
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroporation-based delivery of cell-penetrating peptide conjugates of peptide nucleic acids for antisense inhibition of intracellular bacteria†
Sai
Ma
a,
Betsy
Schroeder
b,
Chen
Sun
a,
Despina Nelie
Loufakis
c,
Zhenning
Cao
a,
Nammalwar
Sriranganathan
b and
Chang
Lu
*c
aSchool of Biomedical Engineering and Sciences, Virginia Tech, Blacksburg, VA 24061, USA
bDepartment of Biomedical Sciences and Pathobiology, Virginia Tech, Blacksburg, VA 24061, USA
cDepartment of Chemical Engineering, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: changlu@vt.edu; Fax: +1 540 231 5022; Tel: +1 540 231 8681
First published on 18th August 2014
Abstract
Cell penetrating peptides (CPPs) have been used for a myriad of cellular delivery applications and were recently explored for delivery of antisense agents such as peptide nucleic acids (PNAs) for bacterial inhibition. Although these molecular systems (i.e. CPP–PNAs) have shown ability to inhibit growth of bacterial cultures in vitro, they show limited effectiveness in killing encapsulated intracellular bacteria in mammalian cells such as macrophages, presumably due to difficulty involved in the endosomal escape of the reagents. In this report, we show that electroporation delivery dramatically increases the bioavailability of CPP–PNAs to kill Salmonella enterica serovar Typhimurium LT2 inside macrophages. Electroporation delivers the molecules without involving endocytosis and greatly increases the antisense effect. The decrease in the average number of Salmonella per macrophage under a 1200 V cm−1 and 5 ms pulse was a factor of 9 higher than that without electroporation (in an experiment with a multiplicity of infection of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Our results suggest that electroporation is an effective approach for a wide range of applications involving CPP-based delivery. The microfluidic format will allow convenient functional screening and testing of PNA-based reagents for antisense applications.
:1). Our results suggest that electroporation is an effective approach for a wide range of applications involving CPP-based delivery. The microfluidic format will allow convenient functional screening and testing of PNA-based reagents for antisense applications.
Insight, innovation, integrationWe demonstrate electroporation as an effective approach for delivering cell-penetrating peptide conjugates of peptide nucleic acids (CPP–PNAs) into complex cellular systems for antisense applications. Delivery of CPP–PNA to inhibit encapsulated bacteria typically has a very low efficiency due to the multiple membrane barriers involved. We show that electroporation significantly improves the bioavailability of CPP–PNA for inhibition of bacterial cells encapsulated in macrophages. Our technology will allow functional screening of CPP–PNAs in realistic cellular environment and study of the antisense effects in vitro or in vivo. |
1. Introduction
Cell penetrating peptides (CPPs) are short (<30–35 amino acid residues), water-soluble peptides with a net positive charges at physiological pH.1,2 CPPs may serve as delivery vectors for large molecules and penetrate cell membranes at micromolar concentrations without causing serious damage to cells. There have been a number of CPPs discovered over the years, including TAT peptide derived from HIV-1,3–5 transportan and its analogue TP-10,6–10 penetratin11 and pVEC.8,12,13 The mechanisms for CPP entry into cells are still in debate. In general, it is believed that most CPPs enter cells via endocytosis while direct penetration may also be involved or even important in certain cases.2 These mechanisms also may vary based on the experimental conditions and the properties of the cargos.2There has been growing interest in using CPPs for delivery of antisense reagents such as siRNAs (small interfering RNAs) and PNAs (peptide nucleic acids, gene-specific artificial oligonucleotide analogs with peptide backbone instead of sugar backbone having high binding strength, specificity, and resistance to protease and nuclease degradation).14,15 Antisense technology permits silencing a selected gene by binding to the mRNA produced by the gene and effectively inactivating the gene.16 Endosomal escape following endocytotic entry is a critical step for these CPP conjugates to enter cytosol/nucleus.17,18 The low efficiency in this step also forms the most important roadblock for intracellular delivery of CPP conjugates.19 There have been demonstrations of improving the endosomal release by photodynamic treatment, Ca2+ treatment and chloroquine treatment.20–22 However, there has not been any quantitative assessment in the literature on the efficiency of the endosomal release of CPP conjugates and how much such process affects the eventual gene silencing result.
Antisense CPP–PNA conjugates have been used to inhibition of bacterial growth.23–26 Delivery of naked PNAs into bacteria was typically limited by their outer-membrane lipopolysaccharide (LPS) layer.27,28 However, CPPs such as (KFF)3K could effectively breach the LPS layer and enable delivery of PNAs that target gene with sequence specificity for essential mRNA or rRNA sequences, inhibiting gene expression and thereby cell growth.26,29,30
In spite of the success with using CPP–PNA conjugates on bacterial culture, there are additional layers of complexity when such strategy is applied to infected humans and animals. Pathogens like Salmonella, Brucella, Listeria and Mycobacterium have the unusual ability to not only survive in a host, but also subvert host immune responses. They can invade and live inside macrophages that are normally programmed to kill them.31 Not surprisingly, CPP–PNAs such as (KFF)3K-O-PNA conjugates showed much lower activity against bacteria encapsulated in membrane vesicles inside mammalian cells (e.g. macrophages) compared to direct use on bacterial culture, presumably due to the additional barriers formed by the plasma membrane and more importantly the endosomal membrane.32–34 Thus novel delivery methods are in high demand for enhancing the antisense effects of CPP–PNAs toward bacteria that are encapsulated inside host cells. So far there has been no demonstration of physical delivery methods for this application.
In this work, we delivered (KFF)3K-O-PNA conjugates into murine macrophages J774.A cells that were infected with Salmonella enterica serovar Typhimurium LT2 (S. typhimurium LT2). Such infected systems are typical in animals and humans.35–41 We found that the application of electroporation dramatically improved the bioavailability of (KFF)3K-O-PNA conjugates inside the macrophages and produced substantially more inactivation of the encapsulated bacteria (up to a factor of 9) than the experiments without electroporation. Since electroporation is a technique that allows both in vitro and in vivo applications,42 we believe that the use of electroporation for CPP–PNA conjugates delivery will be an effective strategy for a wide range of applications. Our microfluidic electroporation device will permit functional screening of CPP–PNA systems based on their effectiveness for inhibition of intracellular bacteria.
2. Experimental section
2.1 Microfluidic channel fabrication
The Salmonella infected macrophages were electroporated in a microfluidic channel, which permitted counting of the viable macrophages before and after the treatment. The devices were made by standard soft lithography.43,44 Photomasks were designed by Freehand MX (Macromedia, San Francisco, CA) and printed on 4000 dpi film. SU-8 2025 (MicroChem Corp., Newton, MA) was spincoated at 500 rpm for 10 s and 1500 rpm for 30 seconds in sequence on a 3 inch silicon wafer. The thickness of SU-8 was around 55 μm. SU-8 layer on the silicon wafer was patterned under UV light and then developed.Once the SU-8/silicon wafer master was fabricated, a two component silicone elastomer polydimethylsiloxane mixture (PDMS, GE RTV 615) with 10:1 ratio was poured on the master in a petri dish. The PDMS was vacuumed for 1 h and cured in an oven for 1 h at 80 °C. Then the PDMS structure was peeled off from the master and access holes were punched. The PDMS structure and a pre-cleaned glass slide were then oxidized in a plasma cleaner (Harrick Plasma, Ithaca, NY) and brought into contact immediately after oxidization for irreversible bonding to form closed channels. After fabrication, the channels were immediately filled by 20 μl PBS buffer containing fibronectin (10 μg ml−1) in order to keep the channel hydrophilic and facilitate cell adherence.
2.2 CPP–PNA
(KFF)3K-O-PNA (H-KFFKFFKFFK-o-cataagacggt-NH2, PNA Bio Inc, CA) targeting an RNA polymerase sigma 70 (sigma D) factor gene (rpoD) demonstrated enhanced inhibition of Salmonella growth in vitro and insufficient inhibition in infected macrophages.32 The PNA is conjugated with the CPP, (KFF)3K, via O linker which enhances the solubility. (KFF)3K-O-PNA is heated at 60 °C for 10 min before each use.2.3 In vitro CPP–PNA inhibition assay
S. typhimurium LT2 (ATCC 700720) was cultured in Tryptic Soy Broth (TSB) overnight at 37 °C. The CPP–PNAs (0, 10, 20 and 30 μM) were incubated with 106 cfu ml−1Salmonella in TSB for 8 h in a 96-well plate in triplicate. Samples were serially diluted and the colony forming units (CFUs) number of the surviving bacteria after each treatment were determined by subsequent plating onto Tryptic Soy Agar (TSA) plates.2.4 Macrophage culture, infection and CPP–PNA treatment
A murine macrophage cell line (J774A.1) was cultured in DMEM medium supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin Streptomycin (PS). Macrophages were seeded at a density of 5 × 105 cells per ml in a 6-well plate in 2 ml. J774A.1 cells were infected with S. typhimurium LT2 following the previously reported procedures.14,32 Briefly, overnight broth culture of Salmonella was centrifuged, resuspended in DMEM medium. Macrophages were infected at the multiplicity of infection (MOI) of 1:1 to 10:1 for 20 min. After 20 min infection, infected macrophages were washed with fresh DMEM containing 30 μg ml−1 gentamicin to kill extracellular Salmonella. The medium was replaced with fresh DMEM containing (KFF)3K-O-PNA and 10 μg ml−1 gentamicin at 1 h post infection (h.p.i.). Infected macrophages were incubated at 37 °C, under 5% CO2. Macrophages were counted and lysed with PBS containing sodium deoxycholate (0.5%) at 4 h.p.i. The sample was then serially diluted, and the number of Salmonella was determined by plating onto TSA for cfu counting.
2.5 Electroporation-assisted CPP–PNA treatment of infected macrophage cells
Electric pulses with a defined duration were generated by a relay (5501-05-1, Coto Technology, Kingstown, RI) controlled by a LabVIEW program via a data acquisition card (NI USB Digital I/O Device USB 6501) and a power supply (Bio-Rad, Hercules, CA) that provided a constant dc voltage (Fig. 1).45 The pulse duration was measured by an oscilloscope (B&K precision 2530, IL). | ||
| Fig. 1 Experimental setup for conducting electroporation-assisted CPP–PNA delivery. Electric pulse(s) was applied across a PDMS microfluidic channel via a pair of platinum (Pt) electrodes inserted in the reservoirs at both ends of the channel. The pulse duration was set by LabVIEW through DAQ and relays (supported by a 5 V power supply). The field intensity of the pulse was determined by the voltage output from the high voltage power supply and the channel dimensions. The dimensions of each channel were 3.8 mm × 0.6 mm × 55 μm. Each channel contained 500–700 macrophages. The scale bar in the phase contrast image is 200 μm. | ||
J774A.1 cells were infected with Salmonella as described above. Infected macrophages were harvested and suspended in fresh DMEM containing 30 μg ml−1 gentamicin at a density of 107 cell per ml. This cell density ensured high seeding density in the microfluidic channel, at about 500–700 cells in each channel.46 20 μl macrophage cell suspension was loaded into each microfluidic channel that was pre-treated with fibronectin. The microfluidic chip was then incubated at 37 °C, 5% CO2 for 1 h in an incubator to allow attachment of macrophages to the glass substrate. Image were taken to count macrophages in the microfluidic channel. The number of seeded macrophages in each channel was counted under a microscope. At 1 h.p.i., the infected cells were washed using 20 μl of electroporation buffer (1 mM MgSO4, 8 mM Na2HPO4, 2 mM KH2PO4, 250 mM sucrose). Then the channel was filled with the electroporation buffer containing (KFF)3K-O-PNA at a concentration of 30 μM. Two platinum electrodes were inserted into the two reservoirs at the end of the channel (Fig. 1) and electric pulses of various durations and field intensities were applied to infected macrophages. After electroporation, the medium inside the channel was replaced with fresh DMEM containing 10 μg ml−1 gentamicin and CPP–PNA. The reservoirs were sealed using microseal adhesive for PCR plates (Bio-Rad, Hercules, CA) to avoid evaporation. The chip was incubated at 37 °C, 5% CO2 for 4 h. PBS buffer was then flowed gently under gravity through the channel to flush dead cells out. Images were taken again to count the viable macrophages that had healthy appearance and adherence to the substrate.47,48 The number of Salmonella cells was determined by lysing macrophages and conducting cfu plate count.
3. Results and discussion
Electroporation (i.e. the exposure of cells to an external electric field with a higher-than-threshold intensity for a short duration) breaches cell membranes by creating nanoscale pores and destabilizing the membrane structures.43,49–51 Macromolecules in the solution such as nucleic acids or proteins become bound to the breached membrane during electroporation process and they move into the cytosol or nucleus driven by a combination of electrophoresis and diffusion.24,49–52 Electroporation process does not involve formation of endosomes and endocytosis as a part of delivery. In comparison, although the mechanism for (KFF)3K-O-PNA conjugates delivery is not completely understood, such processes are believed to involve endocytosis which involves encapsulation of the molecules in vesicles.21,34 The release of the conjugates from the endosomes is critical for the bioavailability and antisense efficiency for bacterial inhibition. The comparison of the two mechanisms is shown in Fig. 2. | ||
| Fig. 2 Postulated mechanisms for (KFF)3K-O-PNA conjugate delivery via electroporation and endocytosis into macrophages with lysosomal membrane encapsulated bacterial cells. (KFF)3K-O-PNAs are encapsulated in endosomes after endocytosis into the macrophage cells. A fraction of the CPP–PNAs get released from endosomes inside the macrophage and then enter the encapsulated bacterial cells after penetrating the membrane and create antisense effects. In comparison, naked (KFF)3K-O-PNAs enter the macrophage cell through electropores created by electroporation. They then move in the macrophage by diffusion and eventually enter the bacterial cells. | ||
We used (KFF)3K-O-PNA (H-KFFKFFKFFK-o-cataagacggt-NH2, PNA Bio Inc, CA) targeting an RNA polymerase sigma 70 (sigma D) factor gene (rpoD) in our experiments. KFFKFFKFFK, (KFF)3K, is a synthetic peptide rich in cationic lysine and hydrophobic phenylalanine residues. It sensitizes gram-negative bacteria by creating disorganization of the outer membrane and weak damage to the cytoplasmic membrane.53RpoD gene is an initiation factor that promotes attachment of RNA polymerase. The designed sequence of the PNA is complementary to a region of the rpoD gene's mRNA that is accessible to ribosome assembly.54,55 This conjugate shows effective inhibition on Salmonella growth.32
Fig. 3 shows the significant difference in the antisense activity of (KFF)3K-O-PNA toward Salmonella culture in vitro and Salmonella after infection of macrophages. To prove CPP–PNA can enter and inhibit Salmonella effectively, off-chip inhibition assay was applied. In vitro Salmonella culture and infected macrophages were treated with (KFF)3K-O-PNA targeting rpoD.32 For in vitro bacterium, various concentrations of the conjugate were applied for 8 h in 96-well plates. The CPP–PNA conjugate had efficient antisense effect. Compared to the control (without application of the antisense reagent), the bacterial concentration was lower by 7 log reduction at 20 μM CPP–PNA concentration. In comparison, we applied the same antisense reagent to Salmonella infected J774A.1 macrophages and then examined the change in the bacterium/macrophage ratio at 4 h.p.i. (i.e. the number of Salmonella cells per macrophage cell). Fig. 3 shows that the increase in the CPP–PNA concentration from 0 to 45 μM decreased the Salmonella/macrophage (a metric for evaluating the infection level of mammalian cells56) ratio from 3.7 to 1.3. The antisense effect of CPP–PNA (anti-rpoD) matches that in the literature very well under similar conditions.32 The increase in the concentration beyond 30 μM did not appear to decrease the ratio further. This result suggests that the dependence on the endocytic pathways puts a limit to the maximum delivery efficiency. Our results confirm that the encapsulation of Salmonella with endosomal membrane inside macrophages greatly decreases the bioavailability of CPP–PNA toward bacterial inhibition.
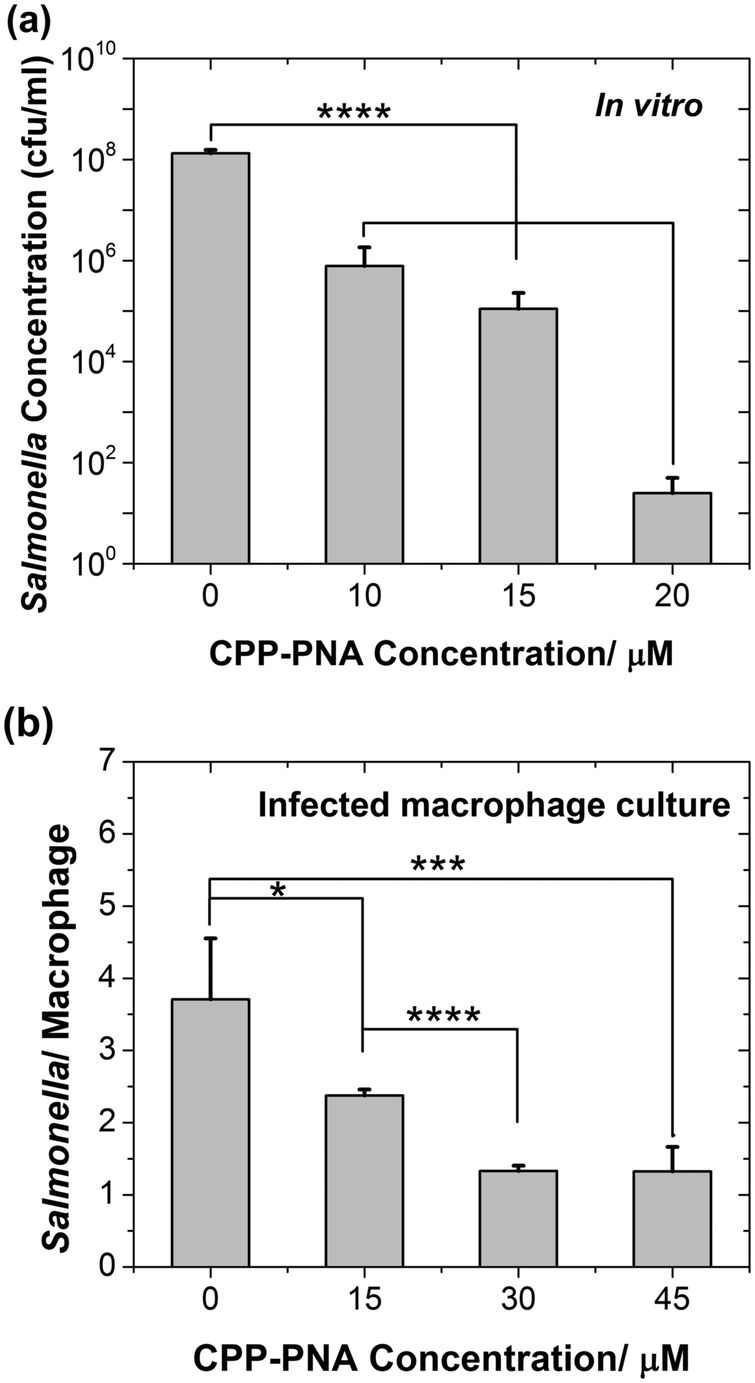 | ||
| Fig. 3 Antisense effect of (KFF)3K-O-PNAs in vitro (i.e. treating Salmonella culture directly) (a) and toward infected macrophages (b). (KFF)3K-O-PNA designed to target rpoD gene was used in all experiments. The macrophage cells were infected with multiplicity of infection (moi) of 4:1. Each data point represents 3–4 independent trials. Student's t tests were performed to determine the significance of the difference. The statistical significance level was defined as P < 0.001 (****), P < 0.005 (***), P < 0.01 (**) or P < 0.05 (*). | ||
We applied single-pulse electroporation of various field intensities to macrophages infected with various amounts of Salmonella, while having a CPP–PNA concentration of 30 μM. Fig. 4 shows that electroporation significantly improves the bioavailability of CPP–PNA inside macrophages. Fig. 4 shows that under electroporation (one single pulse of 800 V cm−1 and 5 ms), the average number of Salmonella per macrophage cell decreased consistently and drastically (from 1.7 at 0 μM to 0.2 at 30 μM) while CPP–PNA concentration increased. The data indicates that the CPP–PNA delivery was not saturated at the concentrations 20–30 μM under the electroporation condition. In comparison, when there was no electroporation applied, there was only a modest decrease (∼21%) in the Salmonella/macrophage ratio when the CPP–PNA concentration increased up to 30 μM. Fig. 5 shows that the increase in the field intensity of the pulse (from 0 to 1200 V cm−1) also created decrease in the Salmonella/macrophage ratio when CPP–PNA was delivered, due to increased delivery efficiency. In comparison, there was increase in the Salmonella/macrophage ratio with higher field intensity when CPP–PNA was not present. This was due to increased macrophage death under more intensive electroporation conditions (as shown in ESI,† Fig. S1) while the majority of Salmonella released by dead macrophages survived in the medium. The comparison between Fig. 5 and ESI,† Fig. S1 reveals that Salmonella was not killed by the electric field at 0–800 V cm−1 because the death rates of the macrophages (shown in ESI,† Fig. S1a) solely contributed to the increase in the Salmonella/macrophage ratio (shown in Fig. 5, without CPP–PNA). However, roughly 23% of the total Salmonella died due to electroporation under 1200 V cm−1 pulse. This threshold for electric lysis of Salmonella is similar to the one we previously reported for E. coli.57
 | ||
| Fig. 4 The effect of the CPP–PNA concentration on the antisense activity of (KFF)3K-O-PNA. The average number of Salmonella per macrophage under various (KFF)3K-O-PNA concentrations before and after electroporation (with a single pulse of 800 V cm−1 and 5 ms) are presented. MOI of 1:1 was used in the infection. Roughly 1000 macrophages were evaluated in each trial. Each data point represents 3–4 independent trials. Student's t tests were performed to determine the significance of the difference. The statistical significance level was defined as P < 0.001 (****), P < 0.005 (***), P < 0.01 (**) or P < 0.05 (*). | ||
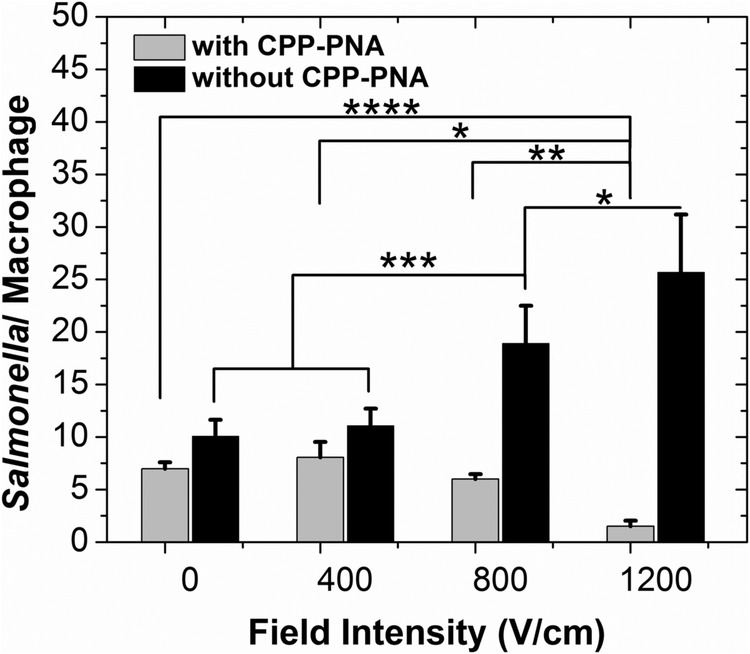 | ||
| Fig. 5 The effect of the pulse intensity on the antisense activity of (KFF)3K-O-PNA. The average number of Salmonella per macrophage under electroporation of various intensities (with a single pulse of 5 ms) with and without (KFF)3K-O-PNA (at a concentration of 30 μM) are presented. MOI of 10:1 was used in the infection. Roughly 1000 macrophages were evaluated in each trial. Each data point represents 3–4 independent trials. Student's t tests were performed to determine the significance of the difference. The statistical significance level was defined as P < 0.001 (****), P < 0.005 (***), P < 0.01 (**) or P < 0.05 (*). | ||
In Fig. 6, we examine the decrease in the average Salmonella per macrophage due to the antisense inhibition (i.e. the difference in the Salmonella/macrophage ratio between the cases of with and without CPP–PNA) under various infection and electroporation conditions. Electroporation is effective for the antisense inhibition in macrophage infected by various amounts of Salmonella. The antisense inhibition increased with higher field intensity used in the electroporation. Furthermore, the more heavily infected macrophages experienced more significant reduction in the average bacterial cell number per macrophage cell.
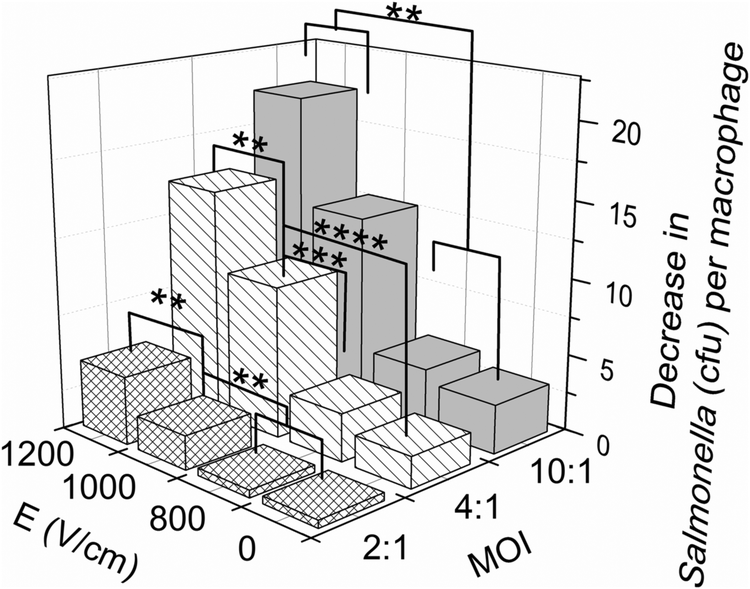 | ||
| Fig. 6 The antisense effect (measured by the decrease in the average Salmonella number per macrophage due to the addition of (KFF)3K-O-PNA) under various electroporation and infection conditions. Macrophages with various multiplicity of infection (2:1 to 10:1) were treated with a single 5 ms electric pulse and 30 μM (KFF)3K-O-PNA. The decrease in the average Salmonella per macrophage was calculated by examining the difference in the ratio Salmonella/macrophage between the test with (KFF)3K-O-PNA application and the one without its application. Each data point represents 3–4 independent trials. Student's t tests were performed to determine the significance of the difference. The statistical significance level was defined as P < 0.001 (****), P < 0.005 (***), P < 0.01 (**) or P < 0.05 (*). | ||
4. Conclusions
To summarize, we found that electroporation dramatically increases the bioavailability of (KFF)3K-O-PNA for inhibiting intracellular Salmonella inside macrophages. More intense electroporation (i.e. higher field intensity and longer duration) leads to more effective antisense activity but also higher macrophage death rate. We postulate that the improvement in the antisense effect of CPP–PNAs by electroporation was due to elimination of endocytic delivery that involves cargo encapsulation inside endosomes. We believe that electroporation can be widely applied to improve CPP–PNA delivery into cells, especially in complex cellular systems involving multiple barriers. Microfluidic electroporation permits the screening of these antisense reagents on a convenient platform.Acknowledgements
We acknowledge financial support from Virginia Tech ICTAS NanoBio Thrust, NSF CBET grants 1016547 and 0967069.References
- P. Lundberg and U. Langel, J. Mol. Recognit., 2003, 16, 227–233 CrossRef CAS PubMed.
- F. Madani, S. Lindberg, U. Langel, S. Futaki and A. Graslund, J. Biophys., 2011, 2011, 414729 Search PubMed.
- C. C. Berry, Nanomedicine, 2008, 3, 357–365 CrossRef CAS PubMed.
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS PubMed.
- M. Silhol, M. Tyagi, M. Giacca, B. Lebleu and E. Vives, Eur. J. Biochem., 2002, 269, 494–501 CrossRef CAS.
- M. Pooga, M. Hallbrink, M. Zorko and U. Langel, FASEB J., 1998, 12, 67–77 CAS.
- L. Fisher, U. Soomets, V. C. Toro, L. Chilton, Y. Jiang, U. Langel and K. Iverfeldt, Gene Ther., 2004, 11, 1264–1272 CrossRef CAS PubMed.
- N. Nekhotiaeva, A. Elmquist, G. K. Rajarao, M. Hallbrink, U. Langel and L. Good, FASEB J., 2004, 18, 394–396 CAS.
- M. Anko, J. Majhenc, K. Kogej, R. Sillard, U. Langel, G. Anderluh and M. Zorko, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 915–924 CrossRef CAS PubMed.
- R. B. G. Arrighi, C. Ebikeme, Y. Jiang, L. Ranford-Cartwright, M. P. Barrett, U. Langel and I. Faye, Antimicrob. Agents Chemother., 2008, 52, 3414–3417 CrossRef CAS PubMed.
- M. Pooga, U. Soomets, M. Hallbrink, A. Valkna, K. Saar, K. Rezaei, U. Kahl, J. X. Hao, X. J. Xu, Z. Wiesenfeld-Hallin, T. Hokfelt, T. Bartfai and U. Langel, Nat. Biotechnol., 1998, 16, 857–861 CrossRef CAS PubMed.
- A. Elmquist, M. Lindgren, T. Bartfai and U. Langel, Exp. Cell Res., 2001, 269, 237–244 CrossRef CAS PubMed.
- A. Elmquist and U. Langel, Biol. Chem., 2003, 384, 387–393 CrossRef CAS PubMed.
- M. N. Seleem, N. Jain, N. Pothayee, A. Ranjan, J. S. Riffle and N. Sriranganathan, FEMS Microbiol. Lett., 2009, 294, 24–31 CrossRef CAS PubMed.
- S. Shakeel, S. Karim and A. Ali, J. Chem. Technol. Biotechnol., 2006, 81, 892–899 CrossRef CAS PubMed.
- S. T. Crooke, Antisense drug technology: principles, strategies, and applications, CRC Press, 2010 Search PubMed.
- J. J. Turner, G. D. Ivanova, B. Verbeure, D. Williams, A. A. Arzumanov, S. Abes, B. Lebleu and M. J. Gait, Nucleic Acids Res., 2005, 33, 6837–6849 CrossRef CAS PubMed.
- M. Mae, S. El Andaloussi, T. Lehto and U. Langel, Expert Opin. Drug Delivery, 2009, 6, 1195–1205 CrossRef PubMed.
- P. Lundberg, S. El-Andaloussi, T. Sutlu, H. Johansson and U. Langel, FASEB J., 2007, 21, 2664–2671 CrossRef CAS PubMed.
- F. Salomone, F. Cardarelli, M. Di Luca, C. Boccardi, R. Nifosì, G. Bardi, L. Di Bari, M. Serresi and F. Beltram, J. Controlled Release, 2012, 163, 293–303 CrossRef CAS PubMed.
- T. Shiraishi and P. E. Nielsen, FEBS Lett., 2006, 580, 1451–1456 CrossRef CAS PubMed.
- T. Shiraishi and P. E. Nielsen, Nat. Protoc., 2006, 1, 633–636 CrossRef CAS PubMed.
- M. Hatamoto, A. Ohashi and H. Imachi, Appl. Microbiol. Biotechnol., 2010, 86, 397–402 CrossRef CAS PubMed.
- H. Bai, G. Sang, Y. You, X. Xue, Y. Zhou, Z. Hou, J. Meng and X. Luo, PLoS One, 2012, 7, e29886 CAS.
- H. Bai, X. Xue, Z. Hou, Y. Zhou, J. Meng and X. Luo, Curr. Drug Discovery Technol., 2010, 7, 76–85 CAS.
- L. Good, S. K. Awasthi, R. Dryselius, O. Larsson and P. E. Nielsen, Nat. Biotechnol., 2001, 19, 360–364 CrossRef CAS PubMed.
- L. Good, R. Sandberg, O. Larsson, P. E. Nielsen and C. Wahlestedt, Microbiology, 2000, 146, 2665–2670 CAS.
- L. Good and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2073–2076 CrossRef CAS.
- M. Vaara and M. Porro, Antimicrob. Agents Chemother., 1996, 40, 1801–1805 CAS.
- N. Bendifallah, F. W. Rasmussen, V. Zachar, P. Ebbesen, P. E. Nielsen and U. Koppelhus, Bioconjugate Chem., 2006, 17, 750–758 CrossRef CAS PubMed.
- J. E. Galan and D. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8754–8761 CrossRef CAS.
- M. A. Soofi and M. N. Seleem, Antimicrob. Agents Chemother., 2012, 56, 6407–6409 CrossRef CAS PubMed.
- S. Abes, D. Williams, P. Prevot, A. Thierry, M. J. Gait and B. Lebleu, J. Controlled Release, 2006, 110, 595–604 CrossRef CAS PubMed.
- P. E. Nielsen, Q. Rev. Biophys., 2005, 38, 345–350 CrossRef CAS PubMed.
- R. M. Tsolis, A. J. Bäumler and F. Heffron, Infect. Immun., 1995, 63, 1739–1744 CAS.
- S. W. Lindgren, I. Stojiljkovic and F. Heffron, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4197–4201 CrossRef CAS.
- N. A. Buchmeier and F. Heffron, Infect. Immun., 1991, 59, 2232–2238 CAS.
- J. Rengarajan, B. R. Bloom and E. J. Rubin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8327–8332 CrossRef CAS PubMed.
- R. K. Ernst, T. Guina and S. I. Miller, J. Infect. Dis., 1999, 179, S326–S330 CAS.
- G. J. Nau, J. F. L. Richmond, A. Schlesinger, E. G. Jennings, E. S. Lander and R. A. Young, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1503–1508 CrossRef CAS PubMed.
- J. E. Graham and J. E. Clark-Curtiss, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11554–11559 CrossRef CAS.
- M. P. Rols, C. Delteil, M. Golzio, P. Dumond, S. Cros and J. Teissie, Nat. Biotechnol., 1998, 16, 168–171 CrossRef CAS PubMed.
- J. Wang, Y. Zhan, V. M. Ugaz and C. Lu, Lab Chip, 2010, 10, 2057–2061 RSC.
- T. Geng, Y. H. Zhan, J. Wang and C. Lu, Nat. Protoc., 2011, 6, 1192–1208 CrossRef CAS PubMed.
- Y. Zhan, Z. Cao, N. Bao, J. Li, J. Wang, T. Geng, H. Lin and C. Lu, J. Controlled Release, 2012, 160, 570–576 CrossRef CAS PubMed.
- F. Wang, H. Wang, J. Wang, H. Y. Wang, P. L. Rummel, S. V. Garimella and C. Lu, Biotechnol. Bioeng., 2008, 100, 150–158 CrossRef CAS PubMed.
- H. Y. Wang and C. Lu, Biotechnol. Bioeng., 2008, 100, 579–586 CrossRef CAS PubMed.
- Y. Zhan, C. Sun, Z. Cao, N. Bao, J. Xing and C. Lu, Anal. Chem., 2012, 84, 8102–8105 CrossRef CAS PubMed.
- T. Geng and C. Lu, Lab Chip, 2013, 13, 3803–3821 RSC.
- J. B. Li and H. Lin, Bioelectrochemistry, 2011, 82, 10–21 CrossRef CAS PubMed.
- J. C. Weaver and Y. A. Chizmadzhev, Bioelectrochem. Bioenerg., 1996, 41, 135–160 CrossRef CAS.
- M. Yu, W. C. Tan and H. Lin, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2494–2501 CrossRef CAS PubMed.
- M. Vaara and M. Porro, Antimicrob. Agents Chemother., 1996, 40, 1801–1805 CAS.
- L. C. Rasmussen, H. U. Sperling-Petersen and K. K. Mortensen, Microb. Cell Fact., 2007, 6, 24 CrossRef PubMed.
- R. Dryselius, S. K. Aswasti, G. K. Rajarao, P. E. Nielsen and L. Good, Oligonucleotides, 2003, 13, 427–433 CrossRef CAS PubMed.
- E. Giacomini, E. Iona, L. Ferroni, M. Miettinen, L. Fattorini, G. Orefici, I. Julkunen and E. M. Coccia, J. Immunol., 2001, 166, 7033–7041 CrossRef CAS.
- H. Y. Wang, A. K. Bhunia and C. Lu, Biosens. Bioelectron., 2006, 22, 582–588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. See DOI: 10.1039/c4ib00172a |
| This journal is © The Royal Society of Chemistry 2014 |