
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the synthesis of glycosylated dihydrochalcone natural products using glycosyltransferase-catalysed cascade reactions†
Alexander
Gutmann
a,
Linda
Bungaruang
a,
Hansjoerg
Weber
b,
Mario
Leypold
b,
Rolf
Breinbauer
b and
Bernd
Nidetzky
*ac
aInstitute of Biotechnology and Biochemical Engineering, Graz University of Technology, NAWI Graz, Petersgasse 12, 8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at; Fax: (+43)-316-873-8434; Tel: (+43)-316-873-8400
bInstitute of Organic Chemistry, Graz University of Technology, NAWI Graz, Stremayrgasse 9, 8010 Graz, Austria
cAustrian Centre of Industrial Biotechnology, Petersgasse 14, 8010 Graz, Austria
First published on 25th July 2014
Abstract
Regioselective O-β-D-glucosylation of flavonoid core structures is used in plants to create diverse natural products. Their prospective application as functional food and pharmaceutical ingredients makes flavonoid glucosides interesting targets for chemical synthesis, but selective instalment of a glucosyl group requires elaborate synthetic procedures. We report glycosyltransferase-catalysed cascade reactions for single-step highly efficient O-β-D-glucosylation of two major dihydrochalcones (phloretin, davidigenin) and demonstrate their use for the preparation of phlorizin (phloretin 2′-O-β-D-glucoside) and two first-time synthesised natural products, davidioside and confusoside, obtained through selective 2′- and 4′-O-β-D-glucosylation of the dihydroxyphenyl moiety in davidigenin, respectively. Parallel biocatalytic cascades were established by coupling uridine 5′-diphosphate (UDP)-glucose dependent synthetic glucosylations catalysed by herein identified dedicated O-glycosyltransferases (OGTs) to UDP dependent conversion of sucrose by sucrose synthase (SuSy; from soybean). The SuSy reaction served not only to regenerate the UDP-glucose donor substrate for OGT (up to 9 times), but also to overcome thermodynamic restrictions on dihydrochalcone β-D-glucoside formation (up to 20% conversion and yield enhancement). Using conditions optimised for overall coupled enzyme activity, target 2′-O- or 4′-O-β-D-glucoside was obtained in ≥88% yield from reactions consisting of 5 mM dihydrochalcone acceptor, 100 mM sucrose, and 0.5 mM UDP. Davidioside and confusoside were isolated and their proposed chemical structures confirmed by NMR. OGT-SuSy cascade transformations present a green chemistry approach for efficient glucosylation in natural products synthesis.
Introduction
Flavonoids are a large and structurally diverse group of natural polyphenols.1,2 They are widely distributed in edible plants and therefore constitute an important part of the human diet.3,4 Some flavonoids are strong antioxidants and thus serve as powerful inhibitors of lipid peroxidation.5,6 Dietary intake of flavonoids has been correlated with reduced risk of chronic diseases, coronary heart disease in particular.7 Dihydrochalcones present a major sub-class of flavonoids and are characterized structurally by two phenolic rings connected through a flexible open-chain three-carbon linker (Table 1).8 Many dihydrochalcones, such as the common phloretin (1), which is abundantly present in the leaves and peel of apples,9,10 exhibit a wide spectrum of interesting and pharmacologically relevant bioactivities.11 Next to a general antioxidative property,121 shows antithrombotic13 and hepatoprotective properties,14 potential suppression of metabolic carcinogen activation15 and effects intracellular drug accumulation.16 Davidigenin (2), the 4′-deoxy analogue of 1, is broadly distributed among different plant families.17 As an antispasmodic compound present in or derived from traditional medicines it is applied to the treatment of intestinal disorders and asthma.172 also exhibits weak antibacterial activity18 and shows antidiabetic activity.19|
|
||||
|---|---|---|---|---|
| Dihydrochalcone | R1 | R2 | R3 | R4 |
| Phloretin (1) | H | H | H | OH |
| Davidigenin (2) | H | H | H | H |
| Phlorizin (3) | Glucose | H | H | OH |
| Trilobatin (4) | H | H | Glucose | OH |
| Davidioside (5) | Glucose | H | H | H |
| Confusoside (6) | H | H | Glucose | H |
| Nothofagin (7) | H | Glucose | H | OH |
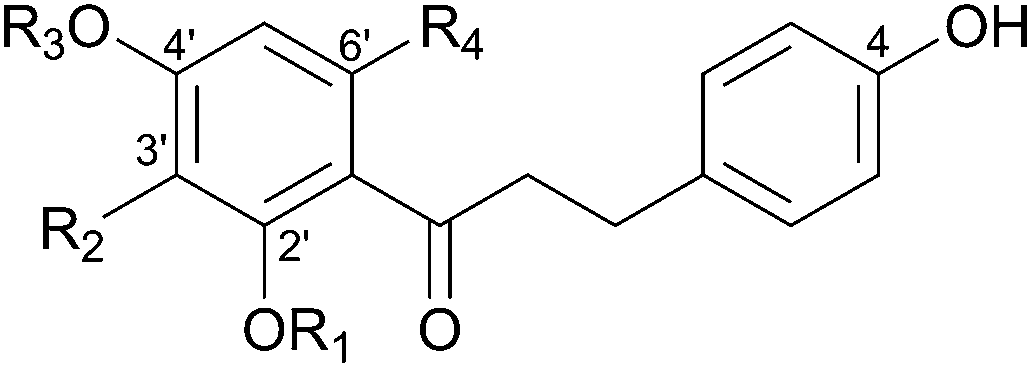
Glycosylation is an important biological mechanism for the structural and functional diversification of natural flavonoids.20,21 Generally, the attachment of sugar residue(s) to a flavonoid core increases compound bioavailability due to strong water solubility enhancement.22,23 Furthermore, physiological and pharmacological properties are often beneficially altered as consequence of glycosylation.24,25 Taste properties (e.g. sweetness, bitterness) and colour can also be modified by glycosylation.26,27 Interestingly, compared to other flavonoid sub-classes that show broadly diverging and often complex glycosylation patterns, glycosylated dihydrochalcones occur primarily as mono-β-D-glucopyranosides.8 Sugar attachment to dihydrochalcones often results from a phenolic O-glycosidic bond, although aromatic C-glucosylation is also found in certain compounds.8,28 Phloretin is naturally present mainly as the 2′-O-β-D-glucoside, commonly referred to as phlorizin (3).29 Due to inhibition of cellular glucose transport 3 was considered for type II diabetes treatment.29 The corresponding 4′-O-β-D-glucoside, which is called trilobatin (4), is less common.30 Davidioside (5) is the 2′-O-β-D-glucoside of 2, while the 4′-O-β-D-glucoside is called confusoside (6). Compounds 5 and 6 were previously isolated from plant extracts and characterized structurally.31,32 However, except for a low yielding total synthesis of 6, preparation of glucosides of 2 was not reported.33
Due to their various prospective applications as fine chemicals, pharmaceutical ingredients and food additives, dihydrochalcone glucosides 3–6 are interesting targets for large-scale preparation.29,34,35 However, their isolation from natural material is complicated by occurrence in complex multi-component mixtures and at relatively low abundance.30–32,36 Selective glucosylation of 1 and 2 would provide convenient access to 3–6 through bottom-up synthesis. Process chemistry for this purpose is not well developed, though, and elaborate procedures involving a substantial amount of protecting group chemistry are required for precise instalment of the glucosyl group.37 Alternative biotransformations using cultured plant cells were successfully applied for direct glucosylation of a variety of phenolic compounds including several flavonoids.38,39 However, presumably due to a mixture of native plant glucosyltransferases, control of regioselectivity was difficult and typically mixtures of various glucosides were obtained. Isolated enzymes in contrast have earned green credentials for being able to cope with issues of reactivity and selectivity in glycosylation reactions.40,41 Glycosyltransferases (GTs) are widely applied for glycosylation of flavonoids, whereas use of glycoside hydrolases, glycosynthases and transglycosidases is so far limited by a narrow substrate range and typically low yields.26,41,42 GTs utilise an activated donor substrate, typically a nucleoside diphosphate (NDP)-sugar, for transfer of a glycosyl residue onto certain position(s) of the acceptor molecule.43 The impressive flexibility displayed by several promiscuous GTs allowed glycosylation of a wide range of acceptors, but distinction between different phenolic hydroxyls in flavonoid acceptors still presents a notable challenge.44–47
Practical enzyme catalysts for selective glucosylation of 1 or 2 are to be established. Up to now only one GT from apple (Malus × domestica) and its ortholog from pear (Pyrus communis) (99% sequence identity) were reported to selectively produce analytical amounts of phlorizin (3) by glucosylation of 1.48,49 Other recently reported glucosylations of 1 by plant GTs yielded mixtures of monoglucosides.49,50 An even more promiscuous bacterial GT glucosylated 1 at 2′-, 4′- and 4-OH, producing a complex mixture of glucosidic compounds including two di- and a triglucoside.51 Therefore, this emphasises that selectivity-based GT enzyme selection is of key importance for successful development of a biocatalytic synthesis process. Furthermore, dedicated reaction engineering is necessary to solve the problem of cost-effective supply of the NDP-sugar substrate.52
Herein we present identification of GT biocatalysts for selective transformation of 1 into 3, and of 2 into 5 or 6. A parallel GT cascade reaction (Scheme 1) was established to enable dihydrochalcone glucosylation from sucrose (8) as a highly expedient and inexpensive glucosyl donor substrate. The overall bi-enzymatic transformations were carried out in the presence of catalytic amounts of uridine 5′-diphosphate (UDP) (9) to generate and constantly recycle in situ the UDP-glucose (10) utilised for glucoside synthesis. Applying reaction conditions optimised for enzyme activity and designed to overcome thermodynamic limitations, target O-β-D-glucoside was obtained as single transfer product in ≥88% yield and could be isolated readily. We propose Scheme 1 to be a generally applicable green chemistry approach for efficient glucosylation in natural products synthesis.
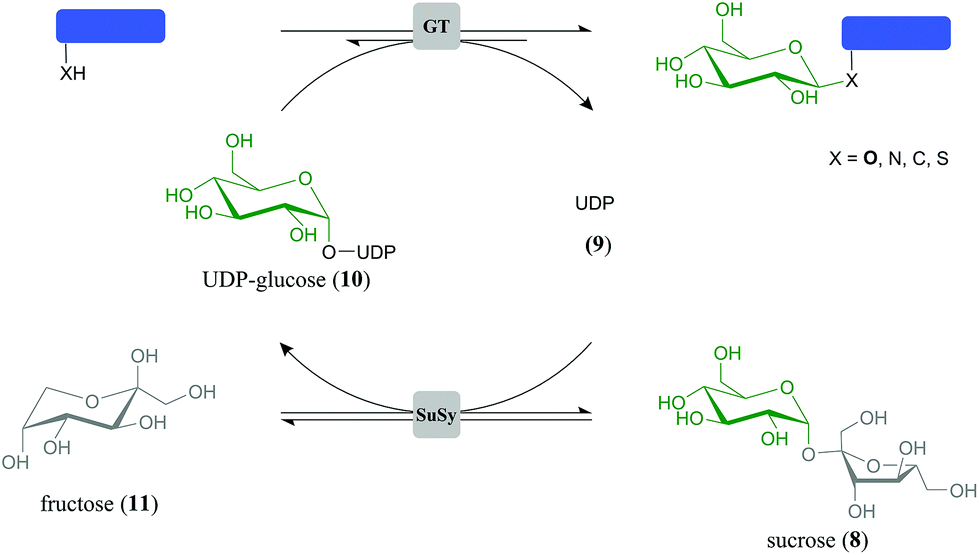 | ||
| Scheme 1 GT-catalysed cascade reaction for synthesis of natural product glucosides. Coupling of a “synthetic” GT with SuSy in one pot enables cost-effective in situ formation of glucosyl donor substrate 10 from 8 and catalytic amounts of 9. End-product inhibition by 9 is obviated. | ||
Results and discussion
Identification of GT enzymes catalysing regioselective glucosylation of phloretin (1) and davidigenin (2)
A GT from pear (Pyrus communis; PcOGT) was recently reported to be highly selective for transferring the glucosyl moiety of UDP-glucose (10) to the 2′-OH of 1, producing 3.49,53 However, evidences were only from thin layer chromatography49 or from initial-rate experiments carried out at low conversion (≤25%) of both donor and acceptor substrate.53 Furthermore, the concentrations of 1 used (300 and 100 μM, respectively) were too low to be of synthetic relevance. A number of studies show that GT regioselectivity may change substantially depending on degree of conversion or on initial substrate concentration.46,53 Therefore, this necessitated rigorous evaluation of PcOGT as catalyst for synthesis of 3 at elevated substrate concentration.Using recombinant PcOGT (0.1 U mL−1) purified from an Escherichia coli overexpression culture (ESI,† Fig. S1), transformation of 10 and 1 (5 mM each, pH 6.5) into phloretin glucoside(s) was examined in dependence of reaction progress until attainment of apparent equilibrium (∼70% conversion of 1). Product analysis was done using a reversed-phase HPLC method in which the monoglucosidic regio-isomers 3 and 4 were baseline separated (Fig. 1a). Only 3 and no 4 was detected at all times (Fig. 1b). Moreover, close balance between 1 consumed and 3 formed was consistent with enzymatic reaction exclusively at the acceptor's 2′-OH.
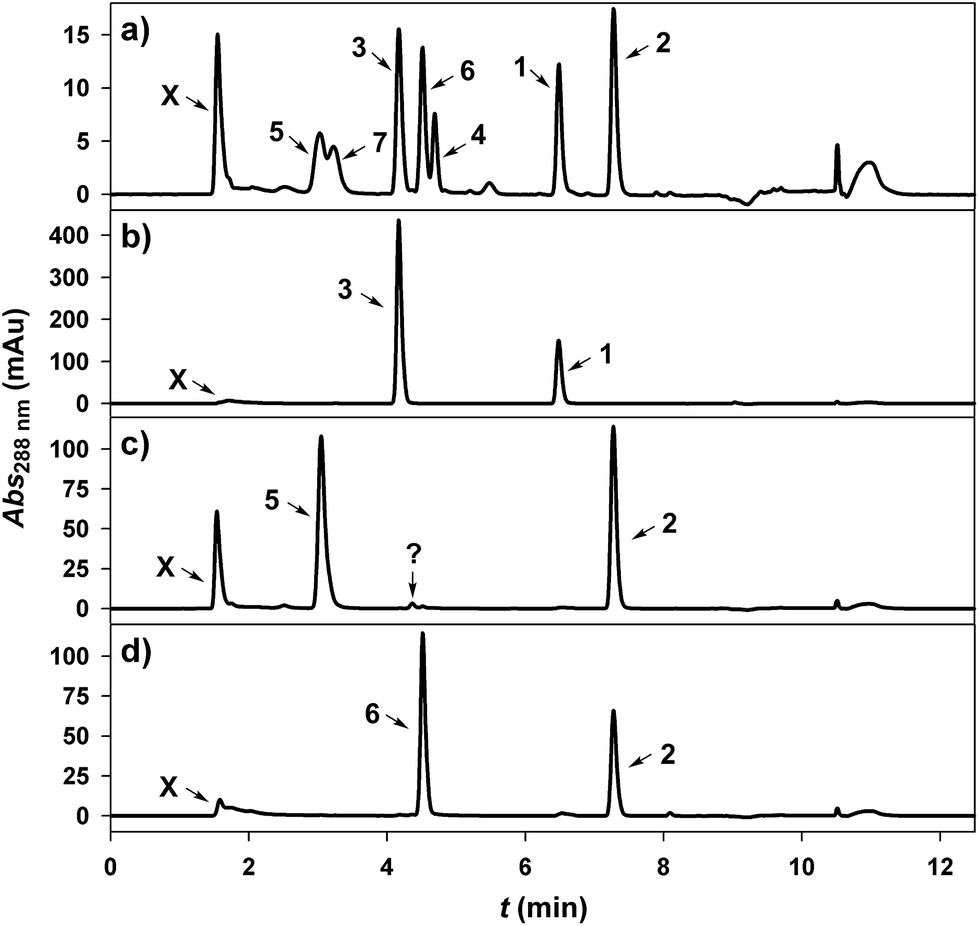 | ||
| Fig. 1 Reversed-phase C-18 HPLC-analysis of dihydrochalcone (glucosides) (X = unbound compounds including 9 and 10). (a) Separation of glucosides of 1 (3, 4, 7) and 2 (5, 6) is achieved. (b) Conversion of 1 by PcOGT. (c) Conversion of 2 by PcOGT (? = unknown by-product). (d) Conversion of 2 by OsCGT I121D. | ||
PcOGT was then examined for glucosylation of 2 from 10 (each 5.0 mM, pH 7.5). UV absorbance traces from HPLC analysis of samples taken at different times revealed gradual consumption of 2 with concomitant appearance of a major and a minor signal eluting at positions consistent with glucosidic products (Fig. 1c). According to HPLC peak areas around 80% of 2 were converted to the major glycoside and less than 2% of the by-product were formed when equilibrium was obtained. NMR data recorded from the reaction mixture gave preliminary evidence that the 2′-OH of 2 had been glycosylated, thereby indicating that regioselectivity was maintained upon exchange of acceptor substrate. Specific activities of PcOGT related to glucosyl acceptor substrate were determined from the time courses of consumption of 1 and 2, and 1 (1.76 U mg−1 protein) was an about 500-fold better substrate than 2.
In search of a differently regioselective GT catalysing glucosyl transfer to the 4′-OH of 1 and 2, we tested a recently described variant of a C-glycosyltransferase from rice (Oryza sativa; OsCGT) that had its Ile121 replaced by Asp.53 Highly purified mutant enzyme was used in all experiments (ESI,† Fig. S1). Wild-type OsCGT was shown to catalyse glucosyl transfer from 10 to the aromatic 3′-C of 1, thus producing the C-β-D-glucoside nothofagin (7).53,54 The particular residue substitution in OsCGT causes change in reaction selectivity such that O-glucosylations of the 2′-OH and 4′-OH occur next to the native C-glucosylation of 1.53 Using 1 at different concentrations between 2.5 μM and 2.5 mM, it was found that the enzymatic conversion resulted invariably in a mixture of glucosidic products that contained 3 or 7 as the main constituent whereas 4 was present only as a by-product (≤15% of total). It was not possible to enhance the relative abundance of 4 by running the reaction at different pH in the range 6.2–10.5. Ability of the OsCGT variant to synthesise substantial amounts of 4 is nevertheless remarkable, and the question of whether isolation of 4 from the enzymatically prepared mixture of phloretin glucosides would still be practical was left for consideration in the future. Notably, though, the OsCGT variant was complementarily regioselective to PcOGT in the glucosylation of 2, as shown in Fig. 1d. The main PcOGT glucosylation product, likely 5, was completely absent from the reaction sample obtained with the mutated OsCGT. However, a single new product peak appeared. Its area increased proportionally to the decreasing peak area of the 2. Interestingly, the retention time was very similar to that of the trace product formed from 2 by PcOGT, however HPLC analysis of a mixture of both conversions clearly reveals them to be distinct (ESI,† Fig. S2). Based on preliminary NMR data of the reaction mixture it was assumed, and will be confirmed later, that 6 was formed in the reaction of the GT mutant. Specific activity of the OsCGT variant for glucosylation of 2 was determined as 22 ± 3 mU mg−1.
In summary, therefore, GT catalysts for selective glucosylation of 1 and 2 have been identified, and they were used further for synthesis of 3, 5 and 6.
Thermodynamic analysis of glucosylation of phloretin (1)
Glucosylation of 1 or 2 from 10 is an equilibrium-controlled process.55 Thermodynamic restrictions on the synthesis of dihydrochalcone β-D-glucosides therefore required clarification. Fig. 2 displays results of time-course analyses for synthesis and degradation of 3, catalysed by PcOGT at pH 6.5 and 30 °C. All compounds present in the reaction were quantified (1, 3, 9, 10). Close balance for the proposed overall conversion, 1 + 10 ↔ 3 + 9, was obtained at each time and in each direction of reaction. Therefore, this indicated absence of enzyme-catalysed or spontaneous side reactions, such as hydrolysis of 10 for example. Reactions run in forward and reverse direction levelled out at exactly the same end concentrations of 1 and 3 within limits of experimental error (Fig. 2a), clearly suggesting that the true thermodynamic equilibrium had been attained. It was affirmed that addition of fresh enzyme to a reaction mixture at (apparent) equilibrium in Fig. 2 did, as expected, not induce further concentration changes. An equilibrium constant (Keq) of 4.6 (±0.7) was calculated from the data. Under the conditions used, therefore, only about 70% of substrate 1 can be converted to product 3. Development of a glucosyl transfer cascade (Scheme 1) that involved in situ generation of the UDP-sugar substrate 10 from sucrose (8) and UDP (9) was highly useful to overcome these thermodynamic restrictions.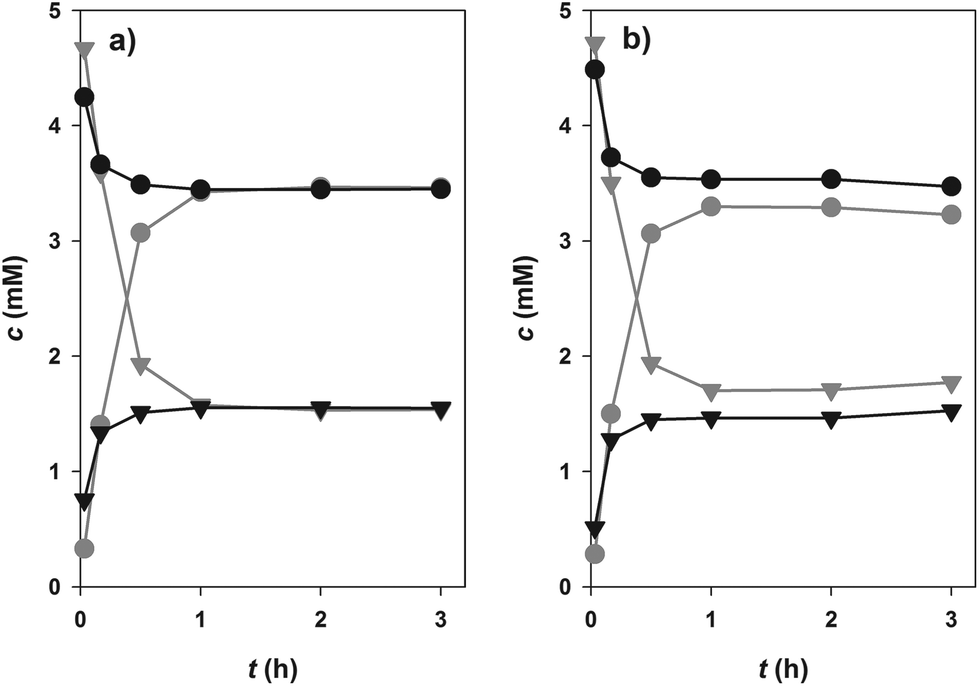 | ||
| Fig. 2 Phlorizin (3) synthesis (grey, 5 mM 1 and 10) and degradation (black, 5 mM 3 and 9) by PcOGT (pH 6.5) level out at the same equilibrium concentrations. (a) 1 (triangles), 3 (circles); (b) 10 (triangles), 9 (circles). | ||
Glucosyl transfer cascade for efficiency-enhanced glucosylation of phloretin (1)
The general principle of the glucosyl transfer cascade is shown in Scheme 1. Two parallel glucosyl transfer reactions are connected in a one-pot biotransformation via their common 10/9 substrate/product pair. Conversion of 8 and 9 is catalysed by sucrose synthase (SuSy) and yields 10 and D-fructose (11). The SuSy reaction, which represents the invariant part of the proposed cascade, is flexibly coupled to different GT-catalysed glucosylations of target acceptors under utilisation of 10 as the donor substrate. Benefit of performing synthetic glucosylations through SuSy-GT cascades as compared to single GT reactions is manifested in significant improvement of key parameters of overall transformation efficiency. First of all, cost-effective supply of 10 is accomplished using 8 as a highly expedient and inexpensive donor substrate. Only catalytic amounts of 9 as compared to stoichiometric amounts of 10 are required in the process. By using 8 in suitable excess over the acceptor substrate, thermodynamic restrictions on acceptor glucosylation are overcome effectively (see later). Equilibrium of the SuSy reaction (Keq ≥ 0.5) favours formation of 10 in a wide pH range (pH ≤ 7.5).52 Finally, problems of pronounced end-product inhibition by 9, which have severely restricted direct synthetic use of several flavonoid glycosyltransferases in the past, are brought under control.56,57 Continuous removal of 9 due to formation of 10 decreases the steady-state concentration of 9 in the reaction to a value even smaller than the one established from the low (catalytic) amount that was initially added.To optimise conditions for synthesis of 3, the coupled reaction of SuSy and PcOGT was studied at different pH values in the range 5.7–8.5. Recombinant SuSy from soybean (Glycine max; GmSuSy) purified from an E. coli expression culture was used (ESI,† Fig. S1). The applied concentrations of 1, 8 and 9 were 5.0 mM, 100 mM and 0.5 mM, respectively. Despite addition of 20% DMSO as co-solvent, poor aqueous solubility of 1 restricted use of concentrations higher than about 5 mM. The time course of 3-formation was measured at each pH (ESI,† Fig. S3). The data was used for calculation of initial production rates of 3 (r3), final yields at reaction equilibrium (24 h) and space-time yields (STY) for a target product concentration of 4.0 mM (Fig. 3). The optimum pH range for high r3 was between pH 6.2 and 7.0 (Fig. 3a). Except for pH 5.7 and 8.5 where r3 was small and reaction equilibrium was therefore not attained within the 24 h timespan of the experiment, the final concentrations of 3 were around 4.4 mM, equivalent to a yield of ∼90% based on conversion of 1 (Fig. 3b). With increasing pH a marginal improvement of equilibrium concentrations of 3 from 4.36 (pH 6.2) to 4.46 (pH 7.8) was observed. However, STY for formation of 4 mM 3 (Fig. 3a) confirmed that pH dependency of initial rates is predominantly determining efficiency of conversions. A clear optimum of STY was found around pH 6.5 with more than 50% loss at pH 6.2 and 7.1, respectively. Overall, compared to synthesis of 3 directly from 1 and 10 (each 5 mM), introduction of the SuSy-GT cascade represented a substantial yield enhancement from ∼70 to ∼90%. The effect of “thermodynamic push” from 8 on glucoside product formation is noted. It is explained from the net reaction of the glucosyl transfer cascade, which is sucrose (8) + phloretin (1) ↔ phlorizin (3) + D-fructose (11).
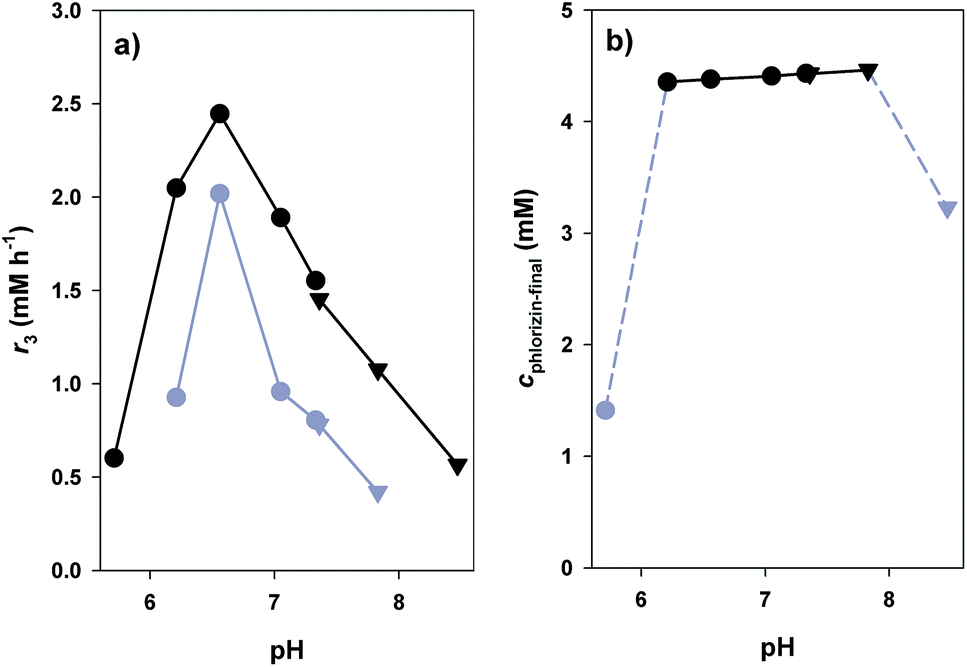 | ||
| Fig. 3 Influence of pH on efficiency of PcOGT-GmSuSy cascade conversions using BisTris (circles) and TAPS (triangles) buffers (5 mM 1, 0.5 mM 9, 100 mM 8). (a) Initial rate of formation of 3 (black), STY for formation of 4 mM 3 (grey). (b) Final equilibrium concentrations of 3 after 24 h (grey: equilibrium was not reached). | ||
To further examine the role of 8 in driving the glucosylation of 1, the cascade reaction was performed at pH 6.5 under conditions where both 8 (100 mM) and 10 (5 mM) were present as donor substrates for glucosylation of 1 (5 mM). Fig. 4 shows the full time course of the conversion along with the corresponding time courses from the coupled reaction without supplementation of 10 (but 0.5 mM 9) and the direct glucosylation in the absence of GmSuSy. Final conversions, initial rates and space time yields are summarised in Table 2. Product formation in the early reaction phase of cascade reactions benefited somewhat (∼25% enhancement of r3) from the presence of external 10, probably because the in situ produced steady-state concentration of 10 was not sufficient for PcOGT to become fully saturated with glucosyl donor substrate. Use of 5 mM 10 instead of 0.5 mM 9 in SuSy-GT cascade reactions furthermore caused moderate gain in final conversion (∼3%) and a more significant improvement of STY for production of 4.0 mM of 3 by about 35%. However, in our opinion a roughly 30% reduced reaction time to obtain a very similar product concentration is not compensating additional costs of replacing UDP with 10-times higher concentrations of the more expensive 10. Both SuSy-PcOGT cascade reactions clearly outperformed direct glycosylation of 1 by PcOGT. By addition of GmSuSy the final concentration of 3 was increased from 3.61 mM to 4.44 (0.5 mM 9) and 4.60 mM (5 mM 10), respectively.
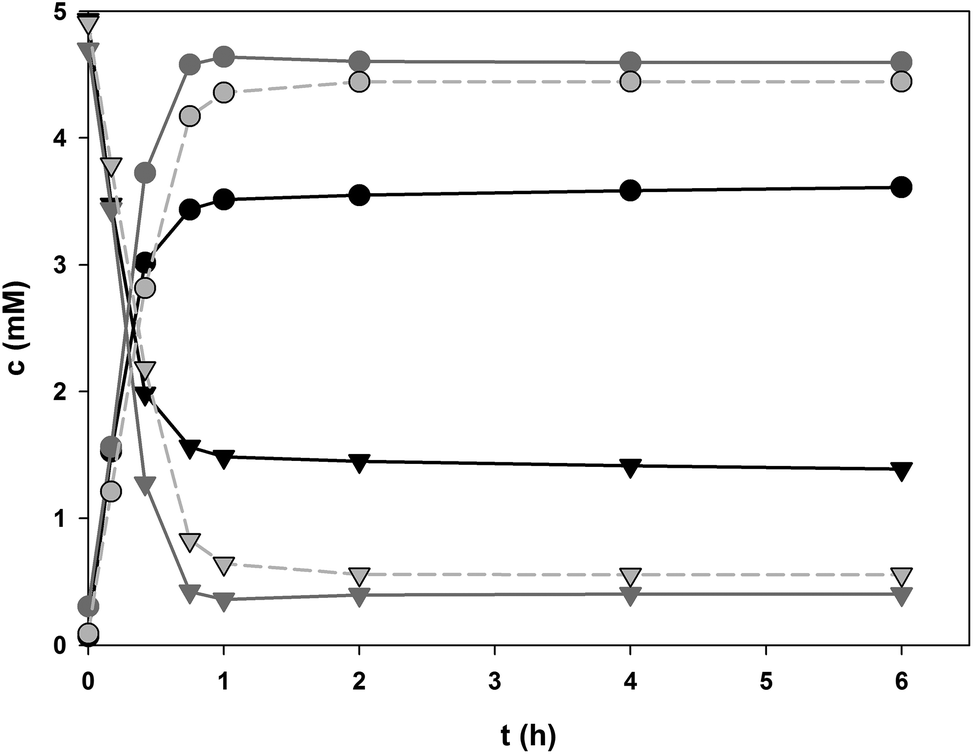 | ||
| Fig. 4 PcOGT catalysed formation of 3 (circles) from 5 mM 1 (triangles) at pH 6.5: black: without GmSuSy, 5 mM 10; dark grey: GmSuSy, 5 mM 10; light grey (dashed): GmSuSy, 0.5 mM 9. | ||
| 3 | 5 | 6 | ||
|---|---|---|---|---|
| a Data extracted from Fig. 4 and 5. b 0.1 U mL−1PcOGT (0.1 U mL−1GmSuSy), pH 6.5, 5 mM 1. c 0.1 U mL−1PcOGT, (0.1 U mL−1GmSuSy), pH 7.5, 5 mM 2. d 0.04 U mL−1OsCGT I121D (0.04 U mL−1GmSuSy), pH 7.5, 5 mM 2. e At equilibrium after 6 (3), 24 (5) or 20 (6) h of conversion. f Rate of glucoside formation during initial 25 (3) or 60 min (5, 6). g STY for formation of 4 mM glucoside (nd: less than 4 mM product). | ||||
| GT 5 mM 10 | Conversione (%) | 72.2 | 79.6 | 88.3 |
r
glucoside![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f (mM h−1) f (mM h−1) |
6.9 | 1.3 | 1.9 | |
| STYg (mM h−1) | nd | nd | 1.2 | |
| GT-GmSuSy 5 mM 10 | Conversione (%) | 91.9 | 91.4 | 95.3 |
|
r
glucosidef (mM h−1) |
8.2 | 1.9 | 2.2 | |
| STYg (mM h−1) | 7.6 | 0.8 | 1.4 | |
| GT-GmSuSy 0.5 mM 9 | Conversione (%) |
88.9 | 88.0 | 91.7 |
|
r
glucosidef (mM h−1) |
6.5 | 1.7 | 1.3 | |
| STYg (mM h−1) | 5.6 | 0.7 | 1.0 | |
Synthesis of davidoside (5) and confusoside (6) via glucosyl transfer cascade reaction
In order to compensate in some degree the 200-fold decrease in specific activity of PcOGT caused by change of acceptor substrate from 1 to 2, the pH of the cascade reaction was raised to 7.5 where PcOGT is optimally active and the specific 2 glucosylation rate was therefore enhanced around 2.5-fold as compared to pH 6.5. The Ile121 → Asp variant of OsCGT is also best active at a pH of around 7.5.53 Loss in specific activity of GmSuSy caused by the rise in pH was negligible in comparison (∼35%). The optimum pH of conversion of 8 by GmSuSy was recently determined to be about 6.0.52Relevant time courses for glucosylation of 2 by PcOGT and OsCGT variant are shown in Fig. 5 and summarised in Table 2. The substrate concentrations used were the same as before in the conversions of 1. The concentration of 2 (5.0 mM) was set according to aqueous solubility in the water-DMSO solvent used. Coupled enzyme reactions carried out in the presence and absence of added 10 are compared to the single enzyme reaction. Confusoside (6) was always obtained as single glucosyl transfer product in conversions with OsCGT I121D (Fig. 5b). However, in PcOGT catalysed conversions besides davidioside (5) a second compound, distinct from 6, accumulated during the first 4 h and diminished again upon prolonged incubation (Fig. 5a). Very low concentrations (∼0.08 mM, <2% of total products) prevented structural analysis of the by-product by NMR. However, most likely the by-product is the third potential mono O-glucoside of 2, resulting from glucosylation at the 4-OH. Its decline in favour of formation of 5 at longer incubation times is explained by reversibility of the glycosylations.55 This causes accumulation of the thermodynamically favoured product (here 5) at equilibrium conditions. Final mixture of the conversion of 2 contained the main product 5 in about 50-fold excess over the by-product, clearly demonstrating that highly regiospecific glucosylation of 2 can be achieved by PcOGT. Comparison of the different reaction conditions used revealed that the initial conversion of 2 proceeded slightly faster and the overall conversion was ∼3.5% higher in GT-GmSuSy cascade reactions that contained external 10. Benefit of coupling the synthetic glucosylation to conversion of 8 was again manifested primarily in the final product concentration, which was enhanced by 12% (5) and 7% (6), respectively in the SuSy-GT cascade reaction as compared to the single GT reference reaction. Product yields in coupled enzyme conversions of 2 were 88% or higher. The STYs of glucosylations of 2 were 5.5 (OsCGT variant) and 8 times (PcOGT) lower than those for glucosylation of 1.
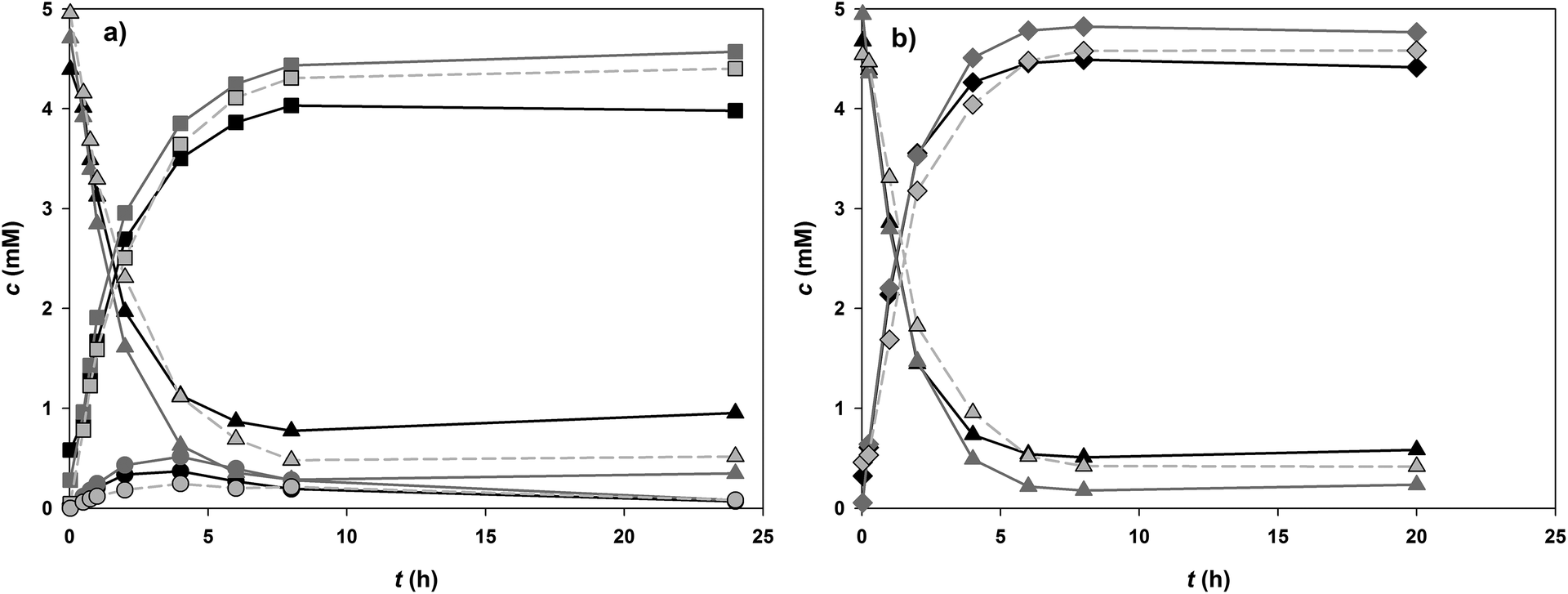 | ||
| Fig. 5 Glucosylation of 5 mM 2 (triangles) at pH 7.5: black: without GmSuSy, 5 mM 10; dark grey: GmSuSy, 5 mM 10; light grey (dashed): GmSuSy, 0.5 mM 9; (a) PcOGT forms 5 (squares) and an unknown compound (circles). (b) OsCGT I121D only produces 6 (diamonds). | ||
Product isolation and NMR spectroscopic characterization
The two products synthesised by glucosylation of 5 mM 2 were isolated from 5 mL reaction mixtures using preparative reversed phase HPLC. 5 and 6 were obtained as white powder after freeze-drying and recovered in ≥80% yield. Identities of 5 and 6 were unambiguously assigned from results of a detailed NMR spectroscopic characterization that involved besides 1H and 13C NMR, HMQC (only 5), COSY and HMBC. Results are summarized in Table S1 and Scheme S1 (ESI†). Although separation was achieved, concentrations of the by-product of 2 glucosylation by PcOGT were too low for NMR analysis. Davidioside (5) and confusoside (6) were recovered from glucosylation of 2 by PcOGT and OsCGT variant as single compounds at purities >98% based on HPLC peak areas (ESI,† Fig. S4).Conclusions
Glucosyltransferases catalysing highly regiospecific β-D-glucosyl-transfer from UDP-glucose (10) to the 2′-OH of the dihydrochalcones 1 and 2, and to the 4′-OH of 2 were identified. So far no glycosyltransferase for selective synthesis of trilobatin (4) by glucosylation of the 4′-OH of 1 was found, but it presumably exists in Malus trilobata which only forms 4 but not 3.30 Maximum glucoside product concentrations were mainly restricted by aqueous solubility of the comparably hydrophobic dihydrochalcone acceptors. Optimised use of co-solvent,58 running the reaction in an aqueous-organic two-phase system59 and feeding the acceptor substrate in accordance to reaction progress52 are all viable strategies to enhance the effective acceptor concentration in the reaction. However, detailed reaction engineering for targeted “de-bottlenecking” of an enzymatic production was beyond the scope of this study. Alternative isolation of single flavonoids from complex mixtures of phenolic compounds in plant materials involves time consuming multi-step procedures and large quantities of organic solvents. With the notable exception of highly abundant 3, no yields above 5 mg g−1 dry weight were reported for extraction of 4–6 from plant leaves.30–32,36 Due to simplification of work-up typical yields of the herein described GT-SuSy cascade reactions of around 2 g L−1 (∼4.5 mM) seem sufficient to compete with extraction from natural sources. Furthermore, natural availability is no longer a bottleneck on supply of aglycons 1 and 2 because they are readily obtained by high yielding single step Friedel–Crafts acylation.60 A two-enzyme one-pot glucosyl transfer cascade for convenient synthesis of the corresponding dihydrochalcone β-D-glucosides (3, 5, 6) was developed. Coupling of the synthetic enzymatic glucosyl transfer to conversion of 8 and 9 catalysed by SuSy presents a generally applicable strategy to provide UDP-glucose (10) donor substrate in a cost-effective manner and to cope with issues of unfavourable thermodynamics and UDP (9) product inhibition. It can be applied as a universal approach for green enzymatic synthesis of flavonoid glucosides.Experimental
Materials
Unless otherwise indicated, all chemicals were from Sigma-Aldrich (Vienna, Austria) in the highest purity available. Phloretin (98%) was from AK Scientific (Union City, CA, US) and phlorizin dihydrate (≥98%), from Carl Roth (Karlsruhe, Germany). Strep-Tactin® Sepharose® and desthiobiotin were from IBA (Goettingen, Germany). BCA Protein Assay Kit was from Thermo Scientific (Waltham, MA, US).Synthesis of davidigenin (2)
Synthesis of 2 was done in a single step from 1,3-benzenediol (resorcinol) and 3-(4-hydroxyphenyl)propanoic acid (phloretic acid) using an adopted Friedel–Crafts acylation method.60 Phloretic acid (6 mmol, 990 mg) and resorcinol (6 mmol, 661 mg) were stirred in 4 mL BF3Et2O (32 mmol) at 90 °C for 120 min under argon. Complete conversion of resorcinol was verified by TLC. The mixture was poured into 200 mL 10% aqueous NaOAc and stirred for 2 h at room temperature. The solution was 3 times extracted with 200 mL EtOAc. Combined EtOAc was washed with 40 mL water and 40 mL brine before drying over MgSO4. The solvent was evaporated under reduced pressure and the residue was chromatographed over silica gel column using cyclohexane–EtOAc mixtures. 2 (61% isolated yield) was confirmed by 1H- and 13C-NMR spectroscopy (Bruker AVANCE III 300 spectrometer).Enzyme production
Escherichia coli BL21-Gold (DE3) expression strains for PcOGT (UGT88F2; GenBank: FJ854496),53OsCGT (GenBank: FM179712) Ile121 to Asp mutant53 and GmSuSy (GenBank: AF030231)52 were described elsewhere in detail. Enzyme expression in lysogeny broth (LB)-medium as N-terminal Strep-tag II fusion proteins and enzyme purification by affinity chromatography on Strep-Tactin® Sepharose® columns was also reported elsewhere.52 Expected molecular mass and purity of enzymes were affirmed by SDS polyacrylamide gel electrophoresis (PAGE). Aliquots of final preparations were stored at −70 °C and thawed only once prior to their use.Activity assays
Sucrose cleavage by GmSuSy was measured spectrophotometrically using a discontinuous two-step enzymatic assay. Oxidation of 10, formed from 8 (100 mM) and 9 (0.5 mM), to UDP-α-D-glucuronic acid is coupled to reduction of two NAD+ molecules to NADH by human UDP-α-D-glucose 6-dehydrogenase (hUGDH) as described elsewhere in detail.52 In short, reactions were started by GmSuSy addition and samples of 150 μL were stopped by heating (95 °C, 5 min). Precipitated protein was removed by centrifugation (13200 rpm, 20 min) and 100 μL of the supernatant were mixed with 400 μL of measuring solution (2.5 mM NAD+, 0.05% TritonTM X-100 in 100 mM HEPES, pH 8.0) in a Half Micro Cuvette. Absorbance at 340 nm was measured before and after incubation with 1.5 mU of hUGDH and concentration of 10 was calculated from the increase of absorbance (εNADH = 6220 M−1 cm−1). Typically 4 distinct measurements were used to calculate linear initial rates. One unit of GmSuSy was defined as the amount of enzyme producing 1 μmol 10 (2 μmol NADH) per minute under following conditions: 30 °C, pH of cascade reactions (6.5 or 7.5) 0.5 mM 9, 100 mM 8 in 50 mM BisTris, 13 mM MgCl2, 50 mM KCl, 0.13% BSA and 20% DMSO.
An reversed phase HPLC-based assay for quantification of dihydrochalcones (1, 2) and their glycosides (3–6) was used to determine activities of PcOGT and the OsCGT variant.53 In short, conversions of 5 mM dihydrochalcone with 0.5 mM 10 were started by GT addition and conducted under agitation (400 rpm) and temperature control (30 °C) using a thermomixer. Typically 4 aliquots of 100 μL were taken within 60 min to determine linear initial rates. Reactions were stopped by mixing with 100 μL acetonitrile. Precipitated protein was removed by centrifugation (13200 rpm, 20 min) before applying 5–10 μL of supernatant to HPLC analysis. An Agilent 1200 HPLC equipped with a Chromolith® Performance RP-18e column (100 × 4.6 mm) was used at 35 °C, and UV-detection at 288 nm was applied. A water (A) to acetonitrile (B) gradient (0.1% trifluoroacetic acid each) was applied for elution: 20–47.5% B (7.5 min, 1 mL min−1), 47.5–100% B (0.05 min, 1 mL min−1), 100% B (1.45 min, 1.5 mL min−1), 100–20% B (0.05 min, 1.5 mL min−1), 20% B (2.45 min, 1.5 mL min−1). One unit of GT activity was defined as the amount of enzyme glucosylating 1 μmol 1 per min under following conditions: 30 °C, 5 mM 1, 0.5 mM 10 in 50 mM BisTris, pH 6.5 (PcOGT) or 7.5 (OsCGT I121D) containing 13 mM MgCl2, 50 mM KCl, 0.13% BSA and 20% DMSO.
Equilibrium constant (Keq) of glucosylation of 1 by PcOGT
K eq of PcOGT was determined by running glucosylation of 1 in forward (5 mM 1, 5 mM 10) and reverse direction (5 mM 3, 5 mM 9) until no further conversion was observed. Reactions were run at 30 °C and started by addition of 100 mU mL−1PcOGT (50 mM BisTris, pH 6.5, 13 mM MgCl2, 50 mM KCl, 0.13% BSA and 20% DMSO). Concentrations of 1 and 3 were determined by the HPLC based GT activity assay.The same samples (10 μL) were applied for quantification of 9 and 10 using an anion exchange HPLC protocol. An Agilent 1200 HPLC system was used for separation on an Agilent ZORBAX SAX (4.6 × 250 mm) column at 30 °C. 9 and 10 were monitored by UV detection at 254 nm. Using 20 and 500 mM potassium phosphate buffer (pH 6.8) as solvent A and B, respectively following gradient was applied at a constant flow rate of 1.5 mL min−1: 0–100% B (7 min), 100% B (2 min), 100–0% B (0.05 min), 0% B (3.95 min).
Formation of 3, 5 and 6 by direct GT and GT-GmSuSy cascade reactions
5 mM 1 and 2 were converted in 50 mM BisTris at pH 6.5 and 7.5, respectively. All reactions contained 13 mM MgCl2, 50 mM KCl, 0.13% BSA and 20% DMSO. As glucose source conversions without GmSuSy contained 5 mM 10 while for those with GmSuSy either 5 mM 10 and 100 mM 8 or 0.5 mM 9 and 100 mM 8 were used. Conversions were started by addition of 100 mU mL−1PcOGT (and 100 mU mL−1GmSuSy) or 40 mU mL−1OsCGT I121D (and 40 mU mL−1GmSuSy). Samples were incubated at 30 °C and the GT HPLC protocol was applied for sampling after distinct incubation times and quantification of 1, 2, 3, 5 and 6.Coupled glucosylation of 5 mM 1 by PcOGT and GmSuSy (100 mU mL−1 each) in presence of 0.5 mM 9 and 100 mM 8 was studied at various pH as described for standard conditions. Buffers were prepared in steps of 0.5 pH units (BisTris pH 5.5–7.5, TAPS pH 7.5–8.5) and added to a final concentration of 50 mM. Actual pH in conversions was calculated as the average of measurements in beginning and at the end of conversions.
Isolation and identification of 5 and 6
5 and 6 from glucosylation of 2 by PcOGT and OsCGT I121D mutant were purified by preparative reversed phase C-18 HPLC on an Agilent 1200 system equipped with a SphereClone 5 μm ODS(2) (250 × 10.0 mm) column. Water was used as solvent A and acetonitrile as solvent B (0.1% formic acid each). Separation of 5 and 6 from other compounds was achieved by step gradients from 10% to 100% B at room temperature. 5 eluted at 25% and 6 at 35% B. After removing acetonitrile under reduced pressure water was removed by freeze drying. Purity and identity were confirmed by HPLC and NMR. 1H-NMR, 13C-NMR, COSY, HMBC and HMQC (5 only) were recorded on a Varian Unity Inova 500 MHz spectrometer.Acknowledgements
Financial support from the Austrian Science Fund (FWF, DK Molecular Enzymology W901-B05) and the EU FP7 project SuSy (Sucrose Synthase as Effective Mediator of Glycosylation) is gratefully acknowledged. L. B. is supported from a scholarship of Ramkhamhaeng University (Bangkok, Thailand).Notes and references
- Ø. M. Andersen and K. R. Markham, Flavonoids: chemistry, biochemistry, and applications, CRC Press, Boca Raton, FL, 2006 Search PubMed.
- E. Grotewold, The science of flavonoids, Springer, New York, 2006 Search PubMed.
- S. Barnes, J. Prasain, T. D'Alessandro, A. Arabshahi, N. Botting, M. A. Lila, G. Jackson, E. M. Janle and C. M. Weaver, Food Funct., 2011, 2, 235–244 CAS.
- C. Manach, G. Williamson, C. Morand, A. Scalbert and C. Rémésy, Am. J. Clin. Nutr., 2005, 81, 230S–242S CAS.
- A. K. Ratty and N. P. Das, Biochem. Med. Metab. Biol., 1988, 39, 69–79 CrossRef CAS.
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Free Radicals Biol. Med., 1996, 20, 933–956 CrossRef CAS.
- M. G. L. Hertog, E. J. M. Feskens, P. C. H. Hollman, M. B. Katan and D. Kromhout, Lancet, 1993, 342, 1007–1011 CrossRef CAS.
- N. C. Veitch and R. J. Grayer, in Flavonoids: chemistry, biochemistry, and applications, ed. Ø. M. Andersen and K. R. Markham, CRC Press, Boca Raton, FL, 2006, ch. 16, pp. 1003–1100 Search PubMed.
- A. Picinelli, E. Dapena and J. J. Mangas, J. Agric. Food Chem., 1995, 43, 2273–2278 CrossRef CAS.
- A. Escarpa and M. C. Gonzalez, J. Chromatogr. A, 1998, 823, 331–337 CrossRef CAS.
- Y. Nakamura, S. Watanabe, N. Miyake, H. Kohno and T. Osawa, J. Agric. Food Chem., 2003, 51, 3309–3312 CrossRef CAS PubMed.
- B. M. Rezk, G. R. M. M. Haenen, W. J. F. van der Vijgh and A. Bast, Biochem. Biophys. Res. Commun., 2002, 295, 9–13 CrossRef CAS.
- V. Stangl, M. Lorenz, A. Ludwig, N. Grimbo, C. Guether, W. Sanad, S. Ziemer, P. Martus, G. Baumann and K. Stangl, J. Nutr., 2005, 135, 172–178 CAS.
- R. B. An, E. J. Park, G. S. Jeong, D. H. Sohn and Y. C. Kim, Arch. Pharmacal. Res., 2007, 30, 674–677 CrossRef CAS.
- C. Pohl, F. Will, H. Dietrich and D. Schrenk, J. Agric. Food Chem., 2006, 54, 10262–10268 CrossRef CAS PubMed.
- H. Nguyen, S. Zhang and M. E. Morris, J. Biopharm. Sci., 2003, 92, 250–257 CrossRef CAS PubMed.
- O. Desire, C. Rivière, R. Razafindrazaka, L. Goossens, S. Moreau, J. Guillon, S. Uverg-Ratsimamanga, P. Andriamadio, N. Moore, A. Randriantsoa and A. Raharisololalao, J. Ethnopharmacol., 2010, 130, 320–328 CrossRef CAS PubMed.
- T. Mencherini, P. Picerno, P. Del Gaudio, M. Festa, A. Capasso and R. Aquino, J. Nat. Prod., 2010, 73, 247–251 CrossRef CAS PubMed.
- S. Logendra, D. M. Ribnicky, H. Yang, A. Poulev, J. Ma, E. J. Kennelly and I. Raskin, Phytochemistry, 2006, 67, 1539–1546 CrossRef CAS PubMed.
- R. W. Gantt, P. Peltier-Pain and J. S. Thorson, Nat. Prod. Rep., 2011, 28, 1811–1853 RSC.
- C. J. Thibodeaux, C. E. Melançon III and H. W. Liu, Angew. Chem., Int. Ed., 2008, 47, 9814–9859 CrossRef CAS PubMed.
- Y. Kaminaga, A. Nagatsu, T. Akiyama, N. Sugimoto, T. Yamazaki, T. Maitani and H. Mizukami, FEBS Lett., 2003, 555, 311–316 CrossRef CAS.
- X. Wang, FEBS Lett., 2009, 583, 3303–3309 CrossRef CAS PubMed.
- C. M. M. Gachon, M. Langlois-Meurinne and P. Saindrenan, Trends Plant Sci., 2005, 10, 542–549 CrossRef CAS PubMed.
- V. Křen and L. Martínková, Curr. Med. Chem., 2001, 8, 1303–1328 CrossRef.
- D. Bowles, J. Isayenkova, E. K. Lim and B. Poppenberger, Curr. Opin. Plant Biol., 2005, 8, 254–263 CrossRef CAS PubMed.
- M. Montefiori, R. V. Espley, D. Stevenson, J. Cooney, P. M. Datson, A. Saiz, R. G. Atkinson, R. P. Hellens and A. C. Allan, Plant J., 2011, 65, 106–118 CrossRef CAS PubMed.
- P. W. Snijman, E. Joubert, D. Ferreira, X. C. Li, Y. Ding, I. R. Green and W. C. A. Gelderblom, J. Agric. Food Chem., 2009, 57, 6678–6684 CrossRef CAS PubMed.
- J. R. L. Ehrenkranz, N. G. Lewis, C. R. Kahn and J. Roth, Diabetes Metab. Res. Rev., 2005, 21, 31–38 CrossRef CAS PubMed.
- C. Gosch, H. Halbwirth and K. Stich, Phytochemistry, 2010, 71, 838–843 CrossRef CAS PubMed.
- S. R. Jensen, B. J. Nielsen and V. Norn, Phytochemistry, 1977, 16, 2036–2038 CrossRef CAS.
- T. Tanaka, K. Kawamura, H. Kohda, K. Yamasaki and O. Tanaka, Chem. Pharm. Bull., 1982, 30, 2421–2423 CrossRef CAS.
- G. D. Winget, S. Izawa and N. E. Good, Biochemistry, 1969, 8, 2067–2074 CrossRef CAS.
- M. Gaucher, T. Dugé de Bernonville, D. Lohou, S. Guyot, T. Guillemette, M. N. Brisset and J. F. Dat, Phytochemistry, 2013, 90, 78–89 CrossRef CAS PubMed.
- H. Q. Dong, M. Li, F. Zhu, F. L. Liu and J. B. Huang, Food Chem., 2012, 130, 261–266 CrossRef CAS PubMed.
- T. Dugé de Bernonville, S. Guyot, J. P. Paulin, M. Gaucher, L. Loufrani, D. Henrion, S. Derbré, D. Guilet, P. Richomme, J. F. Dat and M. N. Brisset, Phytochemistry, 2010, 71, 443–452 CrossRef PubMed.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- K. Shimoda, H. Hamada and H. Hamada, Phytochemistry, 2008, 69, 1135–1140 CrossRef CAS PubMed.
- K. Shimoda, N. Kubota, K. Taniuchi, D. Sato, N. Nakajima, H. Hamada and H. Hamada, Phytochemistry, 2010, 71, 201–205 CrossRef CAS PubMed.
- B. M. de Roode, M. C. Franssen, A. van der Padt and R. M. Boom, Biotechnol. Prog., 2003, 19, 1391–1402 CrossRef PubMed.
- T. Desmet, W. Soetaert, P. Bojarová, V. Křen, L. Dijkhuizen, V. Eastwick-Field and A. Schiller, Chem. – Eur. J., 2012, 18, 10786–10801 CrossRef CAS PubMed.
- M. Plaza, T. Pozzo, J. Liu, K. Z. Gulshan Ara, C. Turner and E. Nordberg Karlsson, J. Agric. Food Chem., 2014, 62, 3321–3333 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- R. W. Gantt, R. D. Goff, G. J. Williams and J. S. Thorson, Angew. Chem., Int. Ed., 2008, 47, 8889–8892 CrossRef CAS PubMed.
- R. P. Pandey, R. B. Gurung, P. Parajuli, N. Koirala, L. T. Tuoi and J. K. Sohng, Carbohydr. Res., 2014, 393, 26–31 CrossRef CAS PubMed.
- M. Zhou, A. Hamza, C. G. Zhan and J. S. Thorson, J. Nat. Prod., 2013, 76, 279–286 CrossRef CAS PubMed.
- M. Weis, E. K. Lim, N. Bruce and D. Bowles, Angew. Chem., Int. Ed., 2006, 45, 3534–3538 CrossRef CAS PubMed.
- H. Jugdé, D. Nguy, I. Moller, J. M. Cooney and R. G. Atkinson, FEBS J., 2008, 275, 3804–3814 CrossRef PubMed.
- C. Gosch, H. Halbwirth, B. Schneider, D. Hölscher and K. Stich, Plant Sci., 2010, 178, 299–306 CrossRef CAS PubMed.
- S. R. Werner and J. A. Morgan, J. Biotechnol., 2009, 142, 233–241 CrossRef CAS PubMed.
- R. P. Pandey, T. F. Li, E. H. Kim, T. Yamaguchi, Y. I. Park, J. S. Kim and J. K. Sohng, Appl. Environ. Microbiol., 2013, 79, 3516–3521 CrossRef CAS PubMed.
- L. Bungaruang, A. Gutmann and B. Nidetzky, Adv. Synth. Catal., 2013, 355, 2757–2763 CrossRef CAS PubMed.
- A. Gutmann and B. Nidetzky, Angew. Chem., Int. Ed., 2012, 51, 12879–12883 CrossRef CAS PubMed.
- M. Brazier-Hicks, K. M. Evans, M. C. Gershater, H. Puschmann, P. G. Steel and R. Edwards, J. Biol. Chem., 2009, 284, 17926–17934 CrossRef CAS PubMed.
- A. Gutmann, C. Krump, L. Bungaruang and B. Nidetzky, Chem. Commun., 2014, 50, 5465–5468 RSC.
- K. Terasaka, Y. Mizutani, A. Nagatsu and H. Mizukami, FEBS Lett., 2012, 586, 4344–4350 CrossRef CAS PubMed.
- D. K. Owens and C. A. McIntosh, Phytochemistry, 2009, 70, 1382–1391 CrossRef CAS PubMed.
- K. De Winter, K. Verlinden, V. Křen, L. Weignerová, W. Soetaert and T. Desmet, Green Chem., 2013, 15, 1949–1955 RSC.
- K. De Winter, T. Desmet, T. Devlamynck, L. Van Renterghem, T. Verhaeghe, H. Pelantová, V. Křen and W. Soetaert, Org. Process Res. Dev., 2014, 18, 781–787 CrossRef CAS.
- V. Siddaiah, C. V. Rao, S. Venkateswarlu and G. V. Subbaraju, Tetrahedron, 2006, 62, 841–846 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Protein gel; HPLC traces: distinction of 2 glucosides, purified 5 and 6; PcOGT-GmSuSy conversions at various pH; NMR data. See DOI: 10.1039/C4GC00960F |
| This journal is © The Royal Society of Chemistry 2014 |