 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The determination of V and Mo by dispersive liquid–liquid microextraction (DLLME) combined with laser-induced breakdown spectroscopy (LIBS)
Amanda M. D.
de Jesus
ab,
Miguel Ángel
Aguirre
b,
Montserrat
Hidalgo
b,
Antonio
Canals
b and
Edenir R.
Pereira-Filho
*a
aDepartment of Chemistry, Federal University of São Carlos, Rodovia Washington Luiz, km 235, Caixa Postal 676, São Carlos, CEP 13565-905, São Paulo, Brazil. E-mail: erpf@ufscar.br
bDepartment of Analytical Chemistry and Food Science and University Institute of Materials, Faculty of Science, University of Alicante, Alicante E-03080, Spain
First published on 25th June 2014
Abstract
Laser-induced breakdown spectroscopy (LIBS) is a promising analytical technique with well-known advantages and limitations. However, despite its growing popularity, this technique has been applied mainly to solid samples and there have been a smaller number of studies devoted to liquid samples. This lack of studies is mainly due to experimental difficulties in the analysis of liquid matrices. Sensitivity can be improved and matrix effects minimized in the LIBS analysis of aqueous samples by using a dispersive liquid–liquid microextraction (DLLME) procedure followed by drying the extract on a suitable surface prior to laser irradiation. The combination of DLLME-LIBS is fast, easy to use, and inexpensive. The small volume of the final extract is sufficient for LIBS analysis, and the procedure generates little waste. It is likely that this combination could be automated during future work. The limits of detection (LOD) and quantification (LOQ) achieved using the proposed method were 30 and 70 μg L−1 for Mo and 5 and 20 μg L−1 for V, respectively. Using this method, we analyzed samples of pharmaceutical, multimineral formulation, soil, mineral water and a reference material NCS ZC 85005 (Beef Liver). In the latter, the concentration of V was below the LOQ, and the recovery of Mo was 103%.
1 Introduction
The laser-induced breakdown spectroscopy (LIBS) technique in analytical chemistry has become popular due to its versatility and simplicity when applied to the multi-element analysis of solid, liquid or gas samples, as it minimizes or eliminates sample pretreatment. In addition, LIBS is a portable technique, permitting field analysis and remote measurements. These factors allow the technique to be safely used in dangerous environments.1The LIBS technique has been successfully used for the determination of elements in different types of samples. These include biological materials,2,3 metal alloys,4,5 polymers,6,7 soil and minerals,8,9 and geological samples,10 among others.11,12 LIBS is applied mainly to solid samples, primarily because the samples can be analyzed directly without further preparation if standards are available.
The determination of V and Mo is generally difficult. This is especially true in the case of aqueous samples. The most common experimental difficulties when using LIBS are the formation of plasma and the generation of bubbles that affect the characteristics of subsequent plasmas.13,14 These drawbacks result in poor sensitivity and reproducibility in aqueous samples.13–16
One practical way to circumvent the limitations of LIBS with aqueous samples is to dry the sample on a suitable surface. We present the use of a microextraction technique followed by the evaporation of the organic phase as one reliable example. Liquid–liquid extraction has been widely used to eliminate interference and increase the sensitivity of analytical procedures. There has been an increase in the use of miniaturized liquid–liquid extraction since the year 2000. Among these techniques is dispersive liquid–liquid microextraction (DLLME), which is in accordance with the principles of green chemistry: it is a simple, fast and inexpensive procedure.17
The use of a single drop of DLLME solvent dried on an aluminum surface combines the benefits of preconcentration by microextraction with the advantages of LIBS, such as multi-element determination. The goal of this study was to combine the DLLME technique with LIBS in the determination of V and Mo.
2 Experimental
2.1 Reagents
All reagents used were of analytical grade. Solutions were prepared using ultrapure water obtained from a Milli-Q® purification system (Millipak-40 Filter Unit 0.22 μm NPT, Bedford, MA, USA) with a resistivity greater than 18.2 MΩ cm.Analytical reference solutions were prepared by diluting stock standard solutions containing 1000 mg L−1 of V and Mo High-Purity Mono Element Standard Solutions (Charleston, USA) with ultrapure water.
The solution of chelating agent 8-hydroxyquinoline (8-HQ) (Vetec, Rio de Janeiro, RJ, Brazil) was prepared daily by dissolving the appropriate amounts of 8-HQ in 10 mL of ethanol and storing these solutions in brown glass flasks. Nitric acid 65% (w/w), H2O2 30% (w/w) and HClO4 65% (w/w) (Merck, Darmstadt, Germany) were used for microwave sample preparation.
2.2 Instrumentation
The LIBS system was composed of a Nd:YAG laser (model HYL-101 Handy-YAG, Q-switched, Quanta System S.P.A., Varese, Italy). We used the fundamental wavelength of the laser (1064 nm) with a pulse energy of 180 mJ (pulse width 6 ns FWHM), operated in single-pulse mode. The laser beam was focused on the sample by a biconvex lens with a focal length of 100 mm. The emitted radiation was collected using a five-furcated optical fiber (5 × 400 μm fibers, model FC5-UV400-2, Avantes, Eerbeek, The Netherlands) and detected using a five-channel spectrometer (model AvaSpec-2048-SPU Avantes) covering the wavelengths from 197.146 to 852.190 nm.A delay system consisting of two pulse generators (delay generator/digital pulse, Model DG 535, Stanford Research Systems, Inc. and 1 Hz to 50 MHz pulse generator, model PM-5715, Philips) was used for synchronizing the firing of the laser and data acquisition. An LG laptop (Intel Core 2, 1.00 GB of RAM and Windows Vista) equipped with AvaSoft© complete software (v. 7.6.1., Avantes) was used for data acquisition.
In order to compare the results obtained, an ICP OES spectrometer (Perkin Elmer, model Optima 4300DV, Norwalk, CT, USA) with dual view capacity but that was operated in the axially viewed plasma mode (radiofrequency power of 1400 W) was used.
2.3 Samples and sample preparation
To demonstrate the applicability of the proposed method, different samples were tested: (1) water, (2) pharmaceutical, (3) multimineral formulation, (4) soil and (5) food samples. Water samples were used without further preparation. The pharmaceutical sample and multimineral formulation were ground manually using an agate mortar and pestle to obtain a homogeneous material. Before the dispersive liquid–liquid microextraction procedure, 500 mg of each samples were weighed and digested using 7.0 mL of HNO3 65% (w/w) and 1 mL of H2O2 30% (w/w). For the soil sample, 250 mg of the sample were weighed and digested using 6 mL of HNO3 65% (w/w), 1 mL of H2O2 30% (w/w) and 1 mL of HClO4 65% (w/w). The digestion procedure was conducted in a microwave (MW) oven (Ethos, Milestone, Italy). The MW digestion program used for the pharmaceutical, multimineral formulation and soil samples was composed of only one step: 30 min at 200 °C (in the first 10 min the temperature was increased from room temperature up to 200 °C).A beef liver certified reference material (NCS ZC 85005) was also used. A sample mass of 100 mg was weighed and MW-digested using 10 mL of HNO3 65% (w/w). The digestion program was configured as follows: 20 min at 180 °C (in the first 10 min the temperature was increased from room temperature up to 180 °C). In all cases the microwave power was 1000 W.
2.4 Dispersive liquid–liquid microextraction procedure
The microextraction procedure is summarized in 3 steps: (1) in a glass tube, 15 mL of sample and 166 μL of a 8-hydroxyquinoline complexing agent (8-HQ) solution were added (0.05 or 0.1% w/v) and the pH value was adjusted to 2 or 5 with HNO3 or NH4OH solutions. Then, either 30 or 60 μL of the extraction solvent (1-undecanol) was added, and the mixture was shaken using a vortex shaker for a specified time (2 or 4 min). (2) The solution was centrifuged (2000 or 4000 rpm) for either 4 or 8 min to separate the two phases, with the organic phase containing the analytes at the top. (3) Ten microliters of the organic phase was collected using a microsyringe. During the optimization, a solution containing 500 μg L−1 of both V and Mo was used.2.5 Analysis of extracts from DLLME by LIBS
For LIBS analysis, 10 μL of the solvent containing the analyte was placed on a suitable sample holder. This holder consisted of a piece of thin Al foil in which several cells had been previously molded with a micropipette tip to contain and prevent spreading of the drop. The Al foil was placed on a plate, heated for 5 min on a hot plate to evaporate the organic phase from the microdroplet, and then allowed to cool.18 Once the support was at room temperature the LIBS measurements were carried out. Fig. 1 shows a pictorial diagram of the DLLME and LIBS analysis steps.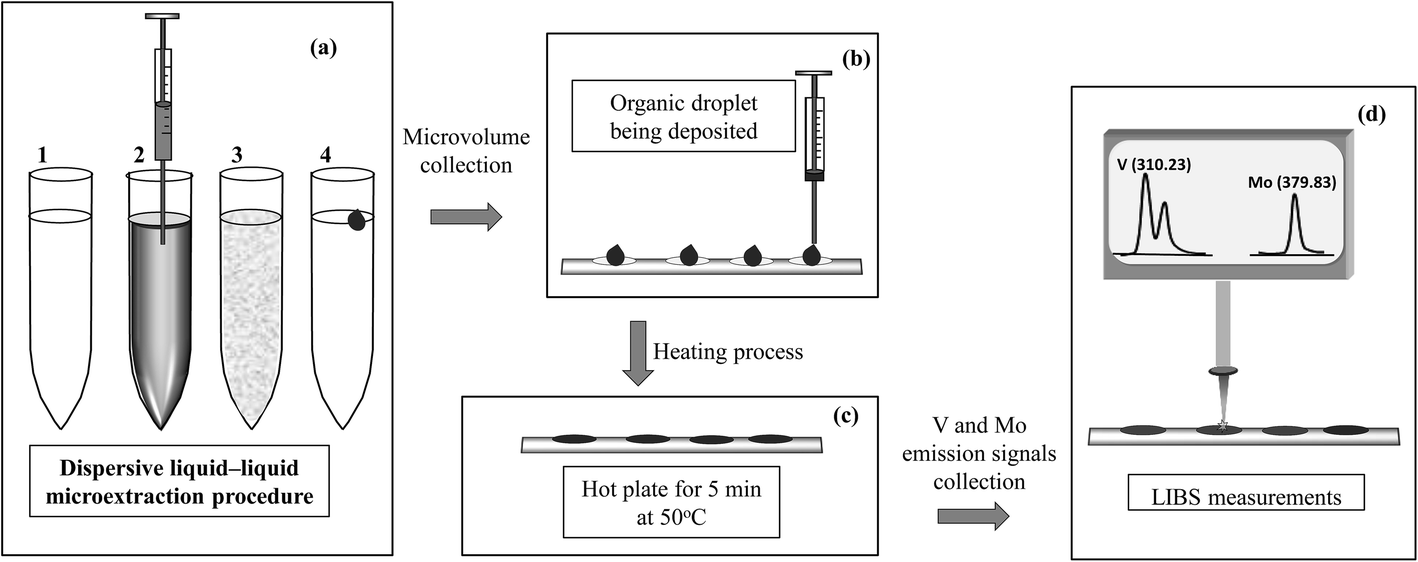 | ||
| Fig. 1 Pictorial description of the steps related to (a) the microextraction procedure (1 – mixture of the sample and 8-HQ solution, 2 – addition of the organic extractant solvent, 3 – vortex shaking and 4 – phase separation), (b) organic microdroplet collection and deposition in the cell, (c) drying process and (d) LIBS analysis of the dried microdroplets deposited on the aluminum support. | ||
3 Results and discussion
3.1 Optimization of dispersive liquid–liquid microextraction procedure
The optimization of the DLLME procedure was divided into two complementary parts. In the first part, a Plackett–Burman design was used to identify the most significant among the 7 variables. In this case, a solution containing both V and Mo at a concentration of 500 μg L−1 was used. The DLLME variables investigated were (a) the concentration (0.05 or 0.1 w/v) of the complexing agent (8-HQ), (b) the volume (30 or 60 μL) of the extractant solvent (1-undecanol), (c) centrifugation time (4 or 8 min), (d) vortexing time (2 or 4 min), (e) pH (2 or 5), (f) the presence or absence of NaCl and (g) centrifuge speed (2000 or 4000 rpm). The variables were studied in two levels (−1 and +1), and 12 experiments were performed.The two variables, the pH value and the volume of the extractant solvent, showed a significant effect on the Plackett–Burman experiment. Microsoft Excel was used in these calculations.
Therefore, a central composite design (CCD) was performed to optimize these two variables. Here the variables were investigated at five levels and the coded values ranged from 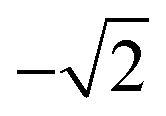 to
to 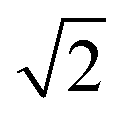 and Microsoft Excel was also used. Table 1 shows the values established in the CCD to investigate the behavior of pH and extractant solvent (SE) volume and the predictive ability of the emission signals obtained for V and Mo. While carrying out the CCD, 12 additional experiments were performed with the V and Mo concentrations fixed again at 500 μg L−1. Four experiments were performed at the central point (variables coded in 0, see experiments 9–12 in Table 1) to calculate the sum of the squares for the pure error and to evaluate the significance of the coefficient models proposed for V and Mo.
and Microsoft Excel was also used. Table 1 shows the values established in the CCD to investigate the behavior of pH and extractant solvent (SE) volume and the predictive ability of the emission signals obtained for V and Mo. While carrying out the CCD, 12 additional experiments were performed with the V and Mo concentrations fixed again at 500 μg L−1. Four experiments were performed at the central point (variables coded in 0, see experiments 9–12 in Table 1) to calculate the sum of the squares for the pure error and to evaluate the significance of the coefficient models proposed for V and Mo.
| Experiment | pH | Extractant solvent volume (SE) | Emission intensity | |||
|---|---|---|---|---|---|---|
| Coded value | Real value | Coded value | Real value (μL) | V | Mo | |
| 1 | −1 | 3.1 | −1 | 40.0 | 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 314 314 |
12758 |
| 2 | 1 | 5.1 | −1 | 40.0 | 22019 |
11299 |
| 3 | −1 | 3.1 | 1 | 80.0 | 40304 |
9812 |
| 4 | 1 | 5.1 | 1 | 80.0 | 3456 | 2140 |
| 5 |
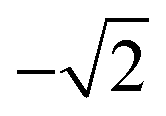
|
2.6 | 0 | 60.0 | 25022 |
16888 |
| 6 | 0 | 4.1 |
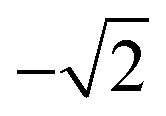
|
31.7 | 28557 |
13247 |
| 7 |
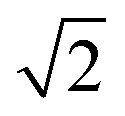
|
5.5 | 0 | 60.0 | 314 | 10 |
| 8 | 0 | 4.1 |
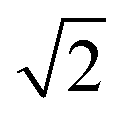
|
88.2 | 35677 |
3305 |
| 9 | 0 | 4.1 | 0 | 60.0 | 32244 |
18073 |
| 10 | 0 | 4.1 | 0 | 60.0 | 38734 |
14505 |
| 11 | 0 | 4.1 | 0 | 60.0 | 37493 |
15008 |
| 12 | 0 | 4.1 | 0 | 60.0 | 44844 |
15707 |
The regression models (only the significant coefficients) proposed for V and Mo are presented as eqn (1) and (2), respectively:
| V (emission intensity) = 38328 − 12886pH − 10985(pH2) | (1) |
| Mo (emission intensity) = 15823 − 4125pH − 3270SE − 3527(pH2) − 3614(SE2) | (2) |
In the case of V, only the linear and quadratic coefficients for pH presented significant values at a confidence level of 95%. In this case, any extractant solvent volume between the evaluated range (32 and 88 μL) can be used. For Mo both linear and quadratic coefficients of pH and extractant solvent volume were significant. Fig. 2 shows the overlapped contour plots for the models obtained for V and Mo. As observed for V (see vertical lines), high signals are obtained when the pH is in the range of 3.0 to 3.8, but the signal is indifferent to the extraction solvent volume in the evaluated range (32–88 μL). For Mo, an optimal condition exists when the pH value lies between 3.0 and 3.8 and the extraction solvent volume is between 48 and 56 μL (see ellipses). For this reason, a compromise condition is necessary to determine both analytes in the same microextraction procedure. Observing the practical operational conditions, a pH of 3.6 and an extraction volume of 50 μL were chosen as optimal conditions for both the variables studied and both the analytes. The other final optimized conditions for the DLLME procedure were: a concentration of 8-HQ of 0.1(%) w/v, a vortex time of 2 (min), a centrifugation time of 8 (min) and a centrifugation speed of 4000 (rpm).
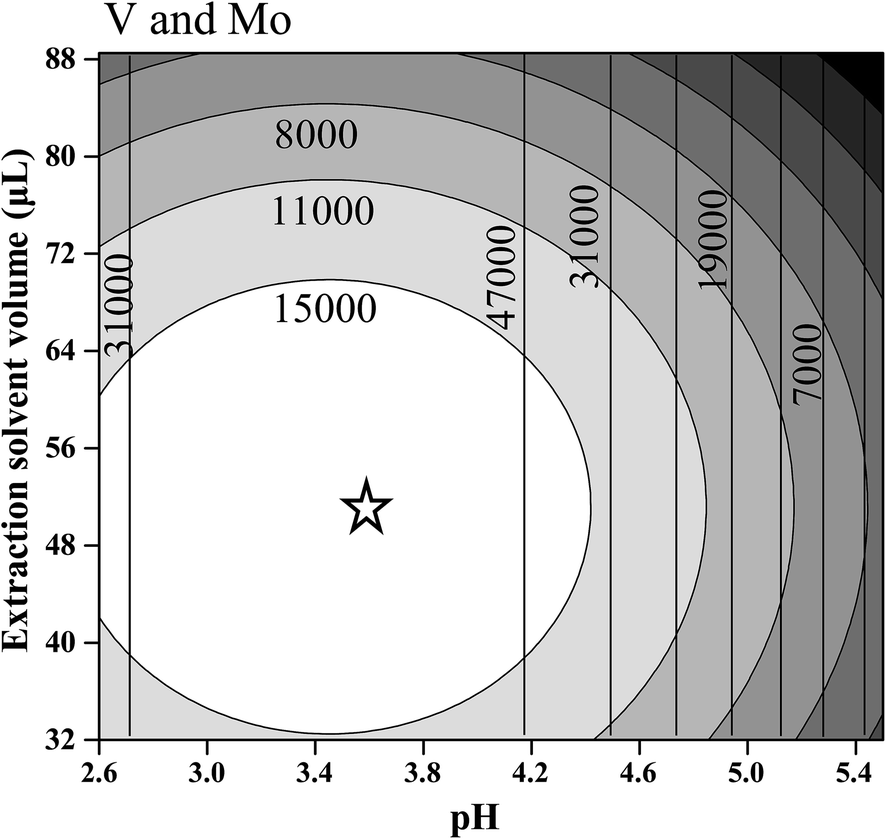 | ||
| Fig. 2 Contour plots overlapped for the regression models proposed for V (vertical lines) and Mo (ellipses). The star shows the optimal conditions. | ||
As mentioned in the experimental section (section 2.5), after the microextraction procedure, a droplet of the organic layer with a volume of 10 μL was dried on an aluminum plate (see details in Fig. 1) and then subsequently analyzed by LIBS.
3.2 Figures of merit
The figures of merit of the developed procedure were evaluated by calculating the limits of detection (LOD) and quantification (LOQ), defined as LOD = 3 σ/s and LOQ = 10 σ/s, where s is the slope (sensitivity) of the analytical curve and σ is the standard deviation of 10 consecutive measurements of the blank.Fig. 3 shows some emission signals obtained for V (Fig. 3a) and Mo (Fig. 3b) when 10 μL aqueous standard solutions were analyzed by only LIBS (40 mg L−1), i.e. without the prior DLLME procedure and by DLLME-LIBS (100 μg L−1). As can be observed when 100 μg L−1 of V and Mo was determined combining DLLME-LIBS it was possible to obtain analytical signals in the same order of magnitude when 40 mg L−1 was determined using only LIBS. The combined method of DLLME-LIBS was linear from 20 to 750 μg L−1 for V and from 70 to 750 μg L−1 for Mo.
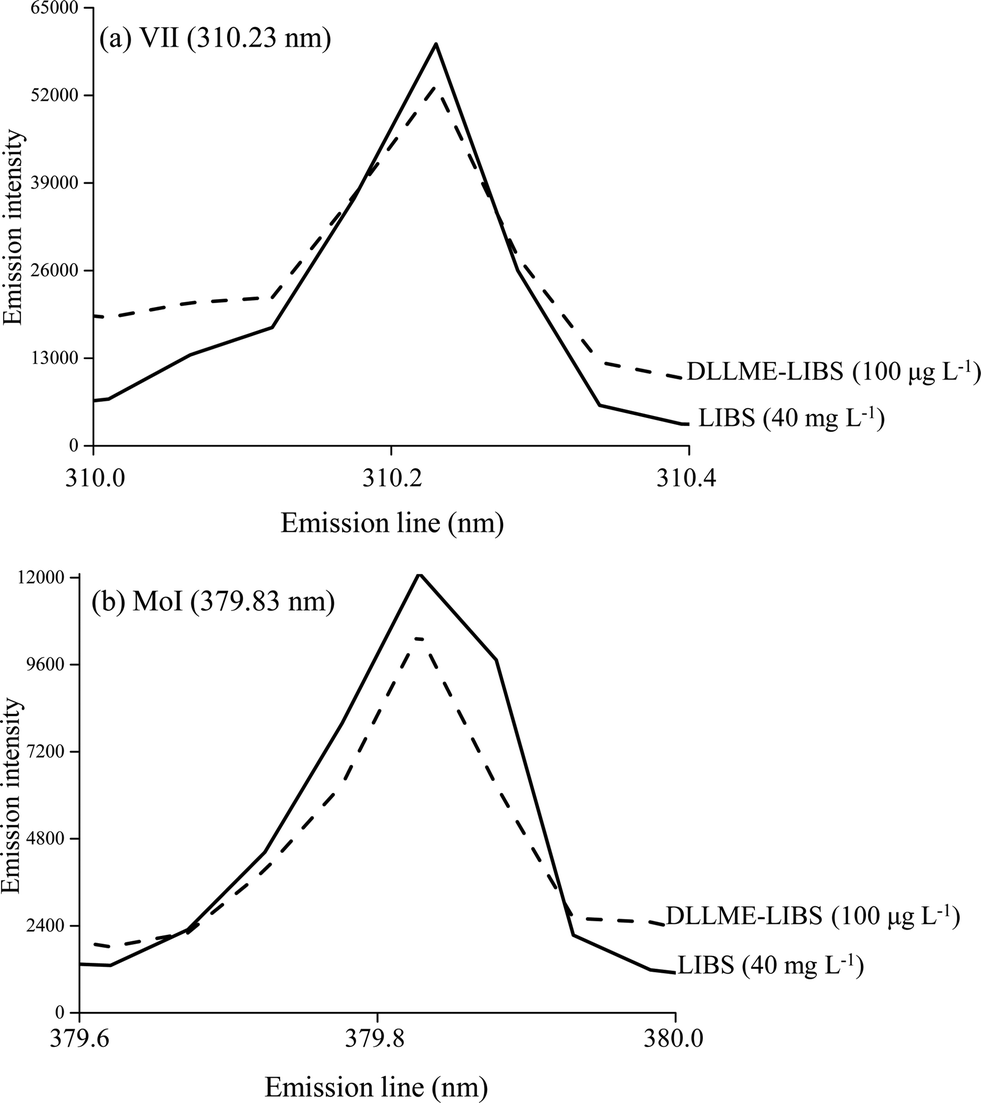 | ||
| Fig. 3 Emission signals of VII (310.23 nm) (a) and MoI (379.83 nm) (b) using LIBS and DLLME-LIBS methodologies. | ||
A comparison of the figures of merit obtained with the proposed method (DLLME-LIBS) and using only LIBS analysis is shown in Table 2. By using two standard calibration curves with microextraction (DLLME-LIBS) and without microextraction (LIBS), it was possible to estimate the preconcentration factors as 12-fold for V and 9-fold for Mo.
| Parameters | VII (310.23 nm) | MoI (379.83 nm) | ||
|---|---|---|---|---|
| LIBS | DLLME-LIBS | LIBS | DLLME-LIBS | |
| a n = 10. b Sensitivity DLLME-LIBS/Sensitivity LIBS. c LOD LIBS/LOD DLLME-LIBS. | ||||
| Linear range (number of calibration points = 5) | 0.2 to 40 mg L−1 | 20 to 750 μg L−1 | 0.5 to 40 mg L−1 | 70 to 750 μg L−1 |
| Correlation coefficient (number of calibration points = 5) | 0.995 | 0.994 | 0.966 | 0.966 |
| Sensitivity (counts L mg−1) | 7575 | 82901 |
1407 | 9810 |
| LOD (μg kg−1) | 60 | 5 | 300 | 30 |
| LOQ (μg kg−1) | 200 | 20 | 500 | 70 |
| Blank signal (mean ± standard deviation) | 145 ± 24 | 158 ± 39 | 387 ± 213 | 245 ± 73 |
| Repeatabilitya (500 μg L−1) (RSD %) | — | 6 | — | 9 |
| Relative sensitivityb | 11 | 7 | ||
| Relative LODc | 12 | 9 | ||
3.3 Application to samples
The recovery of both V and Mo in a sample of mineral water was evaluated by using spiked/recovery assays. The added concentrations of the analyte varied from 506 to 240 μg L−1, and the recoveries ranged from 94 to 105%. The basal concentrations of V and Mo in the sample were below the LOD (see Table 2) for the DLLME-LIBS method.All the digested samples (pharmaceutical, multimineral formulation and soil), including the reference material (food), were analyzed using only the proposed DLLME-LIBS procedure in order to prove experimentally the feasibility of this combination.
Table 3 shows the results obtained for the pharmaceutical, multimineral formulation and soil samples. These results were compared with those obtained from ICP OES analysis. Using these ICP OES results as reference values, the recovery obtained using the DLLME-LIBS methodology ranges from 92 to 104%. As observed from this table, pharmaceutical (vanadium chelate) and multimineral formulation samples were tested. The first has been suggested for the treatment of diabetes, and the second is a multimineral and multivitamin supplement. The V concentration in the chelate was high (3352 mg kg−1), whereas a much lower concentration was found in the multivitamin sample (9.9 mg kg−1). Only Mo was observed in the multimineral at a concentration of 13.2 mg kg−1. In the case of the soil sample, only V was detected with a concentration of 12.0 mg kg−1.
| Samples | Analyte concentration (mg kg−1) | |||
|---|---|---|---|---|
| ICP OES | DLLME-LIBS (recovery, %) | |||
| V | Mo | V | Mo | |
| Pharmaceutical (vanadium chelate) | 3210 ± 92 | <LOD | 3352 ± 748 (104) | <LOD |
| Multimineral formulation | 10.7 ± 2.4 | 13.7 ± 2.7 | 9.9 ± 2.7 (92) | 13.2 ± 4.9 (96) |
| Soil | 12.3 ± 3.0 | <LOQ | 12.0 ± 5.0 (97) | <LOQ |
The analysis of solid samples by digestion + microextraction + LIBS has been made to demonstrate experimentally the feasibility of this combination. In addition, the solid sample digestion makes feasible the comparison with aqueous calibration standards.
The trueness of the proposed procedure was evaluated from the analysis of a certified reference material (CRM), NCS ZC 85005 (Beef Liver). Vanadium and Mo certified values are 0.267 (reference value) and 3.97 ± 0.28 mg kg−1, respectively. The V concentration found was below the LOQ of the proposed method and the Mo recovery was 103%.
4 Conclusions
LIBS can be successfully used in combination with the technique of dispersive liquid–liquid microextraction for the analysis of V and Mo in different types of samples (i.e. solid and liquid). When solid samples are analyzed aqueous standard calibration solutions can be used after the digestion of solid samples.The sensitivity obtained with DLLME-LIBS is approximately 11 and 7 times greater for V and Mo, respectively, than that obtained without DLLME, and the LOD is approximately 12 and 9 times lower for V and Mo, respectively.
This study presents a new step forward in the applicability of LIBS to the analysis of liquid samples. Obviously, further work is required and this is under investigation in our laboratories.
Acknowledgements
The authors are grateful to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-DGU and grants 243/11 and 1267/01-2), the Spanish Government (Projects CTQ2011-23968 and PHB2010-0018-PC), the Regional Government of Valencia (Spain) (ACOMP/2013/072), Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq (grants 304772/2012-7 and 474357/2012-0) and Grants 2012/01769-3 and 2012/50827-6, São Paulo Research Foundation (FAPESP), for the financial support.References
- C. Pasquini, J. Cortez, L. M. C. Silva and F. B. Gonzaga, J. Braz. Chem. Soc., 2007, 18, 463 CrossRef CAS PubMed.
- M. Galiová, J. Kaiser, K. Novotný, J. Novotný, T. Vaculovic, M. Liska, R. Malina, K. Stejskal, V. Adam and R. Kizek, Appl. Phys. A: Mater. Sci. Process., 2008, 93, 917 CrossRef.
- J. Kaiser, K. Novotný, M. Z. Martin, A. Hrdlička, R. Malina, M. Hartl, V. Adam and R. Kizek, Surf. Sci. Rep., 2012, 67, 233 CrossRef CAS PubMed.
- L. Caneve, F. Colao, R. Fantoni and V. Spizzichino, Appl. Phys. A: Mater. Sci. Process., 2006, 85, 151 CrossRef CAS PubMed.
- I. Cravetchi, M. Taschuk, Y. Tsui and R. Fedosejevs, Anal. Bioanal. Chem., 2006, 385, 287 CrossRef CAS PubMed.
- R. J. Lasheras, C. Bello-Gálvez and J. Anzano, Polym. Test., 2010, 29, 1057 CrossRef CAS PubMed.
- K. M. Santos, J. Cortez, I. M. Raimundo Jr, C. Pasquini, E. S. Boa Morte and M. G. A. Korn, Microchem. J., 2013, 110, 435 CrossRef CAS PubMed.
- M. Dell'Aglio, R. Gaudiuso, G. S. Senesi, A. De Giacomo, C. Zaccone, T. M. Miano and O. De Pascale, J. Environ. Monit., 2011, 13, 1422 RSC.
- D. C. Alvey, K. Morton, R. S. Harmon, J. L. Gottfried, J. J. Remus, L. M. Collins and M. A. Wise, Appl. Opt., 2010, 49, 168 CrossRef.
- R. S. Harmon, F. C. DeLucia, C. E. McManus, N. J. McMillan, T. F. Jenkins, M. E. Walsh and A. Miziolek, Appl. Geochem., 2006, 21, 730 CrossRef CAS PubMed.
- M. A. Aguirre, M. Hidalgo, A. Canals, J. A. Nobrega and E. R. Pereira-Filho, Talanta, 2013, 117, 419 CrossRef CAS PubMed.
- Q. Godoy, F. O. Leme, L. C. Trevisan, E. R. Pereira-Filho, I. A. Rufini, D. Santos and F. J. Krug, Spectrochim. Acta, Part B, 2011, 66, 138 CrossRef PubMed.
- D. A. Cremers, L. J. Radziemski and T. R. Loree, Appl. Spectrosc., 1984, 38, 721 CrossRef CAS.
- A. De Giacomo, M. Dell'Aglio and O. De Pascale, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 1035 CrossRef CAS PubMed.
- S. Koch, W. Garen, M. Müller and W. Neu, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 1071 CrossRef CAS.
- X. Fang and S. R. Ahmad, Appl. Spectrosc., 2007, 61, 1021 CrossRef CAS.
- J. M. Kokosa, TrAC, Trends Anal. Chem., 2013, 43, 2 CrossRef CAS PubMed.
- M. A. Aguirre, S. Legnaioli, F. Almodóvar, M. Hidalgo, V. Palleschi and A. Canals, Spectrochim. Acta, Part B, 2013, 79, 88 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |