
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile-prepared sulfonated water-soluble PEPPSI-Pd-NHC catalysts for aerobic aqueous Suzuki–Miyaura cross-coupling reactions†
Rui
Zhong
,
Alexander
Pöthig
*,
Yinkai
Feng
,
Korbinian
Riener
,
Wolfgang A.
Herrmann
and
Fritz E.
Kühn
*
Chair of Inorganic Chemistry/Molecular Catalysis, Catalysis Research Center/Technische Universität München, Ernst-Otto-Fischer Str. 1, D-85748 Garching b. München, Germany. E-mail: fritz.kuehn@ch.tum.de; alexander.poethig@tum.de; Fax: (+49) 89-289-13473; Tel: +49 89-289-13096
First published on 11th August 2014
Abstract
Four sulfonated water-soluble PEPPSI-Pd-NHC complexes (2a–2d) are prepared in a straightforward two-step synthesis. Their activities have been examined in Suzuki–Miyaura cross-coupling reactions in water under air. Complex 2d, bearing a 2,6-diisopropylphenyl substituent, shows the best catalytic activity and a variety of aryl bromides with a catalyst loading of 0.1 mol% can be efficiently activated even at room temperature. The catalyst is recyclable and can be employed in at least four consecutive runs without significant loss in performance. Furthermore, TEM analysis, kinetic studies and mercury poisoning experiments indicate that Pd nanoparticles are formed during the reactions.
Introduction
Over the past few decades, the chemical industry has developed growing interest in inexpensive and environmentally friendly reaction media such as water based on considerations of safety, availability and environmental impact.1 With palladium-catalyzed cross-coupling reactions being widely applied for the synthesis of fine chemicals, functional materials and industrial starting materials,2 the development of water-soluble Pd compounds as highly efficient catalysts for cross-coupling reactions in aqueous reaction media has become an important field of research.3 Since the first example was described by Calabrese and co-workers in 1990,4 considerable efforts have been undertaken to develop water-soluble Pd catalysts for cross-coupling reactions, especially by modifying traditional Pd phosphine catalysts.5 However, most of the phosphine ligands are air and moisture sensitive, restricting the reuse of the catalyst and leading to undesirable residues under aqueous reaction conditions.6 In addition, the syntheses of phosphine ligands are generally demanding and toxic intermediates are involved.7 Therefore, introducing more stable and less toxic ligands is desirable.N-heterocyclic carbenes (NHCs), which have already been employed as supporting ligands for various Pd-catalyzed cross-coupling reactions, are viewed as promising alternatives to phosphines.8 As NHCs have many inherent advantages – e.g., a strong σ-donating ability, thermal and oxidative stability as well as electronic and steric tunability – they appear to be even more versatile in designing water-soluble catalysts than phosphines.6a,9 Accordingly, the development of NHC-based water-soluble catalysts has attracted much attention in the recent ten years.10 The Suzuki–Miyaura cross-coupling reaction is a widely used protocol for organic synthesis2b,11 and the implementation of this reaction in aqueous solution at room temperature using soluble Pd-NHC complexes would significantly reduce the demand of energy and resources. Although a number of water-soluble Pd-NHC catalysts for Suzuki–Miyaura cross-coupling reactions in water have been reported since 2005,6c,12 it remains a challenge to perform this reaction at room temperature. To the best of our knowledge, the only successful example reported so far is a water-soluble polymeric Pd-NHC catalyst, which is able to activate several aryl chlorides in good to excellent yields at room temperature.12j However, the synthesis of the Pd-NHC polymer is relatively complicated and the polymer itself is not easy to characterize. In addition, there are just two more reported catalytic systems, which only give poor coupling yields of aryl halides at room temperature in water.12e,f Thus, the development of easily-prepared and well-defined water-soluble Pd-NHC catalysts for room temperature aqueous Suzuki–Miyaura cross-coupling reactions is still of considerable interest.
In order to develop robust and accessible catalysts for aqueous Suzuki–Miyaura cross-coupling reactions, PEPPSI-type (pyridine, enhanced, precatalyst, preparation, stabilization and initiation) Pd-NHC complexes13 seem to be promising motifs as they have proven to be effective and stable in many cross-coupling reactions while being easy to synthesize.13a,14 In this work, a series of facile-prepared sulfonated water-soluble PEPPSI Pd-NHC complexes (2a–2d) and their catalytic application in aqueous Suzuki–Miyaura cross-coupling reactions are reported. Detailed investigations of complex 2d include its catalytic activity on a broad variety of substrates, and reusability, and it is noteworthy that mercury poison experiments as well as TEM measurements suggest formation of Pd NPs (Pd nanoparticles) during the reaction.
Results and discussion
Synthesis and characterization
Since the nature of the substitution pattern of the employed NHC ligand has a great influence on the catalytic activity,10a four sulfonate-functionalized PEPPSI Pd complexes (2a–2d) with different NHC moieties are prepared in a facile two-step synthesis (Scheme 1). First, sulfonated ligands 1a–1d are synthesized via nucleophilic substitution of sodium 2-bromoethanesulfonate with the corresponding imidazole in good yields ranging from 68 to 82%. Subsequent reaction of the ligand precursors with excess K2CO3 and PdBr2 in neat pyridine under argon affords 2a–2d in 52 to 73% yield according to a modified literature procedure.13a,14g Complexes 2a–2d were characterized by NMR spectroscopy, elemental analysis and mass spectroscopy and exhibit good solubility in water and MeOH.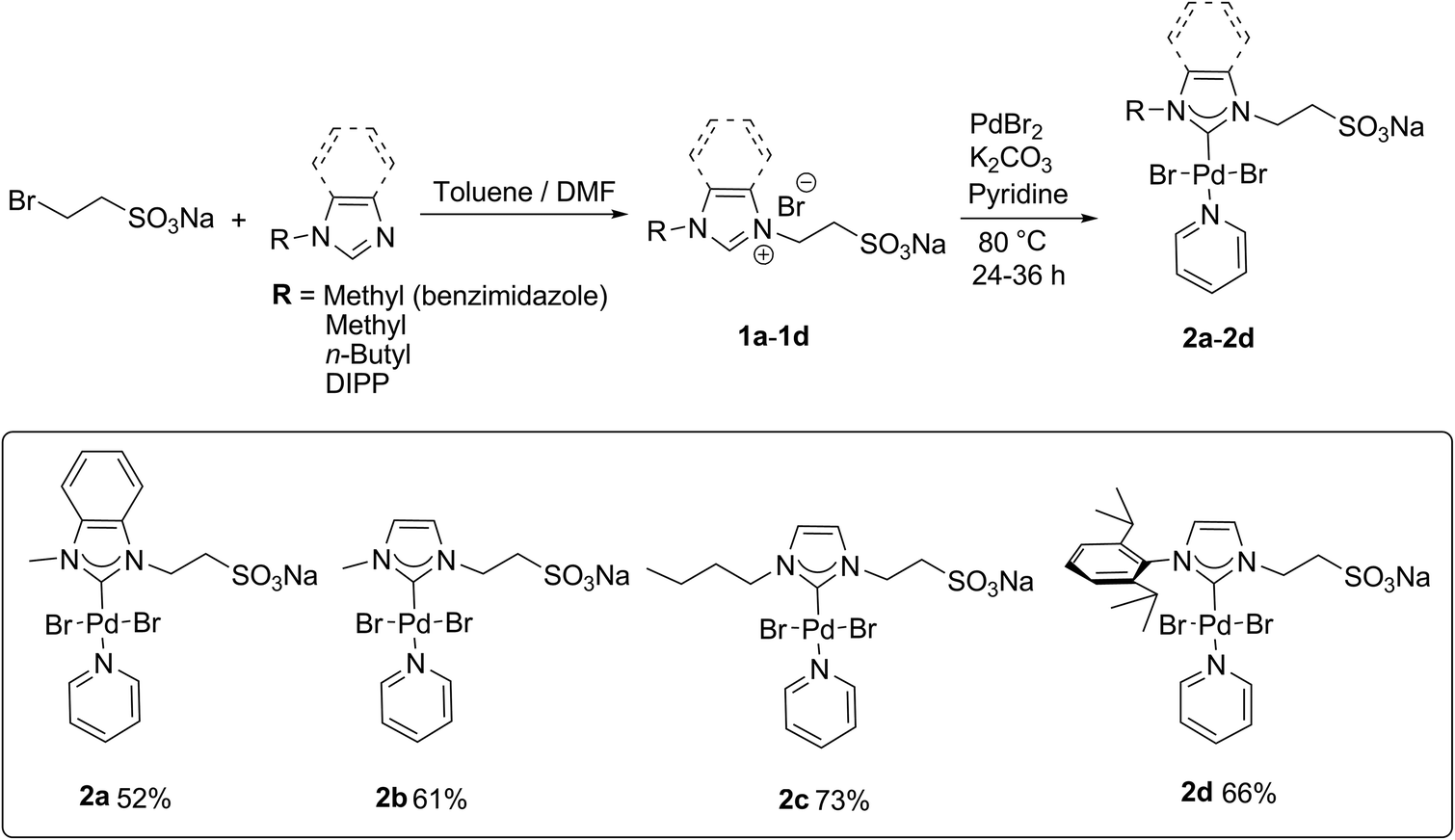 | ||
| Scheme 1 Synthesis of sulfonate-functionalized PEPPSI Pd-NHC complexes 2a–2d. | ||
Catalyst evaluation
In a survey of catalytic activity, complexes 2a–2d were employed in aqueous Suzuki–Miyaura coupling reactions (Table 1). For the evaluation three representative substrates, i.e. 4′-chloroacetophenone, 4′-bromoacetophenone and 4-bromoanisole were used in deionized water under air, employing KOH as a base. As shown in Table 1, complex 2d displays the best catalytic activity, giving excellent yields for all three substrates (entries 4, 8 and 12). The other three complexes (2a–2c) give only moderate yields, while complex 2a shows the lowest coupling yield of all three substrates (entries 3, 7 and 11). Since the solubility of the four complexes in water is higher than 10 mg mL−1, which is more than the catalyst concentration (<4 mg mL−1) required in the reactions, the solubility of the catalysts most probably does not influence the catalytic performance.|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | R (X) | Cat. [mol%] | Temp. [°C] | Yieldb [%] |
| a Reaction conditions: aryl halide (0.5 mmol), phenylboronic acid (0.75 mmol), base (1.0 mmol), catalyst (0.1–1 mol%) and H2O (1 mL) were stirred under air for 24 h at r.t. or 100 °C. b Yields determined by NMR, using 1,3,5-trimethoxybenzene as the internal standard. | |||||
| 1 | 2a | 4-COCH3(Cl) | 1 | 100 | 3 |
| 2 | 2b | 1 | 100 | 13 | |
| 3 | 2c | 1 | 100 | 22 | |
| 4 | 2d | 1 | 100 | 94 | |
| 5 | 2a | 4-OCH3(Br) | 0.1 | 100 | 36 |
| 6 | 2b | 0.1 | 100 | 51 | |
| 7 | 2c | 0.1 | 100 | 56 | |
| 8 | 2d | 0.1 | 100 | >99 | |
| 9 | 2a | 4-COCH3(Br) | 1 | r.t. | 83 |
| 10 | 2b | 1 | r.t. | 95 | |
| 11 | 2c | 1 | r.t. | 84 | |
| 12 | 2d | 1 | r.t. | >99 | |

It is assumed that the different catalytic activities of complexes 2a–2d are attributed to electronic and steric effects of the NHC moieties of the Pd complexes. The relatively poor performance of complex 2a may be associated with the stronger π-accepting ability of the benzimidazolylidene compared to the imidazolylidenes and therefore stronger metal–carbene π-back bonding.15 The 13C NMR resonance originating from the Ccarbene of complex 2a (166.2 ppm) shows a significant downfield shift in comparison to the other three imidazolylidene complexes (2b–2d; 149.7, 149.1 and 148.4 ppm, respectively). This observation supports the assumption of benzimidazolylidene having a stronger electron withdrawing effect on the active Pd center. Since the electronic properties of the imidazolylidene moieties of complexes 2b, 2c and 2d are quite similar, it is reasonable to conclude that the steric influence introduced by the 2,6-diisopropylphenyl group of complex 2d plays a key role in its superior catalytic activity.2d,16 This observation is also consistent with previous studies based on non-aqueous Suzuki–Miyaura cross-coupling reactions,2d,14m as the 2,6-diisopropylphenyl group on the NHC moiety provides ideal electronic properties paired with bulky but flexible surroundings of the metal center, thus leading to increased catalytic performance.
Optimization of the reaction conditions with catalyst 2d
Based on the results described above, further optimization of the reaction conditions was conducted with the best catalyst, namely 2d (Table 2). Initially, three commonly used water-soluble bases were applied for the catalytic coupling of 4′-chloroacetophenone and phenylboronic acid with 0.5 mol% of 2d at 100 °C over a period of 24 h (entries 1–3). KOH proves to be the base of choice, providing the highest coupling yield (81%, entry 1), whereas the other two bases only afford less than 50% coupling yields (entries 2 and 3). Further optimizations were then based on the catalyst loading and reaction temperature. A reduced catalyst loading to 0.1 mol% at 100 °C affords only a moderate yield of 52% after 24 h. It is known that the addition of tetra-n-butylammonium bromide (TBAB) can accelerate the reaction of aqueous Suzuki–Miyaura cross-coupling reactions due to the formation of Bu4NPhB(OH)3.2e,12l,17 A yield of 99% is reached within 2 h even when only 0.1 mol% of 2d is used (entry 5). A further decrease in catalyst loading to 0.01 mol% still leads to a yield of 95% within 24 h (entry 6).|
|
|||||
|---|---|---|---|---|---|
| Entry | R (X) | Base | 2d [mol%] | Time [h] | Yielde [%] |
| a Reaction conditions: aryl halide (0.5 mmol), phenylboronic acid (0.75 mmol), base (1.0 mmol), and 0.1–1 mol% of 2d in H2O (1 mL) at 100 °C. b With the addition of TBAB (1.0 mmol). c At room temperature. d At 45 °C. e Yields determined by NMR, using 1,3,5-trimethoxybenzene as the internal standard. | |||||
| 1 | 4-COCH3(Cl) | KOH | 0.5 | 24 | 81 |
| 2 | KOAc | 0.5 | 24 | 10 | |
| 3 | K2CO3 | 0.5 | 24 | 40 | |
| 4 | KOH | 0.1 | 24 | 52 | |
| 5b | KOH | 0.1 | 2 | >99 | |
| 6b | KOH | 0.01 | 24 | 95 | |
| 7b,c | KOH | 1 | 24 | 40 | |
| 8b,c | KOH | 1 | 72 | 44 | |
| 9b,c | 4-OCH3(Br) | KOH | 1 | 24 | 96 |
| 10b,c | KOH | 0.1 | 24 | 90 | |
| 11b,d | KOH | 0.1 | 24 | >99 | |
| 12b | KOH | 0.1 | 2 | >99 | |
| 13b | KOH | 0.01 | 24 | >99 | |
| 14b | KOH | 0.001 | 24 | >99 | |

Since reactions under ambient conditions are preferred due to the reduced consumption of energy, the reactions were further conducted at room temperature with 1 mol% of 2d. As shown in entries 7 and 8 of Table 2, the obtained yields are less than 50% even after an extended reaction time of 72 h (44%, entry 8). With 4′-chloroacetophenone being a challenging substrate, 4-bromoanisole was then used to further investigate the catalytic performance of 2d in aqueous Suzuki–Miyaura coupling reactions, especially at room temperature with low catalyst loadings. 2d is able to efficiently activate 4-bromoanisole at room temperature and a yield of 96% with 1 mol% of catalyst is reached within 24 h (entry 9). Furthermore, a catalyst loading of 0.1 mol% still allows for 90% yield within the same reaction time. It is worth noting that a slight increment of temperature to 45 °C leads to full conversion to the desired product within 24 h with 0.1 mol% of 2d (entry 11). In addition, further variation of the reaction time and catalyst loading shows that complex 2d can achieve full conversion of 4-bromoanisole at 100 °C with turnovers of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 using a catalyst loading of only 0.001 mol% (entry 14).
000 using a catalyst loading of only 0.001 mol% (entry 14).
Scope and limitations of substrates
Given that complex 2d can efficiently activate 4-bromoanisole at room temperature, it was then attempted to expand the catalytic scope towards various aryl halides and arylboronic acids in aqueous Suzuki–Miyaura reactions at room temperature. As illustrated in Table 3, thirteen different aryl bromides were used and treated with nine phenylboronic acids (entries 1–22). In general, both electron-rich and electron-deficient aryl bromides and arylboronic acids give the desired products in excellent yields using only 0.1 mol% of 2d (entries 1–11, 16, and 19). With respect to the substrates 2-bromotoluene and 1-bromo-4-(trifluoromethoxy)benzene, quantitative yields of the coupling products can also be obtained when the reaction temperature is slightly increased to 45 °C (entries 14 and 15). With respect to substrates such as hindered aryl bromides and aryl chlorides (entries 20–26), activation of 2-bromo-1,3-dimethylbenzene with ortho-substituted arylboronic acids at room temperature proved to be difficult and only traces of the desired tri- and tetra-substituted biaryls are formed (entries 20–22). A moderate yield of 51% is obtained when 2-bromomesitylene is coupled with phenylboronic acid using 1 mol% of 2d at 100 °C and a reaction time of 12 h (entry 23). Regarding the tested aryl chlorides, moderate to excellent yields (57%, 84% and 99%, respectively) can be obtained when coupled with 4-methylphenylboronic acid using 1 mol% of 2d at 100 °C within 12 h (entries 24–26). These results illustrate that complex 2d tolerates functional groups on aryl bromides at room temperature and is capable of activating several deactivated substrates in moderate to good yields at 100 °C.|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 (X) | R2 | 2d [mol%] | Time [h] | Yieldd [%] |
| a Reaction conditions: aryl halide (0.5 mmol), phenylboronic acid (0.75 mmol), KOH (1.0 mmol), TBAB (1.0 mmol), and 2d (0.1 mol%) in 1 mL of H2O, room temperature. b At 45 °C. c At 100 °C. d Yields determined by NMR, using 1,3,5-trimethoxybenzene as the internal standard. | |||||
| 1 | 4-COCH3(Br) | H | 0.1 | 24 | >99 |
| 2 | 4-CHO(Br) | H | 0.1 | 24 | >99 |
| 3 | 2-CHO(Br) | H | 0.1 | 24 | 98 |
| 4 | 4-OMe(Br) | H | 0.1 | 36 | 97 |
| 5 | 4-CO2Me(Br) | H | 0.1 | 36 | 98 |
| 6 | 4-CH3(Br) | H | 0.1 | 24 | 84 |
| 7 | 2-CN(Br) | Me | 0.1 | 24 | >99 |
| 8 | 4-CN(Br) | Me | 0.1 | 24 | 93 |
| 9 | 4-CF3(Br) | Me | 0.1 | 24 | 91 |
| 10 | 4-F(Br) | Me | 0.1 | 24 | 92 |
| 11 | 4-COCH3(Br) | Me | 0.1 | 24 | >99 |
| 12 | 3-CH3(Br) | H | 0.1 | 24 | 67 |
| 13 | 4-CF3O(Br) | Me | 0.1 | 24 | 76 |
| 14b | 3-CH3(Br) | H | 0.1 | 24 | >99 |
| 15b | 4-CF3O(Br) | Me | 0.1 | 24 | >99 |
| 16 | 4-OMe(Br) | 2-F | 0.1 | 36 | 92 |
| 17 | 4-OMe(Br) | 3-COCH3 | 0.1 | 24 | 42 |
| 18 | 4-OMe(Br) | 4-COCH3 | 0.1 | 24 | 73 |
| 19 | 4-OMe(Br) | 4-CF3 | 0.1 | 24 | 89 |
| 20 | 2,6-CH3(Br) | 2-Me | 0.1 | 24 | Trace |
| 21 | 2,6-CH3(Br) | 2-F | 0.1 | 24 | Trace |
| 22 | 2,6-CH3(Br) | 2,6-CH3 | 0.1 | 24 | Trace |
| 23c | 2,4,6-CH3(Br) | H | 1 | 12 | 51 |
| 24c | 4-COCH3(Cl) | Me | 1 | 12 | >99 |
| 25c | 4-CF3(Cl) | Me | 1 | 12 | 84 |
| 26c | 4-F(Cl) | Me | 1 | 12 | 57 |

Kinetic studies and catalyst recycling
The reaction kinetics of the coupling between 4′-bromoacetophenone and phenylboronic acid using 0.1 mol% catalyst loading of 2d at room temperature in the presence and absence of TBAB were also investigated. As the two curves in Fig. 1 indicate, the reaction with TBAB is faster in the beginning than the one without TBAB. The reaction with TBAB reaches quantitative yield after 8 h at room temperature, while the one without it only leads to a yield of 67% after the same time. Furthermore, both reactions show sigmoidal kinetic curves, especially the one with TBAB, and both exhibit the highest catalytic activities during the first two hours. As discussed in earlier work,12e,18 such observations may indicate that both reactions undergo an autocatalytic growth process, where less active Pd nanoparticles (Pd NPs) are formed. | ||
| Fig. 1 Yield–time curve of the reaction of 4′-bromoacetophenone and phenylboronic acid. Reaction conditions: 4′-bromoacetophenone (0.50 mmol) and phenylboronic acid (0.75 mmol) with 0.1 mol% 2d, KOH (1.0 mmol), H2O (1 mL) and TBAB (1.0 mmol, if applicable). | ||
The recyclability of catalyst 2d was further investigated using 4′-bromoacetophenone and 4-bromoanisole, respectively, in aqueous Suzuki–Miyaura coupling with phenylboronic acid (Fig. 2). All reactions were conducted at 100 °C with 1 mol% of 2d. With respect to 4′-bromoacetophenone as the substrate, all four recycling runs lead to full conversion to the coupling product within 1 h, whereas the reaction using 4-bromoanisole as the substrate gives a slightly lower yield of 83% after the second run within 1 h (99% in the first run). However, an increased yield of 94% can be obtained when the reaction time is extended to 4 h in the fourth run, thus suggesting that complex 2d can be used in at least four runs, leading to high yields for both substrates. When the substrate 4′-chlorobenzotrifluoride (coupled with 4-methylbenzeneboronic acid) is used instead, the yield decreases to 35% (from 84% in the first run) after the second run. This indicates that decomposition occurs, resulting in reduced catalytic performance, especially when less active substrates are used for the recycling experiments.
 | ||
| Fig. 2 Recycling experiments with complex 2d in aqueous Suzuki–Miyaura reactions. Reaction conditions: (a) 4′-bromoacetophenone (0.5 mmol) and phenylboronic acid (0.75 mmol) in the presence of KOH (2.0 mmol), complex 2d (1 mol%), TBAB (1.0 mmol), H2O (1.0 mL) at 100 °C for 1 h; (b) 4-bromoanisole (0.5 mmol) and phenylboronic acid (0.75 mmol) in the presence of KOH (2.0 mmol), complex 2d (1 mol%), TBAB (1.0 mmol), H2O (1.0 mL) at 100 °C, the first two runs for 1 h, the third run for 2 h, and the fourth run for 4 h. | ||
Considering the lower activities in the consecutive runs and the formation of a black precipitate, Pd NP formation seemed to be a reason for the observed activity decrease. Therefore, TEM analysis was performed after the first and the fourth run of the coupling reactions between 4-bromoanisole and phenylboronic acid. After the reactions, the black solid was isolated by centrifugation and was washed with methanol (5 × 3 mL) and water (5 × 3 mL) five times each. The black solid was again centrifuged and dispersed in ethanol for TEM measurements. The TEM pictures in Fig. 3 clearly show the presence of Pd NPs both after the first run and the fourth run (Fig. 3, 3a and 3c). It is noteworthy that the Pd NPs form in quite narrow size distributions with approximate sizes of 3.0 ± 0.5 nm in both cases (Fig. 3, 3b and 3d) and originate from the active homogeneous catalyst which has already been reported for comparable catalytic systems.3b,19
 | ||
| Fig. 3 TEM images of Pd NPs generated from complex 2d: (a) and (b) after the first run of the coupling reaction of 4-bromoanisole and phenylboronic acid; (c) and (d) after the fourth run of the coupling reaction of 4-bromoanisole and phenylboronic acid. | ||
In order to get insight into the potential catalytic activity of the particles, a mercury poisoning test of the reaction between 4′-chloroacetophenone and phenylboronic acid at 100 °C using 0.1 mol% of 2d was performed but does not result in decreased activity (99% yield, reaction time: 2 h). Furthermore, the isolated Pd NPs (obtained from 1 mol% of 2d) were directly applied in catalytic coupling of 4′-chloroacetophenone and phenylboronic acid in water at 100 °C over a period of 24 h; however, no conversion is observed. These results clearly strengthen the assumption that the presented transformation is homogeneously catalyzed.
Conclusions
An easily-prepared and effective catalytic system for aqueous Suzuki–Miyaura cross-coupling reactions, working at room temperature with water-soluble Pd-NHC catalysts, has been developed. Four sulfonated water-soluble Pd complexes (2a–2d) ligated with the well-developed PEPPSI NHC system have been prepared via a two-step synthesis in good yields. Complex 2d bearing a 2,6-diisopropylphenyl substituent displays the best catalytic performance and coupling of aryl bromides can even be conducted at room temperature while the catalyst can be recycled and used in at least four consecutive runs. TEM analysis, kinetic studies and mercury poison experiments of 2d reveal that Pd nanoparticles with narrow size distributions are formed during the catalytic reactions.Experimental section
General information
All chemicals were purchased from commercial suppliers and used without further purification. Liquid NMR spectra were recorded on a Bruker Ultrashield 400 (1H NMR, 400.13 MHz; 13C NMR, 100.53 MHz) at 298 K. The spectra were calibrated using the residual solvent shifts as internal standards. Chemical shifts were referenced in parts per million (ppm). Abbreviations for signal multiplicities are as follows: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Spectral assignments are based on HMQC and COSY. Mass spectra were recorded with a Finnigan MAT 311 A and a MAT 90 Spectrometer. Elemental analyses were performed by the microanalytical laboratory of the TUM.General procedure for the preparation of ligands 1a–1c
A mixture of imidazole (1.5 equiv.) and sodium 2-bromoethanesulfonate (300 mg, 1.42 mmol) in toluene (3.0 mL) was stirred at 150 °C in a pressure tube for 48 h. The reaction mixture was filtered and the resulting solid was washed with CH2Cl2 and ether. The product was dried in vacuo to afford the corresponding compound.1a: white solid, yield: 70%. 1H NMR (DMSO-d6 400 MHz): δ 9.72 (s, 1H, Himid), 8.05–7.98 (m, 2H, Ar–H), 7.69–7.66 (m, 2H, Ar–H), 4.74 (t, J = 8.0 Hz, 2H, NCH2CH2), 4.08 (s, 3H, NCH3), 3.04 (t, J = 8.0 Hz, 2H, CH2CH2SO3). 13C NMR (DMSO-d6 100 MHz) δ 33.1 (NCH3), 43.8 (NCH2CH2), 48.7 (CH2CH2SO3), 113.5 (aromatic), 126.2 (aromatic), 126.4 (aromatic), 130.8 (aromatic), 131.6 (aromatic), 143.6 (Himid). Anal. Calcd for C10H12BrN2NaO3S·1.85CH2Cl2: C, 27.89; H, 2.81; N, 6.51. Found: C, 28.01; H, 3.17; N, 6.07.
1b: white solid, yield: 82%. 1H NMR (DMSO-d6 400 MHz): δ 9.17 (s, 1H, Himid), 7.80 (s, 1H, Himid), 7.68 (s, 1H, Himid), 4.40 (t, J = 8.0 Hz, 2H, NCH2CH2), 3.83 (s, 3H, NCH3), 2.99 (t, J = 8.0 Hz, 2H, CH2CH2SO3). 13C NMR (DMSO-d6 100 MHz) δ 35.62 (NCH3), 45.9 (NCH2CH2), 50.2 (CH2CH2SO3), 122.5 (Himid), 123.1 (Himid), 137.1 (Himid). Anal. Calcd for C6H10BrN2NaO3S·NaBr: C, 14.45; H, 2.02; N, 5.62. Found: C, 14.66; H, 2.36; N, 5.16.
1c: white solid, yield: 75%. 1H NMR (DMSO-d6 400 MHz): δ 9.20 (s, 1H, Himid), 7.80 (s, 1H, Himid), 7.74 (s, 1H, Himid), 4.40 (t, J = 8.0 Hz, 2H, NCH2CH2SO3), 4.14 (t, J = 8.0 Hz, 2H, NCH2CH2CH2), 2.98 (t, J = 8.0 Hz, 2H, CH2CH2SO3), 1.74 (sept, J = 8.0 Hz, 2H, CH2CHCH2), 1.25 (sept, J = 8.0 Hz, 2H, CH2CH2CH3), 0.88 (t, J = 8.0 Hz, 3H, CH2CH3). 13C NMR (DMSO-d6 100 MHz) δ 13.3 (CH2CH3), 18.7 (CH2CH3), 31.4 (CH2CHCH2), 46.1 (NCH2CH2), 48.4 (NCH2CH2CH2), 50.1 (CH2CH2SO3), 121.9 (Himid), 122.6 (Himid), 136.7 (Himid). Anal. Calcd for C9H16BrN2NaO3S·1.1CH2Cl2: C, 28.30; H, 4.28; N, 6.55. Found: C, 27.90; H, 4.75; N, 7.05.
Preparation of ligand 1d
A mixture of N-(2,6-diisopropylphenyl)imidazole (343 mg, 1.50 mmol) and sodium 2-bromoethanesulfonate (300 mg, 1.42 mmol) in toluene–DMF (6:1, 3.0 mL) was stirred at 150 °C in a pressure tube for 3 d. The reaction mixture was filtered. The filtrate was removed under vacuum and the residue was triturated by THF to give a white solid. The solid was washed with CH2Cl2 (10 ml × 2) and dried in vacuo to afford the imidazolium salt 1d. White solid, yield: 68%. 1H NMR (DMSO-d6 400 MHz): δ 9.37 (s, 1H, Himid), 8.12 (s, 1H, Himid), 7.98 (s, 1H, Himid), 7.60 (t, J = 8.0 Hz, 1H, Ar–H), 7.43 (s, 1H, Ar–H), 7.42 (s, 1H, Ar–H), 4.54 (t, J = 8.0 Hz, 2H, NCH2CH2), 3.08 (t, J = 8.0 Hz, 2H, CH2CH2SO3), 2.35 (sept, J = 8.0 Hz, 2H, –CH(CH3)2), 1.11 (dd, J = 8.0 Hz, 12H, –CH(CH3)2). 13C NMR (DMSO-d6 100 MHz) δ 23.9 (CH(CH3)2), 24.1 (CH(CH3)2), 27.7 (–CH(CH3)2), 46.6 (NCH2CH2), 49.9 (CH2CH2SO3), 123.4 (Himid), 124.3 (aromatic), 124.5 (Himid), 130.7 (aromatic), 131.3 (aromatic), 138.3 (Himid), 145.5 (aromatic). Anal. Calcd for C17H24BrN2NaO3S·1.85H2O: C, 43.2; H, 5.91; N, 5.93. Found: C, 43.36; H, 6.09; N, 5.98.
Preparation of complexes 2a–2d
2a: A Schlenk tube was charged with PdBr2 (55 mg, 0.2 mmol), 1a (68 mg, 0.2 mmol), K2CO3 (138 mg, 1.0 mmol) and a stir bar under argon. Pyridine (2.0 mL) was then added as the solvent and the reactant. The mixture was heated and stirred for 36 h at 90 °C. After cooling to r.t., the reaction mixture was diluted with CH2Cl2 and passed through a short pad of silica gel covered with a pad of Celite, eluting with MeOH until the product was completely recovered. Further purification was done using flash column (DCM–MeOH) to get pure complexes for analysis and catalysis. Yellow solid, yield: 52%. 1H NMR (MeOD 400 MHz): δ 9.01 (d, J = 8.0 Hz, 2H, Hpy), 7.92 (t, J = 8.0 Hz, 1H, Hpy), 7.70–7.68 (m, 1H, Ar–H), 7.61–7.59 (m, 1H, Ar–H), 7.48 (t, J = 8.0 Hz, 2H, Hpy), 7.39–7.37 (m, 2H, Ar–H), 5.29 (t, J = 8.0 Hz, 2H, NCH2CH2SO3), 4.29 (s, 3H, NCH3), 3.68 (t, J = 8.0 Hz, 2H, CH2CH2SO3). 13C NMR (MeOD 100 MHz) δ 35.5 (NCH3), 45.4 (NCH2CH2), 51.1 (CH2CH2SO3), 111.1 (Caromatic), 111.4 (Caromatic), 124.6 (Caromatic), 125.9 (Cpy), 135.2 (Cimi), 136.3 (Cimi), 139.6 (Cpy), 153.5 (Cpy), 166.2 (Ccarbene). Anal. Calcd for C15H16Br2NaN3O3PdS·0.4H2O: C, 29.31; H, 2.75; N, 6.84. Found: C, 28.86; H, 3.00; N, 6.47. MS (FAB): m/z = 583.8 [M]+, 527.9 [M − Py]+, 504.9 [M − Py − Na]+.2b: This complex was prepared by the same method as 2a starting from 1b (61 mg, 0.2 mmol) and PdBr2 (55 mg, 0.2 mmol). Yellow solid, yield: 61%. 1H NMR (MeOD 400 MHz): δ 8.96 (d, J = 4.0 Hz, 2H, Hpy), 7.89 (t, J = 4.0 Hz, 1H, Hpy), 7.44 (t, J = 4.0 Hz, 2H, Hpy), 7.33 (d, J = 4.0 Hz, 1H, Himid), 7.20 (d, J = 4.0 Hz, 1H, Himid), 4.93 (t, J = 8.0 Hz, 2H, NCH2CH2), 4.04 (s, 3H, NCH3), 3.66 (t, J = 8.0 Hz, 2H, CH2CH2SO3). 13C NMR (MeOD 100 MHz) δ 38.4 (NCH3), 48.1 (NCH2CH2), 52.8 (CH2CH2SO3), 123.8 (Cimid), 124.7 (Cimid), 125.7 (CPy), 139.4 (CPy), 149.8 (Ccarbene), 153.6 (CPy). Anal. Calcd for C11H14Br2N3NaO3PdS·1.9CH2Cl2·2H2O: C, 20.52; H, 2.91; N, 5.57. Found: C, 20.31; H, 2.71; N, 5.79. MS (FAB): m/z = 558.7 [M]+, 454.9 [M − Py − Na]+.
2c: This complex was prepared by a similar method to that for 2a starting from 1c (70 mg, 0.2 mmol) and PdBr2 (55 mg, 0.2 mmol). Yellow solid, yield: 73%. 1H NMR (MeOD 400 MHz): δ 8.94 (d, J = 8.0 Hz, 2H, Hpy), 7.88 (t, J = 8.0 Hz, 1H, Hpy), 7.44 (t, J = 8.0 Hz, 2H, Hpy), 7.33 (d, J = 4.0 Hz, 1H, Himid), 7.22 (d, J = 4.0 Hz, 1H, Himid), 4.97 (t, J = 8.0 Hz, 2H, NCH2CH2SO3), 4.47 (t, J = 8.0 Hz, 2H, NCH2CH2CH2), 3.64 (t, J = 8.0 Hz, 2H, CH2CH2SO3), 2.11 (sept, J = 8.0 Hz, 2H, CH2CHCH2), 1.49 (sept, J = 8.0 Hz, 2H, CH2CH2CH3), 1.06 (t, J = 8.0 Hz, 3H, CH2CH3). 13C NMR (MeOD 100 MHz) δ 14.1 (CH2CH3), 20.9 (CH2CH3), 33.2 (CH2CHCH2), 48.2 (NCH2CH2), 51.9 (NCH2CH2CH2), 52.8 (CH2CH2SO3), 123.6 (Cimid), 123.7 (Cimid), 125.7 (Cpy), 139.3 (Cpy), 149.1 (Ccarbene), 153.5 (Cpy). MS (FAB): m/z = 600.7 [M]+, 498.9 [M − Na − Py]+. Anal. Calcd for C14H20Br2NaN3O3PdS·1.95CH2Cl2·0.4H2O: C, 25.01; H, 3.16; N, 5.48. Found: C, 24.63; H, 3.04; N, 5.88.
2d: This complex was prepared by a similar method to that for 2a starting from 1d (68 mg, 0.2 mmol) and PdBr2 (55 mg, 0.2 mmol). Yellow solid, yield: 66%. 1H NMR (MeOD 400 MHz): δ 8.68 (d, J = 8.0 Hz, 2H, Hpy), 7.76 (t, J = 8.0 Hz, 1H, Hpy), 7.60 (d, J = 4.0 Hz, 1H, Himid), 7.51 (t, J = 8.0 Hz, 1H, Ar–H), 7.37 (d, J = 8.0 Hz, 2H, Ar–H), 7.31 (d, J = 4.0 Hz, 1H, Himid), 7.28 (t, J = 8.0 Hz, 2H, Hpy), 5.25 (t, J = 8.0 Hz, 2H, NCH2CH2), 3.82 (t, J = 8.0 Hz, 2H, CH2CH2SO3), 3.00 (sept, J = 8.0 Hz, 2H, –CH(CH3)2), 1.36–1.03 (dd, J = 8.0 Hz, 12H, –CH(CH3)2). 13C NMR (MeOD 400 MHz) δ 23.8 (CH(CH3)2), 26.7 (CH(CH3)2), 29.6 (–CH(CH3)2), 49.1 (NCH2CH2), 52.8 (CH2CH2SO3), 123.3 (Cimid), 125.1 (Cimid), 125.5 (Cpy), 128.2 (Caromatic), 131.3 (Caromatic), 136.1 (Caromatic), 139.1 (Cpy), 148.4 (Caromatic), 152.5 (Ccarbene), 153.4 (Cpy). Anal. Calcd for C22H28Br2NaN3O3PdS·1.75H2O: C, 35.94; H, 4.32; N, 5.71; S, 4.35. Found: C, 35.79; H, 4.15; N, 5.55; S, 4.36. MS (FAB): m/z = 600.8 [M − Py − Na]−, 521.0 [M − Py − Na − Br]−.
General procedure for Suzuki–Miyaura cross-coupling in water
Pd complex 2d (0.01 or 0.02 mmol), phenylboronic acid (0.75 mmol), base (1.00 mmol) and aryl halide (0.50 mmol) were added to a flask containing a magnetic stir bar. Water (1.0 mL) was then added. The reaction was stirred at the specified temperature for the desired period. Upon completion, the mixture was extracted using ethyl acetate (2 × 5 mL) and filtered. The filtrate was removed in a rotary evaporator (40 °C, 100 mbar) and the crude product was directly dissolved using CDCl3 with a standard (1,3,5-trimethoxybenzene, 0.3 mmol) for 1H-NMR analysis.Recycling of catalyst 2d in aqueous Suzuki–Miyaura cross-coupling
The catalyst 2d (1 mol%, 3.5 mg), phenylboronic acid (120 mg, 1.00 mmol), KOH (56 mg, 1.00 mmol) and the aryl halide (0.50 mmol) were added to a flask containing a magnetic stir bar. Water (1.0 mL) was added to the mixture. The reaction was stirred at 100 °C for a certain period of time. Upon completion, the mixture was extracted using ethyl acetate (2 × 5 mL). The solvent was collected by centrifugation, and removed in a rotary evaporator (40 °C, 100 mbar). Further, the crude product was dissolved using CDCl3 with a standard (1,3,5-trimethoxybenzene, 0.3 mmol) for 1H-NMR analysis to determine the yield of the reaction. The residue from centrifugation was dried in vacuo and used for the next cycle of catalysis.Transmission electron microscopy (TEM)
The samples were prepared by pipetting a drop of an ethanol solution of Pd NPs on copper grids covered with a Quantifoil Multi A holey carbon film and a 2 nm carbon film on top. Electron micrographs were recorded at a nominal magnification of 300000 and 400000 using a JEOL JEM 2011 electron microscope operated at 120 kV. Micrographs were digitized at a resolution of 1500 and 3000 dpi using a FlexTight Precision II (Hasselblad) array scanner.
Acknowledgements
Support from the Fonds der Chemischen Industrie (stipend to K. R.) and the TUM Graduate School is gratefully acknowledged.Notes and references
- (a) C. J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed; (b) M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- (a) R. P. Aravinda, R. A. Babul, R. G. Ramachandra and R. N. Subbarami, J. Heterocycl. Chem., 2013, 50, 1451–1456 CrossRef PubMed; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (c) Y. Cao, Adv. Mater. Res., 2011, 284–286, 2404–2408 CrossRef CAS; (d) M. G. Organ, G. A. Chass, D. C. Fang, A. C. Hopkinson and C. Valente, Synthesis, 2008, 2776–2797 CrossRef CAS PubMed; (e) L. Botella and C. Nájera, Angew. Chem., Int. Ed., 2002, 41, 179–181 CrossRef CAS.
- (a) K. H. Shaughnessy and R. B. DeVasher, Curr. Org. Chem., 2005, 9, 585–604 CrossRef CAS; (b) D. Zhao, Z. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 15876–15882 CrossRef CAS PubMed.
- A. L. Casalnuovo and J. C. Calabrese, J. Am. Chem. Soc., 1990, 112, 4324–4330 CrossRef CAS.
- (a) B. Çetinkaya, N. Gurbuz, T. Seckin and I. Ozdemir, J. Mol. Catal. A: Chem., 2002, 184, 31–38 CrossRef; (b) D. Cauzzi, M. Lanfranchi, G. Marzolini, G. Predieri, A. Tiripicchio, M. Costa and R. Zanoni, J. Organomet. Chem., 1995, 488, 115–125 CrossRef CAS; (c) C. S. J. Cazin, M. Veith, P. Braunstein and R. B. Bedford, Synthesis, 2005, 622–626 CAS; (d) C. S. J. Cazin, C. R. Chim., 2009, 12, 1173–1180 CrossRef CAS PubMed; (e) C. S. J. Cazin, M. Veith, P. Braunstein and R. B. Bedford, Synthesis, 2005, 622–626 CAS; (f) K. H. Shaughnessy, Eur. J. Org. Chem., 2006, 1827–1835 CrossRef CAS PubMed.
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (b) J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS PubMed; (c) A. Azua, S. Sanz and E. Peris, Organometallics, 2010, 29, 3661–3664 CrossRef CAS.
- D. H. Valentine and J. H. Hillhouse, Synthesis, 2003, 2437–2460 CAS.
- (a) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed; (b) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS PubMed; (c) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS PubMed.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS PubMed.
- (a) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC; (b) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC; (c) L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed.
- (a) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (b) J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS; (c) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- (a) D. Schonfelder, O. Nuyken and R. Weberskirch, J. Organomet. Chem., 2005, 690, 4648–4655 CrossRef PubMed; (b) F. Churruca, R. SanMartin, B. Ines, I. Tellitu and E. Dominguez, Adv. Synth. Catal., 2006, 348, 1836–1840 CrossRef CAS PubMed; (c) C. Fleckenstein, S. Roy, S. Leuthaeusser and H. Plenio, Chem. Commun., 2007, 2870–2872 RSC; (d) M. Meise and R. Haag, ChemSusChem, 2008, 1, 637–642 CrossRef CAS PubMed; (e) B. Ines, R. SanMartin, M. Jesus Moure and E. Dominguez, Adv. Synth. Catal., 2009, 351, 2124–2132 CrossRef CAS PubMed; (f) H. Turkmen, R. Can and B. Çetinkaya, Dalton Trans., 2009, 7039–7044 RSC; (g) S. Roy and H. Plenio, Adv. Synth. Catal., 2010, 352, 1014–1022 CrossRef CAS PubMed; (h) T. Tu, X. Feng, Z. Wang and X. Liu, Dalton Trans., 2010, 39, 10598–10600 RSC; (i) F. Godoy, C. Segarra, M. Poyatos and E. Peris, Organometallics, 2011, 30, 684–688 CrossRef CAS; (j) B. Karimi and P. F. Akhavan, Chem. Commun., 2011, 47, 7686–7688 RSC; (k) B. Karimi and P. Fadavi Akhavan, Inorg. Chem., 2011, 50, 6063–6072 CrossRef CAS PubMed; (l) L. Y. Li, J. Y. Wang, C. S. Zhou, R. H. Wang and M. C. Hong, Green Chem., 2011, 13, 2071–2077 RSC; (m) H. Turkmen, L. Pelit and B. Çetinkaya, J. Mol. Catal. A: Chem., 2011, 348, 88–93 CrossRef PubMed; (n) L. Benhamou, C. Besnard and E. P. Kündig, Organometallics, 2013, 33, 260–266 CrossRef; (o) S. Gupta, B. Basu and S. Das, Tetrahedron, 2013, 69, 122–128 CrossRef CAS PubMed; (p) E. L. Kolychev, A. F. Asachenko, P. B. Dzhevakov, A. A. Bush, V. V. Shuntikov, V. N. Khrustalev and M. S. Nechaev, Dalton Trans., 2013, 42, 6859–6866 RSC; (q) T. E. Schmid, D. C. Jones, O. Songis, O. Diebolt, M. R. L. Furst, A. M. Z. Slawin and C. S. J. Cazin, Dalton Trans., 2013, 42, 7345–7353 RSC.
- (a) C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2006, 12, 4743–4748 CrossRef PubMed; (b) M. G. Organ, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien and C. Valente, Chem. – Eur. J., 2006, 12, 4749–4755 CrossRef CAS PubMed.
- (a) M. G. Organ, M. Abdel-Hadi, S. Avola, N. Hadei, J. Nasielski, C. J. O'Brien and C. Valente, Chem. – Eur. J., 2007, 13, 150–157 CrossRef CAS PubMed; (b) C. Valente, S. Baglione, D. Candito, C. J. O'Brien and M. G. Organ, Chem. Commun., 2008, 735–737 RSC; (c) M. G. Organ, S. Calimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CrossRef CAS PubMed; (d) G. T. Achonduh, N. Hadei, C. Valente, S. Avola, C. J. O'Brien and M. G. Organ, Chem. Commun., 2010, 46, 4109–4111 RSC; (e) S. Calimsiz, M. Sayah, D. Mallik and M. G. Organ, Angew. Chem., Int. Ed., 2010, 49, 2014–2017 CrossRef CAS PubMed; (f) M. Dowlut, D. Mallik and M. G. Organ, Chem. – Eur. J., 2010, 16, 4279–4283 CrossRef CAS PubMed; (g) J. Nasielski, N. Hadei, G. Achonduh, E. A. B. Kantchev, C. J. O'Brien, A. Lough and M. G. Organ, Chem. – Eur. J., 2010, 16, 10844–10853 CrossRef CAS PubMed; (h) C. Valente, M. E. Belowich, N. Hadei and M. G. Organ, Eur. J. Org. Chem., 2010, 4343–4354 CAS; (i) S. Calimsiz and M. G. Organ, Chem. Commun., 2011, 47, 5181–5183 RSC; (j) N. Hadei, G. T. Achonduh, C. Valente, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2011, 50, 3896–3899 CrossRef CAS PubMed; (k) K. H. Hoi, S. Calimsiz, R. D. J. Froese, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2011, 17, 3086–3090 CrossRef CAS PubMed; (l) H. N. Hunter, N. Hadei, V. Blagojevic, P. Patschinski, G. T. Achonduh, S. Avola, D. K. Bohme and M. G. Organ, Chem. – Eur. J., 2011, 17, 7845–7851 CrossRef CAS PubMed; (m) C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- G. J. Barbante, P. S. Francis, C. F. Hogan, P. R. Kheradmand, D. J. D. Wilson and P. J. Barnard, Inorg. Chem., 2013, 52, 7448–7459 CrossRef CAS PubMed.
- E. Brenner, D. Matt, M. Henrion, M. Teci and L. Toupet, Dalton Trans., 2011, 40, 9889–9898 RSC.
- D. Badone, M. Baroni, R. Cardamone, A. Ielmini and U. Guzzi, J. Org. Chem., 1997, 62, 7170–7173 CrossRef CAS PubMed.
- K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161–171 CrossRef CAS PubMed.
- (a) C. Zhou, J. Wang, L. Li, R. Wang and M. Hong, Green Chem., 2011, 13, 2100–2106 RSC; (b) D. Saha, K. Chattopadhyay and B. C. Ranu, Tetrahedron Lett., 2009, 50, 1003–1006 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc00986j |
| This journal is © The Royal Society of Chemistry 2014 |