 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-free glucose oxidation using air with supported gold catalysts
Peter J.
Miedziak
a,
Hamed
Alshammari
b,
Simon A.
Kondrat
a,
Tomos J.
Clarke
a,
Thomas E.
Davies
a,
Moataz
Morad
a,
David J.
Morgan
a,
David J.
Willock
a,
David W.
Knight
a,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
bChemistry Department, Faculty of Science, Ha'il University, P. Box 2440, 81451 Ha'il, Saudi Arabia
First published on 10th April 2014
Abstract
We report the selective oxidation of glucose to gluconic acid under mild conditions and show that if a basic support is used then the reaction can be carried out without the addition of sacrificial base or pH control. The use of sol-immobilisation prepared catalysts supported on magnesium oxide facilitates the use of ambient air as an oxidant source. These mild conditions resulted in an excellent selectivity towards gluconic acid. Different heat treatments result in an improvement in the activity of the catalyst, these improvements are discussed in terms of XRD, DRIFTD and TEM analysis of the catalysts, despite significant particle growth and phase segregation occurring during the thermal treatments.
Introduction
The oxidation of glucose has been the focus of considerable attention in recent years. Glucose is a bio-renewable feedstock and its major product of its oxidation, gluconic acid, is used in the pharmaceutical, food, paper and concrete industries. Typically to achieve high conversion of glucose a sacrificial base is added to the reaction and the pH of the solution is maintained by the continual addition of a base, typically sodium hydroxide.1–3 Furthermore, pure oxygen is generally used as the oxidant,4–8 either at raised pressures or by bubbling oxygen through the solution. The preparation of oxygen from air, together with its purification, requires considerable energy which adds significantly to the cost of the process. The removal of the base from the process and its replacement with a catalytic equivalent, together with the utilisation of air as a source of oxygen, would represent a significant advance in the process for glucose oxidation adhering to the principles of green chemistry.Early reports, that have used heterogeneous catalysts for this oxidation, focused on the use of platinum on carbon as a catalyst. However, it was found that these catalysts suffered from marked deactivation, which was reduced when the reaction was carried out at high initial pH; but this deactivation could not be eliminated.9 The deactivation of these catalysts was addressed when the pH of the solution was readjusted to pH 9 during the reaction by continuous addition of base. The use of bimetallic catalysts has also been reported for this oxidation Besson et al.10 who used palladium catalysts supported on carbon promoted with bismuth. The selectivity and conversion were close to 100% when the reaction was carried out at pH 9 with bubbling air as the oxidant and the pH was maintained at a constant level throughout the reaction by addition of NaOH. Although the authors demonstrated five reuses of the catalyst they did not rule out deactivation as the reaction is run under mass transfer control. Gallezot11 also reported the oxidation of bismuth palladium supported on carbon, noting that 3–10 nm particles were more active than 1–2.5 nm particles and suggesting the bismuth prevents the over-oxidation of the products.
Wenkin et al.12 carried out an extensive study into the preparation of carbon-supported bismuth-promoted palladium catalysts using various bismuth precursors. Organic based acetate precursors were found to be the most active precursors, however, bismuth leaching into the reaction solution was detected for all the catalysts tested. Gold catalysts have also been reported for this oxidation, Biella et al.4 proposed the use of gold supported on carbon prepared by the sol-immobilisation method for this oxidation, under various pH conditions, the gold catalyst was found to be more active than platinum, palladium–bismuth or platinum–palladium–bismuth trimetallic catalysts. A pH of 9.5 was reported as the optimum value, however, the gold was also reported to maintain activity at pH values other than the optimised pH. Upon recycling the gold catalyst did display a decrease in its activity, thought to be due to metal leaching. In a further study13 “naked” gold nanoparticles were formed by the colloidal method and tested for the oxidation of glucose, the particles of gold were found to undergo growth during the reaction preventing the reaction from reaching completion but the initial turnover frequencies (TOFs) of the oxidation were extremely high demonstrating the potential of gold as a catalyst for this oxidation. Similar nanoparticles of copper, silver, palladium and platinum were tested but found to be inactive. Önal et al.14 studied different sol preparation methods and different carbon supports at pH 7–9.5 and found the best results were obtained at 50 °C and pH 9.5 using Vulcan-type carbons, Comotti et al.7 also reported the oxidation with H2O2 and Prüße and co-workers published a series of papers investigating the long term stability of gold catalysts on various supports.15–18 Gold on alumina was prepared by the deposition precipitation and incipient wetness methods, the relatively low loaded (0.3% Au) catalysts were shown to be active for this reaction. Further work reported 0.45% Au catalysts supported on titania16 prepared by deposition precipitation and sol methodologies, gave reasonable stability of the catalysts demonstrated over seventeen reuses. They further demonstrated the incipient wetness prepared gold on alumina catalysts in a continuous flow reactor17 with high catalyst stability for 110 h. Different deposition precipitation (DP) methods were studied for gold on alumina18 and urea was reported to be the best precipitating agent for this preparation method.
An interesting comparison between a gold catalyst, supported on carbon and an enzymatic reaction was provided by Rossi and co-workers,19 the work demonstrated that if the activity of the gold catalysts was calculated based on theoretical surface gold atoms, the rate of reaction would be of the same order of magnitude as the enzymatic reaction. Ishida et al.20 used the solid grinding method to prepare gold catalysts which they found gave higher activity than the DP catalyst preparation when supported on alumina or zirconia.
Bimetallic catalysts comprising combinations of gold, platinum, palladium and rhodium were studied by Comotti et al.8 and the gold platinum catalysts were the most active and with the exception of gold rhodium the alloying of the metals improved the activity. The fine-tuning of the ratio of gold to platinum led to an optimised ratio of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt of 2:1 (molar) and with this catalyst at pH 9.5 a TOF of 17600 h−1 was observed.
:Pt of 2:1 (molar) and with this catalyst at pH 9.5 a TOF of 17600 h−1 was observed.
Karski et al.21 used thallium alloyed with palladium on silica catalysts and there was an observable increase in the selectivity when the thallium was alloyed. However, high amounts of thallium were required to observe a significant effect and there was significant leaching of the toxic thallium which would be prohibitive for industrial applications. Witońska et al.6 investigated palladium tellurium on silica catalysts and found that tellurium could also be used as a promoter for this reaction, again however deactivation of the catalysts was reported after ten reuses of the catalyst. Recently Zhang and co-workers have prepared homogeneous colloidal nanoparticles of gold–silver2 and gold–platinum–silver catalysts, once again these catalysts have demonstrated the extremely high TOF's with 20090 h−1.
We have previously shown that alloying gold and palladium can lead to a significant increase in the activity of the catalyst when compared to the gold or palladium monometallic equivalents,22 we have also shown that the sol immobilisation method can give catalysts with a narrow particle size distribution that are extremely active for alcohol oxidations.23 Heat treatment of these sol-immobilised catalysts can remove the polyvinyl alcohol (PVA) ligand but simultaneously leads to metal sintering causing deactivation of the catalyst during CO oxidation.24 Recently we have reported the use of magnesium oxide as a support for gold–palladium and gold platinum for the oxidation of glycerol, we have shown that during catalysts preparation the magnesium is predominately hydrated to form Mg(OH)2. The use of these catalysts facilitated the oxidation of glycerol without the addition of sacrificial base and at lower temperatures than those reported when other support materials are used.25 We now show that by combining the use of magnesium oxide/hydroxide support and the sol immobilisation preparation method we can carry out glucose oxidation under green, base-free conditions, using air at ambient pressure as the source of oxygen with excellent selectivity towards gluconic acid. We have shown that various heat treatments on the support and catalyst have a significant effect of the activity of the catalyst and explain these differences using XRD and TEM analysis.
Experimental
Catalyst preparation
All catalysts were prepared after pre-treatment of the support. The MgO (nanoscale corporation) was calcined at 400 °C for 5 h under static air (5 °C min−1). Au–Pd bimetallic catalysts were prepared using the following sol-immobilisation method: aqueous solutions of PdCl2 (Johnson Matthey, 6 mg in 1 ml) and HAuCl4·3H2O (Johnson Matthey, 12.25 g in 1000 ml) were freshly prepared. Polyvinyl alcohol (PVA) (1 wt% solution, Aldrich, Mw = 10000, 80% hydrolyzed) was added to the solution and stirred with HAuCl4·3H2O for 15 min. 0.1 M of NaBH4 (>96% Aldrich, NaBH4/Au (mol/mol = 5)) was freshly prepared and then added to form a dark brown sol. The mixture was stirred for 30 min and adjusted to pH 1 by the drop-wise addition of sulphuric acid. The pre-treated MgO was added to mixture and the slurry was then stirred for 1 h. Following this the catalyst was recovered by filtration, washed with 2 L distilled water and dried at 110 °C overnight. The 0.5% Au–Pd/Mg(OH)2 catalyst, denoted with the subscript z (zero treatments) was calcined at 450 °C for 5 h under air (20 °C min−1), denoted with the subscript a (air calcination) or treated at 450 °C for 5 h under N2 (20 °C min−1), denoted with the subscript n (nitrogen heat treatment) or subjected to both heat treatments sequentially, denoted with the subscript b (both treatments).
Glucose oxidation
Oxidation of glucose using air was carried out in a glass reactor consisting of a round-bottomed flask (50 ml) fitted with a reflux condenser. Typically, a supported gold catalyst (0.06 g) was suspended in the aqueous glucose solution (5 ml, 0.5 M glucose) at 50 °C. The reaction mixture was stirred for 24 h at atmospheric pressure. A sample was taken from the final reaction mixture (0.5 ml), diluted with 1 ml of distilled water and analysed by HPLC using an Agilent 1260 infinity fitted UV and RI detectors and a metacarb 87H column. Mass balances were based on calibration against known standards (Aldrich). In all experiments the mass balance was observed to be between 96–104%.Catalyst reuse
To reuse the catalyst a reaction was performed under standard conditions then the catalyst was collected by centrifuging the reaction mixture for 20 min. Acetone (30 ml) was added to the catalyst to remove any residues of reactants and products from the catalyst. The catalyst was recovered by decantation and drying (120 °C, 16 h).Catalyst characterisation
Results and discussion
The effect of reaction temperature
Initial experiments were carried out over a range of temperatures (40–70 °C) using Au–Pd/MgO(b) (0.06 g), for 24 h with oxygen from atmospheric air as the oxidant gas. The results are shown in Fig. 1, as expected increasing the temperature leads to a steady increase in conversion. The selectivity towards gluconic acid at the lower temperatures (40–60 °C) was 100% based on products formed with carbon mass balance between 96–104% while at the higher temperature (70 °C) the selectivity decreased below 100% (98%). Subsequently, we carried out a blank reaction test to be sure that the catalyst was having a beneficial effect on the reaction and the results are shown in Table 1. When no catalyst is present there was no conversion of the glucose, the addition of the support leads to a very small amount of conversion (1.3%) and the selectivity towards gluconic acid was 100% without the metals present. The addition of the gold to the support and subsequent calcination however led to a dramatic increase in the conversion of the glucose to 7.3% whilst still maintaining the 100% selectivity to gluconic acid. The use of a bimetallic Au–Pd/Mg(OH)2(z) increased the conversion from 2% to 27% (Table 3) with the same selectivity, suggesting a synergistic effect of combining the metals, an effect we have observed and reported previously which we consider to be due to an electronic effect.26 | ||
| Fig. 1 The conversion of glucose using Au–Pd/MgO(b): reaction conditions: glucose (5 ml, 0.5 M), catalyst (0.06 g), 24 h at atmospheric pressure, selectivity 100% except at 70 °C which was 98%. | ||
| Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Reaction conditions: glucose (5 ml, 0.5 M), catalyst (0.06 g), 60 °C, 24 h at atmospheric pressure. | ||
| No catalyst | 0 | 0 |
| Mg(OH)2 | 1.3 | 100 |
| 0.5% Au/Mg(OH)2(o) | 7.3 | 100 |
The effect of support
Previously we have shown that titania is an effective support for gold–palladium catalysts27 and that during glycerol oxidation, changing the support from graphite to magnesium oxide can lead to a significant increase in the activity in the absence of base25 we therefore prepared gold catalysts supported on magnesium oxide, titania and graphite. The activity of these supported catalysts is shown in Table 2, the reactions were carried out at 60 °C as this is the highest temperature where 100% selectivity to gluconic acid was maintained. Under these conditions the 0.5% Au/graphite(z) catalyst showed no activity for this reaction, the titania-supported catalyst displayed some conversion (0.8%) and the magnesium oxide-supported catalyst had over double the conversion, however this was still significantly lower than the catalytic data reported in Table 1 and within the realms of experimental error. The final catalyst reported in Table 1 had undergone two heat treatments (as detailed in the Experimental section) and it is clear that catalyst pre-treatment can significantly affect the activity.| Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Reaction conditions: glucose (5 ml, 0.5 M), catalyst (0.06 g), 60 °C, 24 h at atmospheric pressure. | ||
| 0.5% Au/graphite(z) | 0 | 0 |
| 0.5% Au/TiO2(z) | 0.8 | 100 |
| 0.5% Au/Mg(OH)2(z) | 2 | 100 |
The effect of heat treatment
To elucidate the reasons for the significant improvement in activity observed for the catalysts in Table 1 as compared with Table 2, we carried out catalytic tests after each heat treatment for the bimetallic catalysts and the results are shown in Table 3. When no heat treatment is applied to the catalyst, the conversion of glucose was 27%, calcination of the catalyst at 400 °C under static air led to almost a doubling of the activity to 50%, heat treatment under an inert atmosphere (nitrogen) led to a similar activity (53%), however, when the two heat treatments are carried out sequentially the conversion is improved further to 62%, again at this temperature, under these conditions the selectivity towards gluconic acid is 100% with all the catalysts tested. We have previously reported the effect of heat treatment on sol catalysts,24 we have shown that calcinations remove the PVA ligand from the metal particles however there is also significant particle growth with heat treatment. In a previous detailed study for a titania-supported gold catalyst24 we showed that the starting colloid has a mean particle size diameter of 3 nm, the 120 °C drying step increases this to 3.7 nm, however, calcination at 200 °C for 3 h significantly increases the particle size to 5.1 nm, and treatment at 400 °C led to much more significant particle growth with an average particle size of 10.4 nm, there was also a significant change in the particle size distribution from a positively skew when the catalyst was dried at 120 °C to a more standard bell-shaped normal distribution after the 400 °C treatment.| Catalyst | Catalyst heat treatment | Conversion (%) | Selectivity (%) | |
|---|---|---|---|---|
| Under air | Under N2 | |||
| a Reaction conditions: glucose (5 ml, 0.5 M), catalyst (0.06 g), 60 °C, 24 h at atmospheric pressure. | ||||
| 0.5% Au–Pd/Mg(OH)2(z) | X | X | 27 | 100 |
| 0.5% Au–Pd/MgO2(a) | √ | X | 50 | 100 |
| 0.5% Au–Pd/MgO2(n) | X | √ | 53 | 100 |
| 0.5% Au–Pd/MgO2(b) | √ | √ | 62 | 100 |
Catalyst characterisation
To try to understand if similar effects were occurring with the present catalyst characterisation by XRD and TEM was carried out. XRD analysis (Fig. 2) of the catalyst prepared with no heat treatment showed only the presence of crystalline Mg(OH)2 (ICDD 00-007-0239) and no reflections associated with the original MgO support. This hydration of the MgO support occurs during the catalyst synthesis and has previously been reported in the preparation of sol-immobilised catalysts used for glycerol oxidation.25 No reflections associated with the cubic Au or Pd phases were observed, though this could be associated with an overlap of the [101] Mg(OH)2 and [111] cubic metal reflections. Catalyst treatment under air resulted in dehydration of Mg(OH)2 back to MgO (ICDD 01-074-1225), with an average crystallite size of 6 nm (determined from [200] reflection). A reflection at 38.61° 2θ was observed which could be associated with the [101] Mg(OH)2 plane. However, no other reflections associated with Mg(OH)2 were observed, of particular note is the absence of the [011] reflection at 39.31° 2θ with a reported relative intensity of 57.6%. This along with the 0.6° shift in reflection position suggests that the observed reflection is more likely to be associated with a cubic metal [111] reflection. The position of this reflection lies between the reflections expected for pure Au (ICDD 00-004-0784) (38.19° 2θ) and pure Pd (ICDD 00-046-1043) (40.12° 2θ) and shows a degree of alloy formation, though the reflection position is lower than that expected for a bulk homogeneous alloy at 39.17° 2θ (ICDD 01-072-5376) suggesting the particles observable by XRD were Au rich. The calculated average crystallite size of the phase was relatively large at 22 nm. | ||
| Fig. 2 XRD patterns of the 0.5% Au–Pd/Mg(OH)2 catalyst after various heat treatments: (a) dried, (b) dried with calcination at 450 °C, (c) dried with heat treatment under nitrogen and (d) dried with both heat treatments. Expanded region shows the position of [111] cubic metal phase reflection. | ||
Treatment of the catalyst under nitrogen also resulted in the dehydration of the Mg(OH)2 to MgO with a crystallite size of 7 nm (Fig. 2). The presence of a broad poorly defined reflection, likely to be associated with a metal phase, was again observed, at 32.29° 2θ. The reflection position is again suggestive of an Au–Pd alloy, though with a unit cell size closer to Pd than that expected for a bulk homogeneous alloy. From the Scherrer equation this phase had an average crystallite size of 10 nm. Interestingly, the sequential heat treatments results in the formation of a metal phase with similar crystallite size to the nitrogen treatment (9 nm) but an apparent unit cell size that is now closer to Au than after the nitrogen treatment but still smaller than the calcined sample. The apparent trend we observe is that calcination, i.e. heating in air, resulted in the presence of an Au–Pd alloy that is possibly Au rich, whereas nitrogen heat treatment a phase that was Pd rich and the combined treatments in a phase that lies between the two single treatments.
TEM images of the unused catalyst are shown in Fig. 4 and associated particle size distributions, based on analysis of at least 200 particles, are shown in Fig. 5. In agreement with previous reports on 1% Au–Pd/MgO for glycerol oxidation25 the sample that is dried only has a narrow size distribution of small nanoparticles (1–10 nm). As we have previously reported for titania-supported catalysts24 the calcination treatment (Fig. 3b and 4b) leads to a growth in the mean particle size, although on magnesium hydroxide this is less evident than when titania is used as the support,24 even at the slightly higher calcination temperature of 450 °C, furthermore the particle size distribution remained positively skewed. The heat treatment under nitrogen at 500 °C also led to a growth in the average particle size although the effect was not as significant as when the calcination regime was applied. Fig. 3d and 4d show the TEM image and associated particle size distribution after both heat treatments have been applied to the catalyst, it is noteworthy that the double heat treated catalyst particle size distribution is most similar in nature to the to the nitrogen heat treated catalyst. The catalytic activity of both the nitrogen heat treated catalyst and the calcined catalyst are very similar suggesting that the particle size is not the single factor that is affecting the activity, we do not know what effect the removal of PVA ligand from the metal particles is having in this case, however it seems apparent that the smallest metal particles are not the most active for this reaction as has been previously suggested by Besson et al.10
 | ||
| Fig. 3 TEM images of the 0.5% Au–Pd/Mg(OH)2 catalyst after various heat treatments: (a) dried, (b) dried with calcination at 450 °C, (c) dried with heat treatment under nitrogen and (d) dried with both heat treatments. | ||
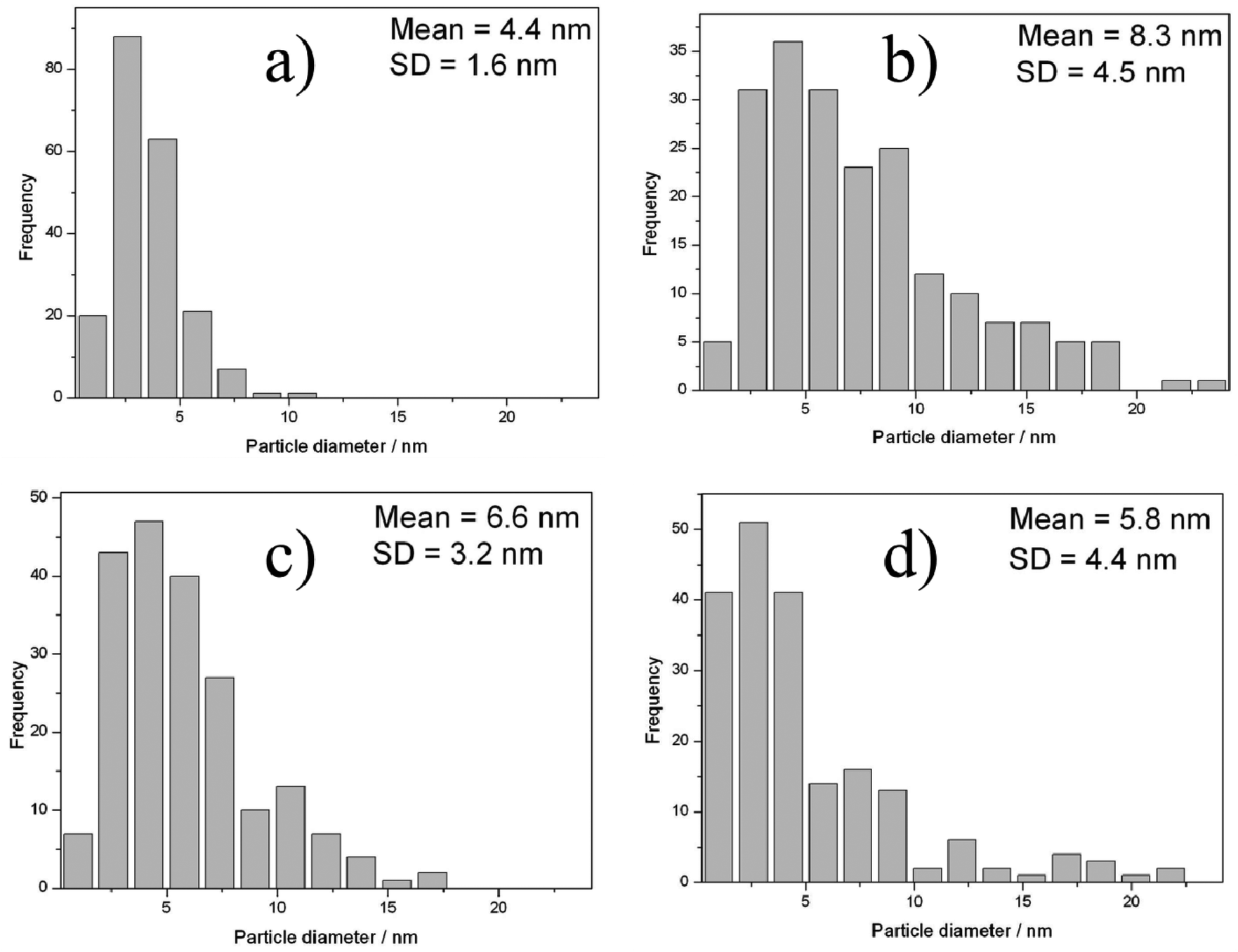 | ||
| Fig. 4 TEM particle size distributions of the 0.5% Au–Pd/Mg(OH)2 catalyst after various heat treatments: (a) dried, (b) dried with calcination at 450 °C, (c) dried with heat treatment under nitrogen and (d) dried with both heat treatments. | ||
 | ||
| Fig. 5 Time on line data for the oxidation of glucose using the 0.5% Au–Pd/MgO catalyst. Reaction condition: glucose (5 ml, 0.5 M), catalyst (0.06 g), 50 °C at atmospheric pressure. | ||
Non-alloyed catalysts
When the TEM data is considered in combination with the XRD data is seems to suggest that a fully homogeneous alloy catalyst is not the most active for this reaction. In fact a catalyst which contains mostly gold or palladium with a small amount of the other metal alloyed appears to be more active for this reaction once a high temperature heat treatment has taken place. To test this theory monometallic gold and palladium catalysts were synthesised and subjected to the same heat treatment regime as the bimetallic catalyst, the results are shown in Table 4, it is clear that the heat treatments lead to a significant increase in the catalytic activity of both monometallic catalysts (57% for Au and 52.7% for Pd). The value for the gold catalysts can be compared to the 7.3% conversion reported in Table 1 where one heat treatment had been carried out on an equivalent catalyst. The gold monometallic catalyst was slightly more active than the palladium and under the mild conditions employed in this work the selectivity towards gluconic acid remained at 100% with both catalysts. Both monometallic catalysts are slightly less active than the bimetallic catalyst supporting the theory that a small amount of alloying is required for maximum activity. For comparison a reaction was carried out with the addition of sodium hydroxide, the result is also shown in Table 4, although the conversion is significantly higher in the presence of base the selectivity to gluconic acid was dropped below 100%.Catalyst deactivation studies
To investigate the reactivity of the catalysts for glucose oxidation the effect of reaction time on the conversion and selectivity was studied with the optimised Au–Pd/MgO(b) catalyst at 50 °C as this temperature has been reported previously in several publications,9,11,14 the results are shown in Fig. 6, it is clear that while the catalyst has a high initial activity the rate of reaction decreases with time and between 24 and 48 h there is only a very small increase in the conversion. As the conversion is still significantly less than 100% at the end of the reaction it is possible that there is catalyst deactivation occurring. Therefore we carried out reuse tests on the catalysts to see if there was deactivation of the catalyst between runs. The results of these tests are shown in Table 5. The activity of the catalyst roughly halved upon reuse therefore we carried out a product inhibition test. As there is only one product in this reaction, gluconic acid, this was added to glucose at the start of the reaction. Gluconic acid (0.1 g, 5.1 × 10−4 mol, glucose:gluconic acid ∼4.9) was added (representing the concentration that would be present at ∼20% conversion) to the standard reaction but otherwise the reaction was carried out as has been described previously. The results of this reaction are also shown in Table 5. The conversion of the reaction after 24 h is about 33% lower than under the standard reaction conditions (41.3% (Table 5) as compared with 62% (Table 3)). With this deactivation apparently caused by the product we heat treated the used catalyst, the heat treatment was carried out at 450 °C after the catalyst had been recovered by the method described in the experimental in the catalyst reuse section. As shown in Table 5, the catalyst heat treatment did lead to an increase in conversion when compared with the untreated reused catalyst but it did not restore the activity to that of the fresh catalyst (Table 6).
 | ||
| Fig. 6 Characterisation of AuPd/MgO catalyst after use in the base free glucose oxidation reaction at 50 °C. Upper figure shows XRD diffraction pattern of catalyst with □ indicating reflections associated with Mg(OH)2. Lower figures show a representative TEM image of metal nanoparticles on the used catalyst (left) and the corresponding particle size distribution. | ||
| Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Reaction conditions: glucose (5 ml, 0.5 M), of 0.5% Au–Pd/MgO(b) (0.06 g), 60 °C, 24 h at atmospheric pressure. | ||
| Fresh | 62.0 | 100 |
| First reuse | 24.4 | 100 |
| First reuse with treatment of catalyst | 33.7 | 100 |
| Second reuse | 18.7 | 100 |
| Second reuse with treatment of catalyst | 23.2 | 100 |
| Catalyst | Gluconic acid | Conversion (%) | Selectivity (%) |
|---|---|---|---|
| a Reaction conditions: glucose (5 ml, 0.5 M), catalyst (0.06 g), 60 °C, 24 h at atmospheric pressure. | |||
| 0.5% Au/MgO(b) | — | 62.0 | 100 |
| 0.5% Au/MgO(b) | 0.1 g | 41.3 | 100 |
Characterisation by DRIFTS, XRD and TEM microscopy of the Au–Pd/MgO(b) catalyst after a 24 h reaction at 50 °C was performed to further investigate catalyst deactivation. XRD analysis (Fig. 6) revealed that the MgO support completely converted to Mg(OH)2 under the aqueous reaction conditions, which could be expected to result in significant agglomeration of the supported nanoparticles. TEM analysis (Fig. 6) did show a modest increase in particle size from a mean of 5.8 nm to 6.5 nm. This small increase in particle size could impact on activity, although it being a significant factor is highly improbable. What is surprising is the minimal change in metal particle size on restructuring of the support. Interestingly DRIFTS of the initial and used catalysts shows, not only the presence of adsorbed organic species, but also the “surface” structure of the catalyst itself (Fig. 7 and Table 7). The spectra of both the initial and used catalysts show strong hydroxyl groups associated with magnesium hydroxide, demonstrating that even the initial Au–Pd/MgO(b) catalyst, shown to be comprised of bulk MgO by XRD, had a hydrated surface. This surface magnesium hydroxide could potentially stabilise the AuPd nanoparticles when the bulk MgO support hydrates to Mg(OH)2 under reaction conditions.
 | ||
| Fig. 7 DRIFTS analysis of AuPd/MgO catalyst (a) before reaction, (b) after base free glucose oxidation reaction at 50 °C, (c) in situ spectra of post reaction sample heated to 115 °C and (d) in situ spectra of post reaction sample heated to 440 °C. In situ spectra were recorded on samples at designated temperature under vacuum. Arrows indicate bands associated with adsorbed organic species. | ||
| Mg(OH)2 | MgCO3 | Fresh catalyst | Used catalyst | Used at 115 °C | Used at 450 °C | Band assignment |
|---|---|---|---|---|---|---|
| 3698 | — | 3763, 3714, 3701, 3620 | 3699, 3646 | 3699, 3646 | 3697, 3646 | ν(Mg–OH) |
| — | — | 3361 | 3400 | 3360 | 3295 | ν(OH) |
| — | — | 2930, 2870, 2845, 2740 | 2930, 2870, 2845, 2740 | 2930, 2870, 2845, 2740 | ν(CH) | |
| — | — | 1647 | — | δ(H2O) | ||
| — | 1635 | 1653 | 1653 | ν(CO) | ||
| — | 1450 | 1454 | — | ν 3(CO3) | ||
| 1396, 1360 | 1405, 1336 | 1420, 1330 | δ(CH)–(OCH) | |||
| 1232 | 1230 | δ(CH) | ||||
| 1130 | 1126 | 1126 | ν(CO) | |||
| — | 1080 | 1070 | ν 1(CO3) | |||
| — | 870 | 830 | ν 4(CO3) | |||
| 787 | 784 | 785 | ||||
| 625 | — | 660 |
DRIFTS of the used catalyst did also show distinct bands, relative to the initial catalyst, that correspond to alcohol groups, C–H and CO bands. Heating of the used catalyst in situ under vacuum up to 450 °C resulted in only a reduction in the broad OH band at 3400 cm−1, associated with physisorbed water. This showed that the adsorbed organic species were exceptionally strongly bound to the catalyst surface and supports the idea that product inhibition is the primary cause of the observed deactivation. Identification of the adsorbate from band assignments of the used and used heat treated samples is complicated due to the range of structural isomers of glucose and gluconic acid. However, the presence of the band at 1653 cm−1 in the used and heated samples is suggestive of a carbonyl group. The band position is at the extreme low end of the range observed for carbonyls and is in a similar range to the expected bending mode of physisorbed water. Yet its retention at 450 °C, strongly suggests that the band in question is not associated with physisorbed water. The ν(OH) band remaining in the heated samples, at 3295 cm−1, can be identified as being associated with a carboxylic acid group. It is therefore probable that the adsorbed species was gluconic acid. Clearly product inhibition represents a key area that needs to be addressed in future studies. Perhaps the use of polar solvents could be beneficial but this negates the green chemistry approach of using a solvent-free reaction system however, our observations do show that there may be limitations to the application of aqueous catalysed reactions.
In addition to the previously mentioned post reaction characterisation, MP-AES analysis was carried out on the reaction mixture after the 24 h reaction had been completed, to determine if metal leaching was occurring during the reaction. Analysis of both gold and palladium revealed that neither metal was present in the post reaction sample within the detection limits of the equipment (standards made between 0.6 and 10 ppm). However, we observed that Mg was present (2890 ppm), showing that the Mg(OH)2 support was partially soluble in the reaction medium. Previous use of MgO as a solid base for base free glycerol oxidation resulted in 77 ppm of Mg2+ being leached into solution, though the reaction time was far shorter at 4 h. In view of the extent of Mg2+ that is leached in the current experiments we have to consider the effect of the homogeneous Mg(OH)2 acting as a sacrificial base. The calculated [OH−] was 2.38 × 10−4 mol ml−1, which is equivalent to a [OH] : glucose ratio of 0.048:1 at the start of the reaction, and a ratio of 0.16:1 at 70% conversion. This suggests that while the catalyst itself is unstable, the dissolved Mg(OH)2 does not provide significant amounts of sacrificial homogeneous base to influence the reaction. To test this we carried out a reaction with a stable catalyst which we have previously reported,22 with and without the addition of sodium hydroxide at the same ratio (i.e. [OH−] : glucose = 0.048:1). Under our reaction conditions we observed conversions of 3.6 and 3.8% in the absence and presence of base respectively, indicating that the dissolved base is not responsible for the high activity we observe with the catalysts we now report. Though we can determine the efficacy of the work in demonstrating the ability to perform base free oxidation of glucose under mild conditions, the instability of the catalyst support shows that MgO is clearly not fully viable for this application. Further work on other more stable basic supports is now required.
Conclusions
We have shown that the oxidation of glucose can be carried out under extremely mild conditions. In particular, the choice of support for the reaction can lead to the removal of sacrificial base from the reaction by using a basic support for the catalytically active metal nanoparticles. We have also shown that by using the sol-immobilisation technique catalysts can be prepared that can utilise atmospheric air as the oxidant gas, rather than using pressurised or bubbling oxygen through the reaction mixture. Furthermore the mild conditions of this reaction are massively beneficial in terms of the selectivity towards gluconic acid.We have demonstrated that the heat treatment of these sol immobilised catalysts can have a marked effect on their activity. XRD and TEM analysis show that there is large particle growth when the catalyst is either calcined, heat treated under nitrogen or undergoes both treatments. There is also apparent phase separation of the metals when both treatments are applied however this is the most active catalyst indicating that only a small amount of alloying is required. Finally we have observed catalyst deactivation with time which we attribute to product inhibition which may be alleviated by using a suitable solvent however support dissolution remains a problem.
Acknowledgements
We would like to thank Cardiff University for funding. Also we would like to acknowledge the UK catalysis hub. TEM data was acquired at the Research Complex at Harwell (UK Catalysis Hub) by Catherine Brooks and Wilm Jones.References
- S. Hermans and M. Devillers, Appl. Catal., A, 2002, 235, 253–264 CrossRef CAS.
- H. Zhang, J. Okuni and N. Toshima, J. Colloid Interface Sci., 2011, 354, 131–138 CrossRef CAS PubMed.
- M. Comotti, P. C. Della, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313–316 CrossRef CAS.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS.
- S. Karski, J. Mol. Catal. A: Chem., 2006, 253, 147–154 CrossRef CAS PubMed.
- I. Witońska, M. Frajtak and S. Karski, Appl. Catal., A, 2011, 401, 73–82 CrossRef PubMed.
- M. Comotti, P. C. Della, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313–316 CrossRef CAS.
- M. Comotti, C. D. Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 89–92 CrossRef CAS PubMed.
- A. Abbadi and B. H. van, J. Mol. Catal. A: Chem., 1995, 97, 111–118 CrossRef CAS.
- M. Besson, F. Lahmer, P. Gallezot, P. Fuertes and G. Fleche, J. Catal., 1995, 152, 116–121 CrossRef CAS.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- M. Wenkin, R. Touillaux, P. Ruiz, B. Delmon and M. Devillers, Appl. Catal., A, 1996, 148, 181–199 CrossRef CAS.
- M. Comotti, P. C. Della, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- Y. Önal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef PubMed.
- C. Baatz and U. Prüße, J. Catal., 2007, 249, 34–40 CrossRef CAS PubMed.
- A. Mirescu, H. Berndt, A. Martin and U. Prüße, Appl. Catal., A, 2007, 317, 204–209 CrossRef CAS PubMed.
- N. Thielecke, K.-D. Vorlop and U. Prüße, Catal. Today, 2007, 122, 266–269 CrossRef CAS PubMed.
- C. Baatz, N. Thielecke and U. Prüße, Appl. Catal., B, 2007, 70, 653–660 CrossRef CAS PubMed.
- M. Comotti, P. C. Della, E. Falletta and M. Rossi, J. Catal., 2006, 244, 122–125 CrossRef CAS PubMed.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and H. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS PubMed.
- S. Karski, Chem. Environ. Res., 2006, 15, 113–122 CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921–1930 RSC.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS PubMed.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS PubMed.
- S. A. Kondrat, G. Shaw, S. J. Freakley, Q. He, J. Hampton, J. K. Edwards, P. J. Miedziak, T. E. Davies, A. F. Carley, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Chem. Sci., 2012, 3, 2965–2971 RSC.
| This journal is © The Royal Society of Chemistry 2014 |