cells/Leukocytes | N/A | Recovery: ∼99% | 100 μl min−1 | 22 | |
| A trachea-inspired bifurcated microfilter | MCF-7 breast cancer cells/Human primary leukocytes | N/A | Recovery: 93% | 100 μl min−1 | 23 | |
| Purity: 59 | ||||||
| Ensemble-decision aliquot ranking (eDAR) | MCF-7 and SKBr-3 breast cancer cells/Blood samle | Patients with stage IV metastatic breast cancer (20)/1–2 ml | Recovery: 93% | 50 μl min−1 | 52 | |
| Purity: 10–50% | ||||||
| Negative enrichment | Size-Selective Microcavity Array | NCI-H358 lung cancer, AGS and SNU-1 gastric cancer, SW620 colon cancer, MCF-7 breast cancer and HS578T ductal breast cancer cells/Blood sample | N/A | Recovery: ∼80% | 200-1000 μl min−1 | 25 |
| 3D microfilter device | LNCaP prostate cancer and MCF-7 breast cancer cells/Blood sample | N/A | Capture efficiency: 86.5 ± 5.3% | 10 ml/3–5 min | 26 | |
| Double spiral microchannel/Size-based dean Flow Fractionation (DFF) | MCF-7 breast cancer and Hela cervical cancer cells/Diluted blood sample | N/A | Recovery: ∼88.5% | 9.33 × 107 cells min−1 | 27 | |
| Spiral microchannel/Size-based DFF | MCF-7 and MDA-MB-231 breast cancer, Hela cervical cancer cells/Blood sample | Metastatic lung cancer patients (20)/Average 6 ml | Recovery: ∼85% | 3 ml h−1 | 28 | |
| Slanted spiral microfluidics/Size based fractionation | MCF-7, MDA-MB-231 breast cancer and T24 bladder cancer cells/Blood sample | Metastatic lung (5) and breast (5) cancer patients/7.5 ml | Recovery: ∼80% | 7.5 ml/8 min | 53 | |
| Purity: ∼4 log depletion of WBCs | ||||||
| Spatially gradated microfluidic chip/Size based separation | Hela cervical cancer cells/RBC-lysed peripheral blood | Liver cancer patients (10)/3 ml | Capture efficiency: ∼95% | 0.5–2 ml h−1 | 54 | |
| Purity: ∼90% | ||||||
| Multi-stage multi-orifice flow fractionation(MS-MOFF)/Size-based fractionation | MCF-7 breast cancer cells/Blood sample | N/A | Recovery: ∼98.9% | 126 μl min−1 | 29 | |
| Enrichment ratio: 163 fold | ||||||
| Parallel MOFF/Size-based fractionation | MCF-7 and MDA-MB-231 breast cancer cells/PBMC | Breast cancer patients (24)/7.5 ml | Recovery: ∼90% | 600 μl min−1 | 30 | |
| Enrichment ratio: ∼120 fold | ||||||
| MOFF + Dielectrophoresis (DEP) | MCF-7 breast cancer cells/Blood sample | N/A | Enrichment ratio: 162.4 fold | 126 μl min−1 | 31 | |
| Optically induced-dielectrophoresis (ODEP) | PC-3 prostate cancer, OEC-M1 oral cancer cells/Leukocytes | N/A | Recovery: ∼61% | 0.1 μl min−1 | 55 | |
| Purity: ∼64% | ||||||
| Acoustophoresis/Acoustic properties and Size based separation | DU145, PC3, LNCaP prostate cancer cells/WBCs | N/A | Recovery: ∼72.5% | 280 μl min−1 | 56 | |
| Purity: ∼79.6% | ||||||
| Microfluidic disk/Immuno-magnetic enrichment | MCF-7 breast cancer cells/Jurkat T cell lymphoblast like cells and mononuclear cells (MNC) from healthy donor | N/A | Detection yield: 60 ± 10% | Within 30 min | 33 | |
| Geometrically activated surface interaction (GASI)-chip/CD45 antibody functionalized chip | MCF-7 and MDA-MB-231 breast cancer cells/Jurkat T cell lymphoblast like cells | Breast cancer (4), gastric cancer (4) and lung cancer (4) patients/1 ml | Recovery: ∼87.43% | 20 μl min−1 | 32 | |
| Enrichment ratio: 130.94 fold | ||||||
| Integrated microfluidic system/Negative selection mode (negCTC-iChip) with CD45 or CD15 antibody conjugated magnetic beads. | MCF10A and MCF10A-LBX1 breast cancer cells/Whole blood | Metastatic breast cancer patients (10)/N/A | Capture efficiency: 96.7 ± 1.9% for MCF10As, 97.0 ± 1.7% for the MCF10A-LBX1 derivatives | 8 ml h−1 (107 cells s−1) | 51 | |
| Purity: >2.5 log purification |
While the HB-chip just increased the interaction frequency between the channel and the cells using a simple microfluidic mixer, Wang et al. combined nanostructured silicon substrates and the herringbone mixer in order to enhance the local topographic interactions between the anti-EpCAM antibody-coated silicon-nanopillar (SiNP) array and the nanoscale components of the cellular surface (e.g., microvilli and filopodia).11 In this platform, more than 95% of the breast cancer cells (MCF-7) were captured, and a substantial number (≥5) of CTCs were detected from 17 of the 26 patients at different stages of prostate cancer. Most affinity-based separation approaches relied on a high surface-to-volume ratio, resulting in higher binding capacity and efficiency. Several research groups have modified the microfluidic channel to have a high surface area by adding a halloysite nanotube12 or by packing micro-beads13 inside the channels. Immuno-magnetic cell separation, in which nano or micro magnetic beads are selectively attached to the cells, is a common technique for CTC enrichment due to its high sensitivity, selectivity, and ability to handle a large range of volumes in continuous microfluidic channels without modification.14–18 Moreover, the magnetic field gradients are created by the micropatterns of the magnetic material and can be used for controlling cell capture.19 An automated motion-controlled microfluidic system was introduced by Huang et al.16 In order to prevent the aggregation of the magnetic nanoparticles near the inlet, Huang et al. inserted a spacer, which made a magnetic field gradient, between the microchannel and the permanent magnet that was close to the front end of the microchannel. In addition, this micro-chip was equipped with a computer-controlled rotational holder that eliminated the negative effects of blood stagnation, such as through mechanical interference and non-specific bindings. In this system, as the blood sample flows through the microchannel on top of the array of permanent magnets, tumor cells bound with magnetic nanoparticles (conjugated with anti-EpCAM) were separated from the blood flow and captured on the substrate of the microchannel. CTCs were successfully isolated from patient blood samples originating from a variety of different cancers, as compared to the CellSearchTM system. However, most immuno-magnetic cell separation techniques, including this micro-chip, require pre-incubation of the samples with the magnetic particles coated with the antibodies in separated tubes, which can inadvertently cause cell loss.
Size-based separation of CTCs has also provided recent and promising applications, owing to the fact that epithelial-derived tumor cells are generally larger than peripheral blood leukocytes. Membrane-based and microfluidic filter systems have demonstrated better recovery rates than others. However, methods of size filtration should be further developed to overcome the innate limitations of having a large number of contaminated hematologic cells in the filter, which hinders efforts to increase purity. In addition, a recent concern has been the fact that the CTCs mimic the circulating leukocytes, potentially causing some CTCs to be smaller than previously thought. The smaller-sized CTCs could be more malignant and pass unhindered through the filters.20 Kim et al. introduced new CTC capture technology utilizing the selective size amplifications of the CTCs. This selective size amplification can be easily performed by anti-EpCAM conjugated microbeads (specifically 3 μm). The selective size-amplifying CTCs have made it possible for these cells to be transformed from ‘often’ larger to always larger, thus allowing size-based separation to achieve a higher recovery rate with purity.20 Various filter designs, such as the multi-obstacle architecture (MOA) filter and the trachea-inspired bifurcated (TRAB) microfilter, have been demonstrated in capturing size-amplified CTCs.20–23 This represents a potentially significant advance towards ensuring higher purity than allowed under conventional filtration.
Although positive enrichment methods can be used to isolate CTCs at a high purity, these methods have significant limitations in that CTCs, heterogeneous by nature, do not all express the same specific antigens. Even with the same origin carcinoma cell lines, such as MCF-7 and MDA-MB-231 cells (i.e. human breast cancer cells), the surface density of EpCAM ranges from 222.1 × 103 binding sites/cell on MCF-7 cells to merely 1.7 × 103 on MDA-MB-231 cells.24 Apart from the down-regulated EpCAM expression level, there is another factor to consider in regard to the EpCAM-based enrichment method. When the needle penetrates the skin of cancer patients to collect blood samples, the skin cells, which have epithelial biomarkers on their surface, may contaminate the sample from the needle puncture site. Even though the first 4–5 ml of blood will be discarded during the analyses, the skin cells may still exist in the collected blood. Moreover, when adherent cells detach from the extracellular matrix (ECM), they will die by a programmed cell death mechanism called anoikis during circulation. However, metastatic tumor cells (presumably smaller-sized) may acquire anoikis resistance, allowing them to survive in the blood stream and to invade other organs.
For these reasons, negative enrichment methods are growing in popularity for the collection of heterogeneous CTCs. Among the various approaches to negative enrichment, such as size-based separation and dielectrophoresis (DEP)-based separation, size-based separation methods—regardless of the EpCAM expression levels—have been demonstrated as potentially efficient, inexpensive, and quick means of CTC enrichment.25,26 Filtration, an intuitive approach that separates cells based on their sizes and deformability, is used to isolate CTCs by exploiting the fact that CTCs, in general, are larger and stiffer than blood cells. Zheng et al. demonstrated a 3D microfilter that can reduce cell damage induced by passage through the filter pores.26 According to their report, the pore positions of the bottom membrane in the device are shifted from the pore positions of the top membrane. Thus, when tumor cells are trapped in the pores of the top membrane, the bottom membrane can generate direct forces in the opposite direction, effectively reducing the concentrated tension stress on the cell plasma membrane.26 The diameters of the pores on the top and bottom were 9 μm and 8 μm, respectively. Using this strategy, spiked MCF-7 cells (breast cancer cell line) were captured from a healthy blood sample with an average capture efficiency of 86.5 ± 5.3%. As mentioned earlier, the presence of CTCs in the blood stream is very rare, and a large volume of blood must be used with a high concentration of normal cells. In this situation, however, clogging of pores in the filtration system is inescapable and may result in locally irregular flow.
Alternatives that exploit inertial microfluidics, such as dean flow fractionation (DFF) and multi-orifice flow fractionation (MOFF), have appeared and are considered to be powerful and promising size-based separation methods.27–31 Hou et al. utilized a spiral microchannel with inherent centrifugal force to continuously separate CTCs from blood.28 In their system, the smaller hematologic cells moved towards the inner wall and then back to the outer wall by the dean vortices, while the larger CTCs were focused along the inner wall by the inertial lift forces, thus achieving separation. Using their optimized device, more than 85% of the spiked cancer cells were separated from the normal blood sample, and the CTCs were successfully isolated from the metastatic lung cancer patients (5–88 CTCs per ml). Jung's group introduced a novel hydrodynamic method that achieves size-based particle separation via multi-orifice flow fractionation (MOFF); in this method, the microparticles were moved laterally according to their size through hydrodynamic inertial force.29–31 This technique is advantageous for CTC separation because of its simple experimental setup and high operational flow rate (100–300 μL min−1). To achieve a high recovery yield, the group designed a multi-stage multi-orifice flow fractionation (MS-MOFF) device.29 This device collected and re-separated non-selected cancer cells from the first separation. The blood cells and MCF-7 cells were efficiently separated up to 93.3% and 98.9%, respectively.
Although hydrodynamic methods show good performance with a high flow rate, the precise isolation of biological samples, such as CTC from blood, is challenging due to the wide variation in cell size. Although dielectrophoresis (DEP), a translational motion of particles in a non-uniform electric field, can discriminate between different cells, the purity decreases if the flow rate increases because the DEP forces have less of a chance to affect the cells. Therefore, Jung's group decoupled the trade-offs between speed and sensitivity by integrating the two independent techniques of hydrodynamic separation (primary) and DEP separation (secondary) for CTC separation. The resulting integrated system was capable of fast CTC separation at a high flow rate (126 μL min−1) with high efficiency (∼99.24%) and without labeling.31 Because human blood is a highly complex mixture of plasma, cells, and proteins, and because the viscosity of blood is three times higher than that of water, blood samples were diluted over 400 times in order to make suitable inter-particle distances for our MOFF system. Therefore, a parallel MOFF chip (p-MOFF) was developed, which is connected by four single MOFF channels, in order to isolate CTCs from cancer patients' blood. Using the breast cancer cell line, the group separated 93.75% of the MCF-7 cells and 91.60% of the MDA_MB_231 cells and eliminated 90.8% of the WBCs from an individual experiment at a 600 μL min−1 inlet flow rate and a 240 μL min−1 outlet flow rate. The p-MOFF device successfully isolated CTCs from breast cancer patients, while achieving high throughput performance (10 ml of suspended cells in PBS per 17 min). When the blood samples of 24 breast cancer patients were subjected to the p-MOFF device, 1 to 21 CTCs were identified in the 19 patients.30 In addition, Jung's group conducted a multi-parameter analysis in order to investigate the EpCAM expression levels of the CTCs. When the group stained the cells using multi-parameters (DAPI for DNA content, phycoerythrin-conjugated pan-Cytokeratin for CTC, APC conjugated EpCAM for EpCAM positive CTC, and FITC conjugated CD45 for WBC), it was observed that 50% of the patients had both EpCAM-positive and -negative CTCs. This result shows the usefulness of label-free microfluidic devices in isolating various cancer cells in an EpCAM-independent manner.
The selectivity of non-labeling methods, such as hydrophoresis and dielectrophoresis, is generally poor in comparison to that of affinity-based separation, given that non-labeling methods exploit the physical properties of the target cells, such as size, density, shape, and deformability. Nevertheless, a significant advantage of non-labeling methods is that intact, circulating rare cells can be obtained, which can then be cultured for down-stream analysis. Although there are several well-known features of CTC isolation, such as EpCAM expression and size, many features of CTCs have not yet been characterized precisely. Furthermore, their correlations with metastatic load have not yet been identified clinically. Therefore, separating CTCs by either one or several criteria may have significant limitations.
Recently, Jung's group introduced a novel negative enrichment strategy with a geometrically activated surface interaction (GASI)-chip.32 In this strategy, only the characteristics of the normal blood cells, such as CD45 expression on the leukocyte surface, were used to isolate the heterogenetic CTCs. There have been several attempts to deplete the leukocytes based on the CD45 expression using immune-magnetic systems. The leukocytes coated with magnetic beads (functionalized with anti-CD45 antibody) can be continuously eliminated by the magnet in the microfluidic channel.33 The pretreatment procedures, however, have the potential to cause considerable cell loss, as described above. As a result, Jung's group developed the GASI-chip using a herringbone shape, functionalizing the microfluidic chip with an anti-CD45 antibody to efficiently capture a large number of leukocytes rather than CTCs.32 Conventional HB-chips, which were developed by Stott et al., are suitable for positive enrichments due to the rarity of the CTCs. In negative enrichment, it is important to capture as many leukocytes as possible inside the microfluidic channel. For this reason, the conventional herringbone design has two problems when applied to the negative enrichment system. One is the insufficient surface interactions between the cell and channel surface, and the other is the unavoidable focusing to the apex of the herringbone structure. In order to resolve these problems, the GASI-chip was designed based on a computational model34 that optimized the geometrical surface interactions in the herringbone, where the apex of the herringbone was increased twofold compared to the conventional HB-chip. Using this device, Jung's group enriched the spiked MCF-7 cells from the Jurkat cells (lymphoblast like cell line) with a high enrichment yield of up to 131-fold. When 12 blood samples from patients with several types of metastatic cancer were subjected to the GASI-chip, at least one CTC was identified, regardless of the type of primary tumor. As standard criteria, CD45 marker-positive cells are regarded as leukocytes and are excluded from CTC characterization. However, several researchers have reported that a significant number of cells appeared as dual-positives for CD45 and cytokeratins.35 Although the identity of these dual-positive cells is not well understood, a significant ratio of dual-positive cells has often been reported, indicating the active and dynamic heterogeneity of CTC.
Although there is controversy around the question of whether positive or negative enrichment is more efficient, it is clear that the latter is more advantageous because the target cells can be captured intact. Novel approaches for negative enrichment (affinity-based methods using antibodies to isolate hematologic cells or non-affinity-based methods, such as dielectrophoresis and hydrophoresis) should be developed, as a negative enrichment microfluidic chip enables the simultaneous isolation of various types of CTCs. The number of intact and heterogeneous CTCs collected continuously by such a device will provide researchers with many opportunities to investigate the molecular nature of these cells.
4. Critical concerns and future directions for research
In the previous sections, we described various approaches for microfluidic CTC enrichment. These approaches are classified as either EpCAM-positive or EpCAM-negative CTC isolation. Although all of the devices aspire to attain high recovery, purity, and throughput while keeping the CTCs alive and intact, each device has so far achieved only one or two of these goals, based on their respective approaches, due to the critical concerns related to CTC enrichment. In microfluidic CTC enrichment systems, the critical concerns are the following: 1) heterogeneity of the CTCs, such as epithelial-mesenchymal transition (EMT) ↔ mesenchymal-epithelial transition (MET) and size overlapping with WBCs; and 2) the necessity for alive and intact CTCs for down-stream analysis. Recent studies have provided evidence that a portion of CTCs undergo a process of EMT in order to enable invasion and dissemination of the CTCs into other organs and circulatory systems. This transition is characterized by events such as a loss of epithelial cell properties (e.g., down-regulated EpCAM level), a stem cell-like phenotype, and anoikis resistance, which is a programmed cell death mechanism.36,37 For this reason, the devices that use the expression of the EpCAM marker as a basis for the isolation of CTCs from normal cells have a limited ability to obtain EpCAM down-regulated CTCs.38 Because the question of what kind of CTCs are aggressive has not yet been answered, the loss of EpCAM-negative CTCs is a crucial issue for various researches, such as cancer prognosis and diagnosis, metastasis investigation, and personalized medicine. To address these EpCAM based technical challenges, antibody mixtures of CTC specific surface markers are used for positive enrichment.39,40 However, it is difficult to optimize the experimental condition including the type and proportion of the antibody mixtures because antigen expression level is variable depending on physiological conditions or subtypes as shown Fig. 3. Moreover, although the cells captured on the EpCAM antibody-coated microfluidic channels could be eluted by shear force or enzymatic digestion for the down-stream molecular analysis, these cells are not intact any longer, which may affect the experimental results. Among the devices that were classified as EpCAM-negative enrichment methods, devices based on the differences in physical properties between CTCs and normal cells cannot avoid the issue of CTC heterogeneity, even though intact CTCs can be obtained. Size overlapping between the CTCs (14–26 μm in diameter) and WBCs (8–20 μm in diameter) indicates that any size-based isolation techniques would result in a loss of a large number of small CTCs.36 The filtration devices enrich the CTCs by utilizing the stiffer and larger size characteristics of the cancer cells compared to the blood constituents.41 However, Cross et al. reported that cancer cells from patients are about 70% less stiff than normal cells, which was determined by using atomic force microscopy (AFM).42 Thus, although it is controversial whether positive enrichment (i.e., specifically capturing the CTCs using their properties) or negative enrichment (i.e., specifically eliminating the normal cells and collecting CTCs regardless of their characteristics) is more efficient, it is clear that the negative enrichment is more suitable for the isolation of heterogeneous and intact CTCs with high recovery. | ||
| Fig. 3 Surface marker expression in malignant and benign pleural effusions. Each of the 5 cell subsets (malignant EpCAM+, EpCAM-/CD45- and CD45+; benign EpCAM-/CD45- and CD45+) was stained with a panel of 35 surface markers by flow cytometry. The absolute number of receptors on the cell surface is shown in the heatmap. Small p-value indicates significant deviation of any one subset from others. Markers with * represents p-value < 0.05 (** p-value < 0.001) based on an analysis of variance (ANOVA) test. Reproduced with permission.57 | ||
Beyond the issue of separating CTCs, achieving a better understanding of the molecular characteristics of CTCs has been regarded as a key issue for advancing personalized treatments and discovering new biomarkers for the detection of CTCs. Sieuwerts et al. performed quantitative mRNA expression profiling specific for CTCs by real-time RT-PCR (reverse transcription polymerase chain reaction) in an environment containing large quantities of contaminating leukocytes.43 However, traditional biological analyses probe large ensembles, on the order of 103–106 cells, thereby revealing only the average genotypic and/or phenotypic characterizations of the population.44 In addition, the way that isolated cells are cultured to expand CTC numbers is not recommended, given that cancer cells have a feature that modifies their characteristics to survive when the surrounding environment is changed. Heterogeneity among CTCs is extremely important, and cell-to-cell variations occur even within a single blood draw.45 The advent of single cell analysis has revealed marked cellular heterogeneity in gene and protein expressions, genetic/genomic alterations, and responsiveness to environmental and chemotherapeutic stimuli.44 Therefore, it is of great importance to accomplish a single CTC analysis on one chip in parallel with the isolation (Fig. 4). A straightforward method to analyze a single CTC is capturing the CTCs in microfluidic channels at a single cell resolution and simultaneously staining them on-chip for specific markers or cytogenetically evaluating them using fluorescence in situ hybridization (FISH).46,47 Recently, Hou et al. introduced the next generation of nanovelcro chips (i.e., poly(lactic-co-glycolic acid) (PLGA)-nanofiber embedded nanovelcro chip), abbreviated PN-nanovelcro chip, which is coated with a melanoma-specific antibody (i.e., anti-CD146). The transparent PN-nanovelcro substrate makes it possible to analyze CTCs at a single cell resolution using laser-assisted micro-dissection. The genomic DNA (gDNA) from each CTC was then amplified using a commercial whole-genome amplification kit, where the amplified gDNA was subjected to Sanger sequencing in order to detect BRAFV600E mutation, which is a key melanoma drug treatment.48 Even though single CTCs can be isolated, there is the limitation that it contains only a pictogram quantity of RNA, which would be insufficient to conduct a reproducible whole genome analysis.45 Therefore, microfluidic single cell analyses are expected to be promising tools for future CTC research, given that microfluidic techniques offer unprecedented capabilities for precisely manipulating small volumes, confining the molecules to a micro-liter volume, reducing diffusion time, and facilitating large-scale integrations and automations.44 As a result, this will make it possible to sensitively analyze a small number of molecules from a single CTC.
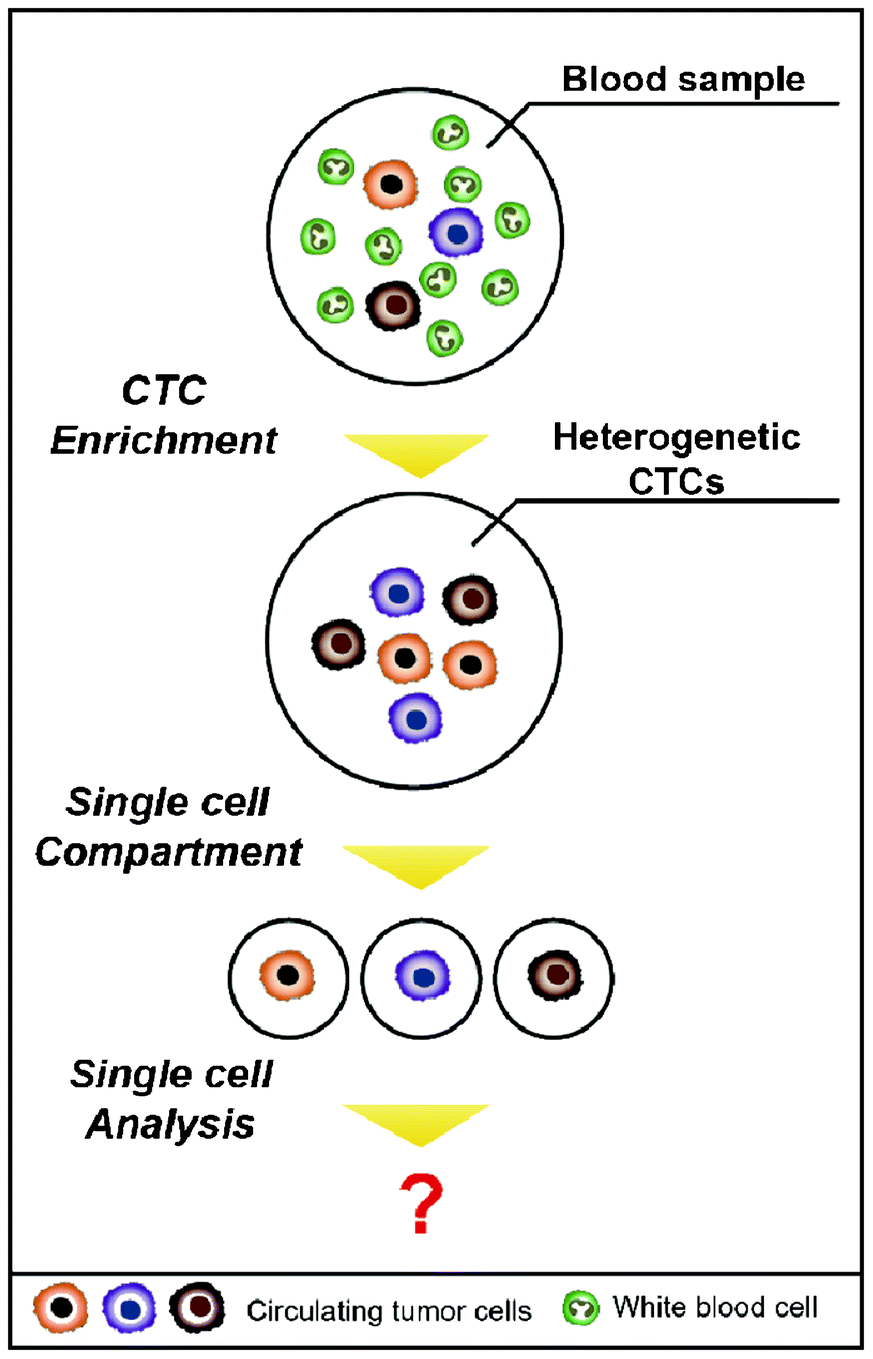 | ||
| Fig. 4 Single cell analysis for future CTC research. After the processing of CTC enrichment, the heterogenetic CTCs are still mixed in the sample. To characterize each of the CTCs, the collected cells should be compartmentalized at a single cell resolution. | ||
Taking this a step further, these microfluidic devices are believed to be capable of providing insights into various types of circulating rare cells (CRCs)—such as circulating endothelial cells (CECs), circulating cancer stem cells (CCSCs), circulating progenitor cells (CPCs), and nucleated red blood cells (nRBCs), including CTCs—which can be good indicators for numerous diseases. Examining the number of intact and heterogeneous CRCs continuously collected by the device is expected to give researchers many opportunities to investigate the molecular nature of rare cells.49 This will open new avenues of research, expanding the research area from CTCs to CRCs.
Acknowledgements
This study was supported in part by a research program of the National Research Foundation of Korea (NRF) (grant no. 2011-0016731) and a grant from the Korea Health technology R&D Project (grant no. A121986), Ministry of Health & Welfare, Republic of Korea.References
- P. Mehlen and A. Puisieux, Nat. Rev. Cancer, 2006, 6, 449–458 CrossRef CAS PubMed.
- K. Pantel and C. Alix-Panabiéres, Trends Mol. Med., 2010, 16, 398–406 CrossRef PubMed.
- C. Aggarwal, N. J. Meropol, C. J. Punt, N. Iannotti, B. H. Saidman, K. D. Sabbath, N. Y. Gabrail, J. Picus, M. A. Morse, E. Mitchell, M. C. Miller and S. J. Cohen, Ann. Oncol., 2013, 24, 420–428 CrossRef CAS PubMed.
- M. Balic, A. Williams, H. Lin, R. Datar and R. J. Cote, Annu. Rev. Med., 2013, 64, 31–44 CrossRef CAS PubMed.
- J. M. Hou, M. G. Krebs, L. Lancashire, R. Sloane, A. Backen, R. K. Swain, L. J. C. Priest, A. Greystoke, C. Zhou, K. Morris, T. Ward, F. H. Blackhall and C. Dive, J. Clin. Oncol., 2012, 30, 525–532 CrossRef PubMed.
- F. C. Bidard, T. Fehm, M. Ignatiadis, J. B. Smerage, C. Alix-Panabières, W. Janni, C. Messina, C. Paoletti, V. Müller, D. F. Hayes, M. Piccart and J. Y. Pierga, Cancer Metastasis Rev., 2012 DOI:10.1007/s10555-012-9398-0.
- K. Pantel, C. Alix-Panabières and S. Riethdorf, Nat. Rev. Clin. Oncol., 2009, 6, 339–351 CrossRef CAS PubMed.
- G. M. Whitesides, Nature, 2006, 44, 368–373 CrossRef PubMed.
- L. Zhu, X. L. Peh, H. M. Ji, C. Y. Teo, H. H. Feng and W. T. Liu, Biomed. Microdevices, 2007, 9, 745–750 CrossRef PubMed.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- S. Wang, K. Liu, J. Liu, Z. T. F. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, L. W. K. Chung, J. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K. F. Shen and H. R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS PubMed.
- A. D. Hughes, J. Mattison, L. T. Western, J. D. Powderly, B. T. Greene and M. R. King, Clin. Chem., 2012, 58, 846–853 CAS.
- J. G. Kralj, C. Arya, A. Tona, T. P. Forbes, M. S. Munson, L. Sorbara, S. Srivastavac and S. P. Forry, Lab Chip, 2012, 12, 4972–4975 RSC.
- M. Takao and K. Takeda, Cytometry, Part A, 2011, 79A, 107–117 CrossRef PubMed.
- J. H. Kang, S. Krause, H. Tobin, A. Mammoto, M. Kanapathipillaia and D. E. Ingber, Lab Chip, 2012, 12, 2175–2181 RSC.
- Y. Y. Huang, K. Hoshino, P. Chen, C. H. Wu, N. Lane, M. Huebschman, H. Liu, K. Sokolov, J. W. Uhr, E. P. Frenkel and J. X. J. Zhang, Biomed. Microdevices, 2012 DOI:10.1007/s10544-012-9718-8.
- S. Kim, S. I. Han, M. J. Park, C. W. Jeon, Y. D. Joo, I. H. Choi and K. H. Han, Anal. Chem., 2013, 85, 2779–2786 CrossRef CAS PubMed.
- K. C. Chen, Y. C. Pan, C. L. Chen, C. H. Lin, C. S. Huang and A. M. Wo, Anal. Biochem., 2012, 429, 116–123 CrossRef CAS PubMed.
- D. W. Inglis, R. Riehn, R. H. Austin and J. C. Sturm, Appl. Phys. Lett., 2004, 85, 5093 CrossRef CAS.
- M. S. Kim, T. S. Sim, Y. J. Kim, S. S. Kim, H. Jeong, J. M. Park, H. S. Moon, S. I. Kim, O. Gurel, S. S. Lee, J. G. Lee and J. C. Park, Lab Chip, 2012, 12, 2874–2880 RSC.
- M. X. lin, K. A. Hyun, H. S. Moon, T. S. Sim, J. G. Lee, J. C. Park, S. S. Lee and H. I. Jung, Biosens. Bioelectron., 2013, 40, 63–67 CrossRef CAS PubMed.
- J. M. Park, J. Y. Lee, J. G. Lee, H. Jeong, J. M. Oh, Y. J. Kim, D. Park, M. S. Kim, H. J. Lee, J. H. Oh, S. S. Lee, W. Y. Lee and N. Huh, Anal. Chem., 2012, 84, 7400–7407 CrossRef CAS PubMed.
- M. S. Kim, J. Kim, W. Lee, S. J. Cho, J. M. Oh, J. Y. Lee, S. Baek, Y. J. Kim, T. S. Sim, H. J. Lee, G. E. Jung, S. I. Kim, J. M. Park, J. H. Oh, O. Gurel, S. S. Lee and J. G. Lee, Small, 2013 DOI:10.1002/smll.201202317.
- N. Prang, S. Preithner, K. Brischwein, P. Göster, A. Wöppel, J. Müller, C. Steiger, M. Peters, P. A. Baeuerle and A. J. da Silva, Brit. J. Cancer, 2005, 92, 342–349 CAS.
- M. Hosokawa, T. Hayata, Y. Fukuda, A. Arakaki, T. Yoshino, T. Tanaka and T. Matsunaga, Anal. Chem., 2010, 82, 6629–6635 CrossRef CAS PubMed.
- S. Zheng, H. K. Lin, B. Lu, A. Williams, R. Datar, R. J. Cote and Y. C. Tai, Biomed. Microdevices, 2011, 13, 203–213 CrossRef PubMed.
- J. Sun, M. Li, C. Liu, Y. Zhang, D. Liu, W. Liu, G. Hu and X. Jiang, Lab Chip, 2012, 12, 3952–3960 RSC.
- H. W. Hou, M. E. Warkiani, B. L. Khoo, Z. R. Li, R. A. Soo, D. S. W. Tan, W. T. Lim, J. Han, A. A. S. Bhagat and C. T. Lim, Sci. Rep., 2013, 3, 1–3 Search PubMed.
- H. S. Moon, K. Kwon, K. A. Hyun, T. S. Sim, J. C. Park, J. G. Lee and H. I. Jung, Biomicrofluidics, 2013, 7, 014105 CrossRef.
- K. A. Hyun, K. Kwon, H. Han, S. I. Kim and H. I. Jung, Biosens. Bioelectron., 2013, 40, 206–212 CrossRef CAS PubMed.
- H. S. Moon, K. Kwon, S. I. Kim, H. Han, J. Sohn, S. Lee and H. I. Jung, Lab Chip, 2011, 11, 1118–1125 RSC.
- K. A. Hyun, T. Y. Lee and H. I. Jung, Anal. Chem., 2013, 85, 4439–4445 CrossRef CAS PubMed.
- C. L. Chen, K. C. Chen, Y. C. Pan, T. P. Lee, L. C. Hsiung, C. M. Lin, C. Y. Chen, C. H. Lin, B. L. Chiang and A. M. Wo, Lab Chip, 2011, 11, 474–483 RSC.
- P. F. Thomas and G. K. Jason, Lab Chip, 2012, 12, 2634–2637 RSC.
- T. M. Gorges, I. Tinhofer, M. Drosch, L. Röse, T. M. Zollner, T. Krahn and O. von Ahsen, BMC Cancer, 2012 DOI:10.1186/1471-2407-12-178.
- T. M. Gorges, I. Tinhofer, M. Drosch, L. Röse, T. M. Zollner, T. Krahn and O. von Ahsen, BMC Cancer, 2012 DOI:10.1186/1471–2407-12–178.
- T. M. Gorges and K. Pantel, Cancer Immunol. Immunother., 2013 DOI:10.1007/s00262-012-1387-1.
- D. J. Burgess, Nat. Rev. Cancer, 2013 DOI:10.1038/nrc3475.
- M. Yu, A. Bardia, B. S. Wittner, S. L. Stott, M. E. Smas, D. T. Ting, S. J. Isakoff, J. C. Ciciliano, M. N. Wells, A. M. Shah, K. F. Concannon, M. C. Donaldson, L. V. Sequist, E. Brachtel, D. Sgroi, J. Baselga, S. Ramaswamy, M. Toner, D. A. Haber and S. Maheswaran, Science, 2013, 339, 580–584 CrossRef CAS PubMed.
- S. D. Mikolajczyk, L. S. Millar, P. Tsinberg, S. M. Coutts, M. Zomorrodi, T. Pham, F. Z. Bischoff and T. J. Pircher, J. Oncol., 2011, 252361 Search PubMed.
- S. J. Tan, L. Yobas, G. Y. H. Lee, C. N. Ong and C. T. Lim, Biomed. Microdevices, 2009, 1, 883–892 CrossRef PubMed.
- S. E. Cross, Y. S. Jin, J. Y. Rao and J. K. Gimzewski, Nat. Nanotechnol., 2007, 2, 780–783 CrossRef CAS PubMed.
- A. M. Sieuwerts, J. Kraan, J. B. de Vries, P. van der Spoel, B. Mostert, J. W. M. Martens, J. W. Gratama, S. Sleijfer and J. A. Foekens, Breast Cancer Res. Treat., 2009, 118, 455–468 CrossRef CAS PubMed.
- Y. Zeng, R. Novak, J. Shuga, M. T. Smith and R. A. Mathies, Anal. Chem., 2010, 82, 3183–3190 CrossRef CAS PubMed.
- A. A. Powell, A. H. Talasaz, H. Zhang, M. A. Coram, A. Reddy, G. Deng, M. L. Telli, R. H. Advani, R. W. Carlson, J. A. Mollick, S. Sheth, A. W. Kurian, J. M. Ford, F. E. Stockdale, S. R. Quake, R. F. Pease, M. N. Mindrinos, G. Bhanot, S. H. Dairkee, R. W. Davis and S. S. Jeffrey, PLoS One, 2012, 7, e33788 CAS.
- J. Chung, H. Shao, T. Reiner, D. Issadore, R. Weissleder and H. Lee, Adv. Healthcare Mater., 2012, 1, 432–436 CrossRef CAS PubMed.
- J. A. Mayer, T. Pham, K. L. Wong, J. Scoggin, E. V. Sales, T. Clarin, T. J. Pircher, S. D. Mikolajczyk, P. D. Cotter and F. Z. Bischoff, Cancer Genet., 2011, 204, 589–598 CrossRef CAS PubMed.
- S. Hou, L. Zhao, Q. Shen, J. Yu, C. Ng, X. Kong, D. Wu, M. Song, X. Shi, X. Xu, W. H. OuYang, R. He, X. Z. Zhao, T. Lee, F. C. Brunicardi, M. A. Garcia, A. Ribas, R. S. Lo and H. R. Tseng, Angew. Chem., Int. Ed., 2013, 52, 3379–3383 CrossRef CAS PubMed.
- K. A. Hyun and H. I. Jung, Electrophoresis, 2013, 34, 1028–1041 CrossRef CAS PubMed.
- Z. Liu, W. Zhang, F. Huang, H. Feng, W. Shu, X. Xu and Y. Chen, Biosens. Bioelectron., 2013, 47, 113–119 CrossRef CAS PubMed.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 4, 179ra47 CrossRef PubMed.
- P. G. Schiro, M. Zhao, J. S. Kuo, K. M. Koehler, D. E. Sabath and D. T. Chiu, Angew. Chem., 2012, 124, 4696–4700 CrossRef.
- M. E. Warkiani, G. Guan, K. B. Luan, W. C. Lee, A. A. S. Bhagat, D. S. W. Tan, W. T. Lim, S. C. Lee, P. C. Y. Chen, C. T. Lim and J. Han, Lab Chip, 2013 10.1039/C3LC50617G.
- P. Lv, Z. Tang, X. Liang, M. Guo and R. P. S. Han, Biomicrofluidics, 2013, 7, 034109 CrossRef.
- S. B. Huang, M. H. Wu, Y. H. Lin, C. H. Hsieh, C. L. Yang, H. C. Lin, C. P. Tseng and G. B. Lee, Lab Chip, 2013, 13, 1371–1383 RSC.
- P. Augustsson, C. Magnusson, M. Nordin, H. Lilja and T. Laurell, Anal. Chem., 2012, 84, 7954–7962 CrossRef CAS PubMed.
- X. Yao, M. Labelle, C. R. Lamb, J. M. Dugan, C. A. Williamson, D. R. Spencer, K. R. Christ, R. O. Keating, W. D. Lee, G. A. Paradis, S. Begum, R. O. Hynes and K. D. Wittrup, Int. J. Cancer, 2013 DOI:10.1002/ijc.28312.
| This journal is © The Royal Society of Chemistry 2014 |