
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tsetse fly attractants from a cashew nut shell extract by isomerising metathesis†
S.
Baader
a,
P. E.
Podsiadly
a,
D. J.
Cole-Hamilton
*b and
L. J.
Goossen
*a
aFachbereich Chemie, Organische Chemie, TU Kaiserslautern, Erwin-Schrödinger-Straße 54, 67663 Kaiserslautern, Germany. E-mail: goossen@chemie.uni-kl.de; Fax: +49 (631) 205-2046; Tel: +49 (631) 205-3921
bEaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews, Fife, KY16 9ST, Scotland, UK. E-mail: djc@st-andrews.ac.uk
First published on 27th August 2014
Abstract
Starting from a purified cashew nut shell extract containing mostly anacardic acid derivatives, the tsetse fly attractants 3-ethyl- and 3-propylphenol were selectively synthesised. The mixture was first converted into 3-(non-8-enyl)phenol in 98% purity via ethenolysis and distillation with concomitant decarboxylation. The olefinic side chain was then shortened by isomerising cross-metathesis with short-chain olefins in the presence of a [Pd(μ-Br)(tBu3P)]2 isomerisation catalyst and a second-generation Hoveyda–Grubbs catalyst, and the synthesis was completed by a hydrogenation step.
Introduction
Cashew nut shell liquid (CNSL) is a by-product of cashew nut processing, and is available on a scale of 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes per year.1 It is an excellent representative of an unused, non-eatable natural resource for which no competition occurs between land use for food or raw material production.2 When CNSL is extracted from the cashew nut shells via cold-press or solvent extraction, it consists predominantly of anacardic acids (1a–d) and other phenolic compounds such as cardanol (3-pentadecenylphenol) and cardol (5-pentadecenylbenzene-1,3-diol).3 Each of these has a 15-carbon side chain with a varying degree of saturation, located in the meta-position relative to the hydroxy group. Anacardic acids (1) are promising substrates for various decarboxylative and decarbonylative transformations.4 Upon distillation, they are known to decarboxylate with formation of cardanol and other phenols. The structurally related kairomones 3-ethyl- (2) and 3-propylphenol (3) are potent tsetse fly attractants (Fig. 1).5
000 tonnes per year.1 It is an excellent representative of an unused, non-eatable natural resource for which no competition occurs between land use for food or raw material production.2 When CNSL is extracted from the cashew nut shells via cold-press or solvent extraction, it consists predominantly of anacardic acids (1a–d) and other phenolic compounds such as cardanol (3-pentadecenylphenol) and cardol (5-pentadecenylbenzene-1,3-diol).3 Each of these has a 15-carbon side chain with a varying degree of saturation, located in the meta-position relative to the hydroxy group. Anacardic acids (1) are promising substrates for various decarboxylative and decarbonylative transformations.4 Upon distillation, they are known to decarboxylate with formation of cardanol and other phenols. The structurally related kairomones 3-ethyl- (2) and 3-propylphenol (3) are potent tsetse fly attractants (Fig. 1).5
 | ||
| Fig. 1 Kairomones used as tsetse fly attractants. | ||
Tsetse flies, the main vector of African sleeping sickness (trypanosomiasis), inhabit sub-Saharan Africa, the region where CNSL is predominantly produced in Africa (Nigeria, Ivory Coast, and Tanzania).6 Traps charged with the kairomones (2 and 3) are used as a sustainable and eco-friendly way to control the tsetse fly population. Both kairomones are worthwhile synthetic targets, because each works best to attract a certain type of vector tsetse fly, 2 for Glossina morsitans morsitans, and 3 for G. pallidipes.7
Various strategies for the synthesis of 3-propylphenol (3) have been reported. These include multi-step reactions with waste-intensive key steps, e.g. Wittig and Grignard reactions of 3-hydroxybenzaldehyde.83 has also been synthesised from cardanol via isomerisation of the double bond into the benzylic position, followed by metathesis with 2-butene.9 However, the isomerisation step resulted in a mixture of isomers, among which the desired product with the double bond conjugated with the aromatic ring made up for only 40%. As a consequence, the overall yield of 3-propylphenol (3) remained unsatisfactory (11% based on cardanol).
More efficient syntheses of 3-propyl- (3) and 3-ethylphenol (2) directly from CNSL would, thus, be highly desirable. We envisioned that isomerising olefin metathesis could be the key technology to access kairomones from CNSL, either as a mixture of 2 and 3 that might directly be usable in the fly traps, or in pure form.
Our strategy was to start with a basic extraction of CNSL, which would serve to remove side components such as the resorcinolic derivative cardol and lead to anacardic acids (1), a mixture of salicylic acids 1a–d with saturated, mono-, di- and tri-unsaturated C15 side chains in the 6-position of the aromatic ring (Scheme 1).3 Since the side-chain double bond closest to the ring is positioned at C8 in all unsaturated isomers, it should be possible to convert this mixture into a reasonably pure single compound by ethenolysis followed by crack distillation with concomitant decarboxylation. Regardless of the number of double bonds on the far side of C8, the ethenolysis would shorten all unsaturated side chains to ω-nonenyl groups. In the distillation, the carboxylate group would be removed and the main product 3-(non-8-enyl)phenol (5) separated from the low-boiling olefins formed during ethenolysis, as well as the higher-boiling saturated cardanol derivative 3-pentadecylphenol (1d).
 | ||
| Scheme 1 Selective conversion of anacardic acids (1) to 3-(non-8-enyl)phenol (5). | ||
Subsequent isomerising ethenolysis of 5 would shorten the olefinic side chain stepwise in a statistical fashion (Scheme 2). Depending on the ethene pressure and catalyst system, the side-chain-length distribution of the resulting product mixture should be adjustable so that a mixture of 6 and 7 is obtained reasonably free of other components, which could be hydrogenated in situ to the kairomones 2 and 3.
 | ||
| Scheme 2 From 3-(non-8-enyl)phenol (5) to the tsetse fly attractants 2 and 3. | ||
An additional, non-isomerising cross-metathesis step with pure ethene or 2-butene prior to hydrogenation should allow directly accessing either product 6 or 7 in pure form, since the thermodynamically favoured conjugated double-bond isomers 6 and 7 should be the main components in the equilibrated isomerising metathesis mixture. Subsequent hydrogenation would give access to pure 2 or 3.
Results and discussion
Ethenolysis of anacardic acids
Based on these considerations, we approached the synthetic procedure starting from the cashew nut shells. An extraction of 400 g of nut shells with diethyl ether yielded 90 g of CNSL (23 wt% based on the nut shells). The acids were separated from other phenolic components by precipitation of their calcium salts using calcium hydroxide followed by acidification, which led to 52 g (58 wt% based on the ethereal extract) of the acid mixture 1.Next, we investigated the ethenolysis step. As a starting point, we used the Ru-1 catalyst, and stirred a solution of 1 and Ru-1 in dichloromethane under 10 bar ethene pressure at 23 °C (Table 1, entry 1). These conditions had previously been shown to work well for the ethenolysis of the decarboxylated derivative cardanol.10 However, for the anacardic acids 1, ethenolysis proceeded with unsatisfactory yields even at a relatively high catalyst loading of 1 mol% Ru-1.
|
|
||||
|---|---|---|---|---|
| Entry | Ru-cat. | Loading/mol% | Yield 4/% | Conversion 1/% |
| Conditions: 0.25 mmol 1, 1 mol% metathesis catalyst, 1 mL DCM, 10 bar ethene, 23 °C, 16 h. Yields were determined by GC analysis. In the GC injector, 4 quantitatively decarboxylates to 5, which was quantified using n-hexadecane as an internal standard.a Preparative-scale reaction (2.50 g, 7.30 mmol). | ||||
| 1 | Ru-1 | 1.0 | 46 | 70 |
| 2 | Ru-2 | 1.0 | 78 | 83 |
| 3 | Ru-3 | 1.0 | 92 | >94 |
| 4 | Ru-4 | 1.0 | 77 | 83 |
| 5 | Ru-5 | 1.0 | 21 | 82 |
| 6 | Ru-6 | 1.0 | 0 | 87 |
| 7 | Ru-7 | 1.0 | 47 | 56 |
| 8 | Ru-3 | 0.5 | 71/89a | 81/99a |
| 9 | Ru-3 | 0.1 | 65 | 76 |
| 10 | Ru-3 | 0.05 | 61 | 71 |

We started optimising the ethenolysis by testing numerous ruthenium-based catalysts (Fig. 2), with a focus on complexes known to promote ethenolysis.11 These include first- and second-generation Grubbs- and Hoveyda-type catalysts (Ru-2,12Ru-3,13Ru-4,14 and Ru-515), and the indenylidene-ruthenium complexes Ru-616 and Ru-7.17
 | ||
| Fig. 2 Isomerisation and metathesis catalysts. | ||
As can be seen from Table 1, other phosphine-ligated catalysts (Ru-2 and Ru-3) were more efficient than Ru-1, and near-quantitative yield was achieved with the first-generation Hoveyda–Grubbs catalyst Ru-3 (entries 2 and 3). Among the NHC-ligated catalysts, the second-generation Grubbs catalyst Ru-4 was most effective, giving 77% yield of 4 (entries 4–7). In contrast, a low yield of the desired product 4 was obtained using Ru-5 (entry 5). Second-generation indenylidene catalysts were inferior (Ru-7; entry 6) or completely inactive (Ru-6; entry 7).
With the best ruthenium catalyst Ru-3, it was possible to reduce the loading to 0.05 mol% and still achieve good yields of the desired product 5 (entries 8–10). The finding that phosphine-based metathesis catalysts outperform modern NHC systems is remarkable but not unprecedented, since Ru-3 has been reported to be optimal also for the ethenolysis of 2-methoxy-6-pentadecenylbenzoic acid methyl esters.10
The results were even better on a preparative scale (2.50 g, 7.30 mmol). In the presence of 0.5 mol% Ru-3, compound 4 was formed in near-quantitative yield. Upon distillation at above 140 °C,184 decarboxylated quantitatively to 3-(non-8-enyl)phenol (5) which was isolated in 89% yield and 98% purity while the saturated compound 1d remained in the distillation residue. This corresponds to an overall yield of 7 wt% of 5 based on the cashew nut shells.
Isomerising ethenolysis of 3-(non-8-enyl)phenol
The key step in our synthesis of the tsetse fly attractants 2 and 3 is the shortening of the olefinic side chains by isomerising cross-metathesis. We have recently demonstrated that isomerising metathesis is a valuable synthetic concept for the valorisation of oleic acid derivatives and of naturally occurring allylarenes.19 In this context, the dimeric palladium(I) complex [Pd(μ-Br)(tBu3P)]2 (Pd-1)20 had proven to be a uniquely effective isomerisation catalyst, fully compatible with state-of-the-art ruthenium metathesis catalysts.21In an isomerising cross-metathesis, the olefinic substrates are continuously converted into an equilibrium mixture of double-bond isomers, which constantly undergo olefin metathesis until olefin blends with a homogeneous chain-length distribution are formed. The mean chain length is determined by the average number of side chain carbons for each substrate weighted with its molar equivalents.
Thus, in the isomerising cross-metathesis of 3-(non-8-enyl)phenol (5) with ethene, an equilibrium mixture of non-volatile aryl alkenes and volatile unsubstituted alkenes with an identical chain length distribution should ideally form. If all double-bond isomers had the same thermodynamic stability and reactivity, their mean chain length should equal ([ethene]·2 + [5]·9)/([5] + [ethene]), calculated from the concentration of each alkene substrate and its respective chain length. This standard formula for the calculation of an arithmetic mean only roughly approximates the reaction outcome. The chain length distribution is more accurately assessed using the simulation program described in ref. 19.
In search for an effective catalyst system, we stirred a THF solution of 3-(non-8-enyl)phenol (5) under 6 bar ethene pressure (corresponding to an ethene/5 ratio of roughly 22:1 and an expected mean chain length of 2.3) with the Pd-based isomerisation catalyst and a Ru-metathesis catalyst for 16 h at 50 °C. For 1.5 mol% Ru-5 and 1.5 mol% Pd-1, a system that had been reported to be optimal for the isomerising ethenolysis of allyl arenes, 24% 3-hydroxstyrene (6) and 53% 3-(prop-1-enyl)phenol (7) were obtained along with minor quantities of longer-chain homologues. However, the mean chain length of 3.2, which was calculated from the GC-integrals, indicates that the reaction did not go to completion (Table 2, entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Yield/% | |||||
| Entry | Ru-cat./mol% | C2H4/bar | 6 | 7 | R ≥ CH3 |
| Conditions: 0.5 mmol 5, 1.5 mol% Pd-1, metathesis catalyst (amount given), 2 mL THF, 50 °C, 16 h. Yields were determined by GC using n-decane as an internal standard.a 4 mL THF.b 8 h. | |||||
| 1 | Ru-5 (1.5) | 6 | 24 | 53 | 23 |
| 2 | Ru-4 (1.5) | 6 | 0 | 17 | 82 |
| 3 | Ru-8 (1.5) | 6 | 22 | 56 | 23 |
| 4 | Ru-8 (2.0) | 6 | 56 | 40 | Traces |
| 5a | Ru-8 (2.0) | 6 | 77 | 23 | Traces |
| 6b | Ru-8 (2.0) | 6 | 37 | 58 | 4 |
| 7 | Ru-8 (2.0) | 2 | 45 | 54 | Traces |
| 8 | Ru-8 (2.0) | 1 | 27 | 67 | 5 |

Phosphine-based first-generation catalysts and NHC-based indenylidene complexes were found to be ineffective, which is understandable from their low activity in non-isomerising styrene cross-metatheses.22 With the second-generation Grubbs catalyst Ru-4, only unsatisfactory yields were obtained, which can be explained by a release of phosphine from the pre-catalyst, which is known to inhibit the isomerisation catalyst Pd-1.19,21 Best results were obtained with the Hoveyda-type NHC catalyst Ru-8 (entries 3–4). After slightly increasing the catalyst loading to 2 mol%, the desired ethenyl- and propenylarenes 6 and 7 were almost exclusively formed within 16 h along with only trace amounts of longer-chain derivatives (entry 4). The mean chain length was found to be 2.4. One would expect that the low solubility of ethene in the reaction solvent leads to an accumulation of more soluble longer-chain olefins in the liquid phase, thus shifting the solution equilibrium towards longer chains.23
Indeed, GC analysis revealed that the relative ratio of ethene/propene/butene is 3.5/2.2/1 in the gas phase and 1/2.8/4.6 in the liquid phase of the equilibrated reaction mixture. Upon increasing the amount of solvent and thereby reducing the gas-phase and increasing the liquid-phase volume, the equilibrium concentration of 6 and 7 changed to 77% and 23%, respectively, which corresponds to a mean chain length of 2.2 (entry 5). Control experiments revealed that it takes more than 8 h until this equilibrium concentration is reached (entry 6).
When lowering the ethene excess to 9:1 (2.0 bar ethene, calculated mean chain length: 2.7), 7 was obtained as the main product and the mean chain length increased to 2.6 (entry 7). At only 6:1 ethene/5 ratio (1.0 bar ethene, calculated mean chain length: 3.0), the mean chain length was found to be 2.8 but the concentration of the longer-chain homologues increased to an undesirably high level (entry 8).
Synthesis of an 3-ethyl- and 3-proylphenol mixture
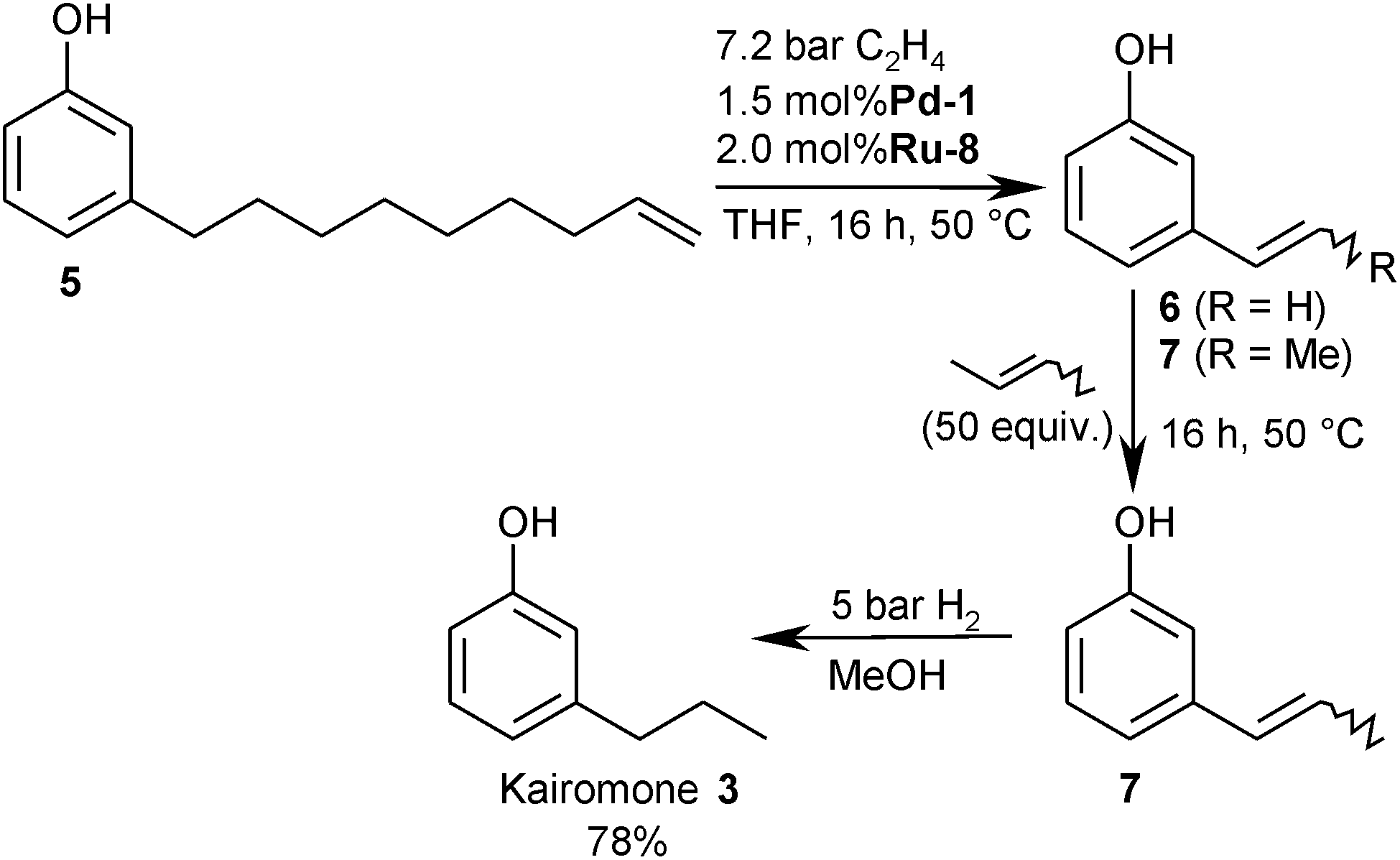
When performing the isomerising ethenolysis using the optimal catalyst system and substrate ratio (1.0 mmol 5, 1.5 mol% Pd-1, 2.0 mol% Ru-8, 4 mL THF, 50 °C, 16 h, approx. 9:1 ethene/5, 5.0 bar ethene),24 a mixture of the ethenylarene 6 (43%) and the propenylarene 7 (56%) was formed. Upon exchanging the olefin atmosphere above the reaction mixture for hydrogen (3.0 bar) and adding charcoal to prevent agglomeration of the metal catalyst, the product mixture was directly hydrogenated to the corresponding saturated kairomones, which were isolated in a combined 85% yield (2/3 = 1:1.3).
Selective synthesis of the tsetse fly attractants 3-propyl- and 3-ethylphenol
Since both kairomones target different tsetse flies, this approach, which produces 2 and 3 in similar amounts, should be advantageous for the application in fly traps. For the selective synthesis of the widely used kairomone 3, an additional butenolysis step is required. Thus, the isomerising ethenolysis of 5 was first allowed to reach its equilibrium. After 16 h, the metathesis catalyst was still fully active, but the isomerisation catalyst had lost most of its reactivity. At this stage, the reaction mixture, which consisted mostly of the conjugated isomers of 3-propenylphenol and of 3-ethenylphenol, was purged with 2-butene (approx. 50:1 ratio 2-butene/6 +7, 2.0 bar) and stirred for another 16 h at 50 °C. This way, 7 was obtained in 92% overall yield and 95% purity.
For a preparative-scale (4.0 mmol), one-pot synthesis of 3-propylphenol (3), the isomerising ethenolysis was performed using only a 7:1 ethene/5 ratio (7.5 bar, the pressure limit of the reactor). After 16 h, the gas phase was first exchanged for 2-butene (approx. 50 equiv., 2.0 bar) and after 16 h for hydrogen (5.0 bar). This way, the desired kairomone 3 was obtained in 78% yield (Scheme 3). This one-pot synthesis of 3-propylphenol (3), which does not require purification of intermediates and delivers the desired product 3 in 69% yield based on anacardic acids (1a–d), compares favourably even with the previously reported isomerisation/butenolysis/hydrogenation sequence.9
 | ||
| Scheme 3 Preparative-scale one-pot synthesis of the kairomone 3. | ||
3-Ethylphenol (2) was also synthesised in pure form from the mixture of 6 and 7 that was obtained in the isomerising metathesis step. The synthetic strategy is identical to the one outlined for the 3-propylphenol (3), only that the 2-butene used in the terminal cross-metathesis step was replaced by ethene. This way, 2 was obtained in an overall yield of 84%.
Isomerising butenolysis of 3-(non-8-enyl)phenol
We also tried whether performing the isomerising metathesis using a vast excess of 2-butene would lead to formation of 3 in satisfactory selectivity. However, even when stirring 5 with approx. 50 equivalents 2-butene (2.0 bar), 3 was formed in only 64% yield along with the higher homologues butenyl- and pentenylphenol. We also attempted to shorten the reaction sequence by performing an isomerising ethenolysis of the anacardic acids (1). Unfortunately, the isomerisation catalyst quickly lost its activity for this rather impure substrate. It could not yet be determined which impurity is responsible for this effect.Experimental
General methods
All reactions were performed in oven-dried glassware containing a Teflon-coated stirring bar. Solvents were purified, dried by standard procedures, and degassed by three freeze–pump–thaw cycles prior to use. All reactions were monitored by GC using n-hexadecane or n-decane as an internal standard. Response factors of the products with regard to n-(hexa-)decane were obtained experimentally by analysing known quantities of the substances. GC analyses were carried out using a HP6890 with an HP-5 capillary column (phenyl methyl siloxane 30 m × 320 × 0.25, 100/2.3-30-300/3) and a time program beginning with 2 min at 60 °C, followed by a 30 °C min−1 ramp to 300 °C, then 3 min at this temperature. Headspace GC analyses were carried out using a Perkin Elmer AutoSystem XL with Supelco packed column (80/100 Poropak, 6 FT × 1/8 In. S.S.). Commercial substrates were used as received unless otherwise stated. The metathesis catalysts used herein are available commercially, for example, from Sigma-Aldrich or Umicore. But-2-ene was obtained from Apollo Scientific Ltd as a mixture of E/Z-isomers (purity: 99% by GC, ratio of E/Z-isomers: 68%/31%) and ethene from Air Liquide GmbH (purity: 99.9%).Extraction of anacardic acids (1) from cashew nut shells
Cashew nut shells (400 g) were ground into ∼1 mm small particles which were than extracted in a Soxhlet apparatus with Et2O (500 mL) for 6 h. Removal of the solvent in vacuo resulted in a highly viscous brown oil (90.0 g, 23 wt%). 40 g of this crude extract were dissolved in 5% aqueous methanol (240 mL), and calcium hydroxide (20.0 g, 259 mmol) was added in portions under stirring. The temperature of the reaction mixture was then raised to 50 °C, and stirring was continued for 2.5 h leading to precipitation of calcium anacardate. The precipitate was filtered off and washed with methanol (200 mL). It was then suspended in distilled water (200 mL), and the anacardic acids were liberated by treatment with HCl (30 mL, 12 M) and stirring for 1 h at RT. The mixture was then extracted with EtOAc (2 × 200 mL) and the combined organic phases were washed with distilled water (100 mL), dried over MgSO4 and concentrated under reduced pressure to yield a mixture of anacardic acids as a viscous, dark brown oil (23.1 g, 58 wt%).Preparative scale synthesis of 3-(non-8-enyl)phenol (5) from anacardic acids (1)
In a glovebox, a PTFA-lined steel autoclave with a stirring bar was charged with the metathesis catalyst Ru-3 (21.6 mg, 36.0 μmol), dichloromethane (30 mL), and the anacardic acid mixture (1) (2.50 g, 7.30 mmol). The autoclave was then purged with ethene three times and pressurised to 10 bar. The resulting mixture was stirred for 16 h at RT. Then, the solvent was removed in vacuo. Crack distillation of the crude product mixture in a Kugelrohr (120 °C, 10−2 mbar) yielded 3-(non-8-enyl)phenol (5) as light yellow oil (1.41 g, 6.46 mmol, 89%, GC-purity: >98%). The analytical data matched those reported in the literature.10Preparative scale synthesis of 3-proplyphenol (3) from 3-(non-8-enyl)phenol (5)
In a glove box, an oven-dried 70 mL Fisher-Porter vessel with a stirring bar was charged with Pd-1 (40.4 mg, 52.0 μmol), Ru-8 (52.5 mg, 80.0 μmol), THF (16 mL) and 3-(non-8-enyl)phenol (5) (950 mg, 4.00 mmol). The vessel was removed from the glovebox and pressurised with ethene (7.5 bar). The mixture was stirred at 50 °C for 16 h. After cooling to −78 °C, the pressure was slowly released under inert conditions. The vessel was warmed to 0 °C and 2-butene (11.2 g, 200 mmol) was condensed in. The mixture was allowed to stir overnight at 50 °C. Subsequently, the reaction mixture was cooled to 0 °C and the pressure was slowly released. Hydrogenation was performed by adding methanol (25 mL) and activated charcoal (300 mg), and stirring the reaction mixture for 3 h at 50 °C under hydrogen pressure (5.0 bar). After slowly releasing the pressure, the mixture was filtered through Celite (1 cm3), the solvent was removed by distillation over a Vigreux column, and the residue was dissolved in Et2O, filtered through a plug of silica gel (5 cm3). Removal of the solvent yielded the desired product 3 as a bright yellow oil (425 mg, 3.12 mmol, 78%). The analytical data matched those reported in the literature.8Synthesis of 3-ethylphenol (2) from 3-(non-8-enyl)phenol (5)
In a glove box, an oven-dried 35 mL Fisher-Porter vessel with a stirring bar was charged with Pd-1 (10.0 mg, 13.0 μmol), Ru-8 (13.2 mg, 20.0 μmol), THF (4 mL) and 3-(non-8-enyl)phenol (5) (250 mg, 1.00 mmol). The vessel was removed from the glovebox and pressurised with ethene (5.0 bar). The mixture was stirred at 50 °C for 16 h. After cooling to −78 °C, the pressure was slowly released. The reaction mixture was then filtered through a plug of silica gel and the solvent removed in vacuo. Under inert conditions, Ru-8 (13.2 mg, 20.0 mmol) and THF (4 mL) were added to the mixture of 3-ethenyl- (2) and 3-(prop-2-enyl)phenol (3) (∼1:1) in a Fisher-Porter vessel, which was pressurised with ethene (4.0 bar). The mixture was allowed to stir overnight at 50 °C. Subsequently, the reaction mixture was cooled to 0 °C and the pressure was slowly released. Hydrogenation was performed by adding methanol (5 mL) and activated charcoal (100 mg), and stirring the reaction mixture for 3 h at 50 °C under hydrogen pressure (5.0 bar). After slowly releasing the pressure, the mixture was filtered through Celite (1 cm3), the solvent was removed by distillation over a Vigreux column, the residue was dissolved in Et2O and filtered through a plug of silica gel (5 cm3). Removal of the solvent yielded the desired product 2 as a colourless oil (101 mg, 0.84 mmol, 84%). The analytical data matched those reported in the literature.25
Conclusions
Isomerising olefin cross-metathesis enabled the synthesis of the tsetse fly attractants 3-ethylphenol (2) and 3-propylphenol (3) from an anacardic acid mixture (1) extracted from cashew nut shells. The key towards providing a uniform substrate is the ethenolysis of the anacardic acids followed by thermal decarboxylation. This way, mono-, di- and tri-unsaturated side chains are all shortened to nine carbons, and the 3-(non-8-enyl)phenol is separated from derivatives with saturated side chains. A one-pot sequence consisting of isomerising ethenolysis followed by an optional butenolysis or ethenolysis and hydrogenation of the olefinic side chain, all effectively mediated by a combination of the isomerisation catalyst [Pd(μ-Br)(tBu3P)]2 and a second-generation Hoveyda–Grubbs metathesis catalyst, furnishes the desired 3-ethylphenol (2) and 3-propylphenol (3) either in pure form or as a mixture. This concise reaction sequence underlines the high potential of isomerising cross-metathesis as a key technology in the chemical valorisation of non-food, renewable resources.Acknowledgements
We thank NanoKat, the Collaborative Research Centre SFB/TRR 88 “3MET”, Carl Zeiss (RCR) and the Deutsche Bundesstiftung Umwelt (fellowship to S.B.) for financial support, and Umicore AG for the donation of chemicals. We also thank Mark Niebergall for technical assistance, Christian Schneider and Denise Scherbl for HPLC analysis.Notes and references
- A. Velmurugan and M. Loganathan, World Acad. Sci. Eng. Technol., 2011, 5, 738–743 Search PubMed.
- V. S. Balachandran, S. R. Jadhav, P. K. Vemula and G. John, Chem. Soc. Rev., 2013, 42, 427–438 RSC; C. S. K. Lin, L. A. Pfaltzgraff, L. Herrero-Davila, E. B. Mubofu, S. Abderrahim, J. H. Clark, A. A. Koutinas, N. Kopsahelis, K. Stamatelatou, F. Dickson, S. Thankappan, Z. Mohamed, R. Brocklesby and R. Luque, Energy Environ. Sci., 2013, 6, 426–464 Search PubMed; R. L. Quirino, T. F. Garrison and M. R. Kessler, Green Chem., 2014, 16, 1700–1715 RSC.
- R. Paramashivappa, P. Phani Kumar, P. J. Vithayathil and A. Srinivasa Rao, J. Agric. Food Chem., 2001, 49, 2548–2551 CrossRef CAS PubMed; P. Phani Kumar, R. Paramashivappa, P. J. Vithayathil, P. V. Subba Rao and A. Srinivasa Rao, J. Agric. Food Chem., 2002, 50, 4705–4708 CrossRef PubMed.
- L. J. Gooßen, F. Collet and K. Gooßen, Isr. J. Chem., 2010, 50, 617–629 CrossRef CAS PubMed; S. Bhadra, W. I. Dzik and L. J. Gooßen, J. Am. Chem. Soc., 2012, 134, 9938–9941 CrossRef PubMed; L. J. Gooßen, L. Winkel, A. Dohring, K. Ghosh and J. Paetzold, Synlett, 2002, 1237–1240 CrossRef PubMed; L. J. Gooßen and J. Paetzold, Adv. Synth. Catal., 2004, 346, 1665–1668 CrossRef PubMed.
- M. L. A. Owaga, Int. J. Trop. Insect Sci., 1985, 6, 561–566 CrossRef CAS; M. L. A. Owaga, A. Hassanali and P. G. McDowell, Int. J. Trop. Insect Sci., 1988, 9, 95–100 CrossRef; M. Okech and A. Hassanali, Int. J. Trop. Insect Sci., 1990, 11, 363–368 CrossRef.
- M. P. Barrett, R. J. Burchmore, A. Stich, J. O. Lazzari, A. C. Frasch, J. J. Cazzulo and S. Krishna, Lancet, 2003, 362, 1469–1480 CrossRef; P. P. Simarro, G. Cecchi, J. R. Franco, M. Paone, A. Diarra, J. A. Ruiz-Postigo, E. M. Fèvre, R. C. Mattioli and J. G. Jannin, PLoS Negl. Trop. Dis., 2012, 6, e1859 Search PubMed; P. G. Kennedy, Lancet Neurol., 2013, 12, 186–194 CrossRef; B. I. Williams, African trypanosomiasis, in The Wellcome Trust illustrated history of tropical diseases, The Wellcome Trust, London, United Kingdom, 1996 Search PubMed; WHO, Fact sheet N°259, http://www.who.int/mediacentre/factsheets/fs259/en (accessed June 2014).
- E. Bursell, A. J. E. Gough, P. S. Beevor, A. Cork, D. R. Hall and G. A. Vale, Bull. Entomol. Res., 1988, 78, 281–291 CrossRef CAS.
- Grignard: I. Ujváry and G. Mikite, Org. Process Res. Dev., 2003, 7, 585–587 CrossRef CAS . Wittig: D. R. Magnin, S. A. Biller, J. K. Dickson, Jr., R. M. Lawrence and R. B. Sulsky, US Pat., 5470845 A1, 1995 CrossRef; Y. L. Bigot, M. Delmas and A. Gaset, Tetrahedron, 1986, 42, 339–350 CrossRef . For further synthetic approaches, see: W. H. Hartung and F. S. Crossley, J. Am. Chem. Soc., 1934, 56, 158–159 CrossRef; S. Landa and J. Macák, Collect. Czech. Chem. Commun., 1958, 23, 1322–1329 CrossRef; G. Ciamician and P. Silber, Ber. Dtsch. Chem. Ges., 1890, 23, 1159–1164 CrossRef PubMed; C. F. Carvalho and M. V. Sargent, J. Chem. Soc., Perkin Trans. 1, 1984, 1621–1626 RSC.
- J. A. Mmongoyo, Q. A. Mgani, S. J. M. Mdachi, P. J. Pogorzelec and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2012, 114, 1183–1192 CrossRef CAS PubMed.
- J. Julis, S. A. Bartlett, S. Baader, N. Beresford, E. J. Routledge, C. S. J. Cazin and D. J. Cole-Hamilton, Green Chem., 2014, 16, 2846–2856 RSC . For recent publication on cardanol ethenolysis, see: T. Shinde, V. Varga, M. Polášek, M. Horáček, N. Žilková and H. Balcar, Appl. Catal., A, 2014, 478, 138–145 CrossRef CAS PubMed.
- C. Thurier, C. Fischmeister, C. Bruneau, H. Olivier-Bourbigou and P. H. Dixneuf, ChemSusChem, 2008, 1, 118–122 CrossRef CAS PubMed; R. M. Thomas, B. K. Keitz, T. M. Champagne and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 7490–7496 CrossRef PubMed; K. A. Burdett, L. D. Harris, P. Margl, B. R. Maughon, T. Mokhtar-Zadeh, P. C. Saucier and E. P. Wasserman, Organometallics, 2004, 23, 2027–2047 CrossRef; G. S. Forman, A. E. McConnell, M. J. Hanton, A. M. Z. Slawin, R. P. Tooze, W. J. van Rensburg, W. H. Meyer, C. Dwyer, M. M. Kirk and D. W. Serfontein, Organometallics, 2004, 23, 4824–4827 CrossRef.
- P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem. Int. Ed., 1995, 34, 2039–2041 CrossRef CAS PubMed.
- J. P. A. Harrity, D. S. La, D. R. Cefalo, M. S. Visser and A. H. Hoveyda, J. Am. Chem. Soc., 1998, 120, 2343–2351 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- S. Monsaert, R. Drozdzak, V. Dragutan, I. Dragutan and F. Verpoort, Eur. J. Inorg. Chem., 2008, 2008, 432–440 CrossRef PubMed.
- F. W. C. Verpoort and C. B. De, WO Pat., 2003062253, 2003 Search PubMed.
- I. Risfaheri, T. T. Irawadi, M. A. Nur and I. Sailah, Indones. J. Agric., 2008, 2, 11–22 Search PubMed.
- D. M. Ohlmann, N. Tschauder, J.-P. Stockis, K. Gooßen, M. Dierker and L. J. Gooßen, J. Am. Chem. Soc., 2012, 134, 13716–13729 CrossRef CAS PubMed; L. J. Gooßen, D. M. Ohlmann and M. Dierker, WO Pat., 2012143067, 2012 CrossRef PubMed; S. Baader, D. M. Ohlmann and L. J. Gooßen, Chem. – Eur. J., 2013, 19, 9807–9810 CrossRef PubMed ; related isomerising functionalisations: D. M. Ohlmann, L. J. Gooßen and M. Dierker, Chem. – Eur. J., 2011, 17, 9508–9519 CrossRef PubMed; L. J. Gooßen, D. M. Ohlmann and M. Dierker, Green Chem., 2010, 12, 197–200 RSC.
- V. Durà-Vilà, D. M. P. Mingos, R. Vilar, A. J. P. White and D. J. Williams, J. Organomet. Chem., 2000, 600, 198–205 CrossRef.
- P. Mamone, M. F. Grünberg, A. Fromm, B. A. Khan and L. J. Gooßen, Org. Lett., 2012, 14, 3716–3719 CrossRef CAS PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed; J. Spekreijse, J. Le Nôtre, J. van Haveren, E. L. Scott and J. P. M. Sanders, Green Chem., 2012, 14, 2747–2751 RSC.
- M. Atiqullah, H. Hammawa and H. Hamid, Eur. Polym. J., 1998, 34, 1511–1520 CrossRef CAS.
- An identical 5/ethene ratio now translates to a different pressure since the same reaction vessels are used as for the 0.5 mmol scale reactions.
- R. Shen, T. Chen, Y. Zhao, R. Qiu, Y. Zhou, S. Yin, X. Wang, M. Goto and L.-B. Han, J. Am. Chem. Soc., 2011, 133, 17037–17044 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General methods and spectroscopic data. See DOI: 10.1039/c4gc01269k |
| This journal is © The Royal Society of Chemistry 2014 |