 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physical properties and hydrolytic degradability of polyethylene-like polyacetals and polycarbonates†
Patrick
Ortmann
,
Ilona
Heckler
and
Stefan
Mecking
*
Chair of Chemical Materials Science, Department of Chemistry, University of Konstanz, Universitätsstrasse 10, D-78457 Konstanz, Germany. E-mail: stefan.mecking@uni-konstanz.de; Fax: +49 7531 885152; Tel: +49 7531 882593
First published on 12th February 2014
Abstract
Long-chain polyacetals and polycarbonates were prepared by polycondensation of α,ω-diols (C18, C19, C23) derived from fatty acids as a renewable feedstock with diethoxymethane and dimethyl carbonate, respectively, in one step. Studies of hydrolytic degradation of the solid polymers show a much higher stability compared to their shorter-chain counterparts. Long-chain polyacetals were found to degrade slowly under acidic conditions, while the long-chain polycarbonates also degraded in a basic environment. To rationalize the impact of acetal and carbonate groups on the thermal and crystalline properties of polyacetals and polycarbonates, additional model polymers with a further reduced and systematically varied functional group density were generated by ADMET copolymerization of the unfunctionalized undeca-1,10-diene with bis(undec-10-en-1-yloxy)methane or di(undec-10-en-1-yl) carbonate, respectively, followed by exhaustive hydrogenation. Long-chain polycarbonates possess polyethylene-like solid state structures. By comparison to polyesters, a given density of carbonate groups in the polymer chain reduces melting and crystallization temperatures significantly more strongly. By contrast, long-chain polyacetals possess more complex non-uniform crystal structures, and only adopt a polyethylene-like structure at very low densities of acetal groups. Also, acetal groups more strongly impact melting and crystallization temperatures vs. carbonates.
Introduction
Recent advances in catalytic conversions of plant oils have provided access to long-chain α,ω-functionalized aliphatic monomers.1 Biotechnological or synthetic approaches like ω-oxidation,2–4 isomerizing alkoxycarbonylation5–8 and olefin metathesis9–11 generate α,ω-dicarboxylic acids and derivatives suitable as monomers for polycondensates like polyesters,7 polyamides7 and polyurethanes.12 The incorporation of the fatty acids’ long-chain methylene sequences into the aliphatic polymer chains results in high degrees of crystallinity and high melting points, advantageous for thermoplastic processing.13 This is due to van der Waals interactions between the hydrocarbon segments, akin to polyethylene.Different from polyethylene, such polycondensates offer the possibility of hydrolytic degradability. This can be a useful property, for example to prevent long-term accumulation of materials lost to the environment. In any case, knowledge of environmental stability or decomposition rates is essential for these new materials. By comparison to shorter chain congeners, such degradation is significantly slower as a result of the high crystallinity and hydrophobicity.14,15 This is illustrated by Heise and coworkers’ study of the hydrolytic degradation of longer-chain polycondensates.16 For aliphatic polyesters containing C14 methylene sequences between the ester functionalities, no mass loss or molecular weight reduction indicative of hydrolysis was observed in slightly basic buffer solution over a period of two years.
By comparison to ester groups, acetal and carbonate functionalities are more amenable to hydrolysis. Polyacetals can be subjected to acid-catalyzed hydrolysis,17–20 leading to formation of the corresponding (monomeric) diols. Their biocompatibility and non-toxicity render such materials unproblematic from this perspective.21 Likewise, polycarbonates are biocompatible and non-toxic,22 and they are sensitive towards acidic as well as basic aqueous environments.23,24 Our preliminary findings20 showed that long-chain polyacetals, generated from isolated α,ω-diacetals, are hydrolyzed slowly. This differs advantageously from their shorter-chain analogues, which are decomposed rapidly upon exposure to an aqueous environment.
We will now give a full account of thermoplastic long-chain polyacetals and polycarbonates including their hydrolytic degradation and physical properties.
Results and discussion
Synthesis of long-chain polyacetals and polycarbonates
In order to provide sufficient access to long-chain polyacetals (PA-18, PA-19, PA-23) and polycarbonates (PC-18, PC-19, PC-23), we established a one-pot synthesis directly from the corresponding diols as the starting material (Scheme 1). Essentially, this follows the procedures for shorter chain (C5 to C12) analogs,17,25,26 slightly adapting reaction conditions to the different melting points and solubilities of the long-chain linear α,ω-diols (Scheme 1).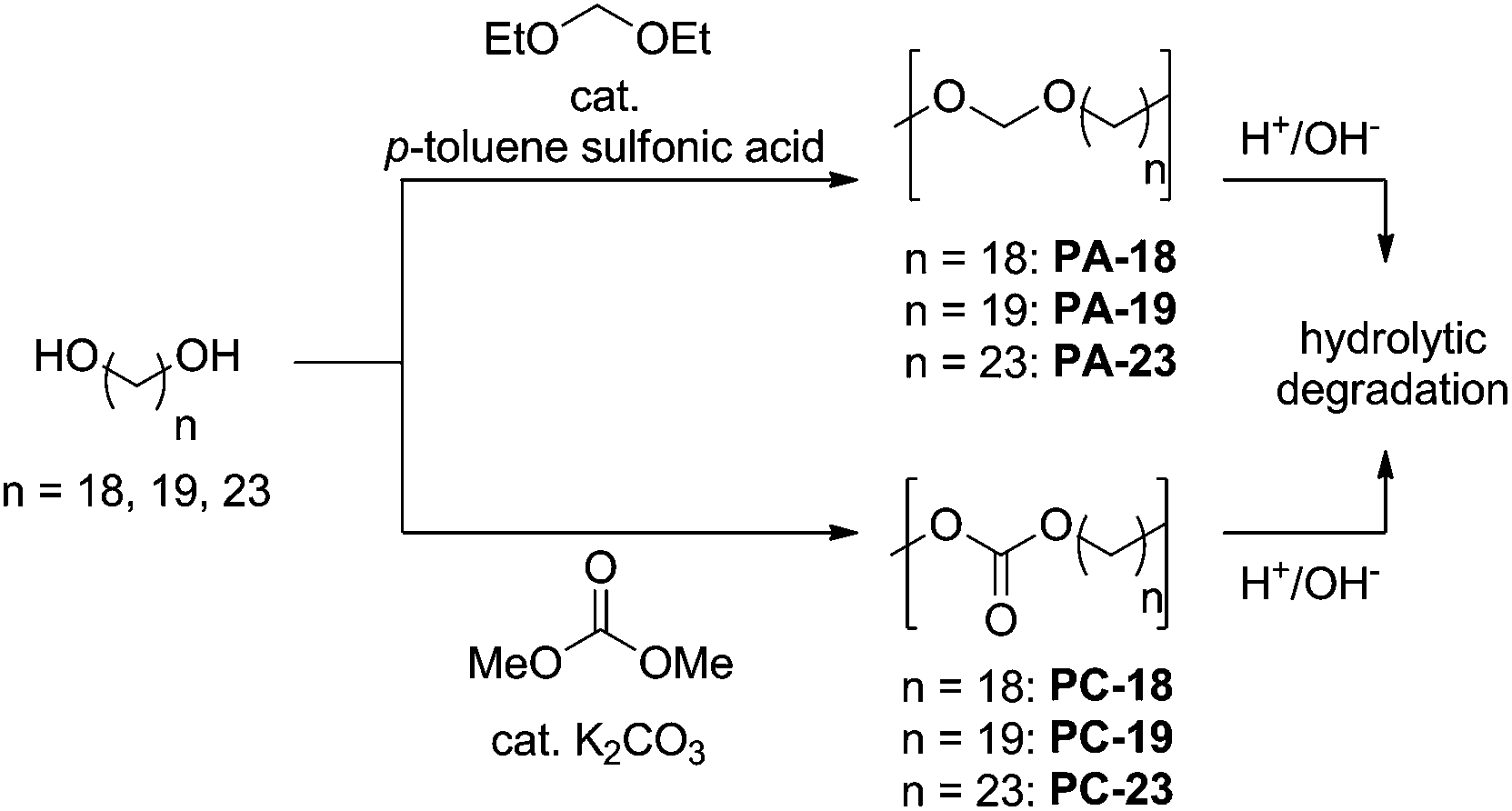 | ||
| Scheme 1 One-pot synthesis of long-chain polyacetals and polycarbonates, together with possible hydrolytic degradation pathways. | ||
The C19- and C23-diol starting materials were generated by catalytic isomerizing alkoxycarbonylation of methyl oleate and ethyl erucate, respectively, followed by catalytic reduction of the ester moieties.7 In the resulting diols the lengths of the linear methylene sequences correspond to the lengths of entire fatty acid chains. The C18-diol was prepared by esterification followed by catalytic reduction of the commercially available C18-diacid (which is generated by enzymatic ω-oxidation of fatty acid feeds).
| Compound |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (g mol−1) (NMR) a (g mol−1) (NMR) |
M
nb (g mol−1) (GPC vs. PE) |
M
w/Mnb |
T
mc (°C) |
T
cc (°C) |
ΔHmc (J g−1) |
|---|---|---|---|---|---|---|
| a Determined by end-group analysis from 1H NMR spectroscopy. b Determined by GPC at 160 °C in 1,2,4-trichlorobenzene versus polyethylene standards. c Determined by DSC with a heating/cooling rate of 10 °C min−1, data collected from the second heating cycle. d Two distinct melting transitions are observed. | ||||||
| PA-18 | 16000 |
n.d. | n.d. | 76/82d | 64 | 147 |
| PA-19 | 14000 |
n.d. | n.d. | 77/83d | 67 | 158 |
| PA-23 | 12000 |
n.d. | n.d. | 87 | 72 | 167 |
| PC-18 | 28000 |
17000 |
2.8 | 89 | 67 | 116 |
| PC-19 | 15000 |
11000 |
5.1 | 89 | 71 | 143 |
| PC-23 | 18000 |
15000 |
7.5 | 97 | 77 | 156 |
Analysis by differential scanning calorimetry (DSC) revealed peak melting temperatures between 82 °C and 87 °C (Table 1) and degrees of crystallinity between 50 and 60%.27 As expected, with increasing diol chain length higher melting points are observed due to increasing van der Waals interactions between the polymer chains. Interestingly, the melting curves of PA-18 and PA-19 clearly display two melting transitions (displayed in the ESI†).
DSC analysis revealed melting temperatures between 89 °C and 97 °C for the long-chain polycarbonates. Different from some of the polyacetals (vide supra), a single well-behaved melting transition was observed for all polycarbonates. Again, with growing aliphatic chain length, the melting points increase as expected.
Hydrolytic degradation studies
Hydrolytic degradation experiments were performed on solid samples in concentrated and diluted acidic and basic aqueous media (on pellets prepared by melting the polymer in a mold, see ESI†). Beyond insights into the behavior towards these media, the two week experiments performed to some extent also allow for an estimation of the behavior on longer terms under milder conditions of pH.| Polymer pelleta | Degradation mediumb | Weight loss after degradation experimentc (%) |
|---|---|---|
| a For pellet preparation and exact weights of the samples used, see ESI. b 5.0 mL of media solution at 40 °C. c For exact weights of the pellets before and after the degradation experiments, see ESI. d Degradation experiments were performed for 16 days. | ||
| PA-18 | 20 wt% aq. NaOHd | 0.6 |
| PA-19 | 0.0 | |
| PA-23 | 0.0 | |
| PA-18 | 3 M aq. HCld | 0.8 |
| PA-19 | 0.6 | |
| PA-23 | 0.7 | |
| PA-18 | Conc. aq. HCld | 5.1 |
| PA-19 | 4.0 | |
| PA-23 | 2.3 | |
All polyacetal pellets were stable in the basic environment. Weight losses of the pellets were within the error of the method.28 Their appearances before (Fig. 1A) and after the degradation experiment were similar (Fig. 1B). The weights of the pellets from the 3 M aq. HCl solution were not altered significantly either, but their appearances varied slightly during the experiments (Fig. 1C). Degradation of polyacetals occurred with an observable weight loss in concentrated aq. HCl solution. The weight loss was up to 5.1% (for PA-18). The extent of degradation increased with decreasing length of the methylene sequences in the polymers. During the degradation experiments all pellets’ surface structures changed significantly (Fig. 1D).
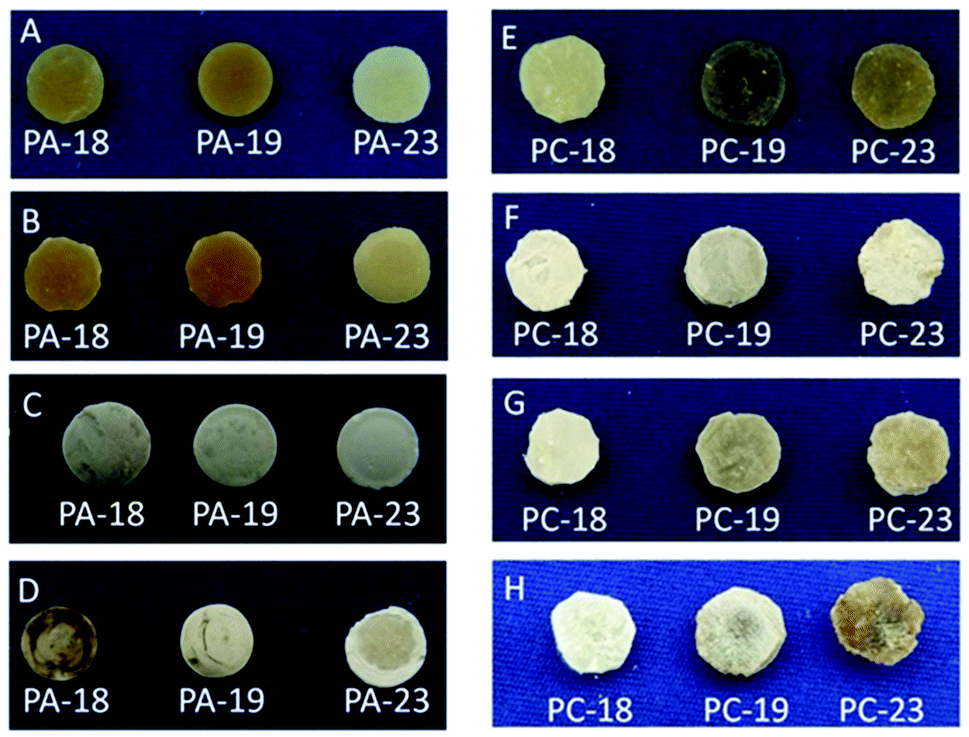 | ||
| Fig. 1 Pellets of polyacetals PA-18, PA-19 and PA-23 before (A) and after hydrolytic degradation in 20 wt% aq. NaOH (B), 3 M aq. HCl (C), and concentrated aq. HCl (D). Pellets of polycarbonates PC-18, PC-19 and PC-23 before (E) and after hydrolytic degradation in 20 wt% aq. NaOH (F), 2 wt% aq. NaOH (G), and concentrated aq. HCl (H). | ||
As a conclusion, long-chain polyacetals are stable in a basic environment, but degrade under acidic conditions. In accordance with prior findings, the degradation rate of the long-chain polyacetals decreases with increasing length of the methylene sequences.20 Mid-chain polyacetals like PA-12 degrade much faster in such acidic environments, losing half of their weight within ca. 15 min.
| Polymer pelleta | Degradation mediumb | Weight loss after degradation experimentc (%) |
|---|---|---|
| a For pellet preparation and exact weights of the samples used, see ESI. b 5.0 mL of media solution at 40 °C. c For exact weights of the pellets before and after the degradation experiments, see ESI. d Degradation experiments were performed for 25 days. e Degradation experiments were performed for 27 days. | ||
| PC-18 | 20 wt% aq. NaOHd | 2.3 |
| PC-19 | 1.2 | |
| PC-23 | 0.9 | |
| PC-18 | 2 wt% aq. NaOHd | 0.4 |
| PC-19 | 0.6 | |
| PC-23 | 0.4 | |
| PC-18 | Conc. aq. HCle | 2.5 |
| PC-19 | 1.6 | |
| PC-23 | 0.5 | |
The weight loss of PC-18, PC-19 and PC-23 in 20 wt% aq. NaOH solution (25 days) was low, but noticeable and depended on the length of the methylene chain length. PC-18 degraded faster than PC-19 and PC-23. Likewise the appearances and surface structure of the pellets changed during degradation (Fig. 1E and F). In 2 wt% aq. NaOH solution, mass losses were less than 1% for all polycarbonates after 25 days, and thus close to the accuracy of the method.28 However, the appearances of the pellets were similar compared to the experiments in 20 wt% aq. NaOH solution after degradation (Fig. 1G).
The experiments performed in concentrated aq. HCl solution resulted in a similar extent of degradation as observed for 20 wt% aq. NaOH. The weight losses of PC-18, PC-19 and PC-23 were again small, but noticeable and depended on the chain length of the diol monomer. PC-18 degraded slightly faster in concentrated aq. HCl solution than PC-19 and PC-23, resembling the trend observed in a basic environment. Significant changes in the shape and surface roughness of the pellets were observed after the degradation experiments (Fig. 1H).
Methanolic NaOH solution (10 M) was studied as a medium at 40 °C to achieve accelerated degradation due to the higher miscibility of the medium with the solid polymer and degradation products by comparison to water. The pellets were removed from the medium after certain intervals over a total period of 15 hours, washed with water and acetone, dried under vacuum, weighed, and returned to the medium. These experiments work out even more clearly the decreased degradation rate with increasing chain length (Fig. 2; PC-18 > PC-19 > PC-23).
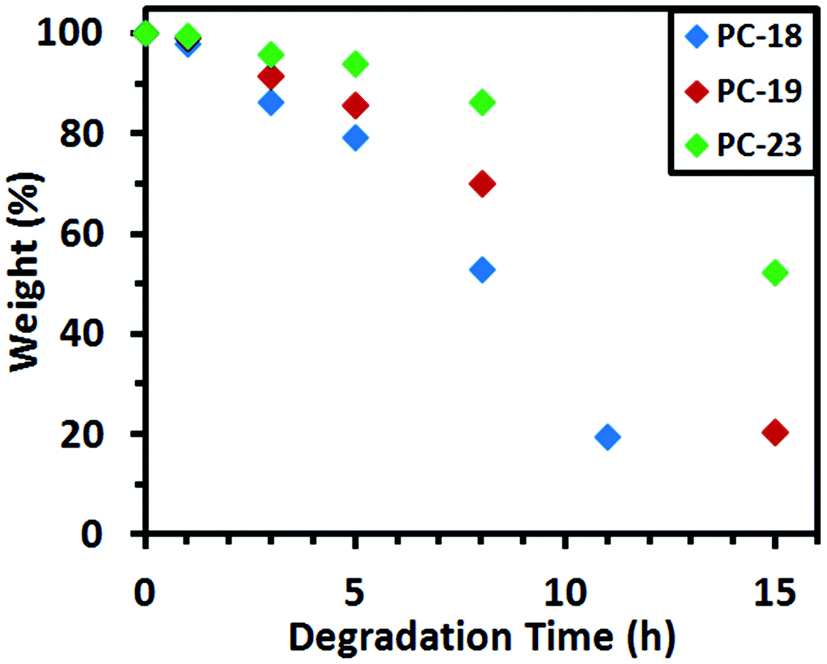 | ||
| Fig. 2 Degradation studies of pellets from different polycarbonates in basic methanolic solution (10 M NaOH) at 40 °C over a period of 15 hours. | ||
Long-spaced aliphatic polyacetals and polycarbonates with further reduced functional group density
The enhanced melting and crystallization points of the long-chain polyacetals and polycarbonates by comparison to their known shorter chain congeners raise the question of how thermal properties evolve with a further reduction of the density of these functional groups along the polymer chain. Eventually, the melting and crystallization behavior must converge toward linear polyethylene. Extending the above synthetic approach to this end would require the preparation of a series of α,ω-diols with an even larger chain length. Other than the rather convenient access to the aforementioned diols directly from fatty acids, this would require more tedious multistep synthesis in each case.Long-chain polyesters like polyester-30,30,29 polyester-44,23 and polyester-38,2330 have in fact been generated by multistep protocols. This doubling of the aliphatic chain length between the ester moieties vs. the long-chain polyesters-X,X (X = 18, 19, 23)7,31 containing methylene sequences originating from one fatty acid molecule resulted in a moderate increase of Tm and Tc. In order to rationalize thermal properties and provide a generic prediction, additional data on polyesters with further reduced ester frequency were required. However, the synthesis of even longer aliphatic compounds becomes more and more challenging with increasing number of methylene units. For the preparation of longer-spaced aliphatic polyesters, this issue was overcome by applying random acyclic diene metathesis (ADMET)32,33 copolymerization in combination with post-polymerization hydrogenation.34 The gap between polyesters from classical A2 + B2 polycondensations and linear polyethylene was overcome in this manner. This approach also offered itself for the polyacetals and polycarbonates of interest here.
To this end, we copolymerized undeca-1,10-diene (1)34 as an aliphatic, non-functionalized monomer with bis(undec-10-en-1-yloxy)methane (2) and di(undec-10-en-1-yl) carbonate (3),35 respectively, to yield unsaturated, linear polyacetals and polycarbonates (Scheme 2).
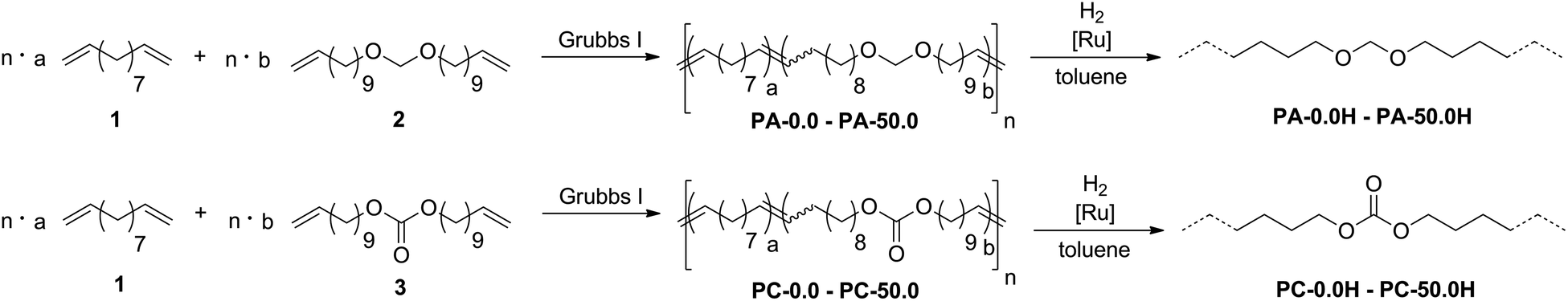 | ||
| Scheme 2 Preparation of long-spaced polyacetals (upper scheme) and polycarbonates (lower scheme) by ADMET copolymerization and postpolymerization hydrogenation. | ||
Exhaustive post-polymerization hydrogenation yielded saturated polymers with a variable density of functional groups randomly distributed along the polymer backbone. Monomer 2 was generated by condensation of 10-undecenol with diethoxymethane using catalytic amounts of methanesulfonic acid to yield the symmetric α,ω-diene with an acetal moiety in the center of the molecule (for synthetic details, see ESI†). The analogous carbonate monomer 3 was prepared by reaction of 10-undecenol with dimethyl carbonate catalyzed by potassium carbonate. Mixtures of defined monomer ratios were copolymerized by 0.5 mol% of [(PCy3)2Cl2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh] (Grubbs first generation catalyst) for two days without solvents under reduced pressure to remove ethylene as a byproduct from the reaction equilibrium. The reaction temperature was raised from room temperature to 65 °C during the first two hours of copolymerization to prevent crystallization of the growing polymer chains. As the catalyst shows the same reactivity for the functionalized and the non-functionalized dienes,36 the acetal and carbonate groups are distributed statistically within the polymer chains. In the case of consecutive incorporation of the functionalized monomers, at least 20 carbon atoms remain between two polar functional groups in the copolymers.
CHPh] (Grubbs first generation catalyst) for two days without solvents under reduced pressure to remove ethylene as a byproduct from the reaction equilibrium. The reaction temperature was raised from room temperature to 65 °C during the first two hours of copolymerization to prevent crystallization of the growing polymer chains. As the catalyst shows the same reactivity for the functionalized and the non-functionalized dienes,36 the acetal and carbonate groups are distributed statistically within the polymer chains. In the case of consecutive incorporation of the functionalized monomers, at least 20 carbon atoms remain between two polar functional groups in the copolymers.
A high degree of conversion could be observed for the unsaturated copolymers by 1H NMR spectroscopy, as evidenced by the virtually complete absence of vinyl proton resonances and the appearance of resonances for internal double bonds (Fig. 3 top, for polyacetals, and Fig. 4 top, for polycarbonates). Molecular weight analysis of the unsaturated copolymers by GPC (at 50 °C in THF versus polystyrene standards) revealed number-average molecular weights Mn around 2 × 104 g mol−1 and molecular weight distributions Mw/Mn around 2 for both polyacetals and polycarbonates as expected for well-behaved polycondensation reactions (Table 4 for polyacetals and Table 5 for polycarbonates). The compositions of the unsaturated copolymers were determined by the ratio of the 1H NMR signal intensities for the methylene groups adjacent to the acetal or carbonate groups for polyacetals and polycarbonates, respectively, to all proton resonances. In all copolymers the compositions were found to be identical to the initial monomer ratios applied for the copolymerization reaction mixtures.
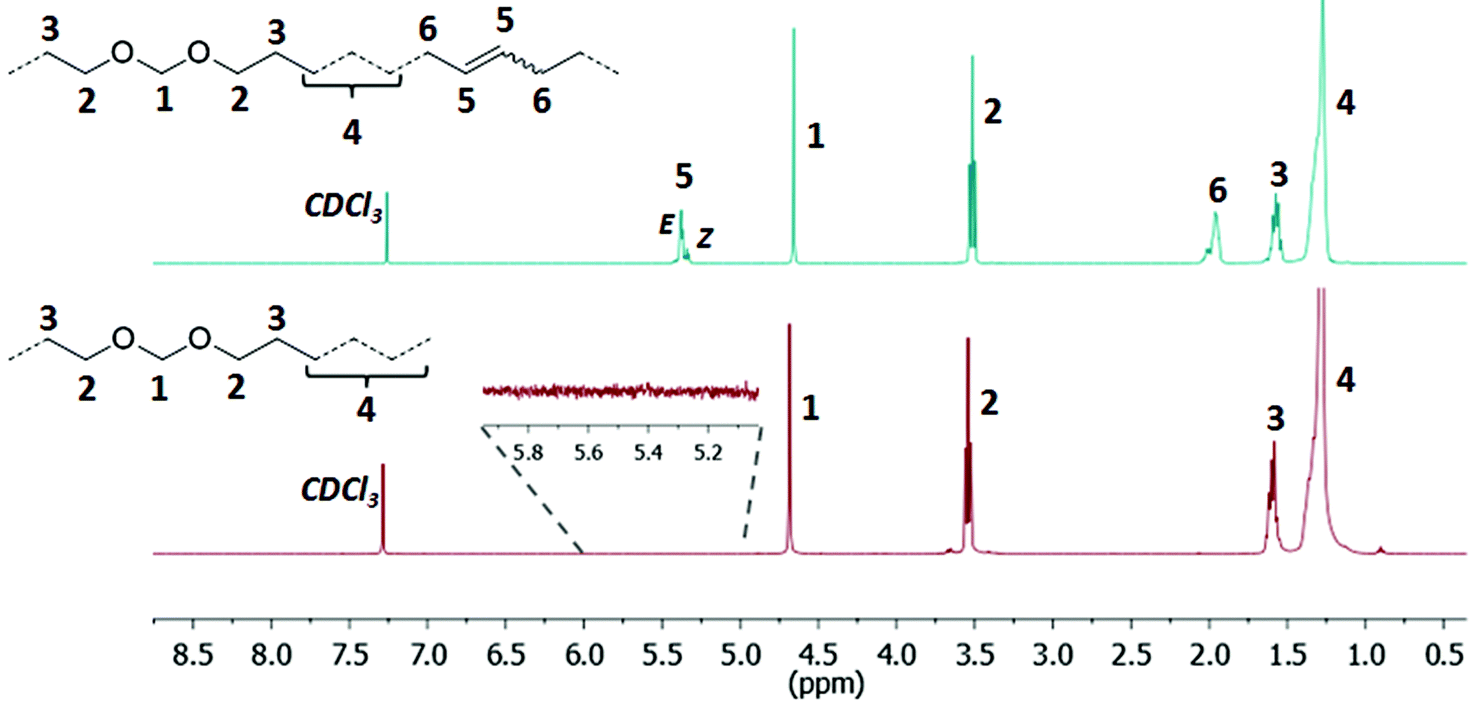 | ||
| Fig. 3 1H NMR spectra of the unsaturated polyacetal PA-50.0 (top, CDCl3, 400 MHz, 25 °C) and the corresponding saturated polyacetal PA-50.0H (bottom, CDCl3, 400 MHz, 25 °C). | ||
 | ||
| Fig. 4 1H NMR spectra of the unsaturated polycarbonate PC-42.9 (top, CDCl3, 400 MHz, 25 °C) and the corresponding saturated polycarbonate PC-42.9H (bottom, CDCl3, 400 MHz, 25 °C). | ||
| Compound | Monomer 1 (mol%) | Monomer 2 (mol%) | Theor. acetal groups per 1000 methylene unitsa | Found acetal groups per 1000 methylene unitsb |
M
nc (g mol−1) (GPC vs. PS) |
M
w/Mnc (GPC vs. PS) |
M
nd (g mol−1) (NMR) |
|---|---|---|---|---|---|---|---|
| a Calculated from the monomer weight ratios applied. b Determined by 1H NMR spectroscopy of the resulting polymers. c Determined by GPC in THF at 50 °C versus polystyrene standards of the unsaturated polymers. d Determined by end-group analysis from 1H NMR spectroscopy of the saturated polymers. | |||||||
| PA-50.0H | 0.0 | 100.0 | 50.0 | 48.4 | 20200 |
2.1 | 13800 |
| PA-39.1H | 38.3 | 61.7 | 39.1 | 38.0 | 24700 |
2.0 | 14100 |
| PA-29.9H | 60.0 | 40.0 | 29.9 | 29.2 | 24800 |
2.0 | 15000 |
| PA-21.3H | 75.0 | 25.0 | 21.3 | 21.0 | 16000 |
2.0 | 10300 |
| PA-9.8H | 90.1 | 9.9 | 9.8 | 10.0 | 14300 |
2.0 | 13000 |
| PA-4.9H | 95.3 | 4.7 | 4.9 | 5.0 | 20100 |
1.9 | 11200 |
| PA-1.5H | 98.7 | 1.3 | 1.5 | 1.5 | 26600 |
1.7 | 10800 |
| PA-0.0H | 100.0 | 0.0 | 0.0 | 0.0 | 16600 |
2.3 | 9200 |
| Compound | Monomer 1 (mol%) | Monomer 3 (mol%) | Theor. carbonate groups per 1000 methylene unitsa | Found carbonate groups per 1000 methylene unitsb |
M
nc (g mol−1) (GPC vs. PS) |
M
w/Mnc (GPC vs. PS) |
M
nd (g mol−1) (GPC vs. PE) |
M
w/Mnd (GPC vs. PE) |
M
ne (g mol−1) (NMR) |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated from the monomer weight ratios applied. b Determined by 1H NMR spectroscopy of the resulting polymers. c Determined by GPC in THF at 50 °C versus polystyrene standards of the unsaturated polymers. d Determined by GPC in 1,2,4-trichlorobenzene at 160 °C versus polyethylene standards of the saturated polymers. e Determined by end-group analysis from 1H NMR spectroscopy of the saturated polymers. | |||||||||
| PC-50.0H | 0.0 | 100.0 | 50.0 | 49.9 | 30500 |
2.0 | 11700 |
2.2 | 23500 |
| PC-42.9H | 27.0 | 63.0 | 42.9 | 42.7 | 30300 |
1.6 | 7500 | 2.1 | 12400 |
| PC-37.5H | 42.5 | 57.5 | 37.5 | 37.5 | 23700 |
1.9 | 7200 | 2.3 | 13600 |
| PC-30.1H | 59.5 | 41.5 | 30.1 | 30.6 | 22100 |
1.8 | 6800 | 2.3 | 13500 |
| PC-20.4H | 76.4 | 23.6 | 20.4 | 21.0 | 22500 |
1.8 | 6600 | 2.0 | 11500 |
| PC-11.5H | 88.1 | 11.9 | 11.5 | 12.0 | 20900 |
1.8 | 6900 | 2.2 | 12000 |
| PC-5.5H | 94.8 | 5.2 | 5.5 | 5.2 | 24200 |
1.7 | 6800 | 2.3 | 13400 |
| PC-1.0H | 99.1 | 0.9 | 1.0 | 1.1 | 24000 |
1.7 | 7200 | 2.3 | 10600 |
| PC-0.0H | 100.0 | 0.0 | 0.0 | 0.0 | 16600 |
2.3 | 10000 |
2.4 | 9200 |
For hydrogenation of the carbon–carbon double bonds, the Fischer carbene formed by quenching Grubbs first generation alkylidene with ethyl vinyl ether was employed. Reaction with hydrogen is known to form [RuHCl(H2)(PCy3)2],37–39 which is very active for the hydrogenation of double bonds. Catalytic hydrogenations of the unsaturated copolymers were performed in toluene in a pressure reactor at 110 °C for 2 days with a hydrogen pressure of 40 bar to yield polyacetals with a content of 39.1 to 1.5 acetal groups and polycarbonates with a content of 42.9 to 1.0 carbonate groups per 1000 methylene units, respectively (PA-39.1H to PA-1.5H40 and PC-42.9H to PC-1.0H, Tables 4 and 5). Full hydrogenation was confirmed by the absence of resonances for unsaturated vinylic protons in the range of 5.00–6.00 ppm in the 1H NMR spectra (Fig. 3 bottom, for polyacetals, and Fig. 4 bottom, for polycarbonates). Furthermore, ADMET homopolymerizations of the monomers 2 and 335 yielded regularly spaced PA-50.0H and PC-50.0H after hydrogenation, respectively. These polymers can also be designated PA-20 and PC-20, analogous to the products of diol polycondensation (vide supra). Polymerization of C20 α,ω-diol with diethoxymethane or dimethyl carbonate, respectively, would yield similar polymers (though exhibiting other chain end groups). Homopolymerization and post-hydrogenation41 of the non-functionalized monomer 1 yielded a defect free linear polyethylene (designated PA-0.0H or PC-0.0H) as previously reported.34
Molecular weights of the saturated polymers were determined by NMR end group analysis for both polyacetals and polycarbonates. High temperature GPC analysis (at 160 °C in 1,2,4-trichlorobenzene versus polyethylene standards) could only be performed on polycarbonates. Polyacetals degraded to a significant extent under the conditions of high temperature GPC analysis, as observed for long-chain polyacetals from α,ω-diol/diethoxymethane polycondensations (vide supra). As expected, GPC vs. polystyrene standards significantly overestimates polymer molecular weights. Due to its more similar structure, linear polyethylene standards represent a better point of reference. Overall, molecular weights from GPC and NMR analyses agree reasonably well with one another and provide a conclusive picture. Number average molecular weights Mn are around 104 g mol−1 and molecular weight distributions Mw/Mn around 2 were observed for saturated polycarbonates.
Thermal and crystalline properties of long-spaced polycarbonates
The thermal properties of long-spaced aliphatic polycarbonates were analyzed by DSC (Table 6 and Fig. 5). The polymers display rather narrow melting and crystallization transitions and crystallinities up to 87% (for all DSC curves, see the ESI†).27 As expected, the melting and crystallization points increase with decreasing number of carbonate functionalities in the polymer and converge towards linear polyethylene (Tm = 134 °C). This qualitative trend is comparable to the behavior of long-spaced polyesters34 and polyethylenes exhibiting methyl branches.42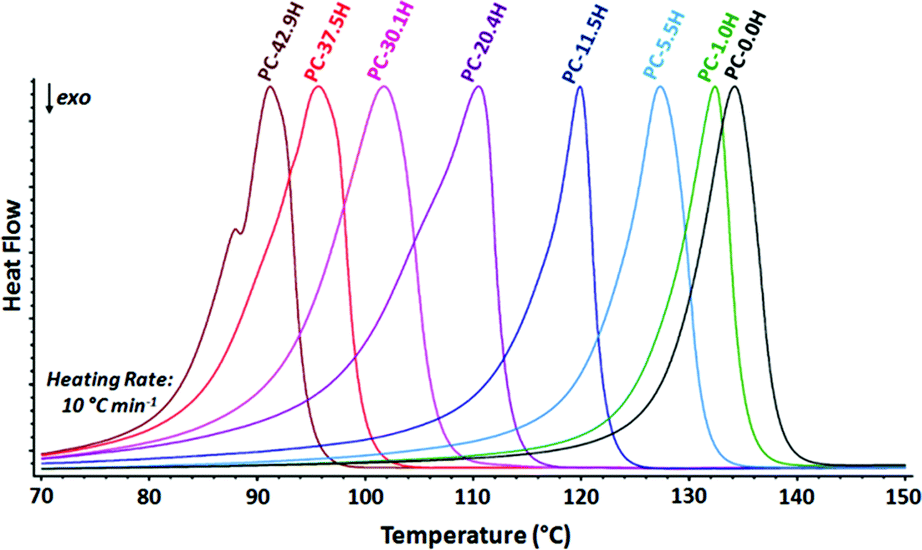 | ||
| Fig. 5 Normalized DSC heating curves of random long-spaced polycarbonates (second heating traces shown). | ||
| Compound | T m (°C) | T c (°C) | ΔH (J g−1) |
|---|---|---|---|
| a Determined at a heating/cooling rate of 10 °C min−1. Peak Tm determined from the second heating cycle. | |||
| PC-50.0H | 92 | 77 | 160 |
| PC-42.9H | 91 | 80 | 190 |
| PC-37.5H | 96 | 83 | 191 |
| PC-30.1H | 102 | 88 | 198 |
| PC-20.4H | 111 | 97 | 186 |
| PC-11.5H | 120 | 107 | 222 |
| PC-5.5H | 127 | 113 | 255 |
| PC-1.0H | 132 | 115 | 246 |
| PC-0.0H | 134 | 117 | 276 |
Wide angle X-ray diffraction (WAXD) patterns of the random long-spaced polycarbonates PC-37.5H and PC-5.5H and of the regular spaced polycarbonates PC-18, PC-19 and PC-23 display dominant reflexes at 2θ angles of 21.5° and 23.9°, which correspond to the 110 plane and the 200 plane of an orthorhombic crystal structure (Fig. 6 and ESI†). For a defect-free ADMET polyethylene (PC-0.0H), the same reflection pattern is found.
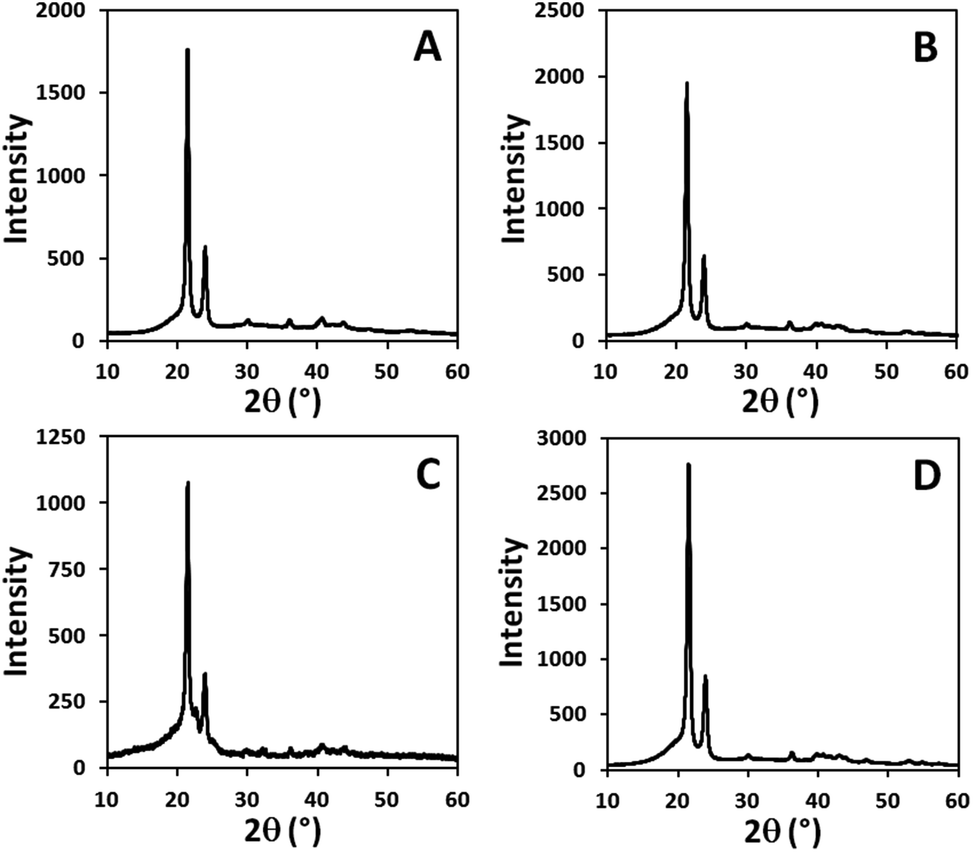 | ||
| Fig. 6 WAXD diagrams of random long-chain polycarbonates PC-37.5H (A) and PC-5.5H (B), regular spaced polycarbonate PC-19 (C) and defect-free linear polyethylene PC-0.0H (D) from ADMET polymerization. | ||
These findings were confirmed by infrared (IR) spectroscopy. Characteristic absorbances are found at 1472–1473 cm−1 and 1463–1464 cm−1 as Davidov splitting for the CH2 scissoring vibration and at 718–720 cm−1 and 730–731 cm−1 for the CH2 rocking vibration, which correspond to a polyethylene-like, orthorhombic crystal modification (Fig. S35†).43,44
As all randomly spaced polycarbonates display the same crystalline structure, the inclusion model for ethylene copolymers of Sanchez and Eby was probed to describe the melting and crystallization behavior.45 While methylene groups (–CH2–) are regarded as repeat units that can crystallize in a lattice, randomly distributed carbonate groups (–OC(O)O–) are considered as non-crystallizable units, which are found in the crystalline regions as defects as well as in the amorphous regions. When the defect units are distributed in a homogeneous fashion in both phases, the melting point depression with respect to the mole fraction of defects and the lamellar thickness should follow the relationship

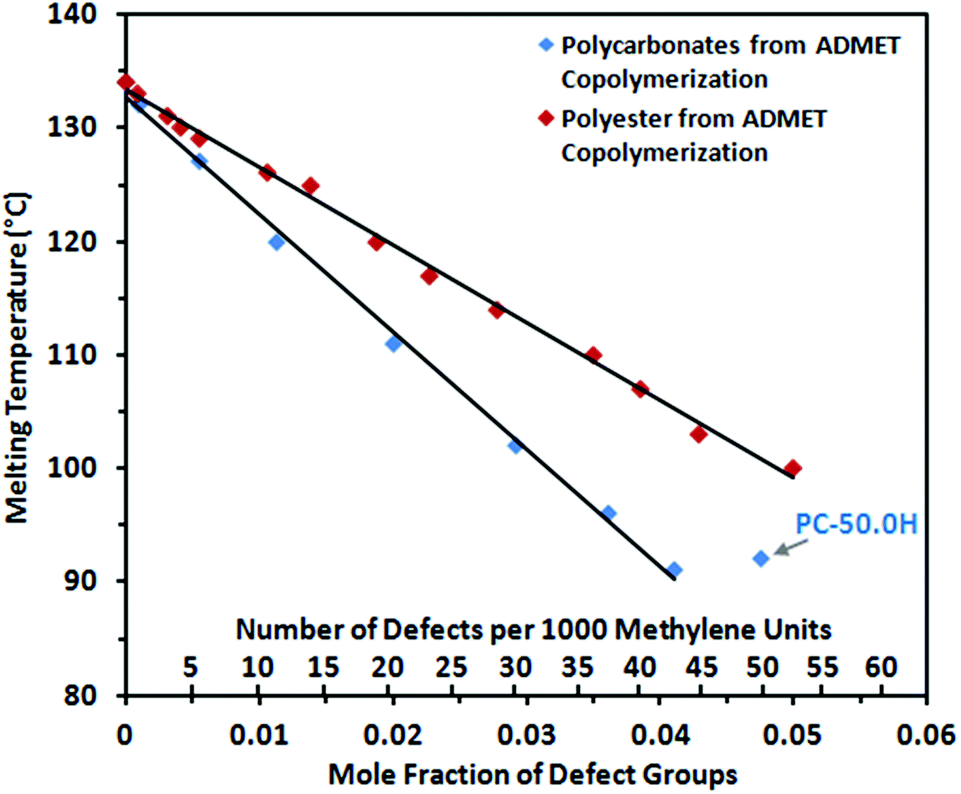 | ||
| Fig. 7 Peak melting points of random long-spaced polycarbonates (blue) and polyesters (red) for comparison34vs. the mole fraction of defect groups (carbonate groups or ester groups, respectively). | ||
 | ||
| Fig. 8 WAXD patterns of regular polyacetals PA-18 (A), PA-19 (B), PA-23 (C) and PA-50.0H (= PA-20) (D) as well as long-spaced random polyacetals PA-39.1H (E), PA-29.9H (F), PA-21.3H (G), PA-9.8H (H), PA-4.9H (I) and PA-1.5H (J). | ||
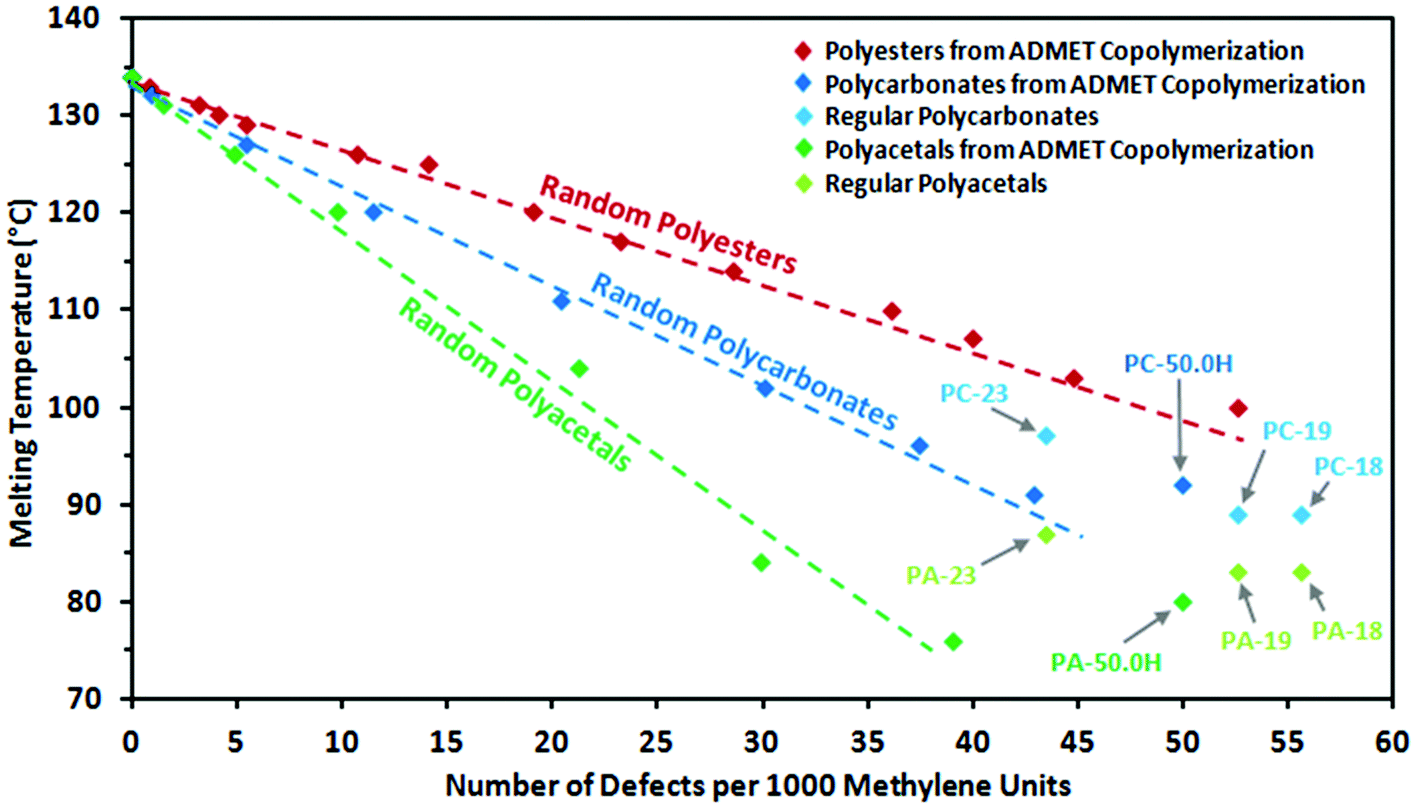 | ||
| Fig. 9 Peak melting points of random long-spaced polyacetals PA-39.1H to PA-0.0H (dark green) and polycarbonates PC-42.9H to PC-0.0H (dark blue) versus number of defects per 1000 methylene units, together with regular spaced polyacetals PA-18, PA-19, PA-23 and PA-50.0H (from homopolymerization of monomer 2) and regular spaced polycarbonates PC-18, PC-19, PC-23 and PC-50.0H (from homopolymerization of monomer 3). Additionally, random long-spaced polyesters from a previous study34 are shown for comparison. The dashed lines are merely a guide to the eye. | ||
Long-spaced polycarbonates display significantly lower melting points and lower crystallinities than the corresponding polyesters with a comparable number of functional groups incorporated into the polymer chain (Fig. 7; for polyesters Tm = (133–683 × XE) °C is found,34 where XE is the mole fraction of ester groups). This lower propensity for crystallization is also reflected by a higher solubility. Polycarbonates containing 37.5 or more carbonate groups (per 1000 methylene units) exhibit sufficient solubility in chloroform at room temperature to allow NMR analytics (Fig. 4 bottom), while comparable polyesters only dissolve at elevated temperatures. This stronger depression of the melting points by carbonate vs. ester groups can be attributed to a lower ability for polar layer formation. Schmidt-Rohr and coworkers investigated the crystalline morphology of polyester-22,4 extensively by solid-state NMR spectroscopy and small angle X-ray scattering (SAXS).49 They observed wide-ranging diester layer formation (layering of –OC(O)CH2CH2C(O)O– units), which controls chain dynamics and the morphology of these polyethylene-like polymers to a noticeable extent. Layer formation of ester groups can be explained by dipole–dipole interactions between the functionalities of neighboring polymer chains,50 resulting in crystalline and conformational packing order by the formation of anisotropic interchain bonding during the crystallization process. Layer formation reduces the disturbing effect of the ester moieties by establishing defect-free areas of crystalline hydrocarbon chains (in contrast to the case of totally random distribution of ester groups throughout the solid). For carbonate groups, comparable interchain bonding and layer formation in polymers can be expected. However, a carbonate group displays less polarity than an ester group51 due to the further electron withdrawing oxygen atom attached to the carbonyl functionality, while the steric demand remains comparable. Consequently, the ability to form carbonate layers in a polyethylene-like crystal lattice is expected to be reduced by comparison to ester containing polymers. This translates to a stronger disturbing effect (and higher energy penalty ε in the Sanchez–Eby model equation) for carbonate groups compared to ester groups in the crystal lattice, leading to less defect-free crystalline areas and reduced van der Waals interactions between the polymer chains. Notably, this is also reflected in an enhanced solubility of polycarbonates compared to polyesters.
Thermal and crystalline properties of long-spaced polyacetals
As for long-spaced polyesters and polycarbonates, the melting and crystallization points of polyacetals increase and resemble linear polyethylene as the number of acetal groups is reduced (Table 7 and Fig. 9, green labels). In contrast to long-spaced polyesters (red labels) and polycarbonates (blue labels), only the melting and crystallization traces of polyacetals with an acetal content of 10 per 1000 CH2 units or less display relatively sharp and distinct melting points (for DSC curves of all polyacetals, see ESI†). For PA-39.1H, PA-29.9H and PA-21.3H very broad thermal transitions are observed, which cover temperature ranges of more than 50 °C. For the regular spaced PA-50.0H (= PA-20) besides the major thermal transitions a second, minor transition is found, resembling the behavior of PA-18 and PA-19.| Compound | T m (°C) | T c (°C) | ΔH (J g−1) |
|---|---|---|---|
| a Determined at a heating/cooling rate of 10 °C min−1. Peak Tm determined from the second heating cycle. b A second, minor thermal transition is found at Tm = 59 °C and Tc = 45 °C. c Broad melting and crystallization transitions are observed. d No distinct melting point can be determined. | |||
| PA-50.0H | 80b | 66b | 162 |
| PA-39.1H | 76c | 63c | 142 |
| PA-29.9H | 84–97c,d | 74c | 171 |
| PA-21.3H | 104c | 93c | 178 |
| PA-9.8H | 120 | 107 | 226 |
| PA-4.9H | 126 | 112 | 233 |
| PA-1.5H | 131 | 114 | 275 |
| PA-0.0H | 134 | 117 | 276 |
To further investigate the crystalline structures, WAXD analysis was performed on regularly and randomly spaced polyacetals (Fig. 8). For the regular polyacetals PA-18, PA-19, PA-23 and PA-50.0H (= PA-20) various reflexes with shifting intensities are found, which cannot be assigned to defined crystal structures. This agrees with the observation of two melting transitions for these polymers. Similarly, for the random polyacetals PA-39.1H and PA-29.9H undefined crystal structures are found, in line with the broad melting and crystallization transitions found by DSC. Eventually, for PA-21.3H and polyacetals with further reduced numbers of acetal units a shift of the WAXD reflexes to 2θ angles of 21.5° and 23.9° with the expected intensity ratios for the 110 and the 200 plane of the orthorhombic, polyethylene-like crystal structure is observed. Concurrent observations resulted from IR spectroscopy (Fig. S36†). For PA-50.0H, PA-39.1H and PA-29.9H no Davidov splitting for the absorbances for the CH2 scissoring vibration (1472–1473 cm−1 and 1463–1464 cm−1) and for the CH2 rocking vibration (718–720 cm−1 and 730–731 cm−1) can be observed. When the number of acetal groups is further reduced, the splitting grows along with the transformation to an orthorhombic crystal structure.
The heterogeneous crystalline morphologies observed for polyacetals (with a higher acetal content) suggest that an inclusion of the acetal moieties into a polyethylene-like crystal is not an appropriate description. Overall, acetal functions impact the melting point even more pronouncedly than carbonates, Tm = (133–1412 × XA) °C (where XA is the mole fraction of acetal groups in the copolymer, see Fig. S37 in the ESI†).
Though the steric demand of acetal groups is not higher than for carbonate and ester groups, acetals obviously impact the polymer crystalline structure more extensively, resulting in more heterogeneous crystalline morphologies for polyacetals with high acetal contents. While the reduced melting temperatures of polycarbonates by comparison to polyesters can be related to different polarities of the functional groups, the polarity of carbonate and acetal units is rather similar.52
However, the conformational characteristics of these functional groups differ significantly. From studies of short-chain acetal and carbonate functionalized molecules like dimethoxymethane53 and dimethyl carbonate,54 the thermodynamically favored conformational arrangements are well understood. Since the rotation around the (OC)–OR bond in carbonate (and ester) functionalities is hindered, only cis- and trans-related conformations are possible. Assuming that the conformational behavior of dimethyl carbonate can be translated to the carbonate groups in polycarbonates, the good agreement with the all-trans zigzag conformation of the CH2 units preferred in polyethylene-like orthorhombic crystal structures becomes obvious. Even the random incorporation of >50 carbonate groups per 1000 methylene units does not lead to significant changes in the crystalline structures (Fig. 6). Differently, in acetal groups a gauche conformation is preferred as a result of the anomeric effect.53 Since the gauche conformation is not compatible with the all-trans CH2 conformation, a high density of acetal units disturbs the formation of an orthorhombic structure, leading to heterogeneous polymer crystals associated with broad melting transitions (Fig. 8). Only for small densities of acetal groups in the polymer chain (<22 acetal groups per 1000 methylene units), orthorhombic crystal structures and more narrow thermal transitions are observed.
Impact of random vs. regular spacing on crystalline and thermal properties
Wagener and coworkers extensively compared precisely and randomly branched or functional-substituted polyethylenes. As shown for alkyl branches with various chain lengths42,55 and polar functionalities,56,57 precisely branched polyethylenes exhibit significantly lower melting points than their randomly branched counterparts with the same overall degree of branching. However, precisely branched polyethylenes exhibit more narrow thermal transitions, which arise from higher ordered and more homogeneous polymer crystals due to more narrow lamellae thickness distributions.58Analogous to the behavior observed for polyesters,34,59 randomly spaced polycarbonates and polyacetals studied here do not follow this behavior (Fig. 9). For the regularly spaced polycarbonates significantly higher melting points are found than expected from linear extrapolation of data from randomly spaced polyacetals. PC-18 and PC-19 (containing 55.6 and 52.6 carbonate groups per 1000 methylene units, respectively) exhibit melting temperatures similar to the randomly spaced PC-42.9H from ADMET copolymerization. This effect is even more pronounced for polyacetals. The regular polymers PA-18 and PA-19 (containing 55.6 and 52.6 acetal groups per 1000 methylene units, respectively) exceed the melting point of PA-39.1H by 6 to 7 °C, despite a significantly higher density of acetal groups in the polymer chain.
Again, the behavior of polycarbonates can be accounted for by an ability of carbonate groups to form layers within the solid semicrystalline polymer. Uniform spacing in the case of regularly spaced polar groups facilitates dipole–dipole interactions and polar layer formation. This in turn promotes the occurrence of defect-free regimes of aliphatic polymer chains in the polymer crystal, resulting in stronger overall van der Waals interactions and higher melting points, consequently.
For polyacetals the regular arrangement of the acetal functionalities does not lead to higher ordered crystalline structures (Fig. 8). Nevertheless, higher melting points than expected are observed. Comparable to polycarbonates a regular spacing between the functional groups favors the formation of defect-free regimes of aliphatic polymer chains, resulting in increased van der Waals interactions.
Conclusions
Long-chain polyacetals and polycarbonates can be obtained conveniently by one-pot procedures via reaction of α,ω-diols with diethoxymethane or dimethyl carbonate. These approaches follow previous detailed studies of shorter chain congeners.17,25,26 Their generic adaptability to higher melting, less soluble long-chain α,ω-diol monomers and corresponding polymers is demonstrated by studies of various diols, ranging from C18 to C23. This is underlined by a meaningful and conclusive analysis of molecular weights by NMR end group analysis and GPC analysis vs. linear polyethylenes as an appropriate standard. Reasonably satisfactory molecular weights of Mn 104 g mol−1 are obtained in these self-polycondensations.In relatively short term studies of hydrolytic degradation of the solid polymers over two to four weeks, the polyacetals only degrade to a significant extent under strongly acidic conditions. By contrast, polycarbonates also degrade in a basic environment. Remarkably, in all cases the length of the long-chain methylene sequence influences the rate of this slow heterogeneous degradation. This appears to be useful for adjusting the balance of a desired long-term slow degradation vs. stability and mechanical performance during the intended lifetime in applications of such novel materials.
Complementary studies of randomly spaced polyacetals and polycarbonates from ADMET copolymerizations in combination with exhaustive post-polymerization hydrogenation decisively illuminate the solid-state structures and allow for far-reaching predictions of the thermal behavior of long-chain polyacetals and polycarbonates in general. Long-chain polycarbonates display polyethylene-like solid state structures. The impact of carbonate moieties on melting and crystallization points is more pronounced than for ester groups. This can be related to the lower polarity of carbonate groups, resulting in a less pronounced propensity for formation of ordered layers of the functional groups. Other than polycarbonates and polyesters, long-chain polyacetals form more complex and less uniform solid state structures and only adopt a polyethylene-like structure at very low contents of acetal groups.
While we have not elaborated application-relevant properties like mechanical behavior and melt processing, the thermal properties and the semicrystalline solid state structures suggest that these polymers may be useful thermoplastic materials. They may be imparted with a tailored slow degradation rate as a specific feature of these hydrophobic largely hydrocarbon polymers.
Acknowledgements
We thank Lars Bolk for DSC and GPC analysis and Daniela Lehr for WAXD measurements. P. O. gratefully acknowledges financial support by the Stiftung Baden-Württemberg (Kompetenznetz funktionelle Nanostrukturen).Notes and references
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- S. Picataggio, T. Rohrer, K. Deanda, D. Lanning, R. Reynolds, J. Mielenz and L. D. Eirich, Nat. Biotechnol., 1992, 10, 894–898 CrossRef CAS PubMed.
- U. Schörken and P. Kempers, Eur. J. Lipid Sci. Technol., 2009, 111, 627–645 CrossRef.
- W. Lu, J. E. Ness, W. Xie, X. Zhang, J. Minshull and R. A. Gross, J. Am. Chem. Soc., 2010, 132, 15451–15455 CrossRef CAS PubMed.
- C. Jiménez-Rodriguez, G. R. Eastham and D. J. Cole-Hamilton, Inorg. Chem. Commun., 2005, 8, 878–881 CrossRef PubMed.
- P. Roesle, C. J. Dürr, H. M. Möller, L. Cavallo, L. Caporaso and S. Mecking, J. Am. Chem. Soc., 2012, 134, 17696–17703 CrossRef CAS PubMed.
- F. Stempfle, D. Quinzler, I. Heckler and S. Mecking, Macromolecules, 2011, 44, 4159–4166 CrossRef CAS.
- D. Quinzler and S. Mecking, Angew. Chem., Int. Ed., 2010, 49, 4306–4308 CrossRef CAS PubMed.
- S. Chikkali and S. Mecking, Angew. Chem., Int. Ed., 2012, 51, 5802–5808 CrossRef CAS PubMed.
- F. Stempfle, P. Roesle and S. Mecking, ACS Symp. Ser., 2012, 1105, 151–164 CrossRef CAS PubMed.
- M. B. Dinger and J. C. Mol, Adv. Synth. Catal., 2002, 344, 671–677 CrossRef CAS.
- M. Unverfreth, O. Kreye, A. Prohammer and M. A. R. Meier, Macromol. Rapid Commun., 2013, 34, 1569–1574 CrossRef PubMed.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- D. S. Muggli, A. K. Burkoth and K. S. Anseth, J. Biomed. Mater. Res., 1999, 46, 271–278 CrossRef CAS.
- A. K. Burkoth and K. S. Anseth, Macromolecules, 1999, 32, 1438–1444 CrossRef CAS.
- I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404–3410 CrossRef CAS PubMed.
- A. G. Pemba, J. A. Flores and S. A. Miller, Green Chem., 2013, 15, 325–329 RSC.
- T. Hashimoto, K. Ishizuka, A. Umehara and T. Kodaira, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4053–4064 CrossRef CAS.
- Y. Wang, H. Morinaga, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 596–602 CrossRef CAS.
- S. Chikkali, F. Stempfle and S. Mecking, Macromol. Rapid Commun., 2012, 33, 1126–1129 CrossRef CAS PubMed.
- S. E. Paramonov, E. M. Bachelder, T. T. Beaudette, S. M. Standley, C. C. Lee, J. Dashe and J. M. J. Fréchet, Bioconjugate Chem., 2008, 19, 911–919 CrossRef CAS PubMed.
- D. Wehrung, S. Sun, E. A. Chamsaz, A. Joy and M. O. Oyewumi, J. Pharm. Sci., 2013, 102, 1650–1660 CrossRef CAS PubMed.
- J. H. Jung, M. Ree and H. Kim, Catal. Today, 2006, 115, 283–287 CrossRef CAS PubMed.
- F.-S. Liu, Z. Li, S.-T. Yu, X. Cui, C.-X. Xie and X.-P. Ge, J. Polym. Environ., 2009, 17, 208–211 CrossRef CAS.
- R. Vanderhenst and S. A. Miller, Green Mater., 2013, 1, 64–78 CAS.
- J. W. Hill and W. H. Carothers, J. Am. Chem. Soc., 1935, 57, 925–928 CrossRef CAS.
- The degree of crystallinity is calculated by dividing the heat of fusion ΔH from DSC by 293 J g−1. This value corresponds to the heat of fusion of fully extended chain crystals of linear polyethylene as a reference for 100% crystalline material; see: B. Wunderlich and G. Czornyj, Macromolecules, 1977, 10, 906–913 CrossRef CAS.
- A maximum error of 1% can be assumed for pellet weight determination.
- I. Cho and K. Lee, Macromol. Chem. Phys., 1997, 198, 861–869 CrossRef CAS.
- F. Stempfle, P. Ortmann and S. Mecking, Macromol. Rapid Commun., 2013, 34, 47–50 CrossRef CAS PubMed.
- H. Mutlu, R. Hosäβ, R. E. Montenegro and M. A. R. Meier, RSC Adv., 2013, 3, 4927–4934 RSC.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- H. Mutlu, L. Montero de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC.
- P. Ortmann and S. Mecking, Macromolecules, 2013, 46, 7213–7218 CrossRef CAS.
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- S. D. Drouin, F. Zamanian and D. E. Fogg, Organometallics, 2001, 20, 5495–5497 CrossRef CAS.
- C. W. Bielawski, J. Louie and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 12872–12873 CrossRef CAS.
- M. Oliván and K. G. Caulton, Inorg. Chem., 1999, 38, 566–570 CrossRef PubMed.
- The additional designation “H” indicates hydrogenated, saturated polymers.
- The hydrogenation reaction to generate linear, defect-free polyethylene was performed in o-xylene at 140 °C.
- J. C. Sworen, J. A. Smith, K. B. Wagener, L. S. Baugh and S. P. Rucker, J. Am. Chem. Soc., 2003, 125, 2228–2240 CrossRef CAS PubMed.
- L. Fontana, M. Santoro, R. Bini, D. Q. Vinh and S. Scandolo, J. Chem. Phys., 2010, 133, 204502 CrossRef PubMed.
- K. Tashiro, S. Sasaki and M. Kobayashi, Macromolecules, 1996, 29, 7460–7469 CrossRef CAS.
- I. C. Sanchez and R. K. Eby, Macromolecules, 1975, 8, 638–641 CrossRef CAS.
- The superscripts “0” refer to linear polyethylene homopolymers.
- B. Crist, Polymer, 2003, 44, 4563–4572 CrossRef CAS.
- M. P. F. Pepels, M. R. Hansen, H. Goossens and R. Duchateau, Macromolecules, 2013, 46, 7668–7677 CrossRef CAS.
- M. G. Menges, J. Penelle, C. Le Fevere de Ten Hove, A. M. Jonas and K. Schmidt-Rohr, Macromolecules, 2007, 40, 8714–8725 CrossRef CAS.
- S. E. Rickert, E. Baer, J. C. Wittmann and A. J. Kovacs, J. Polym. Sci., Polym. Phys. Ed., 1978, 16, 895–906 CrossRef CAS.
- The different polarities of ester and carbonate functionalities can be illustrated by the comparison of experimental values of electric dipole moments of linear short-chain molecules like ethyl butyrate (1.75 D) and diethyl carbonate (0.91 D). Measurements were performed in benzene at 25 °C, cf. A. L. McClellan, Tables of Experimental Dipole Moments, W. H. Freeman and Company, San Francisco and London, 1963 Search PubMed.
- For the dipole moment of diethoxymethane a value of 0.93 D was found, cf. ref. 51.
- K. B. Wiberg and M. A. Murcko, J. Am. Chem. Soc., 1989, 111, 4821–4828 CrossRef CAS.
- H. Bohets and B. J. van der Veken, Phys. Chem. Chem. Phys., 1999, 1, 1817–1826 RSC.
- G. Rojas, B. Inci, Y. Wei and K. B. Wagener, J. Am. Chem. Soc., 2009, 131, 17376–17386 CrossRef CAS PubMed.
- S. E. Lehman Jr., K. B. Wagener, L. S. Baugh, S. P. Rucker, D. N. Schulz, M. Varma-Nair and E. Berluche, Macromolecules, 2007, 40, 2643–2656 CrossRef.
- T. W. Baughman, C. D. Chan, K. I. Winey and K. B. Wagener, Macromolecules, 2007, 40, 6564–6571 CrossRef CAS.
- S. Hosoda, Y. Nozue, Y. Kawashima, K. Suita, S. Seno, T. Nagamatsu, K. B. Wagener, B. Inci, F. Zuluaga, G. Rojas and J. K. Leonard, Macromolecules, 2011, 44, 313–319 CrossRef CAS.
- J. Trzaskowski, D. Quinzler, C. Bährle and S. Mecking, Macromol. Rapid Commun., 2011, 32, 1352–1356 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthetic procedures and degradation studies, characterization methods and DSC, IR and WAXD data for long-chain polyacetals (PA-18, PA-19, PA-23) and polycarbonates (PC-18, PC-19, PC-23) as well as for randomly long-spaced polyacetals (PA-50.0H to PA-0.0H) and polycarbonates (PC-50.0H to PC-0.0H) are given. See DOI: 10.1039/c3gc42592d |
| This journal is © The Royal Society of Chemistry 2014 |