 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Product quality and catalyst deactivation in a four day catalytic fast pyrolysis production run†
Ville
Paasikallio
*a,
Christian
Lindfors
a,
Eeva
Kuoppala
a,
Yrjö
Solantausta
a,
Anja
Oasmaa
a,
Jani
Lehto
a and
Juha
Lehtonen
b
aVTT Technical Research Centre of Finland, P.O. Box 1000, 02044 VTT, Finland. E-mail: ville.paasikallio@vtt.fi
bDepartment of Biotechnology and Chemical Technology, School of Chemical Technology, Aalto University, P.O. Box 16100, 00076 Aalto, Finland
First published on 3rd June 2014
Abstract
Catalytic fast pyrolysis of pine sawdust was successfully carried out in VTT's 20 kg h−1 Process Development Unit using a spray dried HZSM-5 catalyst. Approximately 250 kg of partially deoxygenated pyrolysis oil was produced over a period of four days. The catalytically produced pyrolysis oil had an average moisture content of 8.3 wt%, and average carbon and oxygen contents of 72.0 and 21.5 wt% on a dry basis, respectively. Approximately 24% of the original biomass carbon was present in the pyrolysis oil, whereas 14% of carbon was in the form of aqueous side products, which totaled approximately 600 kg. The pyrolysis oil contained a high amount of lignin derived water-insoluble material, as well as 6.4 wt% of aromatic hydrocarbons. The majority of the carbohydrate derived products, i.e. acids, aldehydes, ketones and sugar-type compounds, were found in the aqueous product fraction. While the quality of pyrolysis oil remained quite stable during the four day experiment, distinct changes were observed in the properties and the behavior of the catalyst. Coke formation was heaviest at the beginning of the experiment, and then subsided over time. Catalyst micropore area and volume also decreased during the experiment. This transformation was accompanied by apparent changes in the crystallinity and the structure of the catalyst. Scanning electron microscope images of the catalyst also revealed clear physical damage to the particles. Biomass alkali metals also deposited on the catalyst, and the spent catalyst contained a total of 1.1 wt% of Ca, K, Mg and P after the experiment. A linear correlation was observed between catalyst alkali metal content and acidity, which indicated that biomass alkalis substituted the proton functionalities of the HZSM-5 acid sites.
1. Introduction
In the recent years, catalytic fast pyrolysis (CFP) of biomass has received a significant amount of attention as a way to produce partially deoxygenated bio-oils.1–7 This is achieved by replacing the inert heat transfer material that is typically used in fast pyrolysis with a solid catalyst. Zeolites such as ZSM-5, β-zeolite, Y-zeolite and mordenite are among the most commonly used catalytic materials for catalytic pyrolysis.6,8,9 In catalytic pyrolysis, the organic vapours released in the primary thermal decomposition reactions come into immediate contact with the catalyst. Oxygen is rejected from the pyrolysis vapours in the form of H2O, CO and CO2 as dehydration, decarbonylation and decarboxylation reactions take place over the catalyst. The intermediate reaction products formed in this stage can then react further on the acid sites of the catalyst to form higher value products such as aromatic hydrocarbons.6,10 As the oxygen content of bio-oil decreases, its chemical composition shifts more towards petroleum-derived hydrocarbon fuels. This change can also be seen in the physicochemical properties of the bio-oil, as the use of catalysts enhances the formation of certain types of compound groups while others are diminished.2,11 Decrease in the concentration of organic acids can result in an increased pH and lower total acid number (TAN).4,12 Conversion of carbonylic compounds,3,7 which are a known precursor13 for the chemical instability of bio-oil, serves to increase the storage stability of bio-oil. Cracking of heavier lignin-derived oligomers into smaller phenolic monomers decreases the average molar mass of bio-oil, and consequently its viscosity. These positive changes in the bio-oil physicochemical characteristics can potentially pave the way for its use in new applications, such as co-processing in existing oil refinery processes.14,15So far, the scale of catalytic pyrolysis experiments presented in open literature has ranged from analytical pyrolysis with a few grams of biomass/catalyst8,16 to continuous fluidized-bed units with biomass processing capabilities of up to approximately 2 kg h−1.4,12 Apart from the results presented by Lappas et al.1,17 the fluidized-bed reactor units employed in catalytic pyrolysis have been of the bubbling fluidized-bed (BFB) variety.2–5,9,11,12 In a BFB reactor, the catalyst is in continuous contact with the biomass feedstock, and thus deactivates over time due to coke formation. If a high activity is to be maintained for the catalyst, it is necessary to periodically regenerate the catalyst. The rate of catalyst deactivation depends on the catalyst that is used. This factor also sets the requirements for the regeneration interval of the catalyst. Mullen et al.4 observed significant differences in product yields and quality when experimenting with five and ten minute regeneration intervals for a β-zeolite related catalyst. In our previous work,18 we identified that catalyst acidity significantly influenced coke formation over a time period of 30 minutes. Coke formation was heavier on more acidic catalysts, and thus they showed a tendency to deactivate more rapidly. A more acidic catalyst would, therefore, require regeneration on a more frequent basis. Because catalyst deactivation and regeneration is such a critical factor, any industrial-scale catalytic fast pyrolysis process would have to be equipped with online catalyst regeneration capabilities. In a conventional BFB pyrolysis reactor, this will require an additional means of removing deactivated catalyst, while simultaneously supplying regenerated catalyst into the reactor.
In contrast to the somewhat problematic nature of the BFB reactor, a circulating fluidized-bed (CFB) reactor offers more flexible operation especially when it comes to the continuous regeneration of the catalyst. The impact of reversible catalyst deactivation becomes less critical, as the catalyst is in contact with the biomass for merely a few seconds at a time. The catalyst is subsequently transported to the regenerator section, where the coke deposits are combusted along with the char and a part of the non-condensable gases. This provides heat for the pyrolysis process itself while simultaneously regenerating the catalyst. The continuous regeneration of the catalyst removes the need to consider how the activity of the catalyst changes over a time span of minutes, which would be a major concern in a BFB process along with the coking phenomenon associated with catalytic fast pyrolysis. When considering the long term stability of the catalyst, there are various factors that need to be taken into account as the catalyst is subjected to both physical and chemical strains. Operation in a CFB reactor can cause catalyst attrition, and thus high mechanical integrity is a very desired property for the catalyst.19 Repeated exposure to both pyrolysis conditions and the oxidative atmosphere of the regenerator can also cause changes in catalyst structure. Brønsted acid sites, which are responsible for the strong acidity of zeolites in their proton form, can be lost due to dealumination of the zeolite framework.20,21 Another potential source for catalyst deactivation are the metals that are found in biomass. While so far only a limited number of studies have discussed this phenomenon,5,22,23 it has been shown that metals present in the biomass feedstock deposit on the catalyst during catalytic pyrolysis. Mullen and Boateng23 reported a correlation between the presence of biomass metals on the HZSM-5 catalyst and its efficiency for producing deoxygenated pyrolysis oil containing aromatic hydrocarbons. Studies in the field of selective catalytic reduction of nitrous oxides have shown that alkali metals, and especially potassium, can decrease the number of Brønsted acid sites.24,25 While this effect is more pronounced for V2O5–WO3–TiO2 catalysts, it has also been observed when using Fe and Cu substituted ZSM-5 catalysts.26,27 One point of interest is, therefore, to see what kind of effect long-term exposure to biomass alkali metals will have on catalyst activity in catalytic pyrolysis of biomass.
Lappas et al. were the first to publish results from catalytic pyrolysis of biomass in a CFB reactor.1 They used two different catalysts in their experiments: a commercial equilibrium FCC catalyst and a ZSM-5 based FCC additive. At that time, the main difference that was detected in the chemical composition of pyrolysis oil was a clear decrease in the concentration of unidentifiable heavy oxygenates. Similar observations were made using both catalysts. High moisture content (37–67 wt%) affected the physical characterization of the pyrolysis oil from the catalytic experiments, but the pyrolysis oils in question showed e.g. better stability and lower micro carbon residue values compared to the non-catalytically produced reference sample. While a CFB reactor could be considered as the conventional choice for a continuous catalytic pyrolysis process with online catalyst regeneration, other reactor types have also been adopted for research purposes. Yildiz et al.28 recently published results where they employed an auger-type reactor for catalytic pyrolysis of pine wood. Fresh ZSM-5 catalyst was continuously fed into the auger reactor along with the biomass, and thus the biomass was always pyrolysed in the presence of a fully active catalyst. As a result, the yield of organic liquids decreased from approximately 35 to 15 wt%. Analysis of the bio-oil organic fraction showed significant decreases in the concentrations of e.g. levoglucosan, acetic acid, hydroxyacetaldehyde, furfural and 1-hydroxy-2-propanone. On the other hand, the concentrations of phenolic monomers, single-ring aromatics and naphthalenes and indenes clearly increased with the use of the catalyst. Jae et al.29 also utilized a similar approach where a ZSM-5 catalyst was continuously added and removed from a BFB reactor in CFP on pine wood. The authors reported a constant yield of aromatic hydrocarbons over a time course of 6 hours. While no online catalyst regeneration was employed in this study, subjecting the catalyst to 30 reaction–regeneration cycles caused the yield of aromatics to decrease from 14.2 to 12.2% (carbon yield). One possible reason for this was a decrease in the Brønsted acidity of the catalyst. Because no changes were observed in the crystallinity of the catalyst, the authors stated mineral impurities in the biomass feedstock could have poisoned the acid sites of the catalyst. Another recent development in the field of pyrolysis reactors was published by Zhang et al.30 The internally interconnected fluidized bed (IIFB) reactor offers the possibility the operate a catalytic pyrolysis process which incorporates continuous catalyst regeneration together with char combustion. Pyrolysing rice husks in this reactor over a HZSM-5 catalyst resulted in a petrochemical (aromatics + olefins) yield of up to 20% on a carbon basis. The duration of the experiment was 3 hours. This, along with the fact that little change was observed in the product distribution as a function of time, indicates that the catalyst is able to retain its activity and good petrochemical yields due to the continuous regeneration process. In contrast to this, the authors stated that clear catalyst deactivation could be seen in just 30 minutes when continuous regeneration of the catalyst was not carried out.
While a few examples of using fresh or continuously regenerated catalyst for catalytic pyrolysis exist in open literature, the duration of these experiments has been limited to a few hours. Thorough physicochemical characterization of pyrolysis oil produced in such a fashion has not yet been carried out either. In this study, catalytic pyrolysis of pine sawdust was carried out in a 20 kg h−1 circulating fluidized-bed reactor with continuous catalyst regeneration. The product yields and bio-oil properties were compared to results obtained in a non-catalytic experiment conducted with the same feedstock and reactor system. Extensive physical and chemical characterisation of the liquid products was carried out in order to better understand how the presence of a catalyst in a CFB reactor affects the physicochemical properties of pyrolysis oil. In addition to analysing the liquid products, characterization of catalyst samples which were taken before, during and after the experiment was conducted. This was done in order to determine how certain catalyst properties change when the catalyst was subjected to both pyrolysis and regeneration conditions over an extended period of time. To the best of our knowledge, this is the first time when results from biomass catalytic fast pyrolysis with continuous catalyst regeneration are publicly reported from a circulating fluidized bed unit of this size and capacity, and when the overall duration of the experiment reached almost one hundred hours.
2. Experimental
2.1. Biomass and catalyst
Pine sawdust was used as the feedstock in the catalytic pyrolysis experiment. The moisture content of the sawdust was 12.0 wt%. The dry biomass contained 0.4 wt% ash, and 51.4 wt%, 5.9 wt% and less than 0.1 wt% of carbon, hydrogen and nitrogen, respectively. Oxygen content, as determined by difference, was 42.2 wt%. Volatiles content was 83.9 wt% and the higher heating value (HHV) 20.4 MJ kg−1. Besides the moisture content, all the aforementioned values are given on a dry basis. Approximately 95 wt% of the sawdust feedstock had a particle size of under 3.15 mm.A spray dried ZSM-5 catalyst powder (CBV 5524G) was obtained from Zeolyst International. The catalyst was supplied in its ammonium form. Calcination of the catalyst, i.e. conversion into its proton form, was carried out in the CFB reactor system during the heat-up phase of the reactor. The SiO2/Al2O3 ratio of the catalyst was 50 and the binder content 50%. Average particle size was 74 μm with 24 vol% of particles being smaller than 40 μm and 3 vol% larger than 150 μm The bulk density of the catalyst was 0.78 g cm−3. The specific surface area, as reported by the catalyst provider, was 235 m2 g−1. The catalyst was chosen based on data from preliminary bench-scale experiments.
2.2. Reactor system
The pyrolysis experiments were carried out using VTT's 20 kg h−1 Process Development Unit (PDU) which employs a circulating fluidized-bed reactor. The system has been previously described in e.g.ref. 31. The feedstock is fed into the reactor using a screw feeder. The initial amount of catalyst in the system was 120 liters, which corresponded to approximately 94 kilograms. The catalyst to biomass ratio, i.e. the ratio of the hourly catalyst circulation rate and biomass feeding rate, was approximately 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 on a weight basis. The pyrolysis temperature was 520 °C and the fluidization velocity in the reactor was approximately 4 m s−1. Upon exiting the reactor, the product vapour/gas stream passes through two cyclones where the catalyst particles and char are separated. The pyrolysis vapours are condensed in two liquid scrubbers where product pyrolysis oil is used for quenching the pyrolysis vapours. The scrubbers were operated at a temperature of 35 °C. The scrubbers are followed by a secondary condensation system where a highly aqueous liquid product, which will be referred to as condensate, was collected. After the liquid products have condensed, a certain part of the non-condensable gases is recycled to the reactor to be used as the fluidization gas, whereas any excess gas is combusted in the regenerator. Char and catalyst coke deposits are also combusted in the regenerator. The regenerator was operated at a temperature of 650–670 °C.
:1 on a weight basis. The pyrolysis temperature was 520 °C and the fluidization velocity in the reactor was approximately 4 m s−1. Upon exiting the reactor, the product vapour/gas stream passes through two cyclones where the catalyst particles and char are separated. The pyrolysis vapours are condensed in two liquid scrubbers where product pyrolysis oil is used for quenching the pyrolysis vapours. The scrubbers were operated at a temperature of 35 °C. The scrubbers are followed by a secondary condensation system where a highly aqueous liquid product, which will be referred to as condensate, was collected. After the liquid products have condensed, a certain part of the non-condensable gases is recycled to the reactor to be used as the fluidization gas, whereas any excess gas is combusted in the regenerator. Char and catalyst coke deposits are also combusted in the regenerator. The regenerator was operated at a temperature of 650–670 °C.
2.3. Pyrolysis oil characterization
All the liquid products were analyzed for water content using Karl Fischer titration according to the standard ASTM E 203-96. The analyses were done using a Metrohm 795 KFT Titrino titrator. Elemental composition (CHN) of the pyrolysis oil was determined according to ASTM D 5291 using an Elementar VARIOMAX CHN analyzer. Higher heating value (HHV) was determined according to DIN 51900 using an IKA Werke C 5000 Control calorimeter. The solids content was analysed according to ASTM D7579. Density of the pyrolysis oil was measured according to ASTM D 4052 at 15 °C using an Anton Paar DMA 4500 M digital density meter. Kinematic viscosity was determined at 40 °C according to ASTM D 445. Total acid number (TAN) was determined potentiometrically according to ASTM D664. Micro carbon residue (MCR) of the bio-oil was analyzed according to ASTM D 4530 using an Alcor Micro Carbon Residue Tester. The ash content of the pyrolysis oil was determined by further combusting the micro carbon residue in a muffle furnace at 775 °C. Total organic carbon (TOC) of aqueous liquid products was measured using a Shimadzu TOC-5000A Total Organic Carbon Analyzer.Several methods were used for characterising the chemical composition of pyrolysis liquids. A general overview for the chemical composition was obtained using the solvent fractionation method,32 where the pyrolysis oil was first separated into water-soluble and water-insoluble (WIS) fractions using water extraction. The water-soluble fraction was then divided into ether-soluble and ether-insoluble parts. The ether-soluble part contained acids, alcohols, aldehydes, ketones and lignin monomers while the ether-insoluble part contained sugar-type compounds such as anhydrosugars and anhydrooligomers. The water-insoluble fraction, on the other hand, was divided into low molecular weight (LMW) lignin and high molecular weight (HMW) lignin fractions. The LMW lignin fraction is defined as the dichloromethane-soluble part of the water-insoluble fraction. In addition to low molecular weight lignin oligomers, this fraction can also contain extractives and aromatic hydrocarbons. Besides the heaviest lignin oligomers, the HMW lignin fraction contains polymerization products and solids.
The water soluble fraction of pyrolysis liquids was also analyzed using gas chromatography. This was done using an Agilent Technologies 7890 A gas chromatograph equipped with an Agilent 19091 N-136 HP-Innowax column. The length of the column was 60 m, inner diameter was 250 μm and thickness of the liquid phase 0.25 μm. The carrier gas was helium. A flame ionization detector (FID) was used at a temperature of 280 °C. The column oven was first heated to 60 °C using a temperature ramp of 3 °C min−1 and then to 230 °C in 30 minutes. Calibration was carried out using 40 water-soluble model compounds, and n-butanol was used as the internal standard. The list of quantified compounds can be seen in ref. 33. The content of aromatic hydrocarbons along with some oxygenated aromatics in the whole pyrolysis oil was quantitatively analyzed using an Agilent Technologies 6890 Series GC system, which was equipped with an Ultra 2 column (length, 23 m; inner diameter, 0.32 mm; film thickness, 0.52 μm. Calibration curves were determined using model compounds which ranged from benzene to pyrene. Dodecane was used as the internal standard. Compound identification was carried out using gas chromatography with a mass selective detector (GC-MSD). Measurements were performed using a Shimadzu GC17A gas chromatograph equipped with a mass selective quadrupole detector and a HP Ultra 1 fused silica capillary column (length: 50 m, inner diameter: 0.32 mm, film thickness: 0.52 μm.
2.4. Gas analysis
Non-condensable gases were sampled at one hour intervals and analyzed online using a Varian CP-4900 micro gas chromatograph. Three separate columns were used for quantifying the individual gas components. The column types were 5 Å molecular sieve (MS-5), porous polymer (PPU) and alumina (Al2O3). The following compounds were quantified: hydrogen (H2), nitrogen (N2), methane (CH4), carbon monoxide (CO), carbon dioxide (CO2), ethane (C2H6), ethene (C2H4), 1,2-propadiene (C3H4), propane (C3H8) and propene (C3H6).2.5. Catalyst characterization
Catalyst samples were taken during the experiment using a sampling system located below the cyclones. Samples were collected before the catalyst–char mixture was fed into the regenerator. The coke content of the spent catalyst samples was measured using temperature programmed oxidation (TPO) with a LECO TGA-601 thermogravimetric analyzer. The samples were first heated to 100 °C to remove any residual moisture, and then the temperature was ramped up to 815 °C using a heating rate of 10 °C min−1. The samples were held at 815 °C for a minimum of 20 min. The coke content was determined in wt% as 100% – TPO residue of dehumidified sample at 815 °C. The specific surface area, micropore area and micropore volume of the catalysts were determined using N2 physisorption. The measurements were performed with a Micromeritics Tristar surface area analyser. The specific surface area was determined at P/P0 0.05–0.3 using the Brunauer–Emmett–Teller (BET) equation. Micropore area and volume were determined at P/P0 0.2–0.5 using the t-plot method where the statistical thickness was calculated using the method of Harkins and Jura. X-ray diffraction (XRD) patterns of the catalysts were recorded with a Philips X'Pert MPD diffractometer using Cu Kα radiation. The samples were scanned for 2θ angles between 5° and 50° with a resolution of 0.02°. Acidity of the catalyst samples was measured using temperature programmed desorption of ammonia NH3 (NH3-TPD). The NH3-TPD spectra of the catalysts were measured using a flow through microreactor equipped with a Balzers GAM-415 quadrupole mass spectrometer. The catalyst sample (400 mg) was saturated with 1 ml pulses of 5 vol% NH3 in He at a temperature of 30 °C. The catalyst was then flushed in He flow for 15 minutes, after which the temperature was ramped at a rate of 5 °C min−1 to 650 °C while monitoring mass number 17. The potassium (K), phosphorous (P), magnesium (Mg) and calcium (Ca) content of the catalyst samples was measured using inductively coupled plasma atomic emission spectroscopy (ICP-AES). The measurements were carried out using a Perkin-Elmer 7100 DV ICP-AES analyzer coupled with a Varian AA240 atomic absorption spectrometer. The catalyst samples were studied with a scanning electron microscope (SEM) JEOL JSM6400 combined with an energy dispersive X-ray analyser PGT Spirit. Samples of the catalyst particles were mounted on an electrically conductive adhesive. The samples were coated with a thin carbon coating using a Polaron TB500 Carbon Coater. Secondary electrons with an acceleration voltage of 12 kV were used in the imaging.3. Results and discussion
3.1. Overview of the experiment
The overall duration of the experiment was approximately 4 days. Samples were collected from both scrubbers at one hour intervals and from the condensate at two hour intervals. The average water content of the pyrolysis oil from scrubbers 1 and 2 was approximately 50 and 30 wt%, respectively. After an initial ten hour stabilization phase at the beginning of the experiment, all the liquid products collected from scrubbers 1 and 2 phase separated into an oil fraction (bottom phase) and an aqueous fraction (top phase). It should be emphasized that phase separation took place even when the water content in scrubber 2 decreased to approximately 20 wt%. This clearly indicates that the chemical composition of the pyrolysis oil had become significantly less polar than what is typically observed for non-catalytically produced oils, which tend to phase separate only when the water content exceeds about 30 wt%.34 Larger samples of the product liquids were collected from the scrubbers at four hour intervals throughout the experiment. The two-phase products were then separated into oil and aqueous fractions using a separating funnel. The water content of the aqueous fraction typically ranged between 60–70 wt%. The oil fraction was analyzed for its water content and elemental composition. The water content varied between 9–11 wt%. The results of the elemental composition analysis are presented in Fig. 1 on a dry basis. The oxygen content of the oil fraction ranged between 22.4–23.7 wt%. Based on this, it is evident that low oxygen content pyrolysis oil was successfully produced in the 20 kg h−1 Process Development Unit over a time course of almost one hundred hours. Another aspect that warrants further attention is the relatively small increase in the oxygen content over time. It clearly indicates that even after almost one hundred hours of time-on-stream, the catalyst still retained a significant part of its initial activity, and could thus efficiently convert part of the primary pyrolysis vapors even at the end of the experiment. Nevertheless, both the carbon and the oxygen content displayed clear trends during the experiment, and it is therefore obvious that some catalyst deactivation did indeed take place. However, because the oxygen content only exhibited a small increase during the course of the entire experiment, all the liquid products were combined to give one overall liquid product. After the combination and the subsequent phase separation, separate organic and aqueous fractions were obtained and subjected to further physicochemical characterization. | ||
| Fig. 1 Oil fraction carbon and oxygen content (wt%, dry basis) as a function of time-on-stream. | ||
In addition to the two-phase product which was collected from the scrubbers, the condensate, which had an overall moisture content of approximately 70–80 wt%, also spontaneously separated into an organic top phase and an aqueous bottom phase. The carbon content of the top phase was high (78.4 wt%, dry basis) and the water content low (2.4 wt%). GC-MSD analysis of the top phase showed that it contained mostly single-ring aromatic hydrocarbons. The four main compounds were toluene, m + p-xylene, ethyl-methylbenzene and ethyl-benzene. In addition to these benzene-derivatives, some indenes and naphthalenes were also identified. Similar aromatic hydrocarbon compounds were also detected in the oil fraction of the scrubber products. The aromatic compounds detected in the condensate top phase are insoluble in water and more volatile than typical lignin derived phenolics, which explains the phase separation and the fact that some aromatic compounds either did not condense, or revolatilized in the scrubbers. Because the top phase of the condensate contained mainly the kind of products which are desirable in catalytic pyrolysis, it was combined with the oil fraction from the scrubbers. It was hypothesized that the addition of these products with solvent characteristics would serve to improve the quality of the overall organic liquid product.
Use of the HZSM-5 catalyst caused clear changes in the pyrolysis product distribution. The product distribution for this experiment and for a typical conventional fast pyrolysis experiment with the same pyrolysis unit and similar biomass feedstock are shown in Table 1. As the yield of char/coke cannot be directly measured in this system, it was determined by difference, i.e. 100% subtracted by the respective yields of organic liquids, pyrolytic water and gases. As it can be seen in Table 1, the use of the catalyst resulted in a significant decrease in the yield of organic liquids. This, and the consequent increase in the yields of pyrolytic water and gases are phenomena typically associated with the use of HZSM-5 catalysts in biomass catalytic pyrolysis. It should, however, be emphasized that not all of the organics were enriched in the oil fraction. Approximately 44 wt% of the overall organics were actually present in the aqueous products, which is something that has to be taken into account when considering the overall efficiency of this process. For the non-condensable gases, the increase in the yield could be mainly attributed to the increased formation of CO (yield 11.5 wt%). This is also typical for catalytic pyrolysis with HZSM-5 catalysts, as the two main routes for oxygen rejection are dehydration and decarbonylation reactions, which produce H2O and CO, respectively. The average molar CO/CO2 ratio was 2.9. The CO/CO2 ratio remained quite stable during the experiment, which was also an indication that the catalyst remained active during the experimental period. Time course gas composition data (see Fig. SM-1 in ESI†) also supports the notion that catalyst deactivation was limited. In addition to the increased CO yield, more light hydrocarbons in the C1–C3 range were also produced. The overall concentration of N2 in the gas stream was approximately 50 vol%. Thus, the product gas stream which was recycled back to the reactor to be used as the fluidization gas, contained 50 vol% of pyrolysis product gases with potential reactivity under CFP conditions. The recirculation of non-condensable gases35 as well as light hydrocarbons, e.g. propene,5 has been shown to be beneficial in catalytic pyrolysis. The overall N2 content remained high because continuous purging streams were fed into e.g. the feed hopper and instrumentation ports.
| CFP pine | FP pine | |
|---|---|---|
| Total liquid product | 51 | 73 |
| Organic liquids | 32 | 63 |
| Pyrolytic water | 19 | 10 |
| Gases | 21 | 9 |
| Char/coke | 27 | 18 |
3.2. Characterization of the overall liquid products
The overall liquid products obtained from catalytic fast pyrolysis were collected and combined in the way which is presented in Fig. 2. Three separate liquid products were obtained for characterization: CFP oil (1), CFP aqueous fraction (2) and condensate aqueous fraction (3). The liquid product from both scrubbers was first combined, and then phase separated in order to obtain separate oil and aqueous fractions. The top phase of the condensate was then added into the scrubber oil fraction. This combined product is referred to as ‘CFP oil’. Typical fast pyrolysis oil produced from pine sawdust is denoted as ‘FP oil’. Characterization of the aqueous fractions is discussed later in the article, where the aqueous fraction obtained from the phase separation of the scrubber products will be referred to as ‘CFP aqueous fraction’.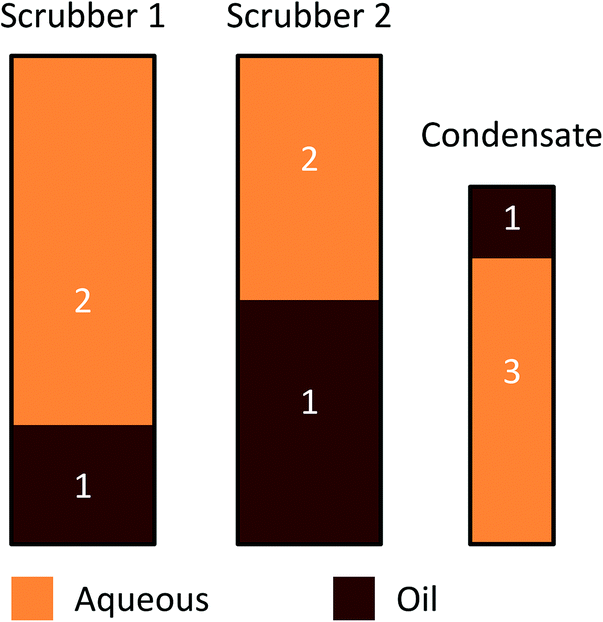 | ||
| Fig. 2 Conceptual representation of overall liquid product collection from catalytic fast pyrolysis: (1) CFP oil (250 kg); (2) CFP aqueous fraction (520 kg); (3) condensate aqueous fraction (90 kg). | ||
The properties of catalytically produced pyrolysis oil differed greatly compared to typical fast pyrolysis oil. The physicochemical properties of the CFP oil and FP oil are shown in Table 2. When considering the properties of the CFP oil, there are two important factors which contributed towards the formation of the product. First was the catalytic effect of the HZSM-5 catalyst. The reactions it catalyzed included dehydration of the pyrolysis vapors, which in turn led to phase separation of the overall liquid product due to the high water content. As a result of the phase separation, the less polar components enriched in the oil fraction, whereas a larger proportion of the polar oxygenates ended up in the aqueous fraction. Thus, the CFP oil exhibited a low water content, a high carbon content and a low oxygen content. The hydrogen content was also low, and the consequently low H/C molar ratio (1.07) suggested that the product would be quite aromatic. The higher heating value was also higher (30.4 MJ kg−1) than that of the pine FP oil (22.2 MJ kg−1), which can be explained by the change in the oil elemental composition. The TAN of CFP oil was distinctly lower compared to FP oil, which suggested that the concentration of the main acidic constituents had decreased significantly. The pH of the CFP oil did not change significantly compared to FP oil.
| Property | CFP oil | FP oil31 |
|---|---|---|
| Water (wt%) | 8.3 | 23.9 |
| Solids (wt%) | 0.76 | 0.01 |
| Ash (wt%) | 0.6 | 0.03 |
| Carbon (wt%, dab) | 72.0 | 53.4 |
| Hydrogen (wt%, dab) | 6.4 | 6.5 |
| Nitrogen (wt%, dab) | 0.02 | 0.1 |
| Oxygen (wt%, dab) | 21.5 | 40.0 |
| Higher heating value (MJ kg−1, dab) | 30.4 | 22.2 |
| Lower heating value (MJ kg−1, dab) | 28.7 | 20.1 |
| Kinematic viscosity (cSt, 40 °C) | 285 | 17 |
| Density (kg dm−3, 15 °C) | 1.183 | 1.206 |
| pH | 2.6 | 2.7 |
| TAN (mg KOH g−1) | 30 | 71 |
| Carbonyls (mmol g−1) | 2.8 | 3.5 |
| Micro carbon residue (wt%) | 29.3 | 20.8 |
The CFP aqueous fraction retained a significant proportion of the overall organic liquids which were produced in the CFP of pine. Physicochemical characteristics of the aqueous products are presented in Table 3. When considering the amount of organics in the CFP aqueous fraction, total organic carbon made up approximately 57% of the total organics. This kind of carbon content would be fairly typical for conventional fast pyrolysis oil. This is an indication that the polar oxygenate molecules which are typical products of biomass fast pyrolysis, enriched in the aqueous fraction that was obtained from catalytic fast pyrolysis. For the condensate aqueous fraction, organic carbon made up only 41 wt% of the overall organics, which suggested that the organics present in this fraction would be highly oxygenated compounds. The pH of both aqueous product fractions was below 3, but the TAN was lower than that of typical fast pyrolysis oil. The overall CFP liquid products contained approximately 39% of the biomass carbon. The carbon yield of the CFP oil was about 24%, whereas the remaining 15% was found in the aqueous product fractions.
| Property | CFP aqueous | Condensate aqueous |
|---|---|---|
| Water (wt%) | 67.7 | 85.6 |
| Organics (wt%) | 32.3 | 14.4 |
| pH | 2.4 | 2.7 |
| TAN (mg KOH g−1) | 47 | 34 |
| TOC (wt%) | 18.4 | 5.9 |
The change in the elemental composition of the pyrolysis oil can be further elucidated by using a Van Krevelen plot, which can be seen in Fig. 3. The lignin-derived water-insoluble (WIS) fraction of FP oil, and the whole CFP oil were actually relatively similar in their elemental composition. The WIS fraction of FP oil was, however, almost solid, whereas the CFP oil had a fairly low viscosity for a pyrolysis oil with low water content (8.3 wt%). Without the addition of the condensate aromatic fraction, the viscosity of the CFP oil was higher (437 cSt at 40 °C). The added condensate aromatic fraction constituted 4.4 wt% of the entire CFP oil, but due to its strong solubilizing properties the relative decrease in viscosity was approximately 35%. The amount of water-insoluble material in the CFP oil was high (76 wt%), which was clearly evident in the solvent fractionation of the CFP liquid products, the results of which are presented in Fig. 4. The high WIS content also explains the small difference in the elemental composition of the whole CFP oil and its respective WIS fraction (see Fig. 3). In addition to the LMW and HMW lignin fractions, the CFP oil contained some water-insoluble volatiles which evaporated during the fractionation process. The presence of these WIS volatiles has been previously correlated with aromatic hydrocarbons that have formed in the catalytic pyrolysis process.36 The amount of water-soluble oxygenates in the CFP oil was low, whereas the CFP aqueous fraction contained significant amounts of sugar-type compounds and ether-soluble light molecular weight oxygenates. If the CFP oil and aqueous fractions are combined into a single product by means of calculation, almost half of this combined product consists of water. This is due to the high moisture content of the feedstock (12 wt%), and the increased water yield originating from the dehydration reactions. If one considers the composition of the overall organic products on a dry basis (data not shown), it could be observed that the total amount of water-insoluble material increased from 27 to 48 wt%, whereas the amount of water-soluble organics decreased from 73 to 52 wt%. Based on this, it was clear that the yield decrease of organic liquids which was observed in CFP, was accompanied by a change in the overall composition of organic liquid products. While the CFP oil fraction exhibited a low content of water-soluble oxygenates (16 wt%), their significant presence in the aqueous fraction clearly necessitates the development of valorization routes for all liquid product fractions.
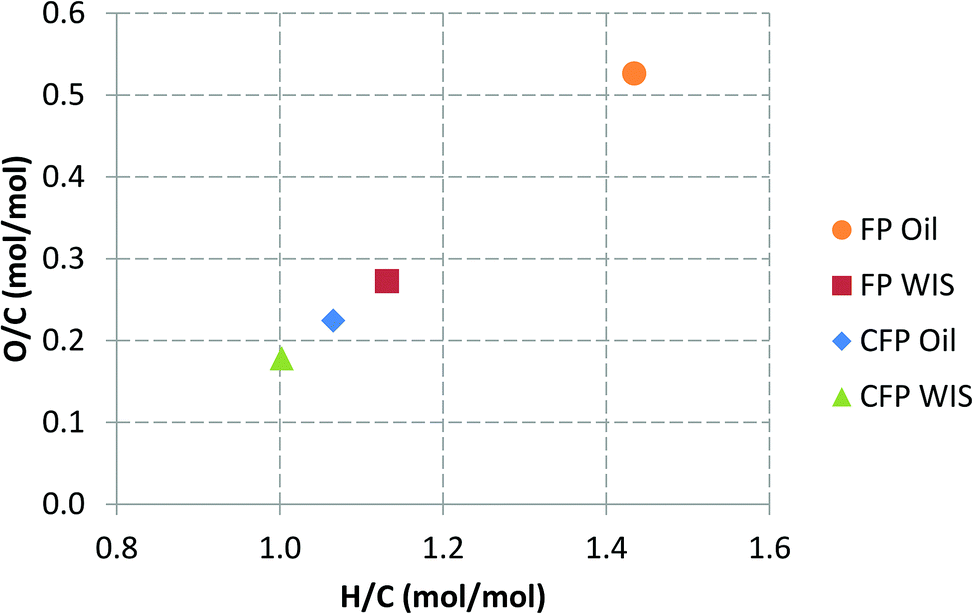 | ||
| Fig. 3 Van Krevelen plot for pyrolysis oils and their water-insoluble (WIS) fractions produced in fast pyrolysis (FP) and catalytic fast pyrolysis (CFP). | ||
 | ||
| Fig. 4 Solvent fractionation of CFP oil, CFP aqueous fraction and typical FP oil from pine. Relative amounts of product fractions are included for the two phase CFP product. | ||
GC-MSD analysis of the CFP oil also revealed clear compound level changes compared to typical fast pyrolysis oils (see Fig. SM-2 in ESI†). Alkylated single-ring aromatic hydrocarbons became dominant, whereas typical cellulose and hemicellulose decomposition products had clearly decreased. Lignin monomers ranging from phenol and cresols to bulkier guaiacyl lignin monomers were also detected. Aromatic hydrocarbon structures larger than naphthalene were not detected in the CFP oil. A certain level of catalyst acidity was previously identified as a requirement for the formation three ring polyaromatic hydrocarbons.18 The absence of such compounds suggests that this particular HZSM-5 catalyst formulation possessed a suitable level of acidity and acid site density for catalytic fast pyrolysis. Overall, the GC-MSD results further support the results of the elemental analysis and solvent fractionation, which both indicated that the catalytically produced pyrolysis oil was highly aromatic and contained relatively small amounts of carbohydrate-derived thermal decomposition products.
Concentrations of certain typical water-soluble pyrolysis products were clearly lower in CFP oil when compared to FP oil. The results of quantitative GC-FID measurements are presented in Table 4, where the various compounds have been combined together into different chemical groups. The aromatic hydrocarbon content was only determined for the CFP oil. Compared to FP oil, the CFP oil contained less carboxylic acids and clearly less GC-detectable aldehydes and ketones. The decrease in the concentration of carboxylic acids is consistent with the observed decrease in the total acid number of the CFP oil. The changes in the concentrations of alcohols and furanics was not remarkable, whereas the concentration of lignin monomers was higher in the CFP oil. The CFP oil also contained 6.4 wt% of aromatic hydrocarbons. Approximately 2/3 of these were single-ring substituted benzenes, whereas the remaining 1/3 was distributed evenly between indenes and naphthalenes. It appears that the process conditions and the catalyst that were employed in this study favoured the formation of the more valuable single-ring aromatics. While aromatic hydrocarbons dominated the GC-MSD analysis (Fig. SM-2 in ESI†), the corresponding quantitative analysis showed that their actual concentration in the pyrolysis oil was still relatively low. For the CFP total product, i.e. all three fractions combined, the decrease in the concentration of acids was clearly smaller when compared to FP oil. In fact, low molecular weight aldehydes and ketones were the only oxygenate compound group which exhibited a significant decrease in the total products. This also highlights the fact that the properties and composition of CFP oil were largely affected by the phase separation, and the consequent enrichment of water-soluble organics in the CFP aqueous fraction.
| Sample | CFP oil | CFP aqueous | Condensate aqueous | CFP total | FP oil |
|---|---|---|---|---|---|
| Acids | 1.8 | 3.9 | 2.9 | 3.2 | 3.7 |
| Aldehydes and ketones | 1.6 | 3.7 | 1.5 | 2.9 | 10.2 |
| Alcohols | 0.2 | 0.5 | 0.9 | 0.5 | 0.7 |
| Furanics | 0.5 | 0.3 | 0.2 | 0.3 | 1.0 |
| Lignin monomers | 3.7 | 1.0 | 0.2 | 1.7 | 1.7 |
| Benzene derivatives | 4.1 | — | — | 1.2 | — |
| Indenes | 1.1 | — | — | 0.3 | — |
| Naphthalenes | 1.1 | — | — | 0.3 | — |
3.3. Catalyst characterization
Spent catalyst samples, which were withdrawn from the pyrolysis unit during the experiment, showed that the catalyst coke content decreased as a function of time-on-stream. The results are shown in Fig. 5. The declining trend in the catalyst coke content as a function of time-on-stream suggested that the catalyst was losing some of its activity in the course of the experiment. The trend is very similar to what was observed in the carbon content of the pyrolysis oil samples (Fig. 1). The decreasing coke content of the catalyst also showed that the temperature and catalyst residence time in the regenerator were both sufficient for effective removal of coke deposits. If the regeneration of the catalyst was not functioning adequately, one would expect to observe an increasing amount of coke on the catalyst. The average coke content of the catalysts was 1.1 wt%. With a biomass feeding rate of 20 kg h−1 and a catalyst-to-biomass ratio of 7:1, this would correspond to a coke yield of approximately 7–8 wt%. It was previously shown18 in catalytic fast pyrolysis bench-scale experiments that coke formation with a HZSM-5 catalyst was highest at the beginning of the experiment, i.e. when the catalyst still retained a larger proportion of its initial activity. French and Czernik37 also used coke formation as a metric to identify high activity catalysts in a catalytic pyrolysis screening study. While catalyst deactivation due to coke formation and the potentially permanent catalyst deactivation that has been observed in this study are different phenomena, it is possible that both could decrease the activity of the catalysts via a similar mechanism.
 | ||
| Fig. 5 Catalyst coke content (wt%) as a function of time-on-stream. | ||
Before any further characterization of the spent catalysts, the samples were regenerated in a laboratory muffle furnace for 5 h at 670 °C. The temperature corresponded to the temperature of the regenerator of the pyrolysis unit. The additional regeneration procedure was carried out in order remove the coke from the catalyst, thus eliminating any effect the coke might have had on the properties of the catalyst. It was envisioned that any potential changes in the properties and the structure of the catalyst could be observed with less ambiguity by utilizing an approach such as this. The results from the physisorption of N2 are reported in Table 5. The results show that the catalyst had already lost a significant part of its specific surface area and micropore volume prior to the start of the experiment, which is represented by the TOS 0 h sample. The actual start of the experiment was preceded by a heat-up period with a duration of approximately two and a half days. During the course of the experiment, the catalyst still continued to lose more of its specific surface area and micropore volume. For the fresh catalyst sample, approximately 80% of the overall specific surface area was contained in the microporous parts of the catalyst. In contrast to this, only 47% of the overall specific surface area was attributable to the micropores for the final catalyst sample (TOS 96 h). The continuous decrease which was observed in the micropore area and micropore volume of the catalyst suggested that some degradation of the zeolite structure could have taken place during the experiment. The catalyst sample which was calcined under laboratory conditions (4 h, 650 °C) and not used in the actual experiment, also exhibited a distinct decrease in its specific surface area and micropore volume. This indicates that the properties of the HZSM-5 catalyst are prone to change during the kind of prolonged heating period, which has typically been employed in VTT's fast pyrolysis Process Development Unit.
| Sample/TOS | BET surface area (m2 g−1) | Micropore area (m2 g−1) | Micropore volume (cm3 g−1) |
|---|---|---|---|
| Fresh catalyst | 212 | 171 | 0.088 |
| Calcined catalyst | 178 | 125 | 0.065 |
| 0 h | 121 | 76 | 0.040 |
| 13 h | 131 | 67 | 0.035 |
| 21 h | 127 | 62 | 0.033 |
| 45 h | 123 | 60 | 0.031 |
| 69 h | 118 | 56 | 0.030 |
| 96 h | 118 | 55 | 0.030 |
Compared to the fresh catalyst, the catalyst which was recovered after the experiment exhibited changes in its XRD pattern (see Fig. SM-3 in ESI†). Two distinct peak groups which are associated with the mordenite inverted framework (MFI) structure of the HZSM-5 catalyst could be observed at approximately 8° and 23°. The HZSM-5 structure and the associated XRD pattern can be found on http://www.iza-structure.org/databases/. The intensity of the latter peak group clearly decreased, which has been previously associated with the removal of framework aluminium in the zeolite structure, as well as decrease in crystallinity.38,39 The intensity of the peak at 23° also exhibited a linear correlation with catalyst micropore area (see Fig. SM-4 in ESI†), which further supports the notion of zeolite structural degradation. The changes in the peak intensities of the zeolite component were accompanied by the disappearance of kaolinite-related peaks (e.g. 12°).40 In addition to this, crystalline quartz with a main peak at approximately 26.5° was also observed in the spent catalyst sample.41 Overall, these observations reflect changes in both the zeolite component and the binder material of the catalyst. However, even with the transformation of the catalyst, the quality of the pyrolysis oil which was produced remained quite consistent throughout the experiment.
Alkali metals, which are present in the biomass feedstock in the form of ash, also deposited on the catalyst. As it can be seen in Fig. 6, the alkali metal content of the catalyst, as determined by ICP-AES, increased linearly over time. With an overall catalyst inventory of 94 kg, the metal content of the spent catalyst corresponded to approximately 1.1 kg of biomass derived contaminant metals. Based on the ash content of the pine feedstock, approximately 7.7 kg of ash were fed into the system during the four day experiment. Thus, the 1.1 kg of metals found on the catalyst would equal approximately 14 wt% of the total feedstock ash. While at this point it is too early to speculate whether the metal content of the catalyst would keep increasing linearly, or whether some kind of saturation would eventually be reached, it is clear that the combined reaction and regeneration conditions of the circulating fluidized bed pyrolysis unit resulted in significant deposition of biomass metals on the zeolite catalyst.
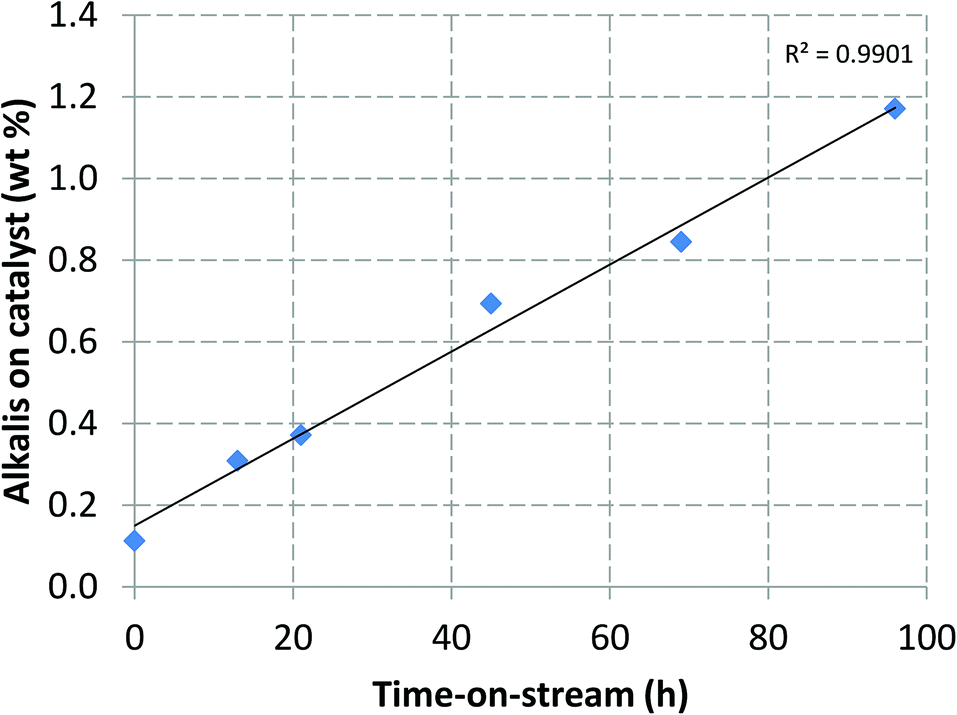 | ||
| Fig. 6 Alkali metal content (K, Ca, P, Mg) of the catalyst as a function of time-on-stream. | ||
While the observed deposition of alkali metals is important in itself, it does not yet explain how these metals affect the properties and ultimately, the activity, of the HZSM-5 catalyst. As it was stated earlier, deposition of alkali metals on the Brønsted acid sites of deNOx catalysts has been previously observed.24–27 While in this catalytic fast pyrolysis experiment the changes in the catalyst properties were not limited only to alkali deposition, a clear correlation between catalyst alkali metal content and acidity could, nevertheless, be observed in Fig. 7. As the content of alkali metals on the catalyst increased over time, the overall acidity exhibited a steady decrease as well. Compared to the acidity of the catalyst sample at TOS 0 h (leftmost data point in Fig. 7), the laboratory calcined catalyst sample had a clearly higher acidity (1.0 mmol g−1). Thus, the catalyst had lost a significant part of its initial acidity during the heat-up phase. The relative decrease in acidity was, however, clearly lower during the actual four day catalytic pyrolysis experiment. These observations coincide with the changes previously seen in the surface area and porosity characteristics of the catalyst in Table 5; all parameters exhibited the largest changes before pyrolysis of the biomass was commenced. This would suggest that the HZSM-5 catalyst used in this experiment possessed a certain level of intrinsic thermal instability, which manifested as transformation of various catalyst properties during the pre-pyrolysis heating period. While the intensity of these so-called transformation phenomena attenuated over time, a clear steady state was not reached during the four day pyrolysis period.
 | ||
| Fig. 7 Catalyst overall acidity (mmol NH3 g−1 of catalyst) as a function of catalyst alkali metal content (wt% of K, Ca, P, Mg). | ||
SEM images of the catalyst samples showed that there was clear damage to the catalyst particles during the experiment. As it can be seen in Fig. 8, the fresh catalyst sample consisted mainly of regularly shaped spherical particles with varying sizes. The catalyst at TOS 0 h already showed some signs of wear and agglomeration, but the surfaces of the particles were still mostly intact. The catalyst sample from the end of the experiment was, however, clearly different compared to the other two. While most of the particles still roughly retained their original spherical shape, there were clear signs of damage and changes in the form fractures and particle agglomeration. The spent catalyst sample still contained smaller particles as well, which shows that catalyst fines were not selectively lost in the circulation process. EDS mapping of Si, Al and Ca was also carried out for a single catalyst particle which had a crater-like cavity (Fig. SM-5 in ESI†). The mapping showed that the zeolite metallic constituents, Si and Al, were distributed evenly across the particle surface, including the cavity. Ca, on the other hand, was more concentrated on the original surface of the catalyst particle. While it is not possible to determine when the fracturing of the particle took place, the images suggest that deposition of Ca inside the cavity may have started after the particle was damaged. These results suggest that Ca mainly deposited on the outer surface of the catalyst, instead of entering the catalyst pores.
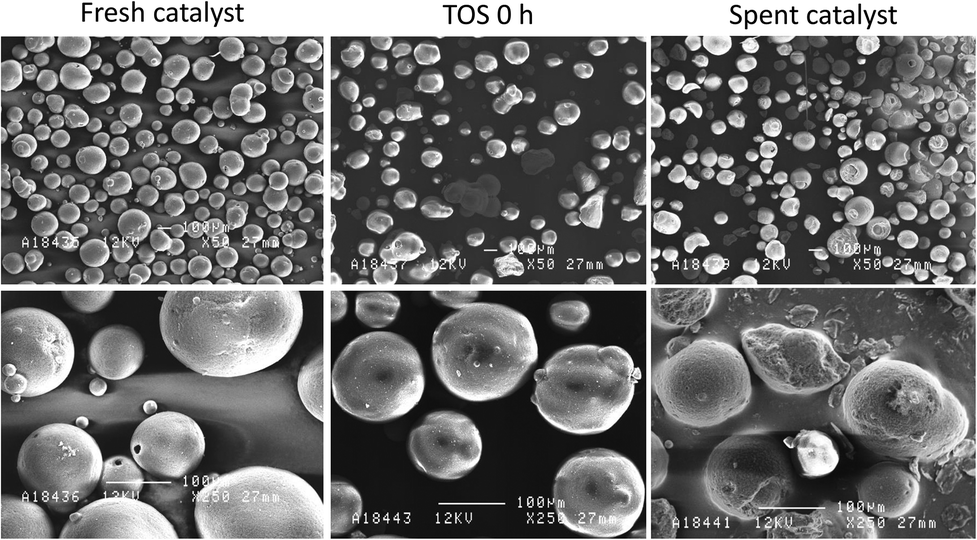 | ||
| Fig. 8 SEM images of the fresh catalyst, the catalyst at the start of the pyrolysis (TOS 0 h) and the spent catalyst sample. | ||
Based on these observations, one clear focal point for further research would be to investigate the potential changes in catalyst properties on a longer time scale. Even though the oxygen content of the pyrolysis oil produced throughout this experiment remained quite low, it is entirely conceivable that after a long enough period of time-on-stream, the permanently deactivated catalyst would no longer be able to produce pyrolysis oil with the desired quality characteristics. The most obvious method for countering this limitation would be to continuously supply fresh catalyst to the catalytic pyrolysis unit while simultaneously withdrawing spent catalyst from the system. This solution would be analogous to what is routinely carried out in oil refinery fluid catalytic cracking units. The catalyst that would be withdrawn from the pyrolysis unit would always be a mixture of catalyst fractions with varying degrees of activity. A suitable rate of catalyst replacement would, therefore, have to be determined to reach an operational equilibrium where product quality remains stable.
4. Conclusions
Catalytic fast pyrolysis in a circulating fluidized bed reactor with continuous catalyst regeneration was shown to be a suitable method for producing partially deoxygenated pyrolysis oil over extended time periods. VTT's 20 kg h−1 Process Development Unit was successfully operated using a spray dried HZSM-5 catalyst, and approximately 250 kg of pyrolysis oil were produced over an operational time of four days. In addition to the low water (8.3 wt%) low oxygen (21.5 wt%) pyrolysis oil, an aqueous product fraction containing the majority of polar carbohydrate-derived oxygenates was also produced. In order to increase the overall efficiency of the catalytic fast pyrolysis process, separate valorization routes must also be devised for the aqueous product fraction. While most of the oxygenate molecules enriched in the aqueous fraction, the organic fraction of pyrolysis oil contained mostly water-insoluble lignin-derived molecules, which were also supplemented by 6.4 wt% of aromatic hydrocarbons. Properties of the HZSM-5 catalyst changed considerably during the initial pre-pyrolysis heating period, and also to a lesser extent during the four day catalytic pyrolysis experiment. Based on this, it appeared that the properties of the catalyst initially transformed due to thermal instability. After this initial deactivation period, the transformation of the catalyst continued as it was further subjected to both thermal stress and the biomass feedstock. Biomass alkali metals deposited on the catalyst, and a direct correlation was observed between this phenomenon and the acidity of the catalyst. SEM images of the also showed clear signs of physical damage to the particles. Although the elemental composition of the pyrolysis oil remained quite stable during the four day experiment, the observed changes in the catalyst properties clearly indicate that catalyst lifetime will in all likelihood become a crucial factor in future commercial scale catalytic fast pyrolysis units.Acknowledgements
Jaana Korhonen, Sirpa Lehtinen and Elina Paasonen are acknowledged for their analytical work. Jouko Kukkonen, Sampo Ratinen, Pekka Saarimäki, Jarmo Juuti, Ilkka Isoksela and Joni Rantala are acknowledged for performing the pyrolysis experiment. Other members of the Catalysis and Synfuels team are acknowledged for their help in parts of the characterization work. Tom Gustafsson is acknowledged for his help with the SEM imaging. Tekes, the Finnish Funding Agency for Innovation, is acknowledged for funding the research project ‘Pilot-scale development of new 2G BTL technologies based on gasification and pyrolysis’ under contract no. 40441/11.References
- A. A. Lappas, M. C. Samolada, D. K. Iatridis, S. S. Voutetakis and I. A. Vasalos, Fuel, 2002, 81, 2087–2095 CrossRef CAS.
- F. A. Agblevor, S. Beis, O. Mante and N. Abdoulmoumine, Ind. Eng. Chem. Res., 2010, 49, 3533–3538 CrossRef CAS.
- H. Zhang, R. Xiao, H. Huang and G. Xiao, Bioresour. Technol., 2009, 100, 1428–1434 CrossRef CAS PubMed.
- C. A. Mullen, A. A. Boateng, D. J. Mihalcik and N. M. Goldberg, Energy Fuels, 2011, 25, 5444–5451 CrossRef CAS.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 CAS.
- D. J. Mihalcik, C. A. Mullen and A. A. Boateng, J. Anal. Appl. Pyrolysis, 2011, 92, 224–232 CrossRef CAS PubMed.
- S. Stefanidis, K. Kalogiannis, E. Iliopoulou, A. Lappas and P. Pilavachi, Bioresour. Technol., 2011, 102, 8261–8267 CrossRef CAS PubMed.
- T. Carlson, G. Tompsett, W. Conner and G. Huber, Top. Catal., 2009, 52, 241–252 CrossRef CAS.
- A. Aho, N. Kumar, K. Eränen, T. Salmi, M. Hupa and D. Y. Murzin, Fuel, 2008, 87, 2493–2501 CrossRef CAS PubMed.
- T. R. Carlson, J. Jae, Y.-C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110–124 CrossRef CAS PubMed.
- O. Mante, F. Agblevor and R. McClung, Biomass Convers. Biorefin., 2011, 1, 189–201 CrossRef CAS PubMed.
- F. A. Agblevor, O. Mante, N. Abdoulmoumine and R. McClung, Energy Fuels, 2010, 24, 4087–4089 CrossRef CAS.
- A. Oasmaa, J. Korhonen and E. Kuoppala, Energy Fuels, 2011, 25, 3307–3313 CrossRef CAS.
- F. A. Agblevor, O. Mante, R. McClung and S. Oyama, Biomass Bioenergy, 2012, 45, 130–137 CrossRef CAS PubMed.
- F. de Miguel Mercader, M. Groeneveld, S. Kersten, N. Way, C. Schaverien and J. Hogendoorn, Appl. Catal., B, 2010, 96, 57–66 CrossRef CAS PubMed.
- C. Torri, M. Reinikainen, C. Lindfors, D. Fabbri, A. Oasmaa and E. Kuoppala, J. Anal. Appl. Pyrolysis, 2010, 88, 7–13 CrossRef CAS PubMed.
- A. Lappas, S. Bezergianni and I. Vasalos, Catal. Today, 2009, 145, 55–62 CrossRef CAS PubMed.
- V. Paasikallio, C. Lindfors, J. Lehto, A. Oasmaa and M. Reinikainen, Top. Catal., 2013, 56, 800–812 CrossRef CAS.
- R. Boerefijn, N. Gudde and M. Ghadiri, Adv. Powder Technol., 2000, 11, 145–174 CrossRef CAS PubMed.
- C. S. Triantafillidis, A. G. Vlessidis, L. Nalbandian and N. P. Evmiridis, Microporous Mesoporous Mater., 2001, 47, 369–388 CrossRef CAS.
- H. Cerqueira, G. Caeiro, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2008, 292, 1–13 CrossRef CAS PubMed.
- Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 124, 1416–1419 CrossRef.
- C. A. Mullen and A. A. Boateng, Ind. Eng. Chem. Res., 2013, 52, 17156–17161 CrossRef CAS.
- Y. Zheng, A. D. Jensen and J. E. Johnsson, Appl. Catal., B, 2005, 60, 253–264 CrossRef CAS PubMed.
- J. Chen and R. Yang, J. Catal., 1990, 125, 411–420 CrossRef CAS.
- P. Kern, M. Klimczak, T. Heinzelmann, M. Lucas and P. Claus, Appl. Catal., B, 2010, 95, 48–56 CrossRef CAS PubMed.
- S. S. R. Putluru, A. Riisager and R. Fehrmann, Appl. Catal., B, 2011, 101, 183–188 CrossRef CAS PubMed.
- G. Yildiz, M. Pronk, M. Djokic, K. M. van Geem, F. Ronsse, R. van Duren and W. Prins, J. Anal. Appl. Pyrolysis, 2013, 103, 343–351 CrossRef CAS PubMed.
- J. Jae, R. Coolman, T. Mountziaris and G. W. Huber, Chem. Eng. Sci., 2014, 108, 33–46 CrossRef CAS PubMed.
- H. Zhang, J. Zheng, R. Xiao, D. Shen, B. Jin, G. Xiao and R. Chen, RSC Adv., 2013, 3, 5769–5774 RSC.
- A. Oasmaa, Y. Solantausta, V. Arpiainen, E. Kuoppala and K. Sipilä, Energy Fuels, 2010, 24, 1380–1388 CrossRef CAS.
- A. Oasmaa, E. Kuoppala and Y. Solantausta, Energy Fuels, 2003, 17, 433–443 CrossRef CAS.
- A. Oasmaa and C. Peacocke, VTT Publications 731: Properties and fuel use of biomass-derived fast pyrolysis liquids. A guide, VTT Technical Report 731, 2010.
- G. Peacocke, P. Russell, J. Jenkins and A. Bridgwater, Biomass Bioenergy, 1994, 7, 169–177 CrossRef CAS.
- O. D. Mante, F. Agblevor, S. Oyama and R. McClung, Bioresour. Technol., 2012, 111, 482–490 CrossRef CAS PubMed.
- V. Paasikallio, F. Agblevor, A. Oasmaa, J. Lehto and J. Lehtonen, Energy Fuels, 2013, 27, 7587–7601 CrossRef CAS.
- R. French and S. Czernik, Fuel Process. Technol., 2010, 91, 25–32 CrossRef CAS PubMed.
- Y. T. Kim, K.-D. Jung and E. D. Park, Microporous Mesoporous Mater., 2010, 131, 28–36 CrossRef CAS PubMed.
- C. Ding, X. Wang, X. Guo and S. Zhang, Catal. Commun., 2008, 9, 487–493 CrossRef CAS PubMed.
- K. G. Bhattacharyya and S. S. Gupta, Sep. Purif. Technol., 2006, 50, 388–397 CrossRef CAS PubMed.
- Y. Ma, C. Yan, A. Alshameri, X. Qiu, C. Zhou and D. Li, Adv. Powder Technol., 2014, 25, 495–499 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc00571f |
| This journal is © The Royal Society of Chemistry 2014 |