 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering ‘water-soluble lignocellulose’ obtained by mechanocatalysis: new insights into the chemical processes leading to deep depolymerization
Mats
Käldström
,
Niklas
Meine
,
Christophe
Farès
,
Ferdi
Schüth
and
Roberto
Rinaldi
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 – Mülheim an der Ruhr, Germany. E-mail: rinaldi@kofo.mpg.de; schueth@kofo.mpg.de
First published on 12th May 2014
Abstract
Recently, the mechanocatalytic depolymerization of lignocelluloses yielding ‘water-soluble lignocelluloses’ was demonstrated. Water-soluble C5 & C6 sugars and sulfur-free lignins are formed through the saccharification of the oligosaccharides, allowing for the fractionation of biomass by simple filtration. Herein, we present an in-depth analysis of water-soluble products obtained from beechwood. The complex nature of the ‘water-soluble beechwood’ is investigated by 2D HSQC NMR, HPLC and gel filtration chromatography. The detailed analysis of the ‘water-soluble beechwood’ lends significant insights into the chemical nature of the lignocellulose depolymerization driven by the mechanical forces.
Introduction
Mechanocatalytic depolymerization of lignocellulose constitutes a new frontier in biorefining.1–4 Through the combination of mechanical and acid-catalyzed chemical processes, deep depolymerization of lignocellulose (i.e. the full conversion of lignocellulose into water-soluble oligosaccharides and lignin fragments) is easily achieved. Scheme 1 illustrates the steps involved in this emerging approach:2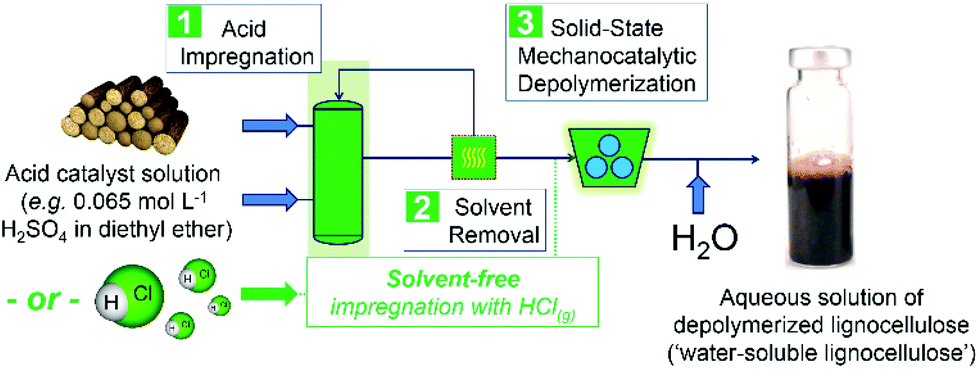 | ||
| Scheme 1 Schematic representation of the method for deep depolymerization of lignocellulose by mechanocatalysis.2 | ||
1. Lignocellulose is impregnated with small quantities of a strong acid (e.g. H2SO4 or HCl). As demonstrated for cellulose depolymerization,2 this process can be performed either by wet impregnation (e.g. in a dilute H2SO4 solution in diethyl ether) or solvent-free impregnation (e.g. by exposing the substrate fibers to gaseous HCl).5
2. In the wet impregnation, the acid is loaded on the substrate surface upon solvent removal. The energy efficiency of the wet impregnation might be debatable due the costs associated with solvent recovery and reuse. Thus, the solvent-free impregnation with gaseous HCl offers a more energy efficient and environmentally friendly alternative to the wet impregnation method.
3. In sequence, the acid-impregnated substrate is processed for relatively short milling durations (e.g. 2 to 3 h),2,5,6 compared with conventional mechanical pretreatment (e.g. days to months).7
We have demonstrated the ‘water-soluble lignocellulose’ to serve as a platform for fractionation of biomass into C5 & C6 sugars in addition to sulfur-free lignins.6 In aqueous solution, the fractionation is obtained by the saccharification of the ‘water-soluble lignocellulose’ at temperatures as low as 140 °C for 1 h. As a result, high yield of sugars (e.g. 88–92% glucose, 3.5–8% glucose dimers and 93–98% xylose relative to the glucan and xylan fractions, respectively) and sulfur-free lignins were obtained from beechwood, pinewood and sugarcane bagasse.6
We also demonstrated the water-soluble oligosaccharides (WSO) can be used as a unique replacement for glucose and xylose, enabling high-yield production of sugar alcohols8 or furfurals.9 Using a similar methodology for the mechanocatalytic depolymerization of cellulose developed by us, Beltramini et al. have confirmed the promising results for the production of sugar alcohols from ‘water-soluble cellulose’.3,10
Despite the great potential of ‘water-soluble lignocelluloses’ as biomass feeds for catalytic valorization, so far there has been little discussion about the chemical nature of the water-soluble lignocelluloses. Aiming to lend detailed insights into the nature of the chemical processes involved in the deep depolymerization, we provide here an in-depth analysis of the chemical nature of ‘water-soluble beechwood.’ This paper is organized as follows. First, the chemical nature of the deep depolymerization (acid-catalyzed vs. radical reaction) is addressed. Next, the effects of the acid type (H2SO4 or HCl) on the mechanocatalytic depolymerization of beechwood are discussed. Surprisingly, unlike cellulose, which is fully converted into water-soluble oligomers either in the presence of H2SO4 or HCl,2 the mechanocatalytic depolymerization of HCl-impregnated beechwood reaches only 74% yield of water-soluble products after milling for a duration of 2 h. The effect of the acid type on the extent of depolymerization of the carbohydrate and lignin fractions is assessed by 2D HSQC NMR, HPLC and gel filtration chromatography. Unexpectedly, the acid type exerts an effect mostly on the depolymerization of lignin. Finally, it is demonstrated by 13C CP-MAS NMR that water-insoluble products obtained from the mechanocatalytic depolymerization of HCl-impregnated beechwood is mostly composed of lignin.
Results and discussion
The chemical nature of the deep depolymerization
Seminal works on the depolymerization of cellulose,11 lignocellulose,12 and lignin models13–16 by mechanochemistry showed that radicals are formed through milling the substrates in absence of acid catalysts. This observation poses an important question: What is the contribution of radicals to the mechanocatalytic depolymerization? To answer this question one should first consider that lignin is a radical scavenger, which is in close contact with cellulose and hemicellulose. As such, if radical processes are of importance for the depolymerization extent, the yield of water-soluble products from lignocellulose will be decreased, compared to that from cellulose. To check this hypothesis, the formation of water-soluble products throughout the course of mechanocatalytic depolymerization of beechwood and α-cellulose (both impregnated with H2SO4 at a loading of 0.9 ± 0.1 mmol H2SO4 per gram of substrate) were compared.Fig. 1 shows similar trends for the formation of water-soluble products obtained from the depolymerization of beechwood and α-cellulose. This observation tends to confirm the chemical nature of the deep depolymerization to be only acid-catalyzed. Nonetheless, one should bring awareness and attention to the fact that the susceptibility of a lignocellulosic substrate to undergo mechanochemical reactions depends also on several factors related to the receptivity of mechanical forces by the substrate, such as chemical composition (e.g. hemicellulose and lignin contents), mechanical properties as well as substrate microstructure. Accordingly, the need for a prolonged milling duration can arise in order to achieve full conversion of a lignocellulosic substrate into ‘water-soluble lignocellulose’. For instance, we recently found that the acid-catalyzed depolymerization of pinewood and sugarcane bagasse is completed within 3 h under the same milling conditions used for the mechanocatalytic depolymerization of beechwood or α-cellulose, which are deep depolymerized in 2 h.
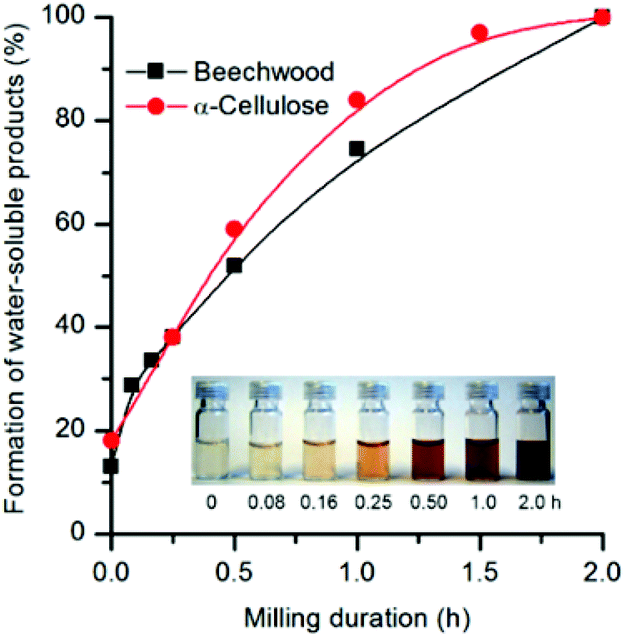 | ||
| Fig. 1 Formation of water-soluble products from beechwood or α-cellulose as a function of milling time. Reaction conditions: H2SO4-impregnated substrate (1 g, 0.8 ± 0.1 mmol H2SO4 per gram of substrate) milled in a planetary mill at 800 rpm. The data was collected in separate experiments, performed at the indicated milling durations. | ||
Conclusive evidence on the acid-catalyzed nature of the process is provided by the fact that the reaction takes place at appreciable rates only when a strong acid (i.e. pKa ≤ −3) is used as a catalyst.2,3,5 This observation clearly indicates that the protonation of the glycosidic O site is also not easy even on the acid-impregnated substrate.5,17 In light of the current results, it is apparent that the contribution of radicals to the overall extent of the mechanocatalytic depolymerization is small.
Interestingly, it is clear from Fig. 1 that directly after impregnation and prior to milling, 13 and 18% of water-soluble products, from beechwood and α-cellulose, were already formed. However, an increase in impregnation duration does not yield improved substrate solubilization.5 This observation agrees with findings by Nevell and Upton.18 They demonstrated the initial depolymerization of cotton cellulose by HCl in benzene as a rapid and random process taking place at the amorphous domains, and contrastingly as a slow and specific process at the chains ends in the crystalline domains. The depolymerization is thus limited to a rather low extent. Hence, partial hydrolysis of the amorphous domains of cellulose and hemicellulose most likely results in ca. 10–20% of the substrate solubilization shortly after acid impregnation.
Finally, Fig. 1 also shows that the color of the solutions containing the water-soluble products from beechwood changed from pale yellow to dark reddish brown with the increase in the milling duration. In contrast, the solution of water-soluble oligosaccharides from α-cellulose is a colorless, clear solution.2 This result is visual evidence that not only the carbohydrates but also lignin undergoes depolymerization rendering water-soluble products.2
Effect of the acid catalyst on the product composition
Since the energy efficiency of the wet impregnation of acid might be debatable due the solvent recovery and reuse, we examined the dry impregnation with gaseous HCl as an alternative to the wet impregnation with H2SO4 solutions. In this section, we will focus our analysis on the depolymerization of H2SO4- and HCl-impregnated beechwood milled for 2 h.By exposing beechwood (6 g) to gaseous HCl for 15 min (1 bar, 25 °C), a loading of 1.0 mmol of HCl per gram of substrate was achieved after evacuation for 1 h (10−3 mmHg). As previously demonstrated by us, this acid loading provides comparable results to those of the depolymerization of a commercial cellulose (α-cellulose) impregnated with 0.9 mmol H2SO4 per gram of substrate. The mechanocatalytic depolymerization of HCl-impregnated α-cellulose yields 100% of water-soluble products within a milling duration of 2 h.2
The H2SO4-processed beechwood is fully soluble in both water and DMSO. In contrast, the HCl-processed beechwood is only partially soluble in water (74%), but still fully soluble in DMSO. To shed light on the effect of acid catalyst on the general composition of chemical entities in the samples, the depolymerized beechwood samples were analyzed in detail by 2D NMR HSQC experiments (Fig. 2 and 3). Since both depolymerized beechwood samples are fully soluble in DMSO-d6, the HSQC NMR analysis thus provides the best way for the characterization of the entities present in the depolymerized beechwood samples.
 | ||
| Fig. 2 Partial HSQC spectra of depolymerized beechwood samples dissolved in DMSO-d6. Spectra “a” (H2SO4-processed beechwood) and “b” (HCl-processed beechwood) display the anomeric region showing the signals for α- and β-reducing end units and that for the 1,4-β internal anomeric H. | ||
 | ||
| Fig. 3 Partial HSQC spectra showing the region of the C3 side-chains of the lignin units (“a” and “b”) and aromatic region of lignin (“b” and “d”) for depolymerized beechwood samples dissolved in DMSO-d6. The spectra “a” and “b” refer to the samples prepared by milling the H2SO4-impregnated substrate, while “c” and “d” the samples obtained by milling the HCl-impregnated substrate. The correlation signals in green correspond to CH and CH3 groups, while those in blue to CH2 groups (Table 1). | ||
Nonetheless, HSQC spectrum data must be interpreted with caution, since the 1JCH dependence of polarization transfer in HSQC experiments is not suppressed in regular HSQC pulse sequences.19 As a result, the absolute intensity of cross peaks is not fully quantitative in the entire spectral range, although considerable progress has been achieved in the field of HSQC NMR that is giving rise to quantitative analysis protocols.19–21 Nevertheless, regular HSQC NMR experiments still offer extremely valuable (direct) semiquantitative information for characterization and comparison of lignins as well as whole plant cell compositions.22–25 Semiquantitative determination of volume integral ratios is possible for 1H–13C pairs in a similar chemical environment (e.g. as for the correlations involving the anomeric H signals, the Cα–Hα signals for the side-chain of lignin units, or the C2–H2 and C6–H6 signals for lignin aromatic units), due to the fact that the 1JCH values for the specific entities are reasonably similar.22,23 Accordingly, for the different regions of the HSQC spectra, semiquantitative analysis was performed separately by integration of 1H–13C pairs of interest.22,23
Fig. 2 shows the HSQC spectra of the depolymerized beechwood in the region of the correlation signals of the pair 1H–13C associated with anomeric H. Through the direct comparison of the spectra from the depolymerized beechwood samples (Fig. 2a and 2b) with that of cellobiose (not shown here), we assigned the correlation signals associated with 1,4-β-glucans: (1) α reducing end units (1-α-H, δC/δH, 91.8/4.9 ppm); (2) β reducing end units (1-β-H, δC/δH, 96.3/4.3 ppm); and (3) 1,4-β internal anomeric H (1,4-β-H, δC/δH, 102.5/4.3).
By using the volume integral of the signals 1-α-H (Vα), 1-β-H (Vβ) and 1,4-β-H (Vi), the degree of polymerization (DP) in anhydroglucose units (AGU) can be estimated by eqn (1):
 | (1) |
Both HCl- and H2SO4-processed beechwood show the same value for the estimated DP (3 AGU). In addition, the DP values by HSQC are in good agreement with the DP values by gel filtration chromatography, which also indicates similar DP values for both samples (DP ∼ 5–6 AGU, as it will be discussed later). We can thus conclude that both H2SO4 and HCl catalysts lead to a similar depolymerization extent in the mechanocatalytic experiments.
This conclusion may seem surprising, considering the total H+ content present in the acid-impregnated samples. The actual H+ content in the H2SO4-impregnated beechwood is about twice as high as that in the HCl-impregnated beechwood, since the acid catalyst loadings were 0.9 mmol H2SO4 and 1.0 mmol HCl per gram of beechwood. Nonetheless, the hydrolysis of the 1,4-β-glycosidic bonds in cellulose requires strong acids (pKa ≤ −3) for taking place at appreciable rates at temperatures lower than 100 °C.2,3,27–29 In effect, for cellulose depolymerization, only half of the H+ content of H2SO4 catalyzes the reaction, since the weakly basic glycosidic O site can selectively distinguish the acidity of the H+ species in H2SO4 (the values of pKa1 and pKa2 are −3 and 1.99, respectively). Thereby, this fact supports the same extent of cellulose depolymerization reached by the processing of the H2SO4- and HCl-impregnated beechwood samples, since the relevant acid content (pKa1 = −3) is similar.
For the purpose of this study, the simple comparison of Fig. 2a and 2b reveals that both samples have identical correlation signals, indicating that both samples show rather similar types of glycosidic linkages in their oligosaccharides. This observation has an important implication for product solubility in water, since the presence of branched oligosaccharides could account for the different solubilities of the HCl and H2SO4 processed beechwood samples, as recently suggested for the water-soluble oligosaccharides obtained from mechanocatalytic depolymerization of cellulose.3
The similar DP of the oligosaccharides in the depolymerized beechwood samples, in addition to their identical patterns of correlation signals in the anomeric region of the HSQC spectra, constitute indirect evidence that the different solubility of the depolymerized beechwood samples are more likely associated with their lignin fractions rather than related to the oligosaccharide fractions formed by mechanocatalytic depolymerization.
To assess the relative distribution of β-O-4, β-5 and β–β linkages still present in the depolymerized beechwood samples, the HSQC spectra were analyzed in the region of C3 side-chains of the lignin units. Fig. 3 displays cross signals of the C3 side-chains of the lignin units (Fig. 3a and 3b) and those of the aromatic region showing the color-coded corresponding structures.
From Fig. 3a and 3c, it is possible to identify the presence of lignin subunit structures A, B and C and thus the corresponding interunit linkage types – β-O-4 (by the presence of the correlation signals, Aα and Aβ), β-5 (Bα and Bβ) and β–β (Cα, Cβ and Cγ). Due to the prominent and overlapping carbohydrate contours, it is very challenging to resolve the correlation signals for the 1H–13C pairs corresponding to Aγ (δC/δH, 59.7/3.6 ppm and 59.4/3.7 ppm) and Bγ (δC/δH, 62.8/3.7 ppm). Worth mentioning, the correlation signal for Bα shows a very low absolute intensity, and is thus hardly seen in the magnification applied to Fig. 3a and 3c. Table 1 lists the ratio of β-O-4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β-5:β–β, which were estimated from the volume integrals of the contours for Aα, Bα and Cα, respectively. The β–β linkage-type was chosen as a “reference” for the comparison of lignins because it represents a class of strong C–C linkages33 and shows a more intense cross signal in the HSQC compared with Bα (β-5).
:β-5:β–β, which were estimated from the volume integrals of the contours for Aα, Bα and Cα, respectively. The β–β linkage-type was chosen as a “reference” for the comparison of lignins because it represents a class of strong C–C linkages33 and shows a more intense cross signal in the HSQC compared with Bα (β-5).
Table 1 indicates that the β-O-4 linkage is still the most abundant in depolymerized wood samples as well as in beechwood lignin extracted by the organosolv process. This result is very interesting since the organosolv pulping is a fairly mild process and, consequently, the isolated lignin retains much of its native β-O-4 interunit linkages, as reported by other studies.36,37 The results from Table 1 suggest that H2SO4-processed beechwood might indeed contain a lignin depolymerized to a slightly higher extent than that in HCl-processed beechwood, as revealed by a lower proportion of β-O-4 linkages found for the H2SO4-processed beechwood (β-O-4:β-5:β–β, 2.0:0.3:1.0) compared with that for HCl-processed beechwood (β-O-4:β-5:β–β, 2.6:0.2:1.0).
Unlike the hydrolysis of cellulose, which requires strong acids (pKa ≤ −3) to proceed at appreciable reaction rates,2,3,27–29 the β-O-4 linkages undergo acidolysis even in the presence of weak acids (e.g., acetic acid).30 This fact may lead to the (false) conclusion that the β-O-4 linkages undergo acidolysis to a much greater extent in the experiments in the presence of H2SO4. However, the problem is not so straightforward because the conjugated base of the acid catalyst markedly affects the hydrolysis rate and the reaction pathway of the acidolysis of β-O-4 linkages, as reported by several studies.38–46
Recently, Yokoyama et al. revisited the mechanism of β-O-4 bond cleavage in the acidolysis of a lignin model.40 They found that the rate of disappearance of veratrylglycerol-β-guaiacyl ether shows rate constants of 3.61 × 10−2 and 8.08 × 10−3 h−1 in the presence of HCl and H2SO4, respectively, when the whole disappearance process in each experiment was approximated as a pseudo-first-order reaction.40 Based on these rate constants, the acidolysis of β-O-4 bond in the processing of H2SO4-impregnated beechwood would be expected to be slower than for the HCl-impregnated substrate. In contrast, the experimental evidence shows that the H2SO4-processed beechwood contains a lignin depolymerized to a slightly higher extent than that in HCl-processed beechwood.
Among the lignin linkages still detectable by HSQC NMR, the β-O-4 is the most thermolabile one, as indicated by its low BDE value and thermolysis experiments.33,34 We propose that the proportion of this linkage in the sample can serve as an internal, natural probe for distinguishing the internal processes driven by mechanical forces from those thermally driven (due to microscopic hot spots formed by the impact of balls in the mill). The slightly lower fraction of β-O-4 found for the processed H2SO4-beechwood sample, in comparison to that found for lignin extracted by the organosolv process at 180 °C, strongly suggests that the mechanocatalytic depolymerization is as mild as the Organosolv process is to the lignin fraction. Accordingly, it is evident that the mechanocatalytic process is driven by the action of mechanical forces on the internal molecular structures of the biopolymers, rather than by the presence of hot spots. If the latter process were the predominant one, the fraction of the β-O-4 linkages would most likely be so low that it could not be detected in the HSQC spectrum – because the β-O-4 ether linkage would be then cleaved to a much greater extent relative to the other, more thermostable linkages β-5 and β–β.
The presence of β-O-4 linkages in the depolymerized beechwood samples thus strongly suggests that the action of mechanical forces is driving the acid-catalyzed depolymerization of the carbohydrate fraction and the acidolysis of the lignin linkages. This notion agrees with a very recent report on the hydrolysis of cellulose by DFT calculations, predicting that both protonation of the glycosidic O site and conformational changes are the rate limiting steps in the mechanism.27 The action of mechanical forces in the lignocellulosic matrix is proposed to facilitate the conformational changes needed for the glycosidic bond activation towards hydrolysis.5,27 In addition, the acid impregnation guarantees high local-concentration of H+-species on the lignocellulosic surface, alleviating the protonation problems.5,27 The one-pot combination of acid catalysis with mechanical forces offers great advantages over the conventional two-step approach, in which the substrate is first milled, to reduce crystallinity only, and next hydrolyzed in an acidic solution at temperatures between 150 and 210 °C, forming moderate yields of fermentable sugars.47
The last important information that can be extracted from the HSQC spectra is the relative composition of lignin in terms of coumaryl (H), coniferyl or guaiacyl (G) and sinapyl (S) units. To obtain the composition of H, G and S units, the aromatic region of the HSQC spectra (Fig. 3b and 3d) was analyzed. The 1H–13C pairs considered in this estimation are H2/6 (δC/δH, 128.0/7.2 ppm, not detected in the beechwood samples studied here), G2/6 and S6 because of their similar chemical environment.22 Hence, by using the half-value of the volume integral of the correlation signals G2/6 (note that they correspond to two 1H–13C pairs), in addition to the entire integral value for S6, the lignin composition can be estimated in terms of S and G units as given by eqn (2) and (3), respectively.
 | (2) |
 | (3) |
As expected for a hardwood, the beechwood samples contain no appreciable amount of coumaryl (H) units, and show a higher content of S units relative to G units. Again, regarding the contents of S and G units, the lignins in H2SO4- and HCl-processed beechwood are very similar. However, the content of S-units found for H2SO4- and HCl-processed beechwood (S/G ratios 1.56 and 1.63, respectively) is lower than that found here for Organosolv beechwood lignin (S/G ratio 1.94). This finding agrees with the results reported by Ragauskas et al. on the structural characterization of ball-milled switchgrass lignins obtained before and after dilute H2SO4 pretreatment.48 They also found a decrease in S/G ratio (from 0.80 to 0.53).48 However, the correlation of this finding with the chemical phenomena occurring in the acid-catalyzed depolymerization of lignocellulose remains unclear.
The data from Table 2 also reveals an important feature of the mechanocatalytic depolymerization of beechwood. The similar values for the relative volume integrals for the 1H–13C pairs – S2/6, G2, and G5 + G6 – suggest that the aromatic rings of lignin in the H2SO4-processed do not undergo sulfonation, which could account for the full water-solubility of the H2SO4-processed substrate in contrast to HCl-processed beechwood. Furthermore, no evidence supporting sulfonation or sulfatation of the processed substrate was detected by capillary electrophoresis experiments on the solutions of ‘water-soluble beechwood’ at different pH values.
Gel filtration chromatography of the water-soluble products
To further assess the composition of the water-soluble products from H2SO4- and HCl-processed beechwood samples, the water-soluble products from depolymerized beechwood were analyzed by gel filtration chromatography (Fig. 4). Table 3 summarizes the yields of glucose dimers, glucose, xylose, HMF and furfural. The yields of glucose dimers, glucose and 5-hydroxymethylfurfural (HMF) were calculated relative to the glucan fraction, while those of xylose and furfural are relative to the xylan fraction in the original sample of beechwood.2 | ||
| Fig. 4 Gel-filtration chromatograms of the water-soluble beechwood samples obtained from milling of H2SO4-impregnated beechwood (chromatograms “a” and “b”) and HCl-impregnated beechwood (chromatograms “c” and “d”). The chromatograms “a” and “c” show the refractive index detector response, “b” and “d” display the UV-Vis DAD response. | ||
The similar product contents listed in Table 3 indicate that the depolymerization of the carbohydrate fraction occurs similarly, irrespective of whether HCl or H2SO4 is used. The chromatogram (refractive index detector) shows the presence of saccharides with DP from 1 to 10 AGU in both samples. The oligosaccharide distributions are both centered around 5–6 AGU. A more intense “lump” background signal is, however, seen in the chromatogram for the HCl-processed sample (Fig. 4c). The chromatogram measured with the UV-Vis diode array detector (Fig. 4b and 4d) indicates the presence of components, which absorb UV light at about 275 nm and co-elute with the oligosaccharides (which do not absorb above 210 nm). Since both, phenols and furfural, show absorption bands at around 275 nm, to distinguish the phenolic species from the furanic ones, the pH of the post-column flow was adjusted to 12 with the co-flow of a 0.1 mol L−1 NaOH solution. At this pH value, the phenolic compounds are fully ionized to phenolates. As a result, bathochromic and hyperchromic shifts occur at the maxima of phenol absorption spectra. In turn, furfural and HMF are not ionized. Thus, no considerable alteration in the spectra of this compound class is expected.
Fig. 5 displays the UV-Vis trace spectra extracted from the DAD chromatogram of HCl-processed beechwood at the retention times 31.3, 43.7, 74.4, 85.5, and 98.7 min, as indicated by yellow-colored arrows in Fig. 4d. It is clear from the bathochromic shifts in the UV-Vis spectra (Fig. 5a) that the co-eluting species sampled at 31.3, 43.7 and 98.7 min correspond to phenolic compounds (lignin fragments), whereas the unchanged UV-Vis spectra collected at 74.4 and 85.5 min confirm the presence of HMF and furfural, respectively. Hence, the intense “lump” background signal shown in Fig. 4a and 4c is most likely due to a response of the refractive index detector to lignin fragments co-eluting with the oligosaccharides. In the chromatogram of the HCl-processed beechwood, the intense absorption band at 280 nm between 20 and 40 min (Fig. 4d) is additional evidence of the presence of large lignin fragments (Mw ∼ 1 to 2 kDa as estimated relative to the eluted saccharides). For comparison, in the chromatogram of the H2SO4-processed beechwood sample, the absorption band at 280 nm shows a less continuous profile, which at higher intensities between 50 and 70 min (Fig. 4c) indicates the presence of lignin fragments with lower molecular weight (∼0.2 to 0.5 kDa). One may thus conclude that a larger quantity of water-soluble lignin fragments with high molecular weight is still present in the solution of HCl-processed beechwood. This is also supported by the trace UV-Vis spectrum at 31.3 min (Fig. 5a). The bathochromic and hyperchromic shifts produced by the post-column adjustment of the pH to 12 causes the appearance of an absorption band at 370 nm (indicated by ‘*’ in Fig. 5a), which is characteristic for lignin-dimeric or larger structures with an α-carbonyl group 1 or even p,p′-stilbene structures 2,49 as depicted in Scheme 2. The presence of 1 was confirmed by HSQC NMR (structure S′, Fig. 3), whereas 2 could not be detected in the spectrum Fig. 3d).
 | ||
| Fig. 5 UV-Vis spectra extracted from the DAD images of the eluates with no post-column pH adjustment and with post-column pH adjusted to 12. (a) Phenolic and (b) furanic components. In Fig. 5a, the absorption band at 370 nm (indicated by ‘*’) is characteristic for lignin-dimeric or larger structures with an α-carbonyl group 1 or even p,p′-stilbene structures 2 (Scheme 2). | ||
 | ||
| Scheme 2 Dimers with an α-carbonyl group 1 and p,p′-stilbene moiety 2. | ||
The chemical nature of the insoluble products
In this section, the chemical nature of the insoluble fraction obtained by milling HCl-impregnated beechwood is presented. In this endeavor, two methods are of importance to elucidate the chemical nature of the insoluble products: saccharification of ‘water-soluble lignocelluloses’ and CP-MAS 13C NMR spectroscopy.Heating a 10% aqueous solution of depolymerized beechwood (pH 1, note that no ‘extra’ acid is required since the mechanocatalytic method does not consume the acid content) the oligosaccharides undergo saccharification.2,6 Simultaneously, a brown precipitate is formed, which shows similar CP-MAS 13C NMR spectra (Fig. 6a and 6b) to that of the lignin extracted by the Organosolv process (Fig. 6d), as it will be discussed next in this section. Upon lignin precipitation, the solution color changes from dark reddish brown to pale yellowish (after removal of the precipitate).6Table 4 summarizes the results obtained from the saccharification experiments with H2SO4- and HCl-processed beechwood.
 | ||
| Fig. 6 CP-MAS 13C NMR spectra of the precipitate obtained from the saccharification of (a) H2SO4- and (b) HCl-processed beechwood samples, from (c) the non-soluble fraction of the HCl-processed beechwood, from (‘d’ and ‘e’, respectively) the organosolv beechwood lignin with and without hemicellulose impurities, (f) α-cellulose and (g) unprocessed beechwood. | ||
| Acid | Sample fraction | Yields relative to the corresponding glucan and xylan fractions (%) | ||||
|---|---|---|---|---|---|---|
| Glucose dimers | Glucose | HMF | Xylose | Furfural | ||
| H2SO4 | Whole | 3.9 | 84 | 1.3 | 89 | 5.3 |
| HCl | Whole | 7.2 | 75 | 0.9 | 88 | 3.6 |
| HCl | Soluble products (74%) | 4.4 | 71 | 0.9 | 71 | 3.6 |
The best sugar yields were obtained by the hydrolysis of the H2SO4-processed beechwood, resulting in 84% glucose, 3.9% cellobiose and 1.3% HMF relative to the glucan fraction in addition to 89% xylose and 5.3% furfural relative to the xylan fraction. The saccharification of the whole sample of HCl-processed beechwood (i.e. insoluble and soluble fractions) led to slightly lower yields compared with that from H2SO4 water-soluble beechwood, i.e. 75% glucose, 7.2% cellobiose, and 0.9% HMF relative to the glucan fraction in addition to 88% xylose and 3.6% furfural relative to the xylan fraction.
Most importantly, the saccharification of the water-soluble fraction of the HCl-processed beechwood released rather similar quantities of glucose (71% vs. 75%) and cellobiose (4.4% vs. 7.1%) in comparison to the saccharification of the whole sample of HCl-processed beechwood. However, the release of xylose distinguishes these samples. While the saccharification of the whole sample produced 88% xylose, the hydrolysis of the water-soluble products led to 71%. These findings indicate that the insoluble part of the HCl-processed sample is composed of lignin associated with a small fraction of hemicellulose fragments.
In order to further assess the chemical nature of the insoluble product obtained by milling HCl-impregnated beechwood, CP-MAS 13C NMR spectra were collected of lignin precipitates, the insoluble fraction of the HCl-processed beechwood, Organosolv beechwood lignin with and without hemicellulose impurities, α-cellulose, and unprocessed beechwood (Fig. 6).
Fig. 6 shows that CP-MAS 13C NMR spectra of the precipitate obtained from the saccharification of H2SO4- and HCl-processed beechwood samples (Fig. 6a and 6b, respectively) and organosolv beechwood lignin (without hemicellulose impurities, Fig. 6d) are similar. The resonances characteristic for lignin are present (Table 5), in addition to a band centered at about 75 ppm, which can be assigned to C2,3,5 of cellulose or C2,3,4 of xylans (Fig. 6f, α-cellulose that comprises 76% glucans and 16% xylans). The existence of this band indicates the presence of residual glucan and xylan in the lignin precipitate, and agrees well with the overall yields of glucan-based products close to 90% and of xylan-based products close to 95%, as obtained by the saccharification at 140 °C for 1 h (Table 4). In addition to the strong signal at about 75 ppm, attributable to the residual content of carbohydrates, both the water-insoluble product of HCl-processed beechwood (Fig. 6c) and the unprocessed beechwood sample (Fig. 6g) show a strong band at around 170 ppm. This signal characterizes several oxidized carboxyl structures, and suggests that galacturonic units be present as carboxylic acid, carboxylate and esters. As the oligosaccharides are hydrolyzed, the intensity of this signal decreases (Fig. 6a and 6b). Altogether, this observation is indicative of a covalent bond through ester linkages between residual hemicellulose and lignin in the water-insoluble residue of HCl-processed beechwood (Fig. 6c).
| 13C shift (ppm) | Assignment |
|---|---|
| 200–160 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in several oxidized structures (e.g. galacturonic units) O in several oxidized structures (e.g. galacturonic units) |
| 153 | C3 and C5 in etherified syringyl units |
| 148 | C3 and C4 in etherified guaiacyl units |
| 135 | C1 in etherified syringyl and guaiacyl units |
| 122 | C6 in guaiacyl units |
| 113 | C2 in guaiacyl units |
| 105–103 | C1 in carbohydrate and C3 and C6 in syringyl units |
| 86–81 | Cβ in β-O-4 linked units |
| 90–70 | C2,3,4,5 in cellulose and C2,3,4 in xylans and Cα in β-O-4 linked units |
| 62–60 | C6 in cellulose, C5 in xylans and Cγ in β-O-4 linked units |
| 56 | Ar–O–CH3 |
| 44 | Cα methyne with aliphatic substitution |
| 35 | Cα in aryl propanol |
| 31 | Alkyl CH2 |
| 20–15 | CH3 group in acetylated xylan and terminal CH3 group |
Conclusion
The in-depth analysis of the ‘water-soluble beechwood’ lends important insights into the chemistry of the mechanocatalytic depolymerization of lignocellulose, as follows:1. The depolymerization is an acid-catalyzed process. The contribution of radical chemistry to the depolymerization rate appears to be negligible. Otherwise, the depolymerization rates found for beechwood would starkly contrast with that of α-cellulose, since lignin is a radical scavenger.
2. The presence of β-O-4 lignin linkages in the depolymerized beechwood samples strongly indicates that the action of mechanical forces, and not the eventual formation of hot spots, is driving the acid-catalyzed depolymerization of the carbohydrate fraction in addition to the acidolysis of the lignin linkages.
3. Either H2SO4 or HCl enables similar process performance with respect to the depolymerization extent of cellulose. Surprisingly, by milling HCl-impregnated beechwood, a 74% yield of water-soluble is achieved. Under similar conditions, the conversion H2SO4-impregnated beechwood leads to a 100% yield of ‘water-soluble beechwood.’ Again, the combination of results from 2D HSQC NMR, HPLC and gel filtration chromatography indicates that lignin is depolymerized to a lesser extent in the presence of HCl.
4. Despite the differences in the extent of lignin depolymerization, the aqueous phase saccharification of the depolymerized samples leads to quite similar yields of glucose and xylose products at 140 °C for 1 h. We found that the insoluble product, obtained by milling HCl-impregnated beechwood for 2 h, is composed mostly of lignin.
5. The solvent-free impregnation of lignocellulose with gaseous hydrogen chloride is showed to be as effective as that with H2SO4 performed in liquid phase. Therefore, the procedure for deep depolymerization can be simplified by contacting lignocellulose with gaseous HCl prior to milling the acid-impregnated substrate, thus eliminating the need of a solvent for the impregnation procedure.
Although the use of gaseous HCl is associated with corrosion problems, it is important to keep in mind that the acid impregnation is performed under ambient conditions (i.e. 25 °C, 1 bar). Under such low-severity conditions is fully enabled the utilization of inexpensive polymeric materials (e.g. PVC) for the construction of the impregnation reactor. With regard to corrosion problems, both the balls and the mill vial (stainless steel) have proven very resistant to corrosion. In fact, no change in the weight of the balls and the mill vial, even after 600 experiments, was detected.
Experimental
General
Sulfuric acid (95–97%, J. T. Baker), hydrogen chloride (99.8%, Air Liquide), α-cellulose (76% glucans, 16% xylans, 6% humidity, 0.1% ash, and 1.9% others, Aldrich) and diethyl ether (99%, Aldrich) were used as received. Pellets from beechwood (41% glucans, 21% xylans, 24% lignin, 5% humidity, 0.4% ash, and 8.4% others) were comminuted with a blender. The sawdust was sieved. Powders with a particle size smaller than 250 μm were collected and used for the mechanocatalytic experiments.Wet impregnation with an H2SO4 solution in diethyl ether
The beechwood or α-cellulose (10 g) was suspended in a diluted H2SO4 solution in diethyl ether (150 mL). Note: To avoid degradation of the substrates upon the prolonged contact with the acidic solution, we chose to use a 0.065 mol L−1 H2SO4 solution for the acid impregnation. The suspension was shaken for 1 h (IKA shaker, KS 130 control, 350 rpm). The organic solvent was removed under reduced pressure at 40 °C. A fine powder with loose particles was obtained. This procedure led to an acid-loading of 0.8 ± 0.1 mmol H2SO4 per gram of substrate. The powder was immediately processed in a ball mill or stored in a closed vial and kept in a freezer (−10 °C) to prevent substrate decomposition that would normally occur to form grayish to black powder after several days of storage at room temperature.9Solvent-free impregnation with gaseous HCl
Beechwood sawdust (5 g) was exposed to a flow of gaseous HCl for 15 min. Next, the substrate was evacuated for 1 h (10−3 mmHg). Again, a fine powder with loose particles was obtained. The powder was immediately processed in a ball mill or stored in a closed vial and kept in a freezer (−10 °C).Determination of acid loading
Typically, 1 g of the acid impregnated substrate was suspended in 40 mL water. Subsequently, titration with a 0.0100 mol L−1 NaOH solution was performed on a Metrohm Titrino Plus 848 automated titrator.Mechanocatalytic depolymerization
The mechanocatalytic depolymerization of lignocellulose was performed in a stainless steel vial (12 mL; 5 stainless steel balls of 4 g each) using a planetary ball mill (Fritsch, Pulverisette P7). The acid-impregnated substrate (1 g) was processed at 800 rpm. The data displayed in Fig. 1 was collected in separate experiments performed at the indicated milling durations. Full conversion of beechwood was achieved at a milling duration of 2 h; pinewood and sugarcane bagasse required a duration of 3 h for full conversion. Under working conditions, the temperature inside the mill did not exceeded 42 °C after milling for 0.5 h. In experiments of longer duration, the mill was switched off every 0.5 h for 10 min to avoid overheating and thermal decomposition of the sample. Note that the experiment duration refers exclusively to the total milling time applied to the sample. The product was then collected and kept in an air-tight vial at −10 °C prior to analysis or saccharification experiments.Extraction of lignin from beechwood by the organosolv process for NMR comparison purposes
Beechwood (16–17 g) was suspended in a 140 mL solution of ethanol–water (1:1, v/v) in a 250 mL autoclave equipped with a mechanical stirrer. The suspension was processed at 180 °C for 3 h. In sequence, the mixture was left to cool down to room temperature. A reddish-brown solution was obtained after filtering off the lignocellulose fibers (pulp). Ethanol was partially evaporated at 60 °C using a rotoevaporator. This procedure leads to lignin precipitation. The solid was collected by filtration and, in sequence, resuspended in hot water in order to remove hemicellulose sugars. Next, the suspension was filtered and the solid washed several times with hot water. Finally, the organosolv beechwood lignin was dried in oven at 40 °C for 1 d.
Determination of the water-soluble products
The processed substrate (0.500 g) was suspended in water (25 mL) and shaken for 5 min. The suspension was centrifuged for 10 min. The residue was washed one more time with water (25 mL), centrifuged and finally dried overnight at 90 °C. The weight of the solid residue was recorded. The solubility was then determined using the difference in weight, as described elsewhere.2Determination of the solubility in DMSO
The depolymerized beechwood sample (50 mg) was dispersed in 1 mL of DMSO and sonicated for 20 min. In sequence, the sample was filtered through a 1.0 μm PTEE membrane (Omnipore membrane filter, JAWP04700, Merck Millipore) and washed with water. The membrane was dried overnight at 60 °C in an oven. The solubility was determined by weight difference.Saccharification of ‘water-soluble beechwood’
The depolymerized beechwood samples (i.e. HCl- and H2SO4-impregnated beechwood samples milled for 2 h) were solubilized in water forming a 10% solution with a pH value of 1. In a closed glass vial, the beechwood solution (9 mL) was heated at the indicated temperatures for 1 h. Upon heating, a solid residue was formed, which was isolated from the sugar solution by centrifugation. The precipitate was washed with 20 mL water six times. The aqueous solutions were combined and set aside for HPLC analysis. In turn, the solid residue was dried in an oven at 60 °C for 24 h. The solid residue was weighed and stored at −10 °C.Sugar and furfural quantification
HPLC analysis was performed on a Shimadzu LC-20 equipped with a column switcher combining two organic acid resin columns (100 and 300 mm in length and 8 mm inner diameter). An aqueous solution of trifluoroacetic acid (2 mmol L−1) was used as the eluent (1 mL min−1). Glucose, glucose-dimers and xylose were analyzed using an RI detector; furfural and HMF, with a UV-Vis detector operating at 280 nm. The yields of glucose, cellobiose and 5-hydroxymethylfurfural are given relative to the glucan content of the unprocessed substrate; the yields of xylose and furfural are given relative to the xylan content.2Gel filtration chromatography
The analyses were performed on a Perkin-Elmer HPLC 200 using a series of four TSKgel G-Oligo-PW columns (6 μm, 7.8 mm I.D. × 30 cm L, Tosoh) and milli-Q water as eluent (0.8 mL min−1) at 80 °C. For detection of the oligosaccharides, a refractive index detector was used. For detection of the lignin fragments, a diode-array detector (DAD) was employed. To differentiate the DAD response of furanic from phenolic eluates, the pH of the post-column flow (0.8 mL min−1) was adjusted to 12 by a co-flow (0.1 mL min−1) of a 0.1 mol L−1 NaOH solution through a secondary HPLC pump (Shimadzu).Solution NMR experiments
The NMR spectra of the samples of depolymerized beechwood (HCl- and H2SO4-impregnated beechwood samples milled for 2 h) were acquired at 25 °C with a Bruker AV spectrometer (400 or 500 MHz 1H frequency) equipped with a BBFO probe head with z-gradient. Spectral widths of 20 ppm were used for the 1D 1H spectrum. The relaxation delay for 1D 1H spectrum was 5.0 s following a 30-degree excitation pulse. For 1D inverse-gated 13C spectrum, the relaxation delay was set to 1.0 s following a 30-degree excitation pulse. 1H-decoupling with the Waltz-16 sequence was applied during acquisition. The number of collected points was 64k for 1H and for 13C. The 1D 1H spectra were processed using an exponential weighting function (lb 0.2 Hz) prior to Fourier transform. The 2D HSQC NMR (Bruker standard pulse sequence “hsqcetgpsi” with delay optimized for 1JCH of 145 Hz) were set up with spectral widths of 20 ppm and 180 ppm for 1H- and 13C-dimensions, respectively. The number of collected complex points was 2048 for 1H-dimension with a recycle delay of 3.13 s (3.0 s relaxation delay and 0.13 s acquisition time). The number of transients for the HSQC spectra was between 12 and 24, and 512 time increments were recorded in 13C-dimension resulting for in an overall experiment time of 6 to 12 h. For HSQC experiments, a squared cosine-bell apodization function was applied in both dimensions, followed by zero-filling to 1024 points in the 13C-dimension prior to Fourier transform. The 1D 1H NMR and 2D HSQC NMR spectra were processed using MestReNova 8.1.1 software.Solid-state 13C CP-MAS NMR experiments
The 13C CP-MAS NMR spectra were measured on a Bruker Avance 500WB spectrometer with a double-bearing standard MAS probe (DVT BL4) at a resonance frequency of 125.8 MHz using 4 mm MAS probe spinning at 10 kHz. The experimental conditions were 2 s recycle delay, between 8000 and 28000 scans, 1 ms contact time, and 4.3 μs 1H π/2 pulse.
Acknowledgements
R.R. is thankful to the Alexander von Humboldt Foundation for the funds provided in the framework of the Sofja Kovalevskaja Award 2010 endowed by the Federal Ministry of Education and Research. The financial support from the Max Planck Society and by an ERC Advanced Grant to F.S. is acknowledged. This work was performed as part of the Cluster of Excellence “Tailor-Made Fuels from Biomass”, which is funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities. We thank A. Deege and H. Hinrichs for HPLC measurements in addition to G. Calvaruso for supporting in some experiments.Notes and references
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- Q. Zhang and F. Jérôme, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed.
- F. Schüth, R. Rinaldi, N. Meine, M. Käldström, J. Hilgert and M. D. K. Rechulski, Catal. Today, 2014 DOI:10.1016/j.cattod.2014.02.019.
- M. Käldström, N. Meine, C. Fares, R. Rinaldi and F. Schüth, Green Chem., 2014, 16, 2454–2462 RSC.
- H. Grohn, J. Polym. Sci., 1958, 30, 551–559 CrossRef CAS.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schueth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- A. Shrotri, H. Kobayashi, A. Tanksale, A. Fukuoka and J. Beltramini, ChemCatChem, 2014, 6, 1349–1356 CAS.
- D. S. Hon, in Developments in Polymer Degradation—7, ed. N. Grassie, Springer, Netherlands, 1987, ch. 5, pp. 165–191 DOI:10.1007/978-94-009-3425-2_5.
- F. Lu and J. Ralph, Plant J., 2003, 35, 535–544 CrossRef CAS.
- D. Y. Lee, M. Matsuoka and M. Sumimoto, Holzforschung, 1990, 44, 415–418 CrossRef CAS.
- D. Y. Lee and M. Sumimoto, Holzforschung, 1990, 44, 347–350 CrossRef CAS.
- D. Y. Lee, S. Tachibana and M. Sumimoto, Cellul. Chem. Technol., 1988, 22, 201–210 CAS.
- Z. H. Wu, M. Sumimoto and H. Tanaka, J. Wood Chem. Technol., 1995, 15, 27–42 CrossRef CAS.
- C. Loerbroks, R. Rinaldi and W. Thiel, Chem. – Eur. J., 2013, 19, 16282–16294 CrossRef CAS PubMed.
- T. P. Nevell and W. R. Upton, Carbohydr. Res., 1976, 49, 163–174 CrossRef CAS.
- S. Heikkinen, M. M. Toikka, P. T. Karhunen and I. A. Kilpeläinen, J. Am. Chem. Soc., 2003, 125, 4362–4367 CrossRef CAS PubMed.
- K. Hu, W. M. Westler and J. L. Markley, J. Am. Chem. Soc., 2011, 133, 1662–1665 CrossRef CAS PubMed.
- K. Cheng, H. Sorek, H. Zimmermann, D. E. Wemmer and M. Pauly, Anal. Chem., 2013, 85, 3213–3221 CrossRef CAS PubMed.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protocols, 2012, 7, 1579–1589 CAS.
- J. Ralph and L. L. Landucci, NMR of lignins, CRC Press, 2010 Search PubMed.
- R. Samuel, M. Foston, N. Jaing, S. Cao, L. Allison, M. Studer, C. Wyman and A. J. Ragauskas, Fuel, 2011, 90, 2836–2842 CrossRef CAS PubMed.
- J.-L. Wen, S.-L. Sun, B.-L. Xue and R.-C. Sun, Materials, 2013, 6, 359–391 CrossRef CAS PubMed.
- M. Bøjstrup, B. O. Petersen, S. R. Beeren, O. Hindsgaul and S. Meier, Anal. Chem., 2013, 85, 8802–8808 CrossRef PubMed.
- C. Loerbroks, R. Rinaldi and W. Thiel, Chem. – Eur. J., 2013, 19, 16282–16294 CrossRef CAS PubMed.
- L. Vanoye, M. Fanselow, J. D. Holbrey, M. P. Atkins and K. R. Seddon, Green Chem., 2009, 11, 390–396 RSC.
- R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits and F. Schüth, ChemSusChem, 2010, 3, 266–276 CrossRef CAS PubMed.
- R. B. Santos, P. W. Hart, H. Jameel and H. M. Chang, BioResources, 2013, 8, 1456–1477 Search PubMed.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 CAS.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- E. Dorrestijn, L. J. J. Laarhoven, I. W. C. E. Arends and P. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54, 153–192 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, in Top Value-Added Chemicals from Biomass, Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, 2007, available from http://www1.eere.energy.gov/bioenergy/pdfs/pnnl-16983.pdf Search PubMed.
- F. Abdelkafi, H. Ammar, B. Rousseau, M. Tessier, R. El Gharbi and A. Fradet, Biomacromolecules, 2011, 12, 3895–3902 CrossRef CAS PubMed.
- E. Adler, J. M. Pepper and E. Eriksoo, Ind. Eng. Chem., 1957, 49, 1391–1392 CrossRef CAS.
- L. H. Hoo, K. V. Sarkanen and C. D. Anderson, J. Wood Chem. Technol., 1983, 3, 223–243 CrossRef CAS.
- T. Imai, T. Yokoyama and Y. Matsumoto, J. Wood Sci., 2011, 57, 219–225 CrossRef CAS PubMed.
- H. Ito, T. Imai, K. Lundquist, T. Yokoyama and Y. Matsumoto, J. Wood Chem. Technol., 2011, 31, 172–182 CrossRef CAS.
- O. Karlsson and K. Lundquist, Acta Chem. Scand., 1992, 46, 283–289 CrossRef CAS PubMed.
- O. Karlsson, K. Lundquist, S. Meuller and K. Westlid, Acta Chem. Scand. Ser. B, 1988, 42, 48–51 CrossRef PubMed.
- K. Lundquist, Acta Chem. Scand., 1973, 27, 2597–2606 CrossRef CAS PubMed.
- K. Lundquist and L. Ericsson, Acta Chem. Scand., 1970, 24, 3681–3686 CrossRef CAS PubMed.
- K. Lundquist and R. Lundgren, Acta Chem. Scand., 1972, 26, 2005–2023 CrossRef CAS PubMed.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, BioEnergy Res., 2008, 1, 205–214 CrossRef.
- S. Y. Lin and C. W. Dence, Methods in Lignin Chemistry, Springer-Verlag, Berlin, Heidelberg, 1992 Search PubMed.
- G. R. Hatfield, G. E. Maciel, O. Erbatur and G. Erbatur, Anal. Chem., 1987, 59, 172–179 CrossRef CAS.
- A. T. Martínez, A. E. González, M. Valmaseda, B. E. Dale, M. J. Lambregts and J. F. Haw, Holzforschung, 1991, 45, 49–54 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |