 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chiral organocatalytic polymer-based monolithic reactor†
Valerio
Chiroli
a,
Maurizio
Benaglia
*ac,
Alessandra
Puglisi
ac,
Riccardo
Porta
a,
Ravindra P.
Jumde
bc and
Alessandro
Mandoli
*bc
aDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy. E-mail: maurizio.benaglia@unimi.it; Fax: +390250314159; Tel: +390250314171
bDipartimento di Chimica e Chimica Industriale, Università di Pisa, via Risorgimento, 35, 56126, Pisa, Italy
cISTM-CNR, via Golgi 19, 20133 Milano, Italy
First published on 5th March 2014
Abstract
Radical copolymerisation of divinylbenzene and a properly modified enantiomerically pure imidazolidinone inside a stainless steel column in the presence of dodecanol and toluene as porogens afforded the first example of a chiral organocatalyst immobilized onto a monolithic reactor. Organocatalyzed cycloadditions between cyclopentadiene and cinnamic aldehyde were performed under continuous-flow conditions; by optimizing the experimental set up, excellent enantioselectivities (90% ee at 25 °C) and high productivities (higher than 330) were obtained, thus showing that a catalytic reactor may work efficiently to continuously produce enantiomerically enriched compounds. The same catalytic reactor was also employed to carry out three different stereoselective transformations in continuo, sequentially, inside the chiral column (Diels–Alder, 1,3-dipolar nitrone-olefin cycloaddition, and Friedel–Crafts alkylation); excellent results were obtained in the case of the former two reactions (up to 99% yield, 93% ee and 71% yield, 90% ee, at 25 °C, respectively). In addition to simplify the product recovery, the monolithic reactor performed better than the same supported organocatalyst in a stirred flask and could be kept working continuously for more than 8 days.
The synthesis of fine chemicals in flow presents new and exciting challenges and opportunities for both homogeneous and heterogeneous transformations;1 in this context great opportunities are offered by the use of immobilized chiral catalysts under flow conditions.2 For decades petrochemical and commodity chemical industries have exploited heterogeneous catalytic processes, performing in flow many important transformations.3 However, the application of continuous-flow methodologies to the stereoselective synthesis of chiral multifunctional molecules is much less developed.4 Although in the fine chemical industry production relies at the present on batch or semi-batch processes, where flexibility and versatility are guaranteed, the possibility in the future to design a multi-purpose plant based on a continuous process is very attractive and full of promise.5 Further opportunities are offered if microreactor-based technologies are taken into consideration. Since HPLC, GC-MS or LC-MS technologies can be easily integrated into microflow systems, the inline analysis of the transformations performed in micro (or mini) reactors can provide reaction information more efficiently than flask reactors.
When the use of a chiral catalyst under continuous-flow conditions is envisaged, the heterogenization of the enantiomerically pure catalytic species plays a crucial point in the development of efficient systems.6 The retention of the catalyst inside the reaction vessel can be achieved by different techniques ranging from ultrafiltration through a MW-selective membrane to immobilization on an organic polymer or an inorganic material like silica gel. According to the method used to incorporate an immobilized catalyst into the microfluidic device, catalytic (micro)reactors7 can be divided into three main typologies: packed-bed, monolithic and inner wall-functionalized. The application of the last mentioned approach is almost unknown for chiral catalysts;8 on the other hand the first one has been widely investigated, with both organic and organometallic systems. Indeed most of the continuous-flow processes utilize reactors with randomly packed catalytic beds; this approach, however, does not withstand requirements from a process and chemical engineering point of view. Major drawbacks of these systems are uncontrolled fluid dynamics, stagnation zones and hot-spot formation, broad residence time distribution, low selectivity and possibly low process efficiency.
Some of these problems may find a solution by the development of monolithic materials, functionalized with a chiral promoter. A monolith is defined as “a block of structured material which consists of continuous substructures of regular or irregular channels”.9 Since monoliths are characterized by a high void volume and a large geometric surface area, the passage of a gas or a fluid occurs without significant pressure drop, and a large contact area of the catalyst with the fluid is guaranteed.10 Monolithic materials may be prepared by copolymerisation of different monomers in the presence of porogens,11 or by polymerisation of a monolithic polymeric phase wedged inside the microchannel pore system of an inert support such as glass.12
While numerous examples of achiral catalytic monoliths are known,13 only very few examples of effective chiral catalysts immobilized onto monoliths have been reported. After early studies on TADDOL derivatives,14 in 2007 Luis, Mayoral and coworkers reported the synthesis of a heterogenized chiral pyridyl bisoxazoline (Pybox) by polymerization of 4-vinyl-pybox in the presence of styrene and divinylbenzene to generate a macroporous monolithic miniflow reactor.15 The corresponding ruthenium/pybox complex was evaluated in the cyclopropanation reaction between styrene and ethyldiazoacetate under flow conditions. A slightly modified monolithic mini-flow reactor containing a supported bisoxazoline-Cu(OTf)2 complex was then prepared and used in the same cyclopropanation reaction.16 Recently the same group described the continuous-flow cyclopropanation reaction promoted by a new supported catalyst with improved efficiency, a pyridineoxazoline (pyox)-copper complex.17
As far as we know, however, chiral organocatalysts immobilized onto a monolithic support are completely unknown; moreover, only a few examples of chiral organocatalysts employed under continuous-flow conditions were reported. After the pioneering work by Lectka with polystyrene-immobilized cinchona alkaloid derivatives,18 Pericas and Massi have studied the use of polymer-supported proline19 and prolinol20 derivatives in mini flow reactors. Lately Fulop21 and Wennemers22 have reported stereoselective Michael additions promoted in continuo by polymer-supported tripeptides. Very recently, Pericas and coworkers reported the synthesis of a polystyrene-supported pyrrolidine-based catalyst23 and a 1,1′-bi-2-naphthol-derived phosphoric acid24 to be used under continuous flow conditions. It should be noted that all these works were almost exclusively limited to the use of packed-bed reactors filled with catalyst supported onto inorganic or gel-type organic materials, with substrate activation via enamine intermediates.
We wish to report here the preparation of the first monolithic material loaded with a chiral imidazolidinone organocatalyst25 and its use to promote stereoselective reactions via substrate-iminium activation.
An ad hoc designed MacMillan type catalyst was easily synthesized starting from (S)-tyrosine methyl ester 1, to give after a single chromatographic purification the imidazolidinone 2 bearing a carbon–carbon triple bond (Scheme 1 and ESI†). The modified MacMillan catalyst 2 was reacted with 4-(azidomethyl)styrene in the presence of CuCl and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA), to afford the corresponding styrene monomer 3.
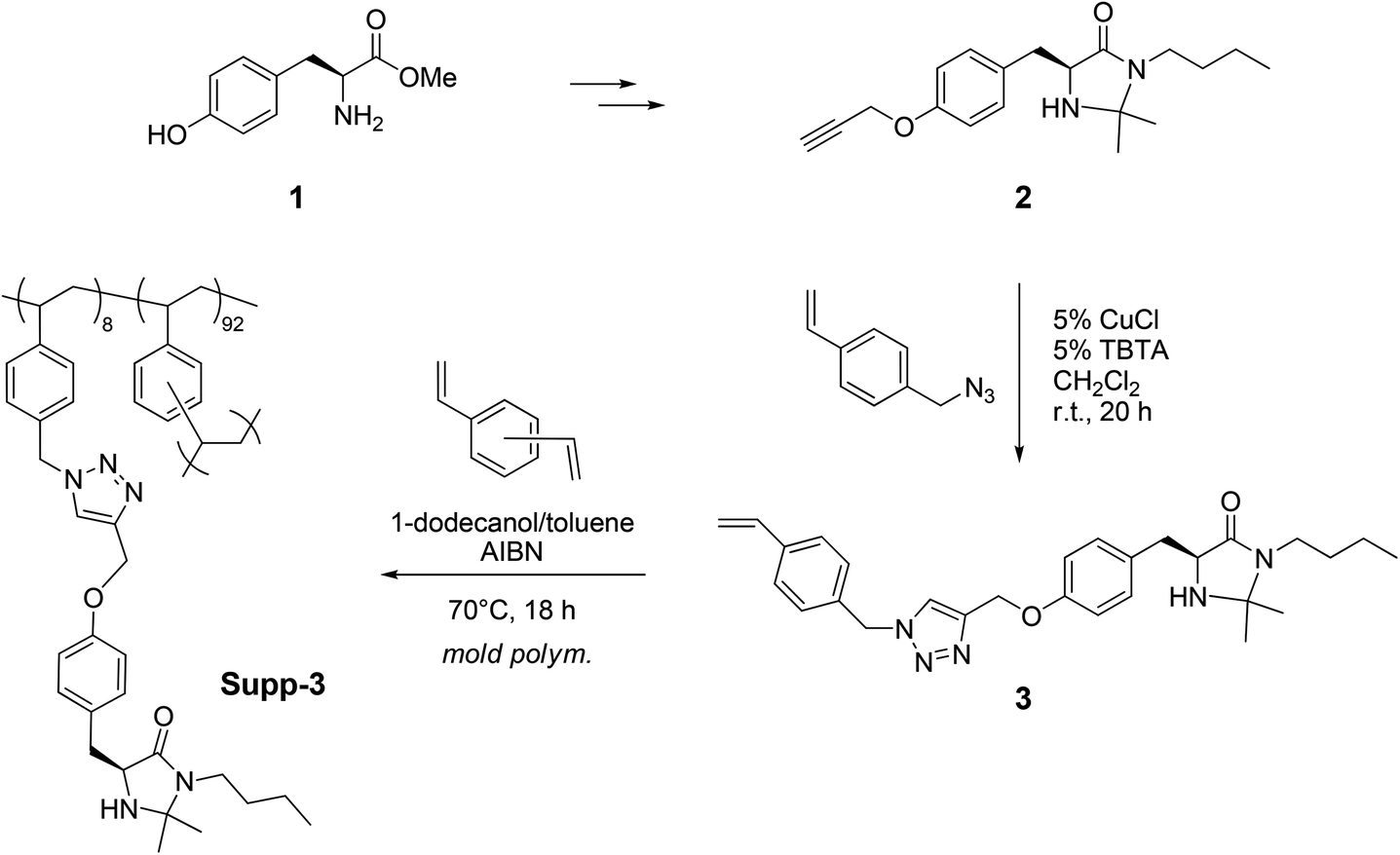 | ||
| Scheme 1 Preparation of the monolithic reactor containing the chiral imidazolidinone catalyst Supp-3. | ||
The monolithic organocatalyst was prepared inside a standard HPLC column (0.46 cm i.d. ×15 cm, 2.49 mL total volume) under Frechét-type conditions for polystyrene materials.11 The AIBN initiated copolymerization of 3 and divinylbenzene (DVB) was carried out at 70 °C in the presence of 1-dodecanol and toluene as the porogenic solvents (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, approx. 60 vol% of the feed mixture). Exhaustive washing of the column with THF and analysis of the eluate indicated a complete incorporation of the monomers into the monolith.26 Based on these results, the loading of MacMillan organocatalyst onto the polymeric support (0.51 mmol g−1 or 25 wt%), the absolute amount of the chiral derivative immobilized inside the flow device (0.48 mmol) and the void volume in the reactor (1.49 ml)27 could be calculated directly from the feed composition.
:1 v/v, approx. 60 vol% of the feed mixture). Exhaustive washing of the column with THF and analysis of the eluate indicated a complete incorporation of the monomers into the monolith.26 Based on these results, the loading of MacMillan organocatalyst onto the polymeric support (0.51 mmol g−1 or 25 wt%), the absolute amount of the chiral derivative immobilized inside the flow device (0.48 mmol) and the void volume in the reactor (1.49 ml)27 could be calculated directly from the feed composition.
For comparative purposes, several monolithic reactors were prepared as described above. The filling polymeric material from two of such columns was removed from the device, crushed, thoroughly washed with MeCN and dried. By this procedure the powder form of the same supported organocatalyst in the monolithic reactors could be obtained, which was used for running batch heterogeneous catalysis reactions as well as for preparing a packed-bed continuous-flow reactor (vide infra).
IR characterization of the material (ESI†)25e confirmed the incorporation and integrity of the chiral units (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1695 cm−1).28 N2 adsorption measurement provided a BET specific surface area (485 m2 g−1) consistent with a texture comprising meso- and micropores.11 At the same time, preliminary flow tests revealed the good fluid dynamic properties associated with the occurrence of convective pores (P < 5 bar at flow-rate ϕ = 300 μL min−1 of THF or MeCN).11 Among the several stereoselective transformations catalyzed by chiral imidazolidinones,29 the Diels–Alder cycloaddition between trans-cinnamaldehyde (4) and cyclopentadiene30 was selected as the first model reaction to study the catalytic behavior.
O = 1695 cm−1).28 N2 adsorption measurement provided a BET specific surface area (485 m2 g−1) consistent with a texture comprising meso- and micropores.11 At the same time, preliminary flow tests revealed the good fluid dynamic properties associated with the occurrence of convective pores (P < 5 bar at flow-rate ϕ = 300 μL min−1 of THF or MeCN).11 Among the several stereoselective transformations catalyzed by chiral imidazolidinones,29 the Diels–Alder cycloaddition between trans-cinnamaldehyde (4) and cyclopentadiene30 was selected as the first model reaction to study the catalytic behavior.
The reaction (Scheme 2 and Table 1) was initially carried out at 25 °C, by pumping a solution of the reagents at ϕ = 5 μL min−1 through the monolithic reactor containing the imidazolidinone organocatalyst Supp-3 activated by treatment with trifluoroacetic acid (TFA).31
 | ||
| Scheme 2 Enantioselective Diels–Alder reactions with different substrates under continuous-flow conditions catalyzed by imidazolidinone supported catalyst Supp-3. | ||
| Entry | Running time [h] | ϕ [μL min−1] | τ [h] | Yieldc [%] | drd | ee endo-5 (ee exo-5)e [%] |
|---|---|---|---|---|---|---|
| a Reactions conditions: 25 °C; HPLC column (0.46 cm i.d. × 15 cm, containing 0.475 mmol of catalyst); cinnamaldehyde (0.195 M, 1 eq.) and cyclopentadiene (1.365 M, 7 eq.) in 95/5 CH3CN–H2O mixture. b Residence time calculated as void volume/flow rate. c Yields determined by 1H NMR and confirmed on isolated products after chromatographic purification. d Endo-5/exo-5 diastereoisomeric ratio, determined by 1H NMR on the crude reaction mixture. e Enantiomeric excess determined by HPLC on the alcohols obtained by NaBH4 reduction of adducts. | ||||||
| 1 | 0–2 | 5 | — | 20 | 46/54 | n.d. |
| 2 | 2–4 | 5 | — | 47 | 44/56 | 86 (85) |
| 3 | 4–6 | 5 | 5 | 54 | 42/58 | 91 (93) |
| 4 | 6–18 | 5 | 5 | 58 | 43/57 | 90 (92) |
| 5 | 18–20 | 5 | 5 | 61 | 43/57 | 91 (90) |
| 6 | 20–48 | 2 | 12.4 | 77 | 44/56 | 90 (88) |
| 7 | 48–120 | 1 | 24.8 | 91 | 43/57 | 87 (86) |
After a conditioning time of 4–6 hours a steady-state regime was reached, which allowed us to produce in continuo the cycloadducts 5 in 54–61% yield. In agreement with the results obtained with the non-supported catalyst,30 the product was obtained as a rough 1:1 mixture of endo/exo isomers, both with enantioselectivities higher than 90% ee (entries 3–5, Table 1). In order to improve the chemical yield, the flow rate was reduced so as to increase the residence time τ. Indeed with ϕ = 2 μL min−1 the product was isolated in 77% yield that was further incremented to 91%, by operating at ϕ = 1 μL min−1 (entries 6–7, Table 1).32
In order to compare the behavior of the polymer-supported catalyst under batch and continuous-flow conditions, the same reaction between 4 and cyclopentadiene was performed in a flask with polymeric powder Supp-3 and trifluoroacetic acid (Table 2). By running the reaction at 25 °C, in the presence of 30% mol amount of the supported catalyst the product was obtained in 60% yield after 24 hours and 85% yield after 48 hours (90% ee, entries 1 and 2, Table 2). The yield did not further increase at longer reaction times, thus suggesting the loss of catalytic activity in a relatively short time with respect to the continuous-flow set-up described above: possibly, the continuous flow efficiently removes trace impurities and minimizes catalyst deactivation due to product inhibition, which helps in preserving the catalytic activity of the chiral species in the flow reactor for longer operation times than in the batch process.
| Entry | Acid additive | t [h] | Yieldb [%] | drc | ee endo-5 (ee exo-5)d [%] |
|---|---|---|---|---|---|
| a Reactions run at 25 °C; for the other conditions, see the ESI. b Yields determined by 1H NMR and confirmed on isolated products after chromatographic purification. c Endo-5/exo-5 diastereoisomeric ratio determined by 1H NMR on the crude reaction mixture. d Enantiomeric excess determined by HPLC on the alcohols obtained by NaBH4 reduction of adducts. e Recovered catalyst was employed. | |||||
| 1 | TFA | 24 | 60 | 46/54 | 90 (90) |
| 2 | TFA | 48 | 85 | 44/56 | 90 (89) |
| 3 | TFA | 72 | 88 | 47/53 | 89 (88) |
| 4e | TFA | 48 | 25 | 42/58 | 71 (75) |
| 5 | HBF4 | 24 | 77 | 45/55 | 90 (86) |
| 6 | HBF4 | 48 | 97 | 43/57 | 90 (88) |
| 7e | HBF4 | 72 | 70 | 44/56 | 85 (83) |
In addition to facilitating the separation of the catalyst from the reaction products, the immobilization on a polymer should allow simple catalyst recovery and recycling. In this case, the separation of the catalyst was obtained by concentrating the reaction mixture under vacuum and adding hexanes/diethyl ether. The polymer-supported catalyst was then isolated by centrifugation and filtration in yields ranging from 50 to 80% and the organic phase was worked-up to obtain the products. The recovered catalyst was then shortly dried under vacuum to remove traces of solvent and recycled.33 However, already after one cycle a marked decrease in the chemical and stereochemical efficiency was observed (25% yield and 71–75% ee after 48 h; entry 4, Table 2). Better results were obtained when the tetrafluoroborate salt of the immobilized MacMillan catalyst was employed (entries 5–7, Table 2). In that case the product was isolated in an almost quantitative yield after 48 hours, and the recovered catalyst performed also somehow better than the corresponding TFA salt. Based on this observation, further experiments were carried out by using HBF4 as the acid additive.
The general applicability of the chiral monolithic reactor was studied next (Scheme 2 and Table 3). With this aim an identical monolithic column, containing polymeric Supp-3 (activated by tetrafluoroboric acid), was used for performing the reaction in continuo between cyclopentadiene and three different aldehydes.
| Entry | Running time [h] | Product | Yieldb [%] | drc | ee endo(ee exo)d [%] |
|---|---|---|---|---|---|
| a Flow rate ϕ = 2 μL min−1; residence time = 12.4 h. Reactions conditions: 25 °C; HPLC column (0.46 cm i.d. × 15 cm, containing 0.475 mmol of catalyst); α,β-unsaturated aldehyde (0.195 M, 1 eq.) and cyclopentadiene (1.365 M, 7 eq.) in 95/5 CH3CN–H2O mixture. b Yields determined by 1H NMR and confirmed on isolated products after chromatographic purification. c Endo/exo diastereoisomeric ratio determined by 1H NMR on the crude reaction mixture. d Enantiomeric excess of the endo diastereoisomer, determined by HPLC (ESI); in parentheses enantiomeric excess of the exo diastereoisomer. e Column washing. | |||||
| 1 | 0–24 | 5 | 75 | 47/53 | 92 (91) |
| 2e | 24–32 | — | — | — | — |
| 3 | 32–48 | 7 | 88 | 49/51 | 90 (91) |
| 4 | 48–60 | 7 | 95 | 47/53 | 91 (90) |
| 5e | 60–72 | — | — | — | — |
| 6 | 72–86 | 9 | 94 | 52/48 | 83 (75) |
| 7 | 86–108 | 9 | 97 | 49/51 | 85 (75) |
| 8e | 108–120 | — | — | — | — |
| 9 | 120–150 | 5 | 73 | 47/53 | 94 (89) |
Expectedly, repetition of the reaction with cinnamic aldehyde afforded product 5 in 75% yield and 90% ee for both isomers (entry 1, Table 3), thus confirming chemical and stereochemical performances comparable to the TFA-activated monolithic catalyst (see entry 6, Table 1).
Then, the column was washed and used in further cycloaddition runs with different aromatic aldehydes. By pumping a 95/5 CH3CN–H2O solution of 2-nitro-cinnamic aldehyde (6) and cyclopentadiene, the catalytic column continuously produced the cycloadduct 7 in up to 94% yield and 90–91% ee for both diastereoisomers (entries 3 and 4, Table 3).
After 3 days of continuous operation the reactor was washed and used for carrying out the reaction between cyclopentadiene and crotonic aldehyde (8). Gratifyingly, also in this third run the expected cycloadduct (9) was obtained in yields higher than 94% and enantioselectivities up to 85% ee (entries 6 and 7, Table 3).
Finally, in order to verify the activity of the system after a prolonged time on stream (TOS) the reactor was washed once more and used to promote again the initial reaction between cyclopentadiene and cinnamic aldehyde. Indeed, after 150 working hours of the catalytic reactor, product 5 was isolated in yields and stereoselectivities totally comparable with those of the first 24 hours of activity (compare entry 9 with entry 1, Table 3).
Based on the data of Tables 1 and 3, it is evident that the long residence time represents a major drawback of the present system, which hampers a really advantageous use of the monolithic reactor in an effective continuous process. Therefore some optimization studies have been performed on the model Diels–Alder reaction in continuo of cinnamic aldehyde with cyclopentadiene, carried out with the tetrafluoroborate salt of the monolithic imidazolidinone Supp-3 (Table 4).
a
| Entry | Running time [h] | ϕ [μL min−1] | τ [h] | Yieldc [%] | drd | ee endo-5 (ee exo-5)e [%] |
|---|---|---|---|---|---|---|
| a Reactions run at 25 °C; Reactions conditions: 25 °C; HPLC column (0.46 cm i.d. × 15 cm, containing 0.475 mmol of catalyst); cinnamaldehyde (0.195 M, 1 eq.) and cyclopentadiene (1.365 M, 7 eq.) in 95/5 CH3CN–H2O mixture. b Residence time. c Yields determined by 1H NMR and confirmed on isolated products after chromatographic purification. d Endo-5/exo-5 diastereoisomeric ratio, determined by 1H NMR on the crude reaction mixture. e Enantiomeric excess determined by HPLC on the alcohols obtained by NaBH4 reduction of adducts. | ||||||
| 1 | 0–4 | 2 | 12.4 | n.d. | — | — |
| 2 | 4–24 | 2 | 12.4 | 75 | 47/53 | 92 (91) |
| 3 | 24–44 | 7.7 | 3.2 | 67 | 48/52 | 90 (90) |
| 4 | 44–55 | 18.8 | 1.3 | 68 | 47/53 | 90 (86) |
| 5 | 55–70 | 18.8 | 1.3 | 73 | 45/55 | 90 (88) |
After the first 24 hours of operation at ϕ = 2 μL min−1, the flow rate was progressively increased up to 18.8 μL min−1 (entries 2–5, Table 4). Much to our delight, under these conditions only a marginal decrease in the chemical yield was observed, but without any change in the enantioselection extent. Indeed, at ϕ = 18.8 μL min−1 the catalytic reactor kept on producing the cycloadducts in 73% yield and 90% ee for both isomers, with a remarkable productivity improvement over previous conditions (for comparative data, see Table 6 below). Although the reasons for the relatively small activity variation on changing the flow-rate are not clear at present, these findings evidence another significant advantage of using HBF4 as the acid additive, instead of TFA of the initial experiments.
For the sake of comparison the packed-bed reactor containing the polymeric catalyst Supp-3 in the powder form was employed to carry out the same model cycloaddition reaction between 4 and cyclopentadiene (Table 5). At 2 μL min−1 flow rate and conditions otherwise identical to those of Table 4, the packed-bed reactor (entries 1 and 2, Table 5) showed comparable results with the monolithic one; however, already when the flow rate was increased to 7.7 μL min−1, the products were formed in lower yield and enantioselectivity (entries 3 and 4, Table 5). The largest difference was observed when the reaction in continuo was performed at 18.8 μL min−1, when the packed-bed reactor produced the cycloadducts in 21% yield only and 87–85% ee (entry 5, Table 5). Compared to 73% yield and 86–90% ee obtained with the monolithic reactor (see entries 2–5, Table 4), these results confirm the higher catalytic activity of monolithic devices over the packed-bed ones, already discussed in the literature.34
a
| Entry | Running time [h] | ϕ [μL min−1] | τ [h] | Yieldc [%] | drd | ee endo-5 (ee exo-5)e [%] |
|---|---|---|---|---|---|---|
| a Reactions conditions: 25 °C; HPLC column (0.46 cm i.d. × 15 cm, containing 0.32 mmol of catalyst); cinnamaldehyde (0.128 M, 1 eq.) and cyclopentadiene (0.896 M, 7 eq.) in 95/5 CH3CN–H2O mixture. b Residence time calculated as void volume/flow rate (void volume = 1.3 ml, determined by picnometry). c Yields determined by 1H NMR and confirmed on isolated products after chromatographic purification. d Endo-5/exo-5 diastereoisomeric ratio, determined by 1H NMR on the crude reaction mixture. e Enantiomeric excess determined by HPLC on the alcohols obtained by NaBH4 reduction of adducts. | ||||||
| 1 | 0–4 | 2 | 10.8 | n.d. | — | — |
| 2 | 4–24 | 2 | 10.8 | 80 | 48/52 | 85 (80) |
| 3 | 24–44 | 7.7 | 2.8 | 65 | 48/52 | 86 (87) |
| 4 | 44–50 | 7.7 | 2.8 | 52 | 47/53 | 89 (86) |
| 5 | 55–70 | 18.8 | 1.2 | 21 | 48/52 | 87 (85) |
It is interesting to compare some results obtained in the organocatalyzed cycloaddition performed with supported catalysts in batch or under continuous-flow conditions, with two different columns (packed-bed and monolithic reactors). The data are collected in Table 6, where the total turnover number (TON) and productivity of the different materials and reaction conditions are reported.
| Entry | Conditionsa | Salt | Time (h) | Productivityb | TONc |
|---|---|---|---|---|---|
| a Reactions run at 25 °C; data taken from Tables 1, 3, 4 and 5. b Productivity is measured in mmol(product) h−1 mmol(catalyst)−1 × 103. c Turn over number is measured in mmol(product) mmol(catalyst)−1. d Flow rate 2 μL min−1 (Table 1 entry 6 and Table 3 entry 1). e Flow rate 18.8 μL min−1 (Table 4 entry 5). f Flow rate 7.7 μL min−1 (Table 5 entry 3). | |||||
| 1 | Batch | TFA | 48 | 59 | 2.8 |
| 2 | Batch | TFA | 120 | 31 | 3.8 |
| 3 | Batch | HBF4 | 120 | 46 | 5.6 |
| 4d | Monolithic flow | TFA | 120 | 38 | 4.6 |
| 5d | Monolithic flow | HBF4 | 120 | 37 | 4.4 |
| 6e | Monolithic flow | HBF4 | 24 | 338 | 8.1 |
| 7f | Packed bed flow | HBF4 | 24 | 120 | 4.4 |
Considering the results obtained with the TFA salt of Supp-3, in batch 1 gram of resin produced 1.4 mmol of the product with 90% ee in 48 hours (85% yield, entry 2, Table 2). According to the data of Table 1, at ϕ = 2–5 μL min−1 the same amount of resin in the monolithic reactor produced 1.6 mmol of cycloadducts in an identical time-frame (48 hours). For longer times, the process in flow offers clearly better performances: by recycling the catalyst, in 120 hours 1.8 mmol of products were obtained in batch (data not shown in Table 2), while operating in flow 3.8 mmol of adducts were produced with high enantioselectivity in the same time (Table 1).
With tetrafluoroborate salt that behaved better in batch, for long operation times the following data were collected: in 120 hours 2.9 mmol of the product was produced in batch, to be compared with the isolation of 4.0 mmol of cycloadducts (90% ee) in the continuous-flow run at ϕ = 2 μL min−1 (conditions of entries 1–4, Table 6).
Productivity and TON of the reactions performed under continuous flow conditions are always better than those for the corresponding reactions in batch, with an improvement of both parameters by a factor 1.4–2.9 (entries 1–5, Table 6). Nevertheless, due to the long residence time, productivity at ϕ = 2 μL min−1 was still quite low (38). However, thanks to the tolerance of the tetrafluoroborate chiral monolithic reactor to flow rate increase, the very remarkable level of productivity of 338 h−1 could be reached at ϕ = 18.8 μL min−1 (entry 6, Table 6). Interestingly, this latter result is larger than attained with powdered Supp-3, either in batch or in the packed-bed reactor (entries 3 and 7, Table 6, respectively).
Finally, a new monolithic reactor containing Supp-3 was prepared and employed in the tetrafluoroborate salt form to sequentially promote three different stereoselective reactions in continuo, with temperature and reaction solvent variations (Scheme 3a–c and Table 7).
 | ||
| Scheme 3 Enantioselective catalytic reactions under continuous-flow conditions with imidazolidinone supported catalyst Supp-3. | ||
| Entry | Running time [h] | T [°C] | Product | Yieldb [%] | drc | eed |
|---|---|---|---|---|---|---|
| a Flow rate ϕ = 2 μL min−1, residence time = 12.4 h; same reaction conditions as in Table 1. b Yields determined by 1H NMR and confirmed on isolated products after chromatographic purification. c Endo/exo diastereoisomeric ratio determined by 1H NMR on the crude reaction mixture. d Enantiomeric excess of the endo diastereoisomer, determined by HPLC (ESI); in parentheses enantiomeric excess of the exo diastereoisomer. e Column washing. | ||||||
| 1 | 0–4 | 25/−5 | 5 | 93 | 47/53 | 90 (90) |
| 2 | 4–28 | −5 | 5 | 90 | 48/52 | 91 (90) |
| 3 | 28–32 | −5/25 | 5 | 97 | 45/55 | 91 (93) |
| 4 | 32–80 | 25 | 5 | 99 | 47/53 | 90 (90) |
| 5e | 80–100 | 25/−15 | — | — | — | — |
| 6 | 100–130 | −15 | 11 | 45 | 94/6 | 90 (n.d.) |
| 7 | 130–180 | 25 | 11 | 71 | 91/9 | 90 (n.d.) |
| 8e | 180–200 | 25 | — | — | — | — |
| 9 | 200–248 | 25 | 13 | 55 | — | 15 |
| 10e | 248–260 | 25/−15 | — | — | — | — |
| 11 | 260–310 | −15 | 13 | 21 | — | 35 |
First the Diels–Alder cycloaddition between 4 and cyclopentadiene was performed under the new set of conditions (Table 7, entries 1–4). As expected on the basis of the results in batch, the catalytic reactor promoted very efficiently the transformation, affording 93% yield and 90% ee for both isomers after only 4 hours (Table 7, entry 1). Cooling to −5 °C seems not to have a decisive effect on the catalytic activity and enantioselectivity (Table 7, entry 2), while at room temperature the monolithic reactor was able to continuously process the reagents and to produce the cycloadducts in essentially quantitative yield and 90% ee for both stereoisomers, even after 80 h time on stream (Table 7, entries 3 and 4).
At this time the column was washed and used in a second reaction, the 1,3 dipolar cycloaddition of N-benzyl-C-phenyl nitrone (10) with crotonic aldehyde.35 By performing the reaction at −15 °C the yield of the transformation was only 45% (Table 3, entry 6), but at 25 °C (Table 3, entry 7) the isooxazoline 11 was isolated in good yield (71%), high diastereoselectivity (91:9) and without compromising the enantioselectivity (90% ee for the major endo-9 isomer).
After 180 hours of continuous operation the column was washed and further used in a third different reaction, the Friedel–Crafts alkylation of N-methyl pyrrole (12) with cinnamaldehyde.36 In this case the results were less satisfactory: the process carried out at 25 °C (Table 3, entry 9) afforded the product 13 in fair yield (55%) and low enantioselectivity (15% ee), whereas at −15 °C (Table 3, entry 11) the stereoselectivity could be only marginally improved (35% ee) at the cost, however, of a significant drop of the yield (21%).
Although these findings clearly indicate the need for further optimization studies, it is worth mentioning that the same reaction performed at 25 °C in batch in the presence of 30% mol amount of powdered Supp-3 and HBF4 afforded the product 13 in equally low enantioselectivity (15% ee) and even lower yield (40%) than in the continuous-flow run (Table 3, entry 9).37 Hence, the worse performance of the monolithic reactor with respect to the soluble MacMillan catalyst38 can be traced back to a negative influence of the macromolecular architecture39 and not to any major degradation of the catalytic material after extended use. However, it is worth mentioning that, also in this case, the reaction performed under continuous-flow conditions led to a cleaner reaction mixture than that obtained in batch, thus greatly simplifying the product isolation procedure.
In conclusion the first example of an enantiomerically pure organocatalyst immobilized into a monolithic reactor was realized: inside the chiral column Diels–Alder reactions were performed under continuous-flow conditions, leading to the products in high yield and excellent enantioselectivity (up to 93% ee at 25 °C).40 The general applicability of the catalytic system was verified, by carrying out Diels–Alder reactions in continuo with three different aldehydes. The versatility of the system was also proven by sequentially performing with the same column, three different reactions in continuo, for a total of more than 300 hours on stream. For the first time stereoselective organocatalyzed Diels–Alder reactions, 1,3-dipolar cycloadditions and Friedel–Crafts alkylations were performed in a monolithic reactor under continuous-flow conditions. Amongst these, sustained catalytic performances were demonstrated for more than 150 hours. Optimization studies allowed to significantly increase the productivity of the monolithic reactor, up to 338. Although the turnover number and reaction rates need to be further improved, it was already demonstrated that the process in continuo may positively compete with the reaction in batch, affording, in the same time, larger amounts of product, in a user-friendly experimental procedure that leads to cleaner crude reaction mixtures and greatly simplified isolation procedures.
For the asymmetric transformations examined in the present study the use of a catalytic monolithic reactor led to less waste materials and better use of a valuable chiral catalyst in comparison with alternative batch and continuous-flow process intensification schemes. Because the advantages disclosed in the present study are expected to be rather general, hopefully the approach described herein will prove a general tool for improving the greenness of many other enantioselective organocatalytic transformations and for addressing the issues that currently hamper their use above the bench-scale.
Acknowledgements
Financial support by Cariplo Foundation (Milano) within the project 2011-0293 “Novel chiral recyclable catalysts for one-pot, multi-step synthesis of structurally complex molecules”, FIRB project RBFR10BF5 V “Multifunctional hybrid materials for the development of sustainable catalytic processes” is gratefully acknowledged. M. B. thanks COST action CM9505 “ORCA” Organocatalysis.Notes and references
- For some recent, selected books and reviews, see: (a) K. Geyer, T. Gustafson and P. H. Seeberger, Synlett, 2009, 2382–2391 CAS; (b) C. Wiles and P. Watts, Chem. Commun., 2011, 6512–6521 RSC; (c) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 4583–4592 RSC; (d) S. Ceylan, L. Coutable, J. Wegner and A. Kirschning, Chem.–Eur. J., 2011, 17, 1884–1893 CrossRef CAS PubMed; (e) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed; (f) C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC; (g) R. M. Myers, K. A. Roper, I. R. Baxendale and S. V. Ley, in Modern Tools for the Synthesis of Complex Bioactive Molecules, ed. J. Cossy and S. Arseniyadis, J. Wiley, NY, 2012, ch. 11, pp. 359–394 Search PubMed; (h) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- (a) Review: A. Puglisi, M. Benaglia and V. Chiroli, Green Chem., 2013, 15, 1790–1813 RSC. For a review on stereoselective reactions in continuous-flow see: (b) K. Geyer, T. Gustafson and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5, 1–11 Search PubMed; See also: (c) C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC ; and; (d) T. Tsubogo, T. Ishiwata and S. Kobayashi, Angew. Chem., Int. Ed., 2013, 52, 6590–6604 CrossRef CAS PubMed.
- (a) M. P. Dudukovic, F. Larachi and P. L. Mills, Chem. Eng. Sci., 1999, 54, 1975–1995 CrossRef CAS; (b) D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS PubMed See also: S. Newton, S. V. Ley, E. Casas Arce’ and D. M. Grainger, Adv. Synth. Catal., 2012, 354, 1805–1812 CrossRef CAS PubMed ; and references cited.
- For the application of flow chemistry for multistep organic synthesis see: (a) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS PubMed; (b) M. Baumann, I. R. Baxendale and S. V. Ley, Mol. Divers, 2011, 3, 613–630 CrossRef PubMed. For some recent, representative contributions see: M. Chen and S. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 4247–4250 CrossRef CAS PubMed ; and I. R. Baxendale, S. V. Ley, A. C. Mansfield and C. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 4017–4021 CrossRef PubMed.
- For a review discussing the merits of batch and micro flow reactors see: R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed ; See also: A. Hartung, M. A. Keane and A. Kraft, J. Org. Chem., 2007, 72, 10235–10238 CrossRef PubMed ; and C. Battilocchio, B. J. Deadman, N. Nikbin, M. O. Kitching, I. R. Baxendale and S. V. Ley, Chem.–Eur. J., 2013, 19, 7917–7930 CrossRef PubMed ; and references cited.
- (a) Handbook of Asymmetric Heterogeneous Catalysts, ed. K. J. Ding and F. J. K. Uozomi, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley and Sons, 2009 Search PubMed; (c) Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis, ed. N. Haraguchi and S. Itsuno, John Wiley & Sons, 2011 Search PubMed.
- (a) Microreactors in Organic Synthesis and Catalysis, ed. T. Wirth, Wiley-VCH, 2008 Search PubMed; (b) V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS PubMed.
- S. Sandel, S. K. Weber and O. Trapp, Chem. Eng. Sci., 2012, 83, 171–179 CrossRef CAS PubMed.
- See: S. Ceylan and A. Kirschning, in Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley and Sons, 2009, ch. 13, pp. 379–403 and Search PubMed; D. Barby and Z. Haq, Z. Eur. Pat., 0060138, 1982 Search PubMed.
- R. M. Heck, S. Gulati and R. J. Farratu, Chem.–Eur. J., 2001, 82, 149–156 CAS . For a few selected recent polymeric monolithic microreactors, see: (a) B. Ngamson, A. M. Hickey, G. M. Greenway, J. A. Littlechild, P. Watts and C. Wiles, J. Mol. Catal. B: Enzym., 2010, 63, 81–86 CrossRef PubMed; (b) H. Lange, M. J. Capener, A. X. Jones, C. J. Smith, N. Nibkin, I. R. Baxendale and S. V. Ley, Synlett, 2011, 869–873 CAS; (c) R. J. Ingham, E. Riva, N. Nibkin, I. R. Baxendale and S. V. Ley, Org. Lett., 2012, 14, 3920–3923 CrossRef CAS PubMed ; and references cited.
- (a) C. Viklund, F. Svec, J. M. J. Frechet and K. Irgum, Chem. Mater., 1996, 8, 744–750 CrossRef CAS; (b) E. C. Peters, F. Svec and J. M. J. Fréchet, Adv. Mater., 1999, 11, 1169–1181 CrossRef CAS; (c) N. Hird, I. Hughes, D. Hunter, M. G. J. T. Morrison, D. C. Sherrington and L. Stevenson, Tetrahedron, 1999, 55, 9575–9584 CrossRef CAS; (d) J. A. Tripp, F. Svec and J. M. J. Fréchet, J. Comb. Chem., 2001, 3, 216–223 CrossRef CAS PubMed; (e) A. Sachse, A. Galarneau, B. Coq and F. Fajula, New J. Chem., 2011, 35, 259–264 RSC.
- (a) U. Kunz, H. Schönfeld, W. Solodenko, G. Jas and A. Kirschning, Ind. Eng. Chem. Res., 2005, 44, 8458–8467 CrossRef CAS; (b) U. Kunz, A. Kirschning, H.-L. Wen, W. Solodenko, R. Cecillia, C. O. Kappe and T. Turek, Catal. Today, 2005, 105, 318–324 CrossRef CAS PubMed.
- See ref. 2c–d; for a recent monolithic achiral organocatalytic system see: O. Bortolini, A. Cavazzini, P. Dambruoso, P. P. Giovannini, L. Caciolli, A. Massi, S. Pacifico and D. Ragno, Green Chem., 2013, 15, 2981–2992 RSC.
- (a) B. Altava, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis and M. J. Vicent, Green Chem., 2006, 8, 717–726 RSC.
- (a) B. Altava, M. I. Burguete, J. M. Fraile, J. I. García, S. V. Luis, J. A. Mayoral and M. J. Vicent, Angew. Chem., Int. Ed., 2000, 39, 1503–1506 CrossRef CAS; (b) M. I. Burguete, A. Cornejo, E. Garcia-Verdugo, M. J. Gil, S. V. Luis, J. A. Mayoral, V. Martinez-Merino and M. Sokolova, J. Org. Chem., 2007, 72, 4344–4350 CrossRef CAS PubMed. For a review see: (c) J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral and E. Pires, Chem. Soc. Rev., 2009, 38, 695–706 RSC.
- M. I. Burguete, A. Cornejo, E. Garcia-Verdugo, J. Garcia, M. J. Gil, S. V. Luis, V. Martinez-Merino, J. A. Mayoral and M. Sokolova, Green Chem., 2007, 9, 1091–1096 RSC.
- C. Aranda, A. Cornejo, J. M. Fraile, E. Garcia-Verdugo, M. J. Gil, S. V. Luis, J. A. Mayoral, V. Martinez-Merino and Z. Ochoa, Green Chem., 2011, 13, 983–990 RSC . For immobilized chiral catalysts see also: J. Lim, S. N. Riduan, S. S. Lee and J. Y. Ying, Adv. Synth. Catal., 2008, 350, 1295–1308 CrossRef CAS PubMed.
- A. M. Hafez, A. E. Taggi, T. Dudding and T. Lectka, J. Am. Chem. Soc., 2001, 123, 10853–10859 CrossRef CAS PubMed.
- (a) E. Alza, C. Rodríguez-Escrich, S. Sayalero, A. Bastero and M. A. Pericàs, Chem.–Eur. J., 2009, 15, 10167–10172 CrossRef CAS PubMed; (b) C. Ayats, A. H. Henseler and M. A. Pericàs, ChemSusChem, 2012, 5, 320–325 CrossRef CAS PubMed.
- (a) E. Alza, S. Sayalero, X. C. Cambeiro, R. Martin-Rapun, P. O. Miranda and M. A. Pericàs, Synlett, 2011, 464–468 CAS; (b) X. Fan, S. Sayalero and M. Pericas, Adv. Synth. Catal., 2012, 354, 2971–2976 CrossRef CAS PubMed; See also: (c) O. Bortolini, L. Caciolli, A. Cavazzini, V. Costa, R. Greco, A. Massi and L. Pasti, Green Chem., 2012, 14, 992–1000 RSC; (d) O. Bortolini, A. Cavazzini, P. Giovannini, R. Greco, N. Marchetti, A. Massi and L. Pasti, Chem.–Eur. J., 2013, 19, 7802–7808 CrossRef CAS PubMed ; and references cited.
- S. B. Otvos, I. M. Mandity and F. Fulop, ChemSusChem, 2012, 5, 266–269 CrossRef PubMed ; and references cited.
- Y. Arakawa and H. Wennemers, ChemSusChem, 2013, 6, 242–245 CrossRef CAS PubMed.
- R. Martín-Rapún, S. Sayaleroa and M. A. Pericàs, Green Chem., 2013, 15, 3295–3301 RSC.
- L. Osorio-Planes, C. Rodriguez-Escrich and M. A. Pericas, Chem.–Eur. J., 2014, 20, 2367–2372 CrossRef CAS PubMed.
- For our previous work with immobilized MacMillan catalysts see: (a) M. Benaglia, G. Celentano, M. Cinquini, A. Puglisi and F. Cozzi, Adv. Synth. Catal., 2002, 344, 149–152 CrossRef CAS; (b) M. Benaglia, G. Celentano, M. Cinquini, A. Puglisi and F. Cozzi, Eur. J. Org. Chem., 2004, 567–573 Search PubMed; (c) S. Guizzetti, M. Benaglia and J. S. Siegel, Chem. Commun., 2012, 3188–3190 RSC; (d) A. Puglisi, M. Benaglia, R. Annunziata, V. Chiroli, R. Porta and A. Gervasini, J. Org. Chem., 2013, 178, 11326–11334 CrossRef PubMed; See also: (e) T. E. Kristensen and T. Hansen, Eur. J. Org. Chem., 2010, 3179–3204 CrossRef CAS PubMed. and references cited; (f) J. Y. Shi, C. A. Wang, Z. J. Li, Q. Wang, Y. Zhang and W. Wang, Chem.–Eur. J., 2011, 17, 6206–6213 CrossRef CAS PubMed; (g) P. Riente, J. Yadav and M. A. Pericas, Org. Lett., 2012, 14, 3668–3671 CrossRef CAS PubMed.
- UV control of the final washings showed a negligible concentration of the chiral derivative into the liquid stream (C < 7 × 10−5 M).
- Void volume could be estimated from the amount of porogenic mixture in the polymerization feed (about 60%), provided that (a) the porogen is completely removed from the reactor after polymerization (as confirmed by the weight of the porogen recovered after column washing); (b) the monolith swelling is negligible. Both conditions are satisfied in the present work.
- According to our previous experience (ref. 25a–c) the reaction conditions used to perform catalyst immobilization do not affect the stereogenic center. Moreover, the integrity of amidic bond (as demonstrated by IR) guarantees the integrity of the imidazolidinone ring.
- G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79–92 CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- For detailed experimental procedures see the ESI.†.
- No traces of imidazolidinone or its degradation was found in the products or after column washing, proving that no leaching of the catalyst occurs.
- Several procedures of recovery and recycle of the catalyst were attempted, including thoroughly washing and drying under high vacuum; no appreciable improvements were obtained.
- See for example: (a) K. A. El, R. Chimenton, A. Sachse, F. Fajula, A. Galarneau and B. Coq, Angew. Chem., Int. Ed., 2009, 48, 4969–4972 CrossRef PubMed; (b) A. Sachse, A. Galarneau, F. Di Renzo, F. Fajula and B. Coq, Chem. Mater., 2010, 22, 4123–4125 CrossRef CAS.
- W. S. Jen, J. J. M. Wiener and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874–9875 CrossRef CAS.
- N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370–4371 CrossRef CAS.
- The reaction performed under continuous-flow conditions led to a reaction mixture much cleaner (TLC and 1H NMR) than that obtained in batch, thus greatly simplifying the product isolation procedure. This observation can partially explain the better yield of the product 13 obtained in the continuous-flow device.
- It must be mentioned that in our hands the alkylation of 12 with 8 catalyzed by commercial MacMillan catalyst under similar experimental conditions, at 25 °C, led to the product 13 in 50% yield and 65% ee.
- The modification of the phenyl ring of the MacMillan catalysts can have a profound influence on the catalytic performance. See: M. C. Holland, S. Paul, W. B. Schweizer, K. Bergander, C. Mück-Lichtenfeld, S. Lakhdar, H. Mayr and R. Gilmour, Angew. Chem., Int. Ed., 2013, 52, 7967–7971 CrossRef CAS PubMed.
- For a recent contribution on stereoselective organocatalyzed reactions performed under continuous-flow conditions in packed-bed reactors see: V. Chiroli, M. Benaglia, F. Cozzi, A. Puglisi, R. Annunziata and G. Celentano, Org. Lett., 2013, 15, 3590–3593 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and 1H NMR, 13C NMR, and IR spectra of new compounds. Preparation, analysis and characterization of polymer-supported chiral catalysts. NMR and HPLC traces of the stereoselective reaction products. See DOI: 10.1039/c4gc00031e |
| This journal is © The Royal Society of Chemistry 2014 |