 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A two-step biocatalytic cascade in micro-aqueous medium: using whole cells to obtain high concentrations of a vicinal diol†
Andre
Jakoblinnert
and
Dörte
Rother
*
IBG-1: Biotechnology, Forschungszentrum Jülich GmbH, Leo-Brandt-Str. 1, 52425 Jülich, Germany. E-mail: do.rother@fz-juelich.de; Fax: +49 2461 3870; Tel: +49 2461 6772
First published on 2nd May 2014
Abstract
Although single- and multi-step biocatalytic approaches show persuasive advantages for the synthesis of especially chiral compounds (e.g. high chemo- and stereoselectivity), their application often suffers from low substrate loads and hence low space-time-yields. We herein present a synthetic cascade approach in which lyophilised, recombinant whole cells are applied in micro-aqueous reaction systems yielding extremely high space-time-yields. As an example we investigated the two-step synthesis of 1-phenylpropane-1,2-diol starting from cheap aldehydes and achieved high selectivities (ee/de > 99%) and high product concentrations. The new concept of running biocatalytic cascades in a mixture of high substrate loads and organic solvents under addition of small amounts of highly concentrated buffer is not only very easy-to-apply, but also exhibits several economic and ecologic advantages. On the one hand the usage of whole, lyophilised cells circumvents time-consuming enzyme purification as well as addition of expensive cofactors (here ThDP and NADPH). Additionally, catalyst and product workup is facilitated by the application of organic solvents (here MTBE). On the other hand, the employment of whole cells very effectively circumvents stability problems of biocatalysts in unconventional media enabling the addition of extremely high substrate loads (up to 500 mM in our example) and is therefore an easy and effective approach for multi-step biocatalysis. After optimisation, the combination of a carboligation step followed by a second oxidoreduction step with whole cell catalysts afforded an efficient two-step cascade for the production of 1-phenylpropane-1,2-diol with space-time yields up to 327 g L−1 d−1 and an E-factor of 21.3 kgwaste kgproduct−1.
1. Introduction
Biocatalysis has become a valuable method for manufacturing chiral synthons for agrochemicals and pharmaceuticals due to its high intrinsic regio-, chemo- and stereoselectivity.1,2 Furthermore, as biocatalysts operate under mild (physiological) conditions they are often compatible with each other rendering them suitable for multi-step cascade processes in one pot.3–5 Cascade reactions are very attractive as yield-reducing intermediate product isolation is not necessary. Additionally, improved step- and atom economy translate into significant economic and environmental benefits.3When biocatalysts are used as isolated enzymes, time- and money-consuming purification is required and sometimes costly cofactors have to be added.6,7 The application of recombinant whole cells instead of isolated enzymes is about ten times cheaper and makes enzyme purification as well as cofactor addition obsolete.6 Additionally, the operational stability of whole cell catalysts is usually higher.8
Apart from a few exceptions, enzymes display the highest activity in buffered systems; however, the majority of industrially relevant substrates are hydrophobic and hence hardly soluble in aqueous buffers. If emulsions or two-phase systems are not suitable for the respective biocatalyst, overall productivity of these biotransformations is limited.8,9 Direct operation in organic solvents, micro-aqueous organic systems, or neat substrate systems can alleviate this drawback but most purified enzymes exhibit reduced activity under these non-natural conditions.10,11
Inactivation can be circumvented when enzymes are operated as whole cells. Here, the implementation of living cells, resting cells or even lyophilised cells is an option. The cell envelope may protect the enzyme from the organic exterior by providing an environment closer to nature.12,13 Often the application of organic solvent also facilitates product isolation as tedious product extraction from an aqueous phase can be omitted. Hence, biocatalytic cascades using whole cells in organic media offer a highly potent alternative to standard chemical syntheses to establish cheap, selective and efficient production processes.
Despite the above-mentioned advantages, up to now only four biotransformations with recombinant whole cells in organic or micro-aqueous (not biphasic) media can be found in the literature, all of them single-step biotransformations. Three asymmetric ketone reductions were reported employing alcohol dehydrogenases (ADHs) overexpressed in various hosts in several organic solvents or in pure organic substrates.14–16 Additionally, an E. coli whole cell catalyst harbouring a hydroxynitrile lyase from Arabidopsis thaliana yielded optically pure cyanohydrins in a micro-aqueous system.13 In all these examples, substrate concentrations were very high (0.5–4 mol L−1, (ESI Table S1†) also enabling access to high product concentrations and thus fulfilling the requirements for translation to industrial application.17,18
Here, we demonstrate that micro-aqueous reaction systems are not only applicable for the combination of whole cell catalysts to more complex products in multi-step reactions, but are also highly effective. A model cascade reaction composed of an asymmetric carboligation (step 1) followed by stereoselective reduction of the intermediate α-hydroxy ketone (step 2) is selected (Scheme 1). In the first step, the carboligation of benzaldehyde and acetaldehyde catalysed by the benzaldehyde lyase (BAL) from Pseudomonas fluorescens yields (R)-2-hydroxy-1-phenylpropan-1-one (2R)-HPP.19,20 The α-hydroxy ketone obtained is further reduced by an alcohol dehydrogenase from Ralstonia sp. (RADH) giving vicinal (1R,2R)-1-phenylpropane-1,2-diol ((1R,2R)-PPD, Scheme 1).21 Oxidised cofactor (NADP+) is recycled by substrate-coupled cofactor regeneration.
 | ||
| Scheme 1 Model two-step reaction cascade composed of asymmetric carboligation by BAL (step 1) and subsequent ketone reduction by RADH (step 2). BAL = benzaldehyde lyase from Pseudomonas fluorescens, RADH = alcohol dehydrogenase from Ralstonia sp., (2R)-HPP = 2-hydroxy-1-phenylpropan-1-one, PPD = 1-phenylpropane-1,2-diol. | ||
Such vicinal chiral diols are important building blocks for pharmaceuticals, for instance, for the synthesis of the anti-angina agent diltiazem.22 By combining different ThDP-dependent enzymes and ADHs, all four possible stereoisomers of 1-phenylpropane-1,2-diol are accessible. This was previously demonstrated in two-step reaction sequences in aqueous media at low substrate concentrations (<35 mmol L−1).23
In the present work, we combine the advantages of (i) asymmetric biocatalysis (mild reaction conditions, high selectivity), (ii) whole cell catalysis (cheap and stable catalyst, no cofactor addition), and (iii) operation in organic solvents (high substrate concentration, simple product isolation) in a one-pot two-step cascade reaction (high yields, shorter reaction times, no intermediate work-up). Process development and optimisation aims for high productivity in terms of space-time yield (STY) at the highest possible stereoselectivity for the production of the vicinal diol (1R,2R)-PPD.
2. Results and discussion
In order to identify optimal reaction conditions for the combination of carboligation and ketone reduction employing recombinant E. coli whole cells as catalysts in micro-aqueous medium in one pot, the influence of several reaction parameters such as organic solvent, substrate concentrations, buffer conditions and reaction modes on conversion and selectivity were investigated. Initially, 100 g L−1 lyophilised cells were applied at 100 mM substrate concentration. No reaction was observed in the dry state, but the reactions were started by the addition of one equivalent of water to the dry catalyst (e.g. 100 μL H2O to 100 mg cells). This amount of water conferred good levels of activity and did not lead to the formation of a continuous aqueous phase (data not shown). First, the carboligation reaction with the BAL catalyst was optimised. Subsequently, the ketone reduction was performed with the RADH catalyst operated under the previously identified optimal conditions for carboligation. Finally, the cascade mode (simultaneous or sequential) was evaluated and optimised with respect to high space-time yield and stereoselectivity. No cofactors were added externally throughout the whole process development.2.1 Solvent screen with the BAL and RADH catalyst
An appropriate organic solvent for both catalysts had to be identified for a cascade reaction with BAL and RADH E. coli cells under micro-aqueous reaction conditions. Therefore, activities were determined in the presence of nine different organic solvents (10% v/v doubly distilled H2O).As depicted in Fig. 1A, the BAL catalyst exhibits the best activity in methyl tert-butyl ether (MTBE) as the organic solvent. It can also work in 2-methyl-tetrahydrofuran (73.1%) and ethyl acetate (41.5%), but was inactive in the presence of 2-propanol, dichloromethane and 1,4-dioxane. No significant changes in chemo- or stereoselectivity were observed (data not shown), which is in agreement with a detailed analysis of solvent effects on purified BAL.24 Dominguez et al. screened twelve solvents with E. coli BAL cells in an aqueous-organic biphasic reaction system and revealed that MTBE preserves carboligation activity most effectively.25 The latter report focused on enzyme stability rather than activity but supports the choice of MTBE as a compatible organic solvent for carboligation reactions with the BAL catalyst.
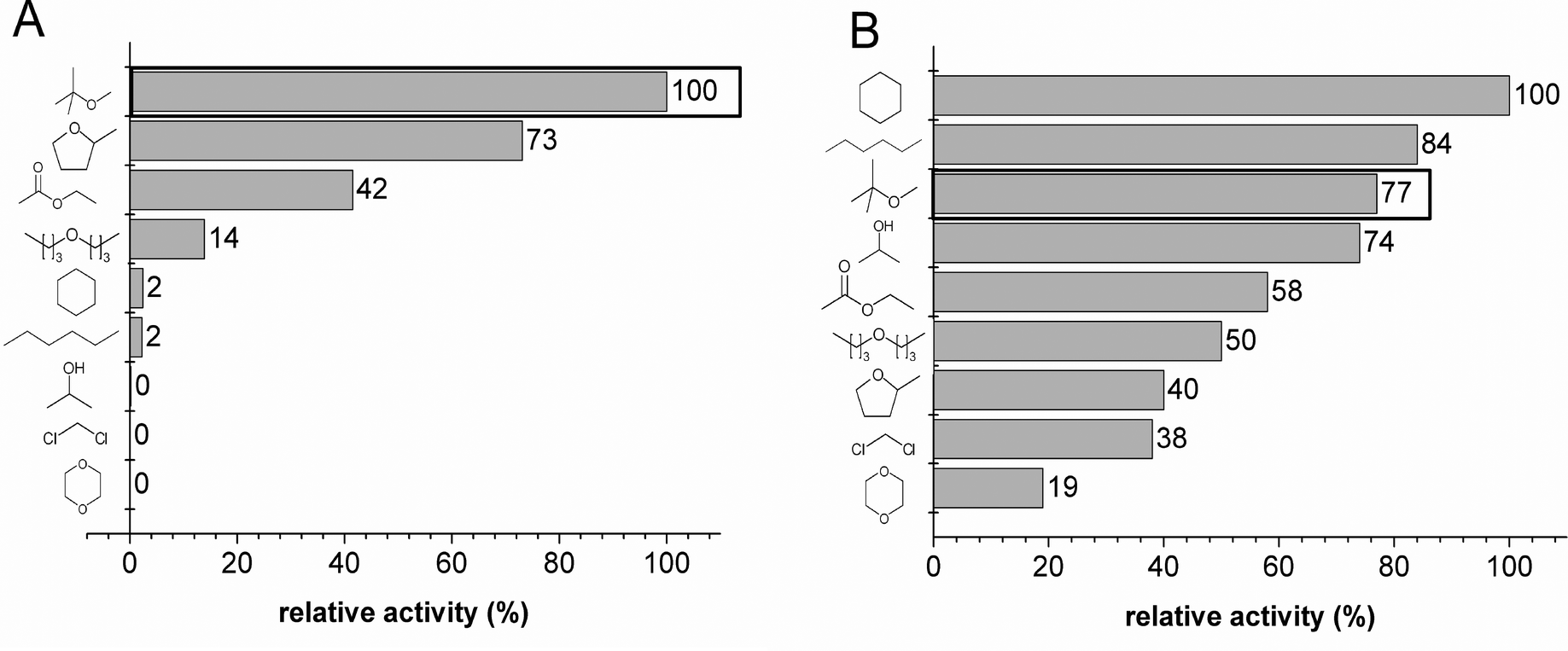 | ||
| Fig. 1 Relative initial rates in the presence of 90% (v/v) organic solvents. A: Carboligation with the BAL catalyst (100% = 11 U gcdw−1); B: Ketone reduction with RADH catalyst (100% = 3 U gcdw−1). MTBE as best compromise for a cascade reaction is highlighted by black boxes. | ||
The tolerance of the RADH catalyst for the organic solvent is rather broad, as shown in Fig. 1B, since it exhibits ketone reduction activity in all nine solvents. The best activities were detected in cyclohexanone (100%) and n-hexane (84%). Most importantly for the cascade, the RADH catalyst is also readily compatible with MTBE (77.3%) as the organic solvent. Notably, RADH is active in the alcohol 2-propanol as solvent, which can also serve as cosubstrate as was shown for a carbonyl reductase overexpressed in E. coli cells.15 Therefore, 2-propanol would represent a very advantageous solvent for the sole application of the RADH catalyst but is not suitable for combination with the BAL catalyst, since the latter shows no activity in 2-propanol (see Fig. 1A). No impact of any organic solvents on the enantiomeric excess was observed (data not shown).
Conclusively, MTBE is the best organic solvent for the present micro-aqueous system since both catalysts showed excellent (BAL: 100%) to good (RADH: 77.3%) relative activity. By using MTBE, the concentration of benzaldehyde can be elevated to levels beyond its solubility limit in aqueous media. Prolonged incubation of the BAL and the RADH catalyst in MTBE as organic solvent did not impair the stereoselectivity of the catalysts (ESI Table S2†). The latter finding is a prerequisite for producing the desired end product (1R,2R)-PPD with high optical purity in a cascade reaction. 2-Methyl-tetrahydrofuran would be a green alternative, since it can be gained from renewable resources, but reactivity for the second reduction step is much impaired compared to MTBE. In a possible industrial scale process it has to be evaluated weather the efficiency or the greenness of the process is the most crucial factor.
2.2 Optimisation of carboligation
Carboligation is as yet the limiting step in the two-step cascade (data not shown). Therefore, the production of the intermediate, (2R)-HPP, has to be optimised first in order to effectively produce (1R,2R)-PPD by combining carboligation and ketone reduction.Applying an excess of benzaldehyde (500 mM) in relation to optimal acetaldehyde (180 mM) would mean that full conversion to (2R)-HPP cannot be achieved. Furthermore, unwanted self-condensation of benzaldehyde leading to the formation of (R)-benzoin is promoted with a benzaldehyde surplus (Scheme 2).27 Thus, in order to obtain high amounts of (2R)-HPP, acetaldehyde had to be dosed to the reaction system.
 | ||
| Scheme 2 Two-step reaction cascade composed of asymmetric carboligation by the BAL catalyst (step 1) and subsequent ketone reduction by RADH (step 2). As a side product (in grey) (R)-benzoin can appear from the carboligation of two benzaldehyde molecules by the BAL catalyst. The (R)-benzoin will be cleaved back to benzaldehyde in the presence of acetaldehyde. Furthermore, benzaldehyde can be reduced by the RADH catalyst to benzyl alcohol. | ||
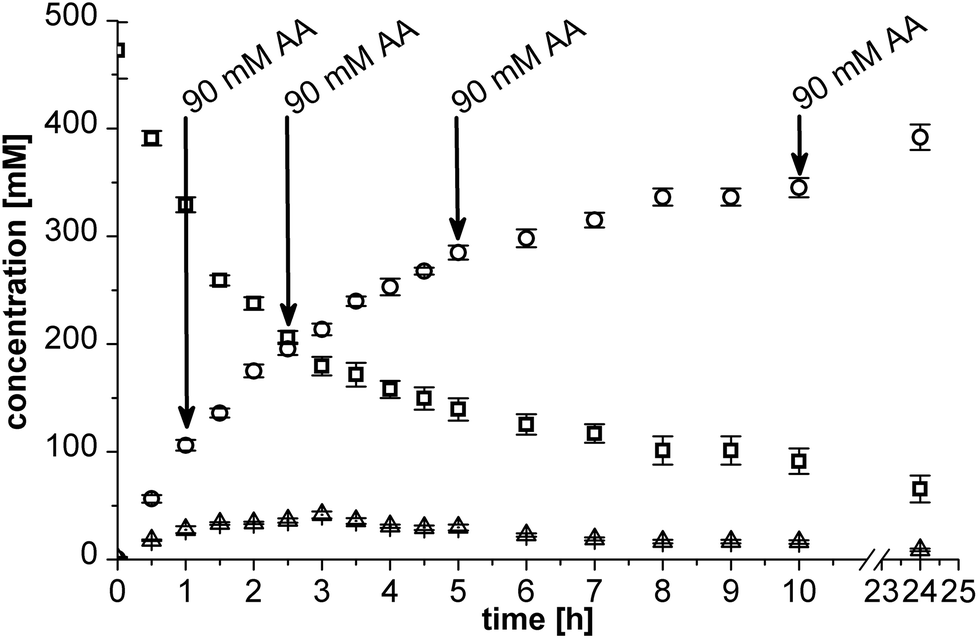 | ||
| Fig. 2 Carboligation of 500 mM benzaldehyde (□) and 180 mM acetaldehyde with the BAL catalyst in micro-aqueous conditions. Production of (2R)-HPP (○) was promoted by portion wise addition of 90 mM acetaldehyde (black arrows), whereas (R)-benzoin (Δ) formation was suppressed. | ||
Altogether, the strategy of portion wise addition of 90 mM acetaldehyde to the system after approximately the same amount had been consumed resulted in a high final conversion towards optically pure (2R)-HPP and a low (R)-benzoin concentration.
2.3 Buffer optimisation for carboligation and ketone reduction under micro-aqueous reaction conditions
As a further optimisation parameter, the aqueous component used to initiate biocatalytic activity in the micro-aqueous system was analysed. The activity and stability of isolated BAL is highly dependent on pH.28,29 In contrast, Dominguez et al. found that BAL in resting E. coli cells operated in a buffer/MTBE biphasic system exhibits similar activity at pH 7.0 and 9.0.25 Hence, we varied pH, buffer concentration and buffer species in order to investigate effects on the BAL catalyst in our micro-aqueous reaction system.Fig. 3A shows a strong effect of pH when water was replaced by 2.5% (v/v) 1 M triethanol amine (TEA) buffer. The cell-specific activity of the BAL catalyst was increased by a factor of 22 when the pH was raised from 7.0 to 10.0. The pH optimum of isolated BAL in buffer is 9.5 for (2R)-HPP formation, which is close to the optimal value obtained here.29 However, the true pH value in the direct environment of the enzyme inside the cell remains undetectable. A pH value of 10 was not exceeded as the buffering capacity of TEA is not optimal in this range anymore.
 | ||
| Fig. 3 Initial rate activity in MTBE with varying buffer conditions. (A) Activity of the BAL catalyst at different pH values in 1 M TEA. (B) Activity of the BAL catalyst at different TEA-buffer concentrations at pH 10. (C) Activity of the BAL catalyst at different buffer agents at 1 M, pH 10. (D) RADH and BAL activities with 2.5% v/v 1 M TEA (pH 10) in comparison to addition of 2.5% v/v pure water. | ||
Interestingly, the BAL catalyst activity was also markedly affected by the buffer concentration. Carboligation activity was elevated by a factor of 3.5 when the TEA concentration was increased from 50 to 1000 mM (Fig. 3B). However, only minor effects on the activity of isolated BAL were observed in aqueous systems when the concentration of potassium phosphate was increased from 10 to 150 mM.30 Conversely, in a different study, BAL activity decreased with increasing ionic strength from 25 to 500 mM in the presence of 30% v/v DMSO.31 As the organic buffer species TEA was extracted to MTBE (data not shown), we assume that in our micro-aqueous system with only 2.5% v/v 1 M TEA buffer solution, such a high buffer concentration is required in order to execute efficient buffering of the BAL enzyme inside the cell.
Finally, besides TEA, three alternative buffer agents were tested for their effect on BAL catalyst activity. As TEA has a pKa of 7.7632 it does not buffer effectively at pH 10.0. Hence, 1 M buffer of borate (pKa 9.2332), glycine (pKa 9.7832) and Tris (pKa 8.0632) at pH 9.5 could be more appropriate. However, with 2.5% v/v of these buffers, the BAL catalyst activity was reduced compared to the addition of 1 M TEA (Fig. 3C). Consequently, the nature of the buffer agent is decisive for the activating effect on the BAL catalyst. Earlier studies on isolated BAL in aqueous systems using different buffer agents such as TEA, Tris or KPi did not exhibit a significant activating effect on benzoin synthesis33 or carboligation of furfuraldehydes.30 The latter findings indicate that this activating effect of 1 M TEA on the BAL catalyst is a unique feature of the micro-aqueous system.
Altogether, the investigation of the buffer influence on carboligation revealed that the addition of 2.5% v/v 1 M TEA at pH 10 is the optimal choice for application of lyophilised BAL cells in our micro-aqueous system. Most experimentalists regard the application of a one-molar buffer to be high, but as only 2.5% v/v is actually used, the overall cost contribution of the buffer agent is still acceptable.
In comparison to the addition of 2.5% v/v pure water the BAL catalyst activity increased 7.4-fold when the optimal buffer was added (Fig. 3D). This strong activating effect directly translates into higher efficiency of the overall process. As a consequence, the BAL catalyst load was reduced from 100 g L−1 to 25 g L−1 (and thus the buffer volume from 10% v/v to 2.5% v/v). To verify the high efficiency of this process, (2R)-HPP was enzymatically synthesised in a preparative 1 litre setup affording 91% conversion within 6 h (see ESI Table S3†).
The asymmetric reduction of (2R)-HPP with lyophilised RADH cells constitutes the second step of the envisaged cascade (Scheme 1). As the carboligation and the ketone reduction will be carried out in one pot in the same reaction medium the effect of 2.5% v/v 1 M TEA at pH 10.0 on the RADH catalyst activity was investigated. Interestingly, (2R)-HPP reduction with RADH in the micro-aqueous system was accelerated by a factor of 3.3 in the presence of 2.5% v/v 1 M TEA at pH 10 (Fig. 3D) in comparison to the addition of pure water. The reported pH optimum for the isolated RADH is rather broad for ketone reduction (pH 6–9), whereas the pH-profile for oxidation is narrow with pH 9.5 as the optimal value.34 In the micro-aqueous system, oxidation and reduction take place simultaneously within the whole cell catalyst as the cofactor regeneration was realised by substrate-coupled cofactor regeneration. From the pH optima of isolated RADH, it can be deduced that both reactions work well at slightly alkaline pH. Hence, it appears reasonable that a buffered system at high pH confers higher activity than pure water at neutral pH, even though the true pH value in the environment of the RADH enzyme within the cell is not known.
2.4 Process engineering of the BAL+RADH cascade
In principle, a two-step cascade reaction can be run in sequential mode (2 steps in 1 pot) or simultaneous mode (both steps simultaneously in 1 pot). Both modes were studied in detail to validate the optimal process conduct mode for the BAL + RADH cascade in micro-aqueous medium.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RADH catalyst ratio was found to be 1:1.32 (e.g. 25 g L−1 BAL: 33 g L−1 RADH), where the (1R,2R)-PPD formation was five times faster than the benzyl alcohol formation (see ESI Fig. S3†).
:RADH catalyst ratio was found to be 1:1.32 (e.g. 25 g L−1 BAL: 33 g L−1 RADH), where the (1R,2R)-PPD formation was five times faster than the benzyl alcohol formation (see ESI Fig. S3†).
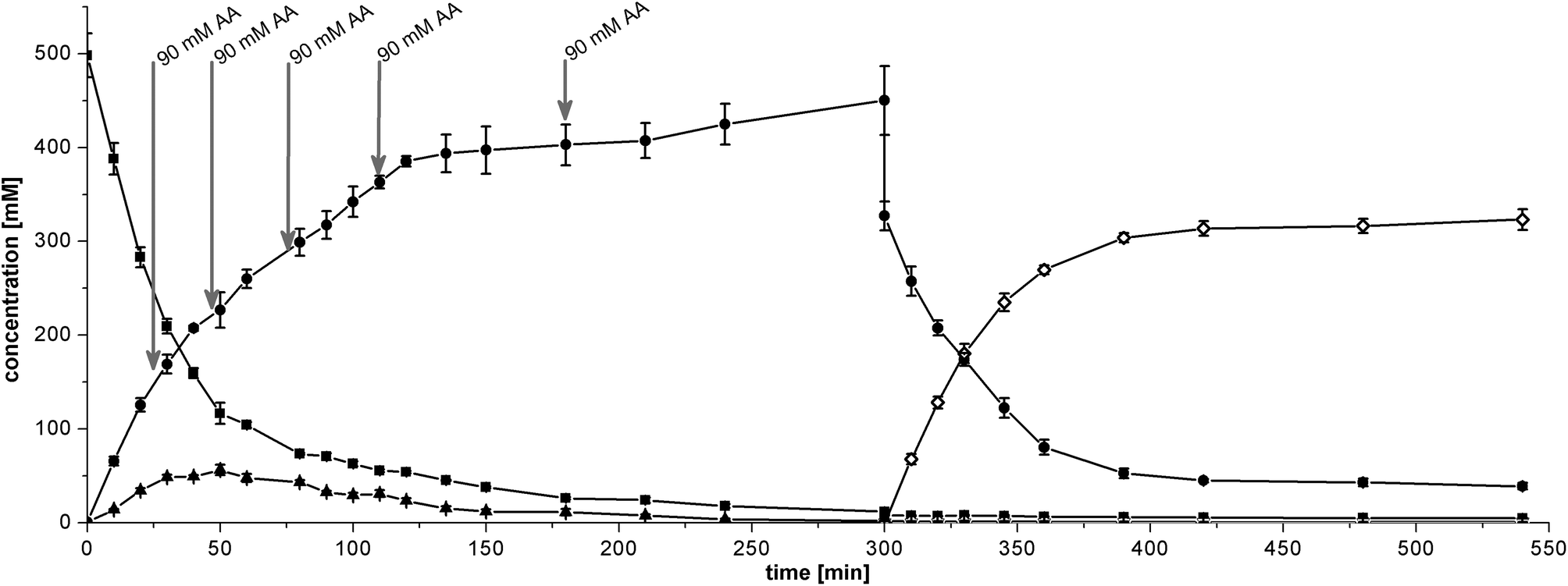 | ||
| Fig. 4 Sequential cascade with 25 g L−1 BAL catalyst and 2.5% v/v 1 M TEA, pH 10 (1st step) + 33 g L−1 RADH catalyst and 3.3% v/v 1 M TEA, pH 10 (2nd step). Starting conditions were 500 mM benzaldehyde and 180 mM acetaldehyde in MTBE. In the first 300 min, benzaldehyde (■) was converted to (2R)-HPP (●) as the main product while (R)-benzoin (▲) appears as a transient side product. Acetaldehyde (AA) was added in 90 mM portions after 25, 45, 75, 110 and 180 min as indicated by arrows. After 300 min, 2.5 M cyclohexanol (27% v/v) and RADH catalyst were added causing a drop in concentration due to the sudden volume increase. (1R,2R)-PPD (◊) was produced from (2R)-HPP as the main product. Benzyl alcohol was only detected in traces (<1 mM) and is therefore not shown. | ||
| (1R,2R)-PPD | (2R)-HPP | Benzyl alcohol | (R)-Benzoin | Benzaldehyde | Sum | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Conc. [mM] | ee [%] | de [%] | Conc. [mM] | ee [%] | Conc. [mM] | Conc. [mM] | ee [%] | Conc. [mM] | Conc. [mM] | |
| Sequential | 440 ± 16 | >99 | 99.6 | 54 ± 5 | 96.2 | 1.0 ± 0.1 | 0.7 ± 0.4 | >99 | 6.5 ± 0.7 | 503 ± 22 |
| Simultaneous | 363 ± 1 | >99 | 99.8 | 45 ± 3 | 99.3 | 39 ± 1 | 1.5 ± 0.5 | >99 | 7.7 ± 0.6 | 457 ± 7 |
The theoretical STY of the overall process is 178 g L−1 d−1 taking the total reaction time of 9 h into account. However, the reaction is virtually complete after 7 h, which translates to a higher STY of 222 g L−1 d−1 if stopped earlier.
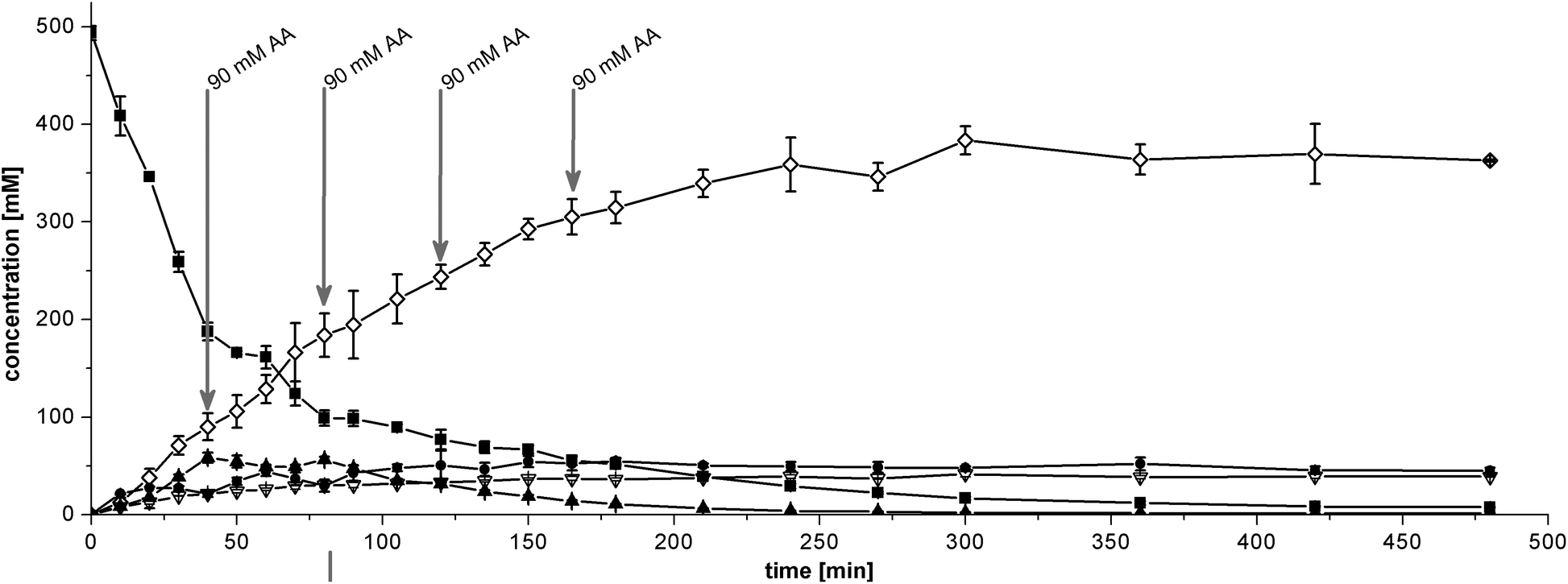 | ||
| Fig. 5 Simultaneous cascade with 25 g L−1 BAL catalyst + 33 g L−1 RADH catalyst and 5.8% v/v 1 M TEA, pH 10. Starting conditions were 500 mM benzaldehyde, 180 mM acetaldehyde and 2.5 M cyclohexanol in MTBE. Production of (1R,2R)-PPD (◊) from benzaldehyde (■) and acetaldehyde with (2R)-HPP (●) as intermediate; (R)-benzoin (▲) and benzyl alcohol (∇) were formed as side products. Acetaldehyde (AA) was added in 90 mM portions after 40, 80, 120 and 165 min as indicated by arrows. | ||
The theoretical STY of the simultaneous cascade is 161.5 g L−1 d−1 considering the total reaction time of 480 min. When the reaction is stopped where an apparent equilibrium has been attained at 240 min, the STY increases to 326.9 g L−1 d−1.
Notably, negative controls with lyophilised E. coli cells carrying only empty plasmid vector produced only traces of benzyl alcohol (3 mM) and no further side products within 20 h (data not shown).
2.5 Preparative (1R,2R)-PPD production in simultaneous cascade mode
The simultaneous mode proved to be simpler to operate and also exhibited higher productivity. Therefore, a preparative batch experiment was performed at 20 mL scale in simultaneous mode. Qualitatively, the reaction course was similar to that obtained on the analytic scale (ESI Fig. S4†). After 360 min the reaction was terminated and the crude product was subjected to chromatographic work-up. The remaining substrates and side products were effectively removed by downstream processing.Finally, 874.4 mg of (1R,2R)-PPD (ee/de > 99/99%) was obtained, corresponding to 58% isolated yield. Spectroscopic analysis of the product supported the identity and purity (1H-spectrum: Fig. S5,†13C-spectrum: Fig. S6†). The overall process afforded an STY of 175 g L−1 d−1 (ESI Table S4†). If the latter value is compared to the STY of a previously published 2-step process where isolated BAL and ADH from Thermoanaerobium species were operated in aqueous media, the increase in productivity is more than 1600-fold (ESI Table S4†).23,27 It is noteworthy that throughout the whole development process the expensive cofactors ThDP (for BAL) and NADPH (for RADH) were never added externally but supplied by the whole cell catalysts itself. In the case of NADPH, the oxidised cofactor was even efficiently recycled. These features render this cascade very cost-efficient in comparison to processes with isolated enzymes where generally cofactors have to be added. Recently, the highest cost contribution in an enzymatic process was attributed to NADP+ even when the cofactor is recycled.35
Additionally, the E-factor, describing the mass of waste per mass of product, of the optimised cascade is 21.3 kgwaste kgproduct−1 (ESI Table S5†) rendering this process very eco-friendly.35–37 Concerning product work-up, laborious and waste-generating solvent extraction was omitted as the product was readily available in MTBE. Including work-up, however, the E-factor increases to 1927 kgwaste kgproduct−1 (ESI Table S5†), which is beyond that desirable in E-factor ranges.37 Nevertheless, for product purification flash chromatography was used, consuming large amounts of solvents. In principle, the solvents can be recycled which would again significantly decrease the E-factor to levels <250. More eco-friendly purification methods such as crystallisation could further reduce the E-factor but where beyond the scope of this study.
3. Conclusion
In our quest to boost the productivity of a biocatalytic 2-step cascade composed of carboligation and ketone reduction, we successfully exploited the advantages of (i) asymmetric biocatalysis (mild reaction conditions, high selectivity), (ii) whole-cell catalysis (cheap and stable catalyst, no cofactor addition), and (iii) operation in organic solvents (high substrate concentration, simple product isolation).We successfully identified MTBE as the best organic solvent, as both whole cell catalysts exhibit good activity and selectivity. Furthermore, optimal substrate concentrations (500 mM benzaldehyde and 180 mM acetaldehyde) were found for carboligation as the first step in the cascade. Thereby, the benzaldehyde concentration is increased by a factor of ten in this micro-aqueous system compared to aqueous systems. Additionally, an efficient acetaldehyde dosing strategy was developed allowing a high conversion of benzaldehyde.
Notably, in the micro-aqueous system a remarkable effect of the buffer on the BAL and RADH catalyst activity was identified. Application of 2.5% v/v 1 M TEA buffer at pH 10 boosted activities 7.4-fold and 3.3-fold, respectively. Therefore, although only little buffer is added to the reaction system, attention should be laid onto buffer agent, pH and concentration in a micro-aqueous reaction system.
Sequential and simultaneous cascades were successfully run under the previously optimised reaction conditions giving access to extremely high product concentrations exceeding 440 mM of (1R,2R)-PPD at excellent stereoselectivity (ee/de > 99/99%). The sequential cascade turned out to be more productive with respect to space-time yield. Finally, a preparative batch in 20 mL scale was performed affording 57.5% isolated yield at a STY of 175 g L−1 d−1. Compared to a previously published process for (1R,2R)-PPD production with isolated enzymes in aqueous medium, the STY is now improved by 1600-fold.
Consequently, the strategy to apply lyophilised, recombinant whole cells under micro-aqueous conditions proved to be highly effective regarding the productivity of the system compared to isolated enzymes operated in aqueous buffers. The cascade developed here is very cost-efficient as not only are whole cell applications cheaper than the application of purified enzymes, but furthermore expensive ThDP and NADPH cofactors were supplied by the whole-cell catalysts. Moreover, the E-factor is low at 21.3 kgwaste kgproduct−1, however, the work-up strategy has to be improved in order to further reduce the E-factor for the complete process (1927 kgwaste kgproduct−1 including work-up using flash chromatography).
In total, a cost-efficient and eco-friendly 2-step cascade for the production of optically pure (1R,2R)-PPD at high concentrations from cheap aldehydes was established. For future work, we envisage the efficient production of all four possible stereoisomers of the vicinal PPD by this newly developed approach to furnish high product concentrations and high STYs.
4. Experimental
4.1 Chemicals
All chemicals purchased were of high chemical purity. Aldehydes and MTBE were obtained from Carl Roth (Karlsruhe, Germany). Other chemicals were purchased from Sigma-Aldrich, unless otherwise specified. Racemic benzoin was purchased from Fluka (Buchs, Switzerland). Benzaldehyde and acetaldehyde were kept under argon gas at 4 °C and −20 °C, respectively.Racemic HPP for the calibration of HPLC analysis was synthesised as described elsewhere.38 Analytical data of HPP were in good agreement with those published.29,39 H-NMR (600 MHz, CDCl3) δ 1.46 (d, 3 H), 3.80 (brs, 1 H), 5.17 (q, 1 H,), 7.51 (mc, 2 H), 7.62 (mc, 1 H), 7.93 (mc, 2 H) ppm. Racemic PPD used for GC calibration was taken from a stock whose synthesis has been described elsewhere.40
4.2 Preparation of lyophilised cells
Lyophilised cells containing BAL were prepared from frozen cell pellets, obtained by high-cell-density fermentation as described elsewhere.41 Recombinant cells with RADH were prepared as described by Kulig et al.40 Frozen pellets were lyophilised for a minimum of two days (Martin Christ GmbH) and stored at −20 °C.4.3 Screening of organic solvents
Lyophilised BAL cells (100 mg) were mixed with 900 μL of organic solvent containing 10 μL of benzaldehyde and 10 μL of acetaldehyde in screw-capped 1.5 mL glass vials. The reaction was initiated by the addition of 100 μL of doubly distilled H2O and agitated at 1000 rpm and RT. Production of (2R)-and (2S)-HPP was recorded by chiral HPLC and the reaction rate was deduced from the linear regime of the progression curve.Lyophilised RADH cells (50 mg) and 50 μL 2-propanol were mixed with 850 μL of organic solvent containing 100 mM (2R)-HPP (1.5 mg). The reaction was initiated by the addition of 100 μL of doubly distilled H2O and agitated at 1000 rpm. The production of PPD was recorded by chiral GC and the reaction rate was deduced from the linear regime of the progression curve.
4.4 Substrate optimisation of carboligation step
In order to determine the optimal benzaldehyde concentration, solutions were prepared as follows: 84.8 mg (100 mM), 212 mg (250 mM), 424 mg (500 mM), 636 mg (750 mM), or 848 mg (1000 mM) benzaldehyde; 80 μL acetaldehyde (180 mM) filled to 8 mL with MTBE.Lyophilised BAL cells (100 mg) were mixed with 900 μL substrate solution in screw-capped 1.5 mL glass vials. The reaction was initiated by the addition of 100 μL of ddH2O and agitated at 1000 rpm and RT. Reactions were carried out in triplicate. Production of (2R)- and (2S)-HPP was recorded by chiral HPLC and the reaction rate was deduced from the linear regime of the progression curve.
In order to determine the optimal acetaldehyde concentration the solutions were prepared as follows: 40 μL (90 mM), 80 μL (180 mM), 100 μL (225 mM), 120 μL (270 mM) or 160 μL (360 mM) acetaldehyde, benzaldehyde 424 mg (500 mM) made up to 8 mL with MTBE. Reactions were conducted in triplicate as described above.
4.5 Dosing mode for acetaldehyde for (2R)-HPP production
Lyophilised BAL cells (500 mg) were added to screw-capped 8 mL glass vials and mixed with 5 mL substrate solution (composed of 265 mg benzaldehyde (500 mM), 50 μL acetaldehyde (180 mM) and 4650 μL MTBE). The reaction was initiated by the addition of 500 μL of doubly distilled H2O and shaken horizontally at 1400 rpm and RT. Reactions were carried out in triplicate and recorded by chiral HPLC. Consumption of acetaldehyde was estimated by the production of HPP neglecting possible evaporation during sampling.A volume of 25 μL ice-cold acetaldehyde (approximately 90 mM) was added portion wise to the reaction mixture when the calculated acetaldehyde concentration dropped below 90 mM. During the total reaction, acetaldehyde was added four times after 1, 2.5, 5 and 10 hours giving a total of 540 mM (180 mM + 4 × 90 mM).
4.6 Buffer optimisation experiments
To investigate the effects of pH, buffer concentration and buffer species on carboligation with BAL cells in MTBE, 25 mg lyophilised BAL cells were mixed with 975 μL substrate solution (500 mM benzaldehyde, 180 mM acetaldehyde in MTBE) in 1.5 mL screw-capped glass vials. The reaction was initiated by the addition of 25 μL buffer and agitated at 1400 rpm and RT. The buffers added to the system varied in the buffer species (1 M TEA, glycine, borate and Tris, pH 10). The best species, namely TEA-buffer, was varied in pH (1 M TEA; pH 7, 8, 9 & 10) and in concentration (50, 100, 250, 500 & 1000 mM TEA pH 10). The pH values of the respective buffers were adjusted with NaOH or HCl and made up to the final volume with doubly distilled H2O. For comparison with water, the buffer was replaced by 25 μL doubly distilled H2O. Production of (2R)- and (2S)-HPP was recorded by chiral HPLC and the reaction rate was deduced from the linear regime of the progression curve.To determine RADH activity in the presence of the optimal buffer (1 M TEA, pH 10), 25 mg lyophilised RADH cells were mixed with 975 μL substrate solution (100 mM (2R)-HPP, 1 M cyclohexanol in MTBE) in screw-capped 1.5 mL glass vials. The reaction was initiated by the addition of 25 μL 1 M TEA, pH 10 and agitated at 1400 rpm and RT. For comparison with water, the buffer was replaced by 25 μL doubly distilled H2O. The production of PPD was recorded by chiral GC and the reaction rate was deduced from the linear regime of the progression curve.
4.7 Tuning of catalyst load
Lyophilised BAL cells (125 mg) were added to screw-capped 8 mL glass vials and mixed with 5 mL substrate solution (265 mg benzaldehyde (500 mM), 50 μL acetaldehyde (180 mM), 1315 μL cyclohexanol and 3385 μL MTBE). Different quantities of lyophilised RADH cells were added (50, 100, 125, 167, 250 and 500 mg).The reaction was initiated by the addition of the corresponding volume of buffer (175, 225, 250, 292, 375 and 625 μL) of 1 M TEA pH 10. The vials were shaken horizontally at 1400 rpm and RT. The production of (1R,2R)-PPD and benzyl alcohol was recorded by chiral GC and the reaction rate was deduced from the linear regime of the progression curve. The ratio of diol and benzyl alcohol formation was calculated from the cell-specific initial reaction rates (U gcdw−1).
4.8 Optimised BAL + RADH cascades
After completion of carboligation, lyophilised RADH cells (167 mg) and 1.315 mL cyclohexanol (2.5 M) were added. The ketone reduction reaction was initiated by the addition of 3.3% v/v (167 μL) 1 M TEA pH 10 and shaken horizontally at 1400 rpm and RT. Benzaldehyde, (2R)-HPP and (R)-benzoin were detected by chiral HPLC, whereas benzyl alcohol and (1R,2R)-PPD formation was recorded by chiral GC.
4.9 Preparative scale: simultaneous cascade
Lyophilised BAL cells (500 mg) and RADH cells (600 mg) were added to a screw-capped 23 mL glass vial and mixed with 20 mL substrate solution (1.06 g benzaldehyde (0.01 mmol), 500 μL acetaldehyde (180 mM), 5.26 mL cyclohexanol (2.5 M) and 13.24 mL MTBE). The reaction was initiated by the addition of 5.8% v/v (1.16 mL) 1 M TEA pH 10 and shaken horizontally at 1400 rpm and RT. Acetaldehyde was added in 90 mM (100 μL) portions after approximately 90 mM acetaldehyde was converted. Product formation was recorded as described above.After 360 min, the reaction was stopped and the crude reaction mixture was dried with MgSO4 and filtered. Cells were washed three times with 20 mL ethyl acetate, filtered and combined with the crude reaction mixture. Excess solvent was removed by evaporation under reduced pressure. Subsequent filter flash chromatography on silica gel (eluent: ethyl acetate–petroleum ether 30/70, Rf (PPD) = 0.18) afforded 58% (847.7 mg) (1R,2R)-PPD diol of ee/de > 99/99%.
4.10 HPLC analysis
Benzaldehyde, 2-HPP, PAC and benzoin were determined via chiral HPLC analysis. Only the organic phase was sampled. 2-Methyl hydroxypropiophenone was used as internal standard and samples were diluted 1000-fold in n-hexane (I: 20 μL 980 μL, II: 50 μL + 950 μL). Reactions were monitored by chiral HPLC at 250 nm using a Dionex Gina 50 autosampler, a Dionex UVD170U detector coupled with a Gynkotek high-precision pump model 480, and a Gynkotek Degasys DG 1310 (Thermo Fisher Scientific, USA). The Chiralpack IC column (4.6 × 250 mm, 5 μm particle size; Daicel Chemical IND., LTD, Japan) was operated with an analytical grade mobile phase of 70% v/v n-heptane and 30% v/v 2-propanol at a flow of 1 mL min−1 at 25 °C. Approximate retention times were 5.0 min for benzaldehyde, 5.8 min for (2R)-HPP, 6.3 min for (2S)-HPP, 6.7 min for (R)-benzoin, and 7.2 min for (S)-benzoin.4.11 GC analysis
Diol and benzyl alcohol formation was monitored using chiral phase GC analysis. Samples taken from the reaction were diluted 30-fold in ethyl acetate supplemented with 1-dodecanol as internal standard. 1 μL of the samples was analysed using a chiral CP-Chirasil-DEX CB column (Varian, Germany; 25 m × 0.25 mm × 0.25 μm) at a constant temperature of 140 °C for 30 min. Detection was undertaken by a flame ionisation detector (FID) using hydrogen as the carrier gas. Typical retentions times were 4.2 min for benzyl alcohol, 24.1 min for (1S,2S)-PPD, 25.9 min for (1R,2R)-PPD, 27.4 min for (1S,2R)-PPD, and 28.3 min for (1R,2S)-PPD.4.12 1H- and 13C-NMR analysis
1H-NMR and 13C-NMR spectra were recorded on a Bruker Advance-DRX 600 spectrometer in CDCl3 with TMS (Me4Si) as internal standard. Chemical shifts are given in ppm relative to the Me4Si (1H, Me4Si = 0 ppm) or relative to the resonance of the solvent (13C, CDCl3 = 77.2 ppm).Acknowledgements
This work was financed by the Helmholtz Association in frame of funding for the Helmholtz Young Investigator Group ‘Synthetic Enzyme cascades’ and by the Helmholtz Alliance on Synthetic Biology.Notes and references
- R. C. Simon, F. G. Mutti and W. Kroutil, Drug Discovery Today: Technol., 2012, 10, 37–44 CrossRef PubMed.
- N. Patel, ACS Catal., 2011, 1, 1059–1074 CrossRef.
- R. A. Sheldon, in Enzyme-Catalyzed Cascade Reactions Multi-step enzyme catalysis: biotransformations and chemoenzymatic synthesis, ed. E. Garcia-Junceda, Wiley-VCH, Weinheim, 2008, pp. 109–135 Search PubMed.
- J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- F. Lopez-Gallego and C. Schmidt-Dannert, Curr. Opin. Chem. Biol., 2010, 14, 174–183 CrossRef CAS PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process. Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- K. Schroer, U. Mackfeld, I. A. Tan, C. Wandrey, F. Heuser, S. Bringer-Meyer, A. Weckbecker, W. Hummel, T. Daussmann, R. Pfaller, A. Liese and S. Lütz, J. Biotechnol., 2007, 132, 438–444 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285 RSC.
- S. Wenda, S. Illner, A. Mell and U. Kragl, Green Chem., 2011, 13, 3007–3047 RSC.
- A. M. Klibanov, Trends Biotechnol., 1997, 15, 97–101 CrossRef CAS.
- N. Doukyu and H. Ogino, Biochem. Eng. J., 2010, 48, 270–282 CrossRef CAS PubMed.
- T. Ishige, K. Honda and S. Shimizu, Curr. Opin. Chem. Biol., 2005, 9, 174–180 CrossRef CAS PubMed.
- K. E. Scholz, D. Okrob, B. Kopka, A. Grünberger, M. Pohl, K. E. Jaeger and U. Krauss, Appl. Environ. Microbiol., 2012, 78, 5025–5027 CrossRef CAS PubMed.
- G. de Gonzalo, I. Lavandera, K. Faber and W. Kroutil, Org. Lett., 2007, 9, 2163–2166 CrossRef CAS PubMed.
- A. Jakoblinnert, R. Mladenov, A. Paul, F. Sibilla, U. Schwaneberg, M. B. Ansorge-Schumacher and P. Dominguez de Maria, Chem. Commun., 2011, 47, 12230–12232 RSC.
- A. Hibino and H. Ohtake, Proc. Biochem., 2013, 48, 838–843 CrossRef CAS PubMed.
- S. Lütz, L. Giver and J. Lalonde, Biotechnol. Bioeng., 2008, 101, 647–653 CrossRef PubMed.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed.
- P. Hinrichsen, I. Gomez and R. Vicuna, Gene, 1994, 144, 137–138 CrossRef CAS.
- B. Gonzalez and R. Vicuna, J. Bacteriol., 1989, 171, 2401–2405 CAS.
- I. Lavandera, A. Kern, B. Ferreira-Silva, A. Glieder, S. de Wildeman and W. Kroutil, J. Org. Chem., 2008, 73, 6003–6005 CrossRef CAS PubMed.
- B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, J. Org. Chem., 2003, 68, 1736–1746 CrossRef CAS PubMed.
- D. Kihumbu, T. Stillger, W. Hummel and A. Liese, Tetrahedron: Asymmetry, 2002, 13, 1069–1072 CrossRef CAS.
- T. Gerhards, U. Mackfeld, M. Bocola, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed.
- P. Dominguez de Maria, T. Stillger, M. Pohl, P. Kiesel, A. Liese, H. Gröger and H. Trauthwein, Adv. Synth. Catal., 2008, 350, 165–173 CrossRef CAS.
- T. Stillger, M. Pohl, C. Wandrey and A. Liese, Org. Process Res. Dev., 2006, 10, 1172–1177 CrossRef CAS.
- A. S. Demir, M. Pohl, E. Janzen and M. Müller, J. Chem. Soc., Perkin Trans. 1, 2001, 633–635 RSC.
- E. Janzen, M. Müller, D. Kolter-Jung, M. M. Kneen, M. J. McLeish and M. Pohl, Bioorg. Chem., 2006, 34, 345–361 CrossRef CAS PubMed.
- P. Domínguez de María, T. Stillger, M. Pohl, S. Wallert, K. Drauz, H. Gröger, H. Trauthwein and A. Liese, J. Mol. Catal. B: Enzym., 2006, 38, 43–47 CrossRef PubMed.
- D. Natalia, Doctoral Thesis, RTWH Aachen University, 2012.
- T. Schmidt, M. Zavrel, A. Spiess and M. B. Ansorge-Schumacher, Bioorg. Chem., 2009, 37, 84–89 CrossRef CAS PubMed.
- R. J. Beynon and J. S. Easterby, Buffer solutions – the basics, IRL Press at Oxford University Press, 1996 Search PubMed.
- T. Stillger, Doctoral Thesis, Heinrich-Heine-University, Düsseldorf, 2006.
- J. Kulig, A. Frese, W. Kroutil, M. Pohl and D. Rother, Biotechnol. Bioeng., 2012, 110, 1838–1848 CrossRef PubMed.
- S. Leuchs, J. Lima-Ramos, L. Greiner, N. Al-Haque, P. Tufvesson and J. M. Woodley, Org. Process Res. Dev., 2013, 17, 1027–1035 CrossRef CAS.
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 2007, 68, 381–388 CrossRef.
- R. A. Sheldon, Chem. Tech. Biotech., 1997, 68, 381–388 CrossRef CAS.
- C. Chen, X. Feng, G. Zhang, Q. Zhao and G. Huang, Synthesis, 2008, 3205–3208 Search PubMed.
- S. B. Sopaci, I. Simsek, B. Tural, M. Volkan and A. S. Demir, Org. Biomol. Chem., 2009, 7, 1658–1664 CAS.
- J. Kulig, R. C. Simon, C. Rose, M. Husain, M. Häckh, S. Lüdeke, K. Zeitler, W. Kroutil, M. Pohl and D. Rother, Catal. Sci. Technol., 2012, 2, 1580–1589 CAS.
- M. Schwarz, Doctoral Thesis, Heinrich-Heine-University, Düsseldorf, 2011.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc00010b |
| This journal is © The Royal Society of Chemistry 2014 |